

Abstract
The standard treatment for metastatic prostate cancer (PCa), androgen deprivation therapy (ADT), is designed to suppress androgen receptor (AR) activity. However, men invariably progress to castration-resistant prostate cancer (CRPC), and AR reactivation contributes to progression in most cases. To identify mechanisms that may drive CRPC, we examined a VCaP PCa xenograft model as tumors progressed from initial androgen sensitivity prior to castration to castration resistance and then on to relapse after combined therapy with further AR targeted drugs (abiraterone plus enzalutamide). AR activity persisted in castration-resistant and abiraterone/enzalutamide-resistant xenografts and was associated with increased expression of the AR gene and the AR-V7 splice variant. We then assessed expression of individual AR-regulated genes to identify those that persisted, thereby contributing to tumor growth, versus those that decreased and may therefore exhibit tumor suppressor activities. The most significantly decreased AR target gene was Dipeptidyl Peptidase 4 (DPP4), which encodes a membrane-anchored protein that cleaves dipeptides from multiple growth factors, resulting in their increased degradation. DPP4 mRNA and protein were also decreased in clinical CRPC cases, and inhibition of DPP4 with sitagliptin enhanced the growth of PCa xenografts following castration. Significantly, DPP4 inhibitors are frequently used to treat type 2 diabetes as they increase insulin secretion. Together these results implicate DPP4 as an AR-regulated tumor suppressor gene whose loss enhances growth factor activity and suggest that treatment with DPP4 inhibitors may accelerate emergence of resistance to ADT.
Keywords: prostate cancer, castration-resistant prostate cancer, androgen receptor, dipeptidyl peptidase 4
Introduction
The standard treatment for metastatic prostate cancer (PCa) is androgen deprivation therapy (ADT) to suppress androgen receptor (AR) activity, but men invariably progress despite castrate androgen levels (castration-resistant prostate cancer, CRPC). AR activity persists in most CRPC, with increased intratumoral androgen synthesis being a major mechanism driving this AR activity (1). AR activity in CRPC can be suppressed by agents such as abiraterone, which further decrease androgen synthesis, or by AR antagonists such as enzalutamide, but patients still invariably progress. A subset of these abiraterone/enzalutamide-resistant tumors express low or undetectable AR and some have neuroendocrine features (2,3), but AR appears to be contributing to progression in most cases. Multiple mechanisms may contribute to persistent AR activity including alterations in the AR (AR gene amplification or activating mutations, expression of constitutively active AR splice variants, or AR posttranslational modifications), further increases in intratumoral androgen synthesis, and activation of multiple signaling pathways or epigenetic alterations that enhance tumor cell growth and may directly or indirectly enhance AR activity. However, the contribution of any single mechanism to resistance is unclear, and multiple mechanisms may contribute to resistance in a single patient.
While most PCa are initially AR-dependent, the critical genes and pathways regulated by AR remain unclear. One basis for this dependence is AR regulation of multiple genes involved in metabolic pathways (4,5). However, in addition to its oncogenic properties, studies in model systems show that AR also can have tumor suppressor activity. The clinical significance of these observations is supported by an inverse relationship between AR activity and cell proliferation in CRPC clinical samples (6), and by recent clinical trials of rapid cycling between high and low serum testosterone concentrations in men with CRPC (7). Possible mechanisms for these responses include AR-mediated DNA damage, AR repression of genes such as MYC, and inactivation of multiple E2F regulated genes through increased recruitment of pRb (8,9). Alternatively, AR may repress growth through increasing expression of multiple genes involved in differentiation, consistent with its normal function in prostate epithelium. Significantly, several studies have indicated that the AR cistrome and transcriptome become reprogrammed during PCa development and progression to CRPC, consistent with selective pressure to block AR’s tumor suppressive functions and potentially acquire new oncogenic functions (10). This study identifies Dipeptidyl Peptidase 4 (DPP4) as an AR-stimulated tumor suppressor gene whose expression is suppressed with progression to CRPC.
Materials and Methods
Cell lines, xenografts, and tissue samples
VCaP and LNCaP cells were from ATCC (Manassas, VA) and used for subcutaneous xenograft injections within 4 passages. VCaP and LNCaP cell identities were confirmed by short tandem repeat (STR) profiling, and Mycoplasma testing was negative. To generate VCaP xenografts, 6 week old male ICR scid mice (Taconic Biosciences) were injected subcutaneously with 5 million VCaP cells in 100% Matrigel. Xenografts were grown until 1000 mm3, then mice were castrated (Cx). For the AER VCaP xenograft model, when tumors in Cx mice exceeded 150% of their nadir volume, they were considered relapsed and mice were started on abiraterone (30mg/kg) + enzalutamide (50mg/kg) in drinking water. Tumors were serial biopsied prior to Cx (Pre-Cx), at tumor relapse (CRPC), and mice were sacrificed when tumors reached 2000 mm3 on dual abiraterone plus enzalutamide treatment (Abi/Enza resistant, AER). For the sitagliptin studies, VCaP, LNCaP, and BID-PC-1 xenografts were grown to ~500 mm3, then mice were castrated and immediately administered daily sitagliptin (120 mg/kg, SelleckChem) in drinking water. The BID-PC-1 xenograft was generated from a metastasis in a patient with BRCA2-deficient CRPC and has been passaged in noncastrated male immundeficient mice. Written informed consent was obtained from patients for the tissue analyses, and all studies involving human materials were carried in accordance with the U.S. Common Rule and approved by the Beth Israel Deaconess Medical Center (BIDMC) Instituional Review Board. All animal studies were approved by the BIDMC Institutional Animal Care and Use Committee.
Knockdown of DPP4 protein expression in VCaP cells was performed using a SMARTpool of ON-TARGETplus siRNAs targeting DPP4 (Dharmacon, Cat.# L-004181–00-0005). Neoadjuvant leuprolide-abiraterone samples were obtained from patients who underwent RP after neoadjuvant treatment in a phase II clinical trial (11). CRPC tissues were obtained from rapid autopsy specimens at BIDMC. Tissue analyses were in accordance with the DF/HCC IRB. A further rapid autopsy TMA was obtained from University of Washington in accordance with their Prostate Cancer Donor Program (6).
Immunohistochemistry
For IHC, 5-mm FFPE sections underwent epitope retrieval using Dako PT Link platform. Staining was on the Dako Link 48 autostainer, with amplification using Envision FLEX rabbit linkers, and visualization using the Envision Flex High-sensitivity visualization system (Dako). Sections were stained for anti-AR (N20, Santa Cruz; 1:1000), anti-ARV7 (RM7, RevMab; 1:100), anti-phospho-AR(S81) (Millipore Sigma, 1:5000), anti-PSA (FLEX polyclonal rabbit anti-human Prostate-specific antigen, IR514, DAKO), and anti-DPP4 (D6D8K, Cell Signaling Technologies, 1:100). DPP4 antibody specificity was confirmed on cultured VCaP cells. After siRNA knockdown of DPP4 mRNA (Supplementary Fig. 1A) we found decreased DPP4 protein levels and expression by IHC (Supplementary Fig. 1B and C, respectively). Anti-DPP4 staining in clinical prostate samples showed strong membrane stainin gin luminal epithelium (Supplementary Fig. 1D). DPP4 positivity was defined by moderate to strong, punctate, membranous and cytoplasmic staining. DPP4 immunointensity was scored as negative (0), weak (1), moderate (2), and strong (3), based on the most predominant intensity pattern. DPP4 percentage score was based on percentage of tumor cells demonstrating the most predominant intensity pattern or stronger as: 0 (negative), 1 (1 – 9 %), 2 (10 – 49%), and 3 (≥ 50%). DPP4 score (0–9) was based on the immunointensity score multiplied by the percentage score.
Gene expression analysis
RNA was isolated from FFPE blocks that contained greater than 90% tumor cell content by cutting approximately ten, 8 mm ribbons from each block and isolating the RNA with the RNeasy FFPE Kit (Qiagen). Quantitative real-time RT-PCR amplification was with TaqMan One-Step RT-PCR reagents (ThermoFisher Scientific) and results were normalized to coamplified β-actin. RNA-seq was performed on 2–3 biological replicates. Sequencing libraries were generated using the NEB Ultra directional RNA library prep kit, and we obtained ~25–30 million paired end reads. Additional gene analysis techniques and RNA-seq analysis methods are in Supplementary Materials and Methods. RNA-seq data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) with the accession code GSE109708.
Statistical analysis
GraphPad Prism 7 Software (GraphPad Software Inc.) was used for all statistical analysis unless otherwise specified. All results are presented as the mean ± the standard error of the mean (SEM). The Mann-Whitney U non-parametric test was used to test the statistical significance between gene expression of controls and experimentally manipulated samples. Values of P<0.05 were considered statistically significant.
Results
AR activity persists in abiraterone/enzalutamide-resistant VCaP xenografts
VCaP xenografts were established and biopsies were taken prior to castration (Pre-Cx), when tumors relapsed following castration (CRPC), and when they again relapsed following combined abiraterone (30 mg/kg/day) and enzalutamide (50 mg/kg/day) treatment (abiraterone/enzalutamide-resistant, AER) (Fig. 1A). Analysis of AR and AR splice variant 7 (AR-V7) mRNA in serial biopsies showed increases as tumors progressed from Pre-Cx to CRPC, with further increases in AR-V7 as tumors progressed to AER (Fig. 1B). IHC similarly showed increased AR-V7 protein in the CRPC and AER tumors, while total AR protein was not substantially altered (Fig. 1C and Supplementary Fig. S2). AR phosphorylation on S81 (an indicator of AR transcriptional activity), also persisted in the CRPC and AER xenografts without significant alteration as tumors progressed (Fig. 1C and Supplementary Fig. S2). Consistent with persistent AR transcriptional activity, CRPC and AER xenografts also expressed levels of the AR target genes KLK3 (PSA) and NKX3.1 that were at least equivalent to the levels in the Pre-Cx tumors (Fig. 1B, C and Supplementary Fig. S2). Moreover, based on RNA-seq and a curated list of 266 AR target genes, the strength of AR signaling was comparable (~8% decrease) between the Pre-Cx and CRPC/AER xenografts (Fig. 1D). There was a similar difference (~12% decrease) in the AR signaling scores in the clinical TCGA (primary PCa) versus the SU2C (CRPC) data sets, while additional clinical CRPC data sets showed AR score decreases ranging between 21–57% relative to primary untreated PCa. These data indicate that VCaP AER tumors had restored AR signaling despite maximal AR blockade.
Figure 1. AER VCaP xenograft tumors have restored AR signaling.
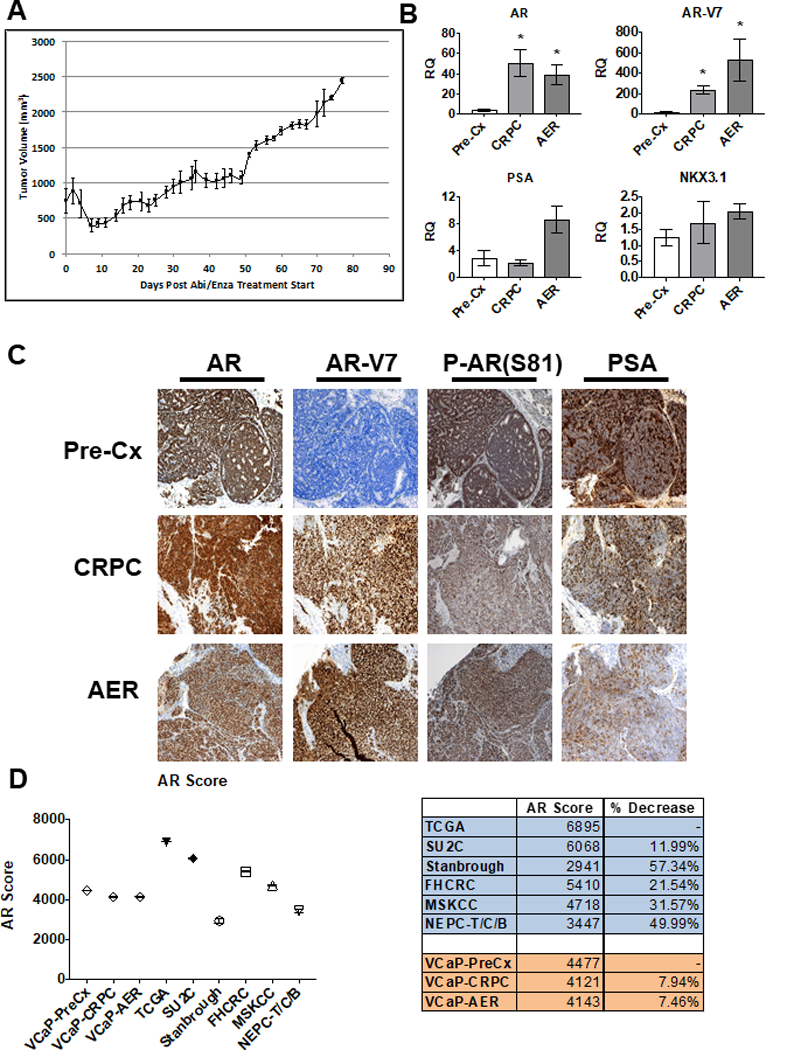
A) VCaP CRPC xenografts are initially sensitive to Abi/Enza, but recover tumor volume by day 30. The initial cohort was 22 mice. B) qRT-PCR of AR, AR-V7, PSA, and NKX3.1 in serial biopsies of Abi/Enza resistant VCaP xenografts. Each column represents xenografts from four mice, with qRT-PCR in technical triplicate. Bars = SEM, * = P<0.03, Mann-Whitney U. RQ = Relative Quantification C) IHC of AR, AR-V7, serine 81 phosphorylated AR (P-AR(S81)), and PSA in serial biopsies of representative tumor. D) AR Score for available Pre-Cx and CRPC data sets (Left). Bars depicting SEM are not visible due to small size. The % decrease in the AR score from Pre-Cx state (TCGA, VCaP-PreCx) to the CRPC state (SU2C, Stanbrough, FHCRC, MSKCC, NEPC-T/C/B, VCaP-CRPC, VCaP-AER) are listed (Right). TCGA –The Cancer Genome Atlas, SU2C – Stand Up 2 Cancer, Stanbrough – from Stanbrough et al. (Supplementary Reference 3), FHCRC – Fred Hutch Cancer Research Center, MSKCC – Memorial Sloan Kettering Cancer Center, NEPC-T/C/B – Neuroendocrine Prostate Cancer-Trento/Cornell/Broad.
Overlapping pathways mediate progression to CRPC and AER
RNA-seq showed that 1441 genes were significantly differentially expressed between AER and Pre-Cx tumors (Fig. 2A, Supplementary Table S1). About half of these were significantly differentially expressed between CRPC and Pre-Cx xenografts (47%, Supplementary Table S2). For most of the remaining genes there was a trend toward altered expression in the CRPC xenografts (Fig. 2B), indicating that progression to AER was driven largely by mechanisms that were already engaged during progression to CRPC. Consistent with this conclusion, comparison of the AER versus the CRPC xenografts identified a much smaller group of genes as being significantly altered (115 genes), with most similarly altered in CRPC (Supplementary Table S3). Moreover, two of the most enriched pathways in the AER versus Pre-Cx tumors were also enriched in the CRPC versus Pre-Cx tumors (axonal guidance signaling and glioblastoma multiforme signaling) (Supplementary Fig. S3A). Finally, hierarchical clustering and principle component analysis both separately grouped the Pre-Cx samples (Supplementary Fig. S3B, C).
Figure 2. Analysis of RNA-seq from serial biopsies.
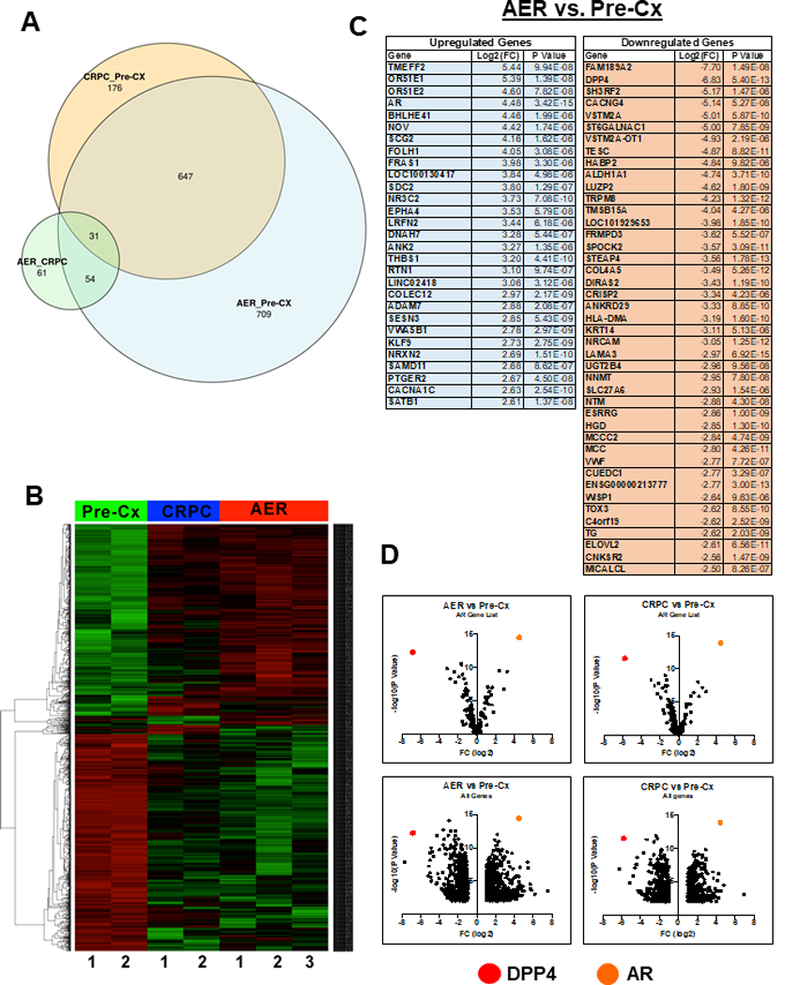
A) Venn diagram depicting the overlap between differentially expressed genes in Pre-Cx, CRPC, and AER serial biopsies of 4 different VCaP xenografts. B) Unsupervised clustering of Pre-Cx, CRPC, and AER xenografts based on differential gene expression. C) Differentially expressed genes between AER and Pre-Cx xenografts that meet the stringent criteria of log2FC > 2.5 and P-Value < 1.00×10-5. D) Volcano plots depicting AR target genes (266 AR gene signature from Mendiratta et al.) (12), supplemented with a selection of DHT-responsive genes from Xu et al. (4) (upper), and all significantly differentially expressed genes (lower) in the AER vs. Pre-Cx and CRPC vs. Pre-Cx comparisons. DPP4 and AR are highlighted.
Using a more stringent cutoff (Log2(FC) ≥ 2.5 and P ≤ 1.00E-05), 72 genes were altered in the AER versus Pre-Cx tumors (Fig. 2C), and these genes were all similarly altered in the CRPC versus Pre-Cx tumors (Supplementary Table S4). As expected, AR was amongst the most highly upregulated genes. Also markedly increased were two olfactory receptor genes (OR51E1 and OR51E2) and the Nephroblastoma Overexpressed (NOV) gene, which have been linked previously to PCa. Interestingly, the only other highly increased nuclear receptor was NR3C2 (Mineralocorticoid Receptor), which we previously found increased in VCaP xenografts treated with single agent abiraterone (12) and in relapsed tumors in men with CRPC being treated with abiraterone plus dutasteride (13).
AR regulated DPP4 gene expression is not restored in CRPC or AER tumors
While AR signaling was substantially restored in the CRPC and AER xenografts, we hypothesized that expression of AR regulated genes that are critical for tumor growth would be most consistently and robustly restored, while those that are less critical (or exhibit growth suppressing effects) may not be restored. Consistent with this hypothesis, volcano plots showed that AR target gene expression in the AER versus Pre-Cx xenografts was not restored in a symmetric fashion, with the AR gene being the most significant outlier amongst genes that are increased in the AER xenografts (Fig. 2D, upper left and Supplementary Table S5). Conversely, the most significantly decreased gene in the AER xenografts was DPP4, which has previously been shown to be an androgen-stimulated gene (4,14,15). We further confirmed that DPP4 gene expression was stimulated by DHT in VCaP and LNCaP cells (Supplementary Fig. 4A), and was decreased by enzalutamide (Supplementary Fig. 4B). A similar pattern was observed when comparing the CRPC versus Pre-Cx xenografts, indicating that AR fails to restore DPP4 expression at this stage as well (Fig. 2D, upper right and Supplementary Table S6). When this AR gene signature list was expanded to include all differentially expressed genes, DPP4 was still amongst the most significantly downregulated genes (Fig. 2D, lower left and right).
The loss of DPP4 mRNA in the CRPC and AER tumors was confirmed by qRT-PCR (Fig. 3A). Moreover, IHC confirmed that DPP4 protein was markedly decreased in the CRPC and AER tumors (Fig. 3B). As DPP4 mediates the degradation of multiple growth factors, we next submitted tumor lysates for reversed phase protein array analysis to determine if there were clear differences in key PCa-related signaling cascades. Indeed, there was increased activation of the PI3K, ERK-MAPK, and p38-MAPK pathways in the CRPC and AER tumors (Fig. 3C). Extending our results to clinical samples, previously published datasets also showed decreased DPP4 mRNA in CRPC (Supplementary Fig. S5).
Figure 3. DPP4 expression is decreased in AER and CRPC VCaP xenografts.
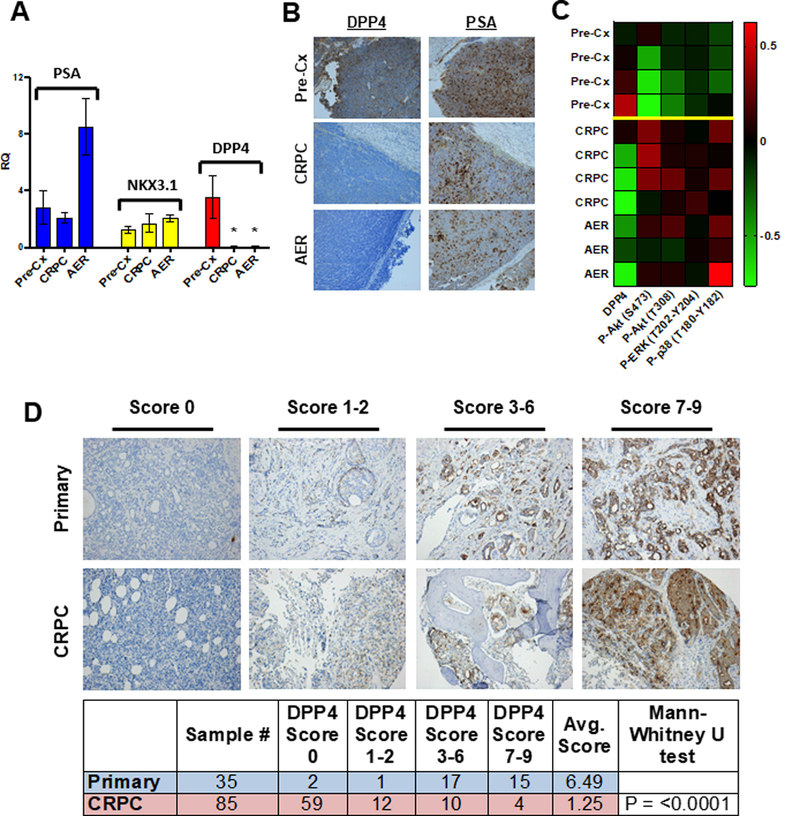
A) qRT-PCR for PSA, NKX3.1, and DPP4 expression in serial biopsies of VCaP xenografts. Each column represents the expression levels of xenograft tumors from four separate mice, with RT-PCR performed on each in technical triplicate. * = P<0.03 B) Representative images of DPP4 and PSA in serial biopsies of VCaP xenografts. C) RPPA from serial biopsies of 4 separate VCaP xenografts (see Supplementary Methods). D) Representative images of DPP4 immunohistochemistry and immunoscoring from a series of hormone-naïve primary PCa and CRPC clinical sections.
DPP4 protein is markedly reduced in CRPC clinical samples
We next performed IHC for DPP4 on untreated primary PCa tissue, residual tumor from a neoadjuvant leuprolide-abiraterone trial (11), and sections of metastatic CRPC. Tumors from the neoadjuvant trial showed markedly decreased DPP4, while metastatic CRPC sections showed near complete absence of DPP4 (Supplementary Fig. S6). We further examined a metastatic CRPC TMA (6) using a DPP4 immunoscore to quantify DPP4 protein expression. Strikingly, 84% (71/85) of CRPC sections had a DPP4 immunoscore ≤ 2, with 69% (59/85) of CRPC sections being negative, while only 9% (3/35) of untreated primary PCa had a score ≤ 2 (Fig. 3D). Overall, CRPC specimens exhibited a much lower average DPP4 immunoscore compared to untreated primary PCa (1.25 and 6.49, respectively, P < 0.0001), further supporting the decreased expression of DPP4 in CRPC.
DPP4 downregulation is mediated by a reversible epigenetic mechanism
Several mechanism of DPP4 downregulation in other contexts have been reported, including DPP4 promoter/early exon 1 methylation (16,17) and downregulation of a lncRNA, lncRNA-OIS1 (18). Bisulfite conversion of DNA from four AER VCaP tumors showed that the DPP4 promoter and early exon 1 regions were unmethylated (Supplementary Fig. S7). Further, there was no difference in lncRNA-OIS1 between the Pre-Cx and AER serial biopsies of 5 tumors (Supplementary Fig. S8). Finally, to determine if DPP4 downregulation is mediated through an irreversible genomic mechanism, we attempted to restore DPP4 expression in CRPC VCaP xenografts by treatment with high-dose testosterone. Treatment of castrated mice bearing CRPC VCaP xenografts with daily intraperitoneal injection of testosterone (200mg/kg) for three days restored DPP4 mRNA to Pre-Cx levels (Supplementary Fig. S9A) and also substantially increased DPP4 protein (Supplementary Fig. S9B and C), indicating that DPP4 downregulation is epigenetic.
Inhibition of DPP4 activity increases in vivo resistance to castration
These findings suggested that DPP4 inhibitors may enhance the growth of PCa after ADT. To test this hypothesis, androgen sensitive VCaP xenografts were grown in intact male mice, followed by castration in combination with a DPP4 inhibitor (sitagliptin, 120 mg/kg/day) or control. Both groups initially responded to castration, but the sitagliptin treated xenografts progressed more rapidly (Fig. 4A). AR expression and activity were comparable in the relapsed control and treated tumors harvested at ~6 weeks, suggesting that DPP4 inhibition was not acting primarily through AR, and consistent with it acting through enhanced growth factor stimulation (Fig. 4B). While DPP4 expression was decreased in both the treated and control tumors, there was a trend towards higher DPP4 expression in the treated versus control xenografts (Fig. 4B-D). This is consistent with decreased selective pressure to downregulate DPP4 expression in mice treated with sitagliptin. There also was a positive correlation between DPP4 protein levels and fold increase in tumor volume over 6 weeks in the treated xenografts (Supplementary Fig. S10), suggesting that tumors expressing the highest levels of DPP4 protein had the greatest increase in growth factors in response to the sitagliptin.
Figure 4. DPP4 inhibitor increases resistance to castration in vivo.
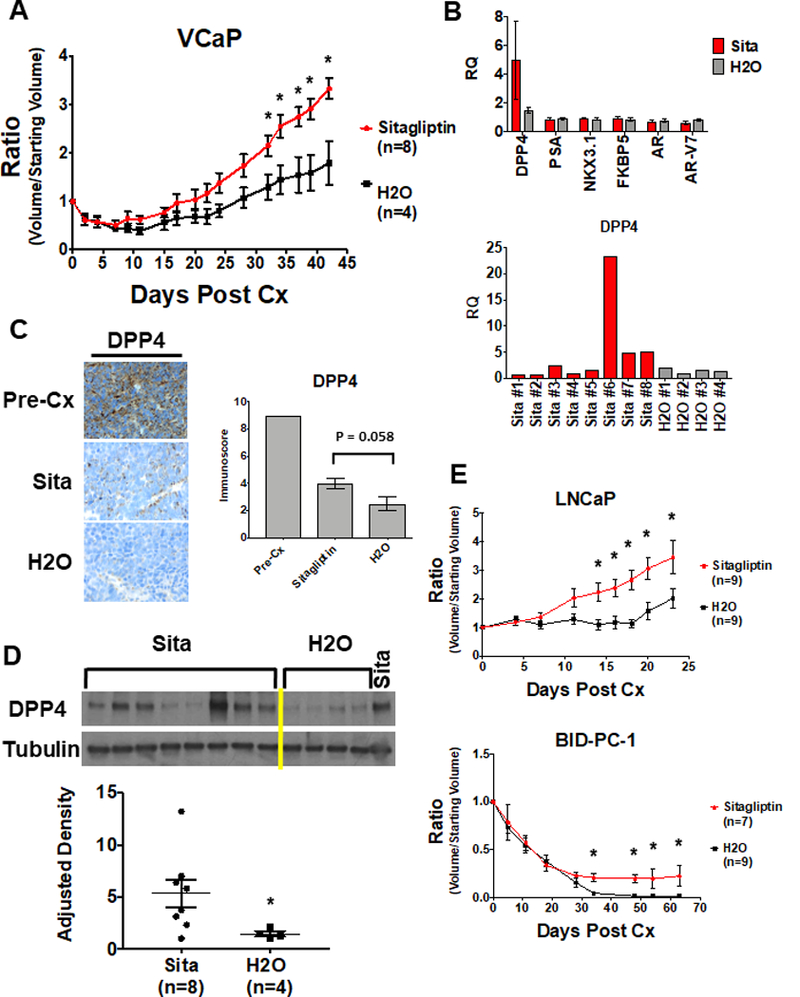
A) VCaP xenografts were grown in intact male mice until 500 mm3, then mice were castrated (Cx) and immediately begun on treatment with sitagliptin (120 mg/kg/day) or vehicle. Y axis is ratio of tumor volume at a given time point divided by starting volume. * = P<0.05. B) qRT-PCR of indicated transcripts in xenografts harvested at Day 42. Each column is expression in tumors from eight mice (Sita) or four mice (H2O), with qRT-PCR performed in technical triplicate. Lower panel is DPP4 mRNA in individual tumors. C) Representative DPP4 immunohistochemistry from Sita and H2O-treated tumors (left) and immunoscoring of DPP4 protein (right). P =0.058. D) Sitagliptin and control tumor lysates probed with anti-DPP4 (above) and densitometric quantification (below). * = P<0.03. E) Mice with LNCaP and BID-PC-1 xenografts were castrated (Cx) and immediately begun on treatment with sitagliptin (120 mg/kg/day) or vehicle. * = P<0.05.
To determine whether these results could be extended to tumors with genomic alterations distinct from those in VCaP (AR amplified, TMPRSS/ERGfusion), we examined the effects of sitagliptin on xenografts generated from LNCaP cells (PTEN-deficient) and on a BRCA2-deficient patient-derived xenograft (BID-PC-1). Similar to the results with VCaP, treatment with sitagliptin decreased the efficacy of castration in the LNCaP xenografts (Fig. 4E, upper). The BID-PC-1 PDX is extremely sensitive to ADT, and castration alone has led to complete responses in all mice examined for up to 12 months. In contrast, 3 of 7 tumors treated with sitagliptin had only partial responses, with one of these progressing by 2 months (Fig. 4E, lower).
Discussion
As a transmembrane protease, DPP4 can target numerous growth factors/cytokines, and may have oncogenic or tumor suppressor properties (19). Its oncogenic functions may be related to suppression of antitumor immune responses, although it may also have an immuostimulatory scaffold function by anchoring adenosine deaminase (20). Previous studies in PCa have indicated that DPP4 may enhance degradation of FGF2 and CXCL12 (21,22), and reduced serum DPP4 activity (due to a low molecular weight inhibitor) was found in men with metastatic PCa (23). However, consistent with our IHC results, DPP4 levels are not decreased in untreated primary PCa (24). Therefore, we hypothesize that there is no selective pressure to downregulate DPP4 in primary PCa, but that the initial decrease in DPP4 and subsequent increase in growth factor levels after ADT is important for tumor cell survival. Strong selective pressure to keep DPP4 levels low would then result in the emergence of CRPC cells with generally restored AR function that have epigenetically silenced the DPP4 gene. Consistent with this hypothesis, treatment with sitagliptin accelerated the progression of PCa xenografts to castration-resistance.
DPP4 inhibitors are used for type 2 diabetes as they block the degradation of GLP-1 and GIP-1, which promotes insulin secretion. Multiple studies have assessed for possible links between DPP4 inhibitors and cancer, but no consistent links have been found (25). This may indicate that DPP4 has only minimal effects on tumor development, or perhaps counterbalancing tumor suppressive (possibly immune) and oncogenic functions. However, while DPP4 inhibition may not have effects on PCa development, previous epidemiological studies have not addressed whether it impairs responses to ADT. This study supports a tumor suppressive function of DPP4 after ADT, and suggests that treatment with DPP4 inhibitors may decrease the efficacy of ADT.
Supplementary Material
Significance.
Findings identify DPP4 as an AR-stimulated tumor suppressor gene that is downregulated during progression to castration-resistant prostate cancer, warning that treatment with DPP4 inhibitors, commonly used to treat type 2 diabetes, may accelerate prostate cancer progression following androgen deprivation therapy.
Acknowledgments
Financial Support: This work was supported by NIH grants P01 CA163227 (S.P. Balk, P.S. Nelson, E.A. Mostaghel, E. Corey), NIH SPORE P50 CA090381 (S.P. Balk, M.E. Taplin), NIH Pacific Northwest SPORE P50 CA097186 (P.S. Nelson, R.B. Montgomery, E.A. Mostaghel, E. Corey), Department of Defense Prostate Cancer Research Program W81XWH-16–1-0431 (S.P. Balk, M. E. Taplin, H.Ye) and Early Investigator Research Award PC170570 (J.W. Russo), the A. David Mazzone Research Awards Program (J.W. Russo), a Research Fellowship from Gunma University Hospital (S.A.), Prostate Cancer Foundation Young Investigator Awards (J.W. Russo, H. Ye), and Prostate Cancer Foundation Challenge Awards (S.P. Balk, P.S. Nelson, R.B. Montgomery, E.A. Mostaghel, E. Corey, M.E. Taplin).
Footnotes
Competing Interests: P.S. Nelson has an advisory role with Janssen. The other authors declare no competing interests.
References
- 1.Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocr Relat Cancer 2016;23:T179–T97 [DOI] [PubMed] [Google Scholar]
- 2.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017;32:474–89 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res 2006;66:7783–92 [DOI] [PubMed] [Google Scholar]
- 5.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 2011;30:2719–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 2016;22:369–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao S, Gao Y, He HH, Han D, Han W, Avery A, et al. Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell reports 2016;17:966–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohammad OS, Nyquist MD, Schweizer MT, Balk SP, Corey E, Plymate S, et al. Supraphysiologic Testosterone Therapy in the Treatment of Prostate Cancer: Models, Mechanisms and Questions. Cancers (Basel) 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009;138:245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taplin ME, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, et al. Intense Androgen-Deprivation Therapy With Abiraterone Acetate Plus Leuprolide Acetate in Patients With Localized High-Risk Prostate Cancer: Results of a Randomized Phase II Neoadjuvant Study. J Clin Oncol 2014;32:3705–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res 2014;20:1590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKay RR, Werner L, Mostaghel EA, Lis R, Voznesensky O, Zhang Z, et al. A Phase II Trial of Abiraterone Combined with Dutasteride for Men with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res 2017;23:935–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al-Deen Ashab H, et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res 2016;76:4948–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chattopadhyay I, Wang J, Qin M, Gao L, Holtz R, Vessella RL, et al. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2017;8:10324–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGuinness C, Wesley UV. Dipeptidyl peptidase IV (DPPIV), a candidate tumor suppressor gene in melanomas is silenced by promoter methylation. Front Biosci 2008;13:2435–43 [DOI] [PubMed] [Google Scholar]
- 17.Turcot V, Bouchard L, Faucher G, Tchernof A, Deshaies Y, Perusse L, et al. DPP4 gene DNA methylation in the omentum is associated with its gene expression and plasma lipid profile in severe obesity. Obesity (Silver Spring) 2011;19:388–95 [DOI] [PubMed] [Google Scholar]
- 18.Li L, van Breugel PC, Loayza-Puch F, Ugalde AP, Korkmaz G, Messika-Gold N, et al. LncRNA-OIS1 regulates DPP4 activation to modulate senescence induced by RAS. Nucleic Acids Res 2018;46:4213–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckenkamp A, Davies S, Willig JB, Buffon A. DPPIV/CD26: a tumor suppressor or a marker of malignancy? Tumour Biol 2016;37:7059–73 [DOI] [PubMed] [Google Scholar]
- 20.Barreira da Silva R, Laird ME, Yatim N, Fiette L, Ingersoll MA, Albert ML. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol 2015;16:850–8 [DOI] [PubMed] [Google Scholar]
- 21.Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res 2005;65:1325–34 [DOI] [PubMed] [Google Scholar]
- 22.Sun YX, Pedersen EA, Shiozawa Y, Havens AM, Jung Y, Wang J, et al. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL12. Clin Exp Metastasis 2008;25:765–76 [DOI] [PubMed] [Google Scholar]
- 23.Nazarian A, Lawlor K, Yi SS, Philip J, Ghosh M, Yaneva M, et al. Inhibition of circulating dipeptidyl peptidase 4 activity in patients with metastatic prostate cancer. Mol Cell Proteomics 2014;13:3082–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson MJ, Ruhland AR, Quast BJ, Reddy PK, Ewing SL, Sinha AA. Dipeptidylpeptidase IV activities are elevated in prostate cancers and adjacent benign hyperplastic glands. J Androl 2000;21:220–6 [PubMed] [Google Scholar]
- 25.Zhao M, Chen J, Yuan Y, Zou Z, Lai X, Rahmani DM, et al. Dipeptidyl peptidase-4 inhibitors and cancer risk in patients with type 2 diabetes: a meta-analysis of randomized clinical trials. Sci Rep 2017;7:8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.