

Abstract
Objective:
Monocytes and macrophages produce interleukin-1β (IL-1β) by inflammasome activation which involves ATP release, pannexin-1 (panx1) channels, and P2X7 receptors. However, IL-1β can also be produced in an inflammasome-independent fashion. Here we studied if this mechanism also involves ATP signaling and how it contributes to inflammasome activation.
Design:
In vitro studies with human cells and randomized animal experiments.
Setting:
Preclinical academic research laboratory.
Subjects:
Wild-type (WT) C57BL/6 and panx1 knockout (KO) mice, healthy human subjects for cell isolation.
Interventions:
Human monocytes and U937 macrophages were treated with different inhibitors to study how purinergic signaling contributes to toll-like receptor (TLR) induced cell activation and IL-1β production. WT and panx1 KO mice were subjected to cecal ligation and puncture (CLP) to study the role of purinergic signaling in IL-1β production and host immune defense.
Measurements and main results:
TLR agonists triggered mitochondrial ATP production and ATP release within seconds. Inhibition of mitochondria, ATP release, or P2 receptors blocked p38 MAPK and caspase-1 activation and IL-1β secretion. Mice lacking panx1 failed to activate monocytes, to produce IL-1β, and to effectively clear bacteria following CLP.
Conclusions:
Purinergic signaling has two separate roles in monocyte/macrophage activation, namely to facilitate the initial detection of danger signals via TLRs and subsequently to regulate NLRP3 inflammasome activation. Further dissection of these mechanisms may reveal novel therapeutic targets for immunomodulation in critical care patients.
Keywords: TLRs, LPS, pannexin-1, adenosine triphosphate, mitochondria, interleukin-1β
INTRODUCTION
IL-1β is a key cytokine with which monocytes and macrophages regulate host immune responses (1, 2). Monocytes and macrophages secrete IL-1β in response to microbial products that bind to pattern-recognition receptors including toll-like receptors (TLRs) (3). TLR4 recognizes bacterial lipopolysaccharides (LPS). TLR4 and CD14 and MD-2 coreceptors bind LPS, resulting in receptor internalization and downstream signaling events that trigger NLRP3 inflammasome activation (4–6). The NLRP3 inflammasome activates caspase-1, which cleaves the precursor molecule pro-IL-1β to the active cytokine (2, 7, 8).
LPS is known to activate the NLRP3 inflammasome via a two-step cascade (7). In a priming phase, TLR4-induced NF-κB signaling causes the transcription of gene products that are needed for NLRP3 inflammasome assembly and pro-IL-1β production. In a second phase, the NLRP3 inflammasome assembles and converts pro-caspase-1 to active caspase-1 that generates IL-1β. Recent work has shown that additional pathways can cause NLRP3 inflammasome activation (8–10). These NLRP3 inflammasome activation pathways involve P2X7 receptors, pannexin-1 (panx1) channels, and ATP release into the extracellular space (8, 11–14). In addition, monocytes can produce IL-1β independently from inflammasome activation (15). However, the mechanisms by which ATP signaling regulates IL-1β production in response to LPS have remained only partially defined and were the objective of this study (7).
Here we show for the first time that TLR agonists trigger rapid ATP release within seconds of TLR stimulation, that mitochondria produce the ATP that is released, and that the released ATP initiates purinergic signaling events needed for cell activation. Our findings show that purinergic signaling has at least two roles in monocyte/macrophage activation, namely to facilitate the initial detection of danger signals and to coordinate subsequent signaling pathways that lead to inflammasome activation and IL-1β secretion.
MATERIALS AND METHODS
Materials
A human TLR1–9 agonist kit (see legend to Fig. 1A) was from Invivogen (San Diego, CA). The panx1 inhibitory peptide 10panx1 was from R&D Systems (Minneapolis, MN) and Rhod-2 AM from Thermo Fisher Scientific (Waltham, MA). Antibodies: mouse anti-human CD14 FITC (clone M5E2; BD Biosciences; San Jose, CA), rabbit anti-human phospho-p38 MAPK, anti-caspase-1 (Cell Signaling; Danvers, MA), rabbit anti-human p38 MAPK (Santa Cruz; Santa Cruz, CA), goat anti-rabbit IgG-peroxidase. All other reagents: from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Figure 1.
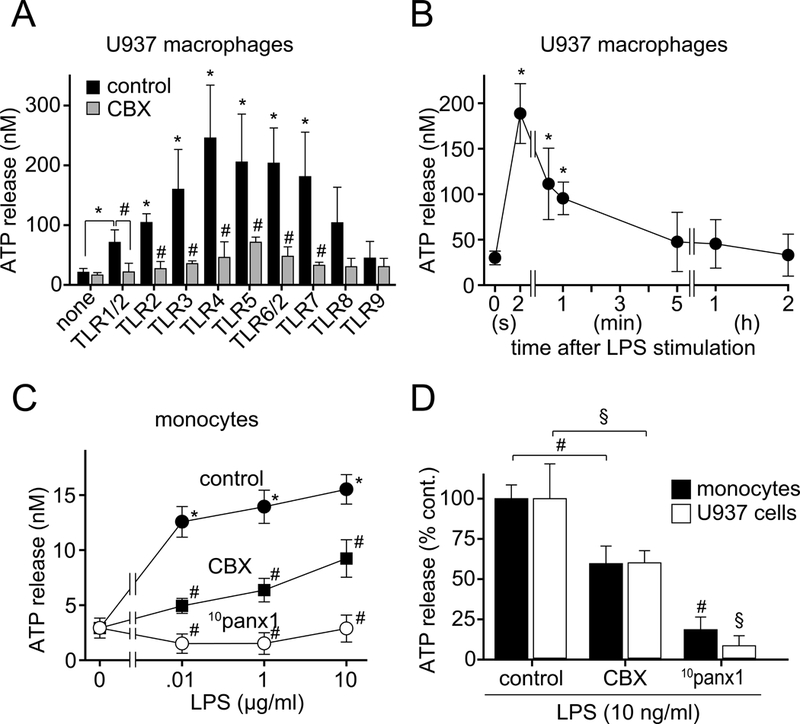
Pannexin-1 channels facilitate ATP release in response to TLR stimulation. A, U937 macrophages were pretreated with the panx1 inhibitor CBX (100 μM) or not for 5 min and then stimulated with different TLR ligands (TLR1/2: Pam3CysSerLys4, 1 μg/ml; TLR2: heat-killed Listeria monocytogenes, 108 cells/ml; TLR3: Poly(I:C), 10 μg/ml; TLR4: LPS, 10 ng/ml; TLR5: flagellin, 10 μg/ml; TLR6/2: FSL-1, 1 μg/ml; TLR7: Imiquimod, 10 μg/ml; TLR8: ssRNA40, 10 μg/ml; TLR9: ODN2006, 5 μM). After stimulation for 5 s, reactions were stopped by chilling in an ice bath. Cells were removed by centrifugation and ATP concentrations in the supernatants determined with a luciferase assay kit. B, U937 macrophages were stimulated with LPS (10 ng/ml) and ATP concentrations in the cell culture supernatants were measured at the indicated time points. C and D, Primary human monocytes or U937 macrophages were pretreated for 5 min with the panx1 inhibitors CBX (100 μM) or 10panx1 (100 μM), stimulated with the indicated LPS concentrations, and ATP release was assessed after 5 s. Data shown are mean values ± SD of n≥3 independent experiments; *P<0.05 vs. unstimulated controls; #, §P<0.05 vs. stimulated cells in the absence of panx1 inhibitors.
Human blood monocytes
The IRB approval was obtained from BIDMC for studies using human subjects; protocols were in accordance with the Declaration of Helsinki. Venous blood was collected from healthy volunteers and peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque (GE, Piscataway, NJ). Monocyte isolation by plastic adherence: PBMCs were suspended in RPMI-1640 media (ATCC, Manassas, VA), placed in 96-well flat-bottom culture dishes (Celltreat, Pepperell, MA) for 2 h at 37°C, and non-adherent cells were removed by washing with fully supplemented RPMI-1640 (10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin; Thermo Fisher Scientific). Viability was assessed with propidium iodide staining (1.5 μM for 10 min) and flow cytometry or fluorescence microscopy.
U937 monocyte cell culture and differentiation
U937 cells (ATCC) were maintained in fully supplemented RPMI-1640 at 37°C in 5% CO2. Prior to use, cells were differentiated to macrophages with phorbol myristate acetate (10 ng/ml) for 48 h (16).
ATP measurements
Cells were incubated in fresh culture medium at 37°C for 30 min, treated with different inhibitors for 5 min, stimulated with LPS or other TLR agonists, and reactions were stopped on ice. Supernatants were obtained by centrifugation (5 min, 400 x g, 0ºC) and ATP was measured with a bioluminescence assay kit (Invitrogen, Carlsbad, CA).
Mitochondrial Ca2+ and ATP release
U937 cells were differentiated in fibronectin-coated glass bottom dishes (Lab-Tek, Rochester, NY) as described above. Human PBMCs were cultured in the same dishes and lymphocytes were removed by vigorous washing. Over 90% of the remaining cells were monocytes as verified by CD14 staining. Cells were stained with the mitochondrial Ca2+ indicator Rhod-2 AM (1 μM) for 10 min or resuspended in HBSS containing 1 μM of the cell membrane-targeting fluorescent ATP probe 2–2Zn (17). Time-lapse imaging was performed with a Leica DMI6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with a temperature-controlled (37°C) stage incubator (Ibidi, Fitchburg, WI) and a DFC365 FX camera. Cells were stimulated with LPS in the presence or absence of different inhibitors and changes in mitochondrial Ca2+ levels or ATP were monitored by capturing images at 1 s intervals using a 100x oil (NA 1.3) objective, a TRITC or FITC filter set, and LAS microscope imaging software. Images were analyzed with NIH ImageJ software.
Immunoblotting
Immunoblotting was performed as described previously (18). MAPK p38 and caspase-1 activation were assessed using antibodies that recognize the phosphorylated form of MAPK p38 and the active p20 caspase-1 fragment, respectively. Pro-IL1β levels were measured in cell lysates using mouse-anti human IL-1β antibody (R&D Systems). Total MAPK p38 was assessed as loading control.
Determination of IL-1β levels
IL-1β was determined with ELISA kits (R&D Systems).
Mouse model of sepsis and infection
Animal experiments were approved by the IACUC of BIDMC and done as per NIH guidelines. C57BL/6 wild-type (WT) mice were from Charles River Laboratories (Wilmington, MA). Panx1 knockout (panx1 KO) mice were from The Knockout Mouse Project Center for Comparative Medicine (University of California, Davis, CA) and backcrossed into a C57BL/6 background. Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) as described (19). In some experiments, mice received i.p. injections of live E. coli (108) and remaining bacteria in the peritoneal cavity were determined after 2 h (19). Monocyte activation was assessed by flow cytometry using anti-CD11b and anti-Ly6C antibodies. Briefly, blood was collected by cardiac puncture, red blood cells lysed, leukocytes treated with Fc blocker (BD Biosciences), and labeled with PE-anti- Gr1 (clone: RB6–8C5), PerCP-anti-Ly6C (clone: HK1.4), and APC-anti-CD11b (clone: M1/70) antibodies (Thermo Fisher Scientific). Monocytes were identified by gating on Gr1-CD11b+Ly6C+ cells.
Statistical analysis
Values are expressed as mean ± standard deviation (SD) Unpaired two-tailed Student’s t-tests was used to compare two groups and one-way analysis of variance (ANOVA) was used for multiple group comparisons. Differences were considered significant at p<0.05.
RESULTS
TLR receptor stimulation triggers rapid ATP release via panx1 channels
Extracellular ATP release from damaged tissues has been shown to stimulate P2X7 receptors and to induce inflammasome activation and IL-1β secretion (13–14). Most previous studies evaluated ATP release hours after cell stimulation at times that coincide with NLRP3 inflammasome activation long after TLR stimulation with LPS or other agonists. However, little is known about whether ATP release is needed for the initial steps associated with TLR stimulation.
We used primary human monocytes freshly isolated from the peripheral blood of healthy donors and human U937 cell cultures that were differentiated to macrophages as previously described (16). Measuring ATP concentrations in the cell supernatant using an ATP bioluminescence assay, we found that both cell types released significant amounts of ATP within 5 s of TLR stimulation with most TLR agonists studied (Fig. 1A; Suppl. Fig. 1A, Supplemental Digital Content 2). Pretreatment with the gap junction inhibitor CBX attenuated TLR-induced ATP release. Next, we studied the kinetics of ATP release in response to TLR4 stimulation with LPS. Both cell types released ATP immediately after LPS stimulation (Fig. 1B; Suppl. Fig. 1B, Supplemental Digital Content 2). Extracellular ATP peaked within 2 s of LPS stimulation and returned to baseline levels in 1 min, probably as a result of ATP breakdown by ectonucleotidases or TLR4 receptor internalization (5). To analyze ATP release in more detail, we stained monocytes with a novel membrane-anchoring fluorescent ATP probe (17) and monitored ATP release from single cells in response to LPS stimulation using live-cell fluorescence microscopy. We found that about half of all monocytes responded with a burst of ATP release within seconds of LPS stimulation (Suppl. Fig. 2, A and B, Supplemental Digital Content 3; Suppl. Video 1, Supplemental Digital Content 7). The dose-dependent ATP release in response to LPS could be reduced with CBX and completely prevented with the panx1-specific inhibitor 10panx1 (Fig. 1, C and D). These data indicate that panx1-dependent ATP release is an early signaling event that precedes IL-1β secretion in TLR-responsive monocytes and macrophages.
Purinergic signaling is required for TLR4-induced p38 MAPK activation
IL-1β expression requires p38 MAPK activation, which is a hallmark of the cell signaling pathway triggered by LPS (20, 21). We studied whether ATP signaling is necessary for LPS-induced p38 MAPK activation in U937 macrophages. We found that LPS but also ATP, UTP, and the non-hydrolysable ATP analog ATPγS activated p38 MAPK in 30 s (Fig. 2A). LPS and UTP had similar effects. Because UTP and ATP are both agonists of P2Y2 receptors, these findings suggest that autocrine stimulation of P2Y2 receptors is involved in the response of cells to LPS. This notion is supported by the finding that inhibition of LPS-induced ATP release with CBX or activation of P2 receptors with the antagonist suramin prevented p38 MAPK activation in response to LPS (Fig. 2B; Suppl. Fig. 1C, Supplemental Digital Content 2).
Figure 2.
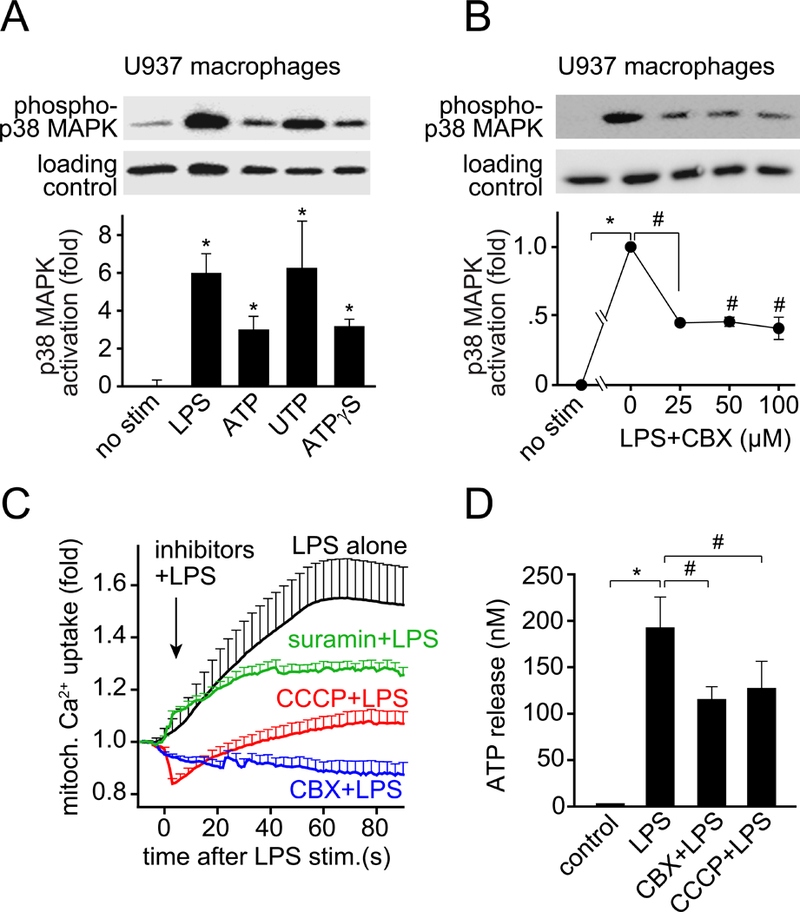
Autocrine purinergic signaling is required for LPS-induced p38 MAPK activation and for the upregulation of mitochondrial activity. A, U937 macrophages were stimulated with LPS (1 μg/ml), ATP (100 μM), UTP (100 μM) or the non-hydrolysable ATP analog ATPγS (100 μM). Reactions were stopped after 30 s by chilling in an ice bath and p38 MAPK activation was assessed by immunoblotting and comparing the ratio between phosphorylated p38 MAPK and total p38 MAPK (loading control). B, U937 macrophages were pretreated with the indicated concentrations of CBX for 5 min, stimulated with LPS (1 μg/ml) for 30 s, and p38 MAPK activation was determined. C, U937 cells were plated onto glass coverslips, differentiated, and loaded with the mitochondrial Ca2+ probe Rhod-2-AM. U937 macrophages were stimulated with LPS (10 ng/ml) with or without inhibitors that block ATP release (CBX, 100 μM), mitochondrial ATP production (CCCP, 10 μM), or P2 receptor activation (suramin, 100 μM). Rhod-2 fluorescence was recorded and plotted over time. Data shown are mean values ± SEM of n≥15 cells derived from 3 independent experiments. D, U937 macrophages were pretreated with CBX (100 μM) or CCCP (10 μM) for 5 min, stimulated with LPS (10 ng/ml) for 5 s, and ATP release was determined as described above. Data in A, B and D represent mean values ± SD of n=3 independent experiments; *P<0.05 vs. unstimulated controls; #P<0.05 vs. LPS-stimulated control without inhibitors.
Mitochondria fuel the panx1/ATP/P2 receptor signaling mechanisms triggered by LPS
Because LPS-induced cell activation required rapid ATP release, we tested whether mitochondria are needed to produce the ATP that is released. Mitochondria produce ATP by oxidative phosphorylation, which requires the influx of Ca2+ into mitochondria (22, 23). Using the mitochondrial Ca2+ probe Rhod-2, we found that mitochondrial Ca2+ uptake increased immediately after the stimulation of U937 macrophages with LPS (Suppl. Fig. 2, C and D, Supplemental Digital Content 3; Suppl. Video 2, Supplemental Digital Content 8). Intracellular ATP levels increased with similar kinetics, suggesting that mitochondria generate the ATP that fuels the purinergic signaling mechanisms triggered by LPS (Suppl. Fig. 2E, Supplemental Digital Content 3). Pretreatment of U937 macrophages with CBX or with the mitochondrial inhibitor CCCP reduced LPS-induced ATP release, indicating that mitochondria are a major source of the ATP released in response to LPS (Fig. 2D). Pretreatment of U937 cells with CBX, suramin, or CCCP also impaired mitochondrial Ca2+ uptake in response to LPS, which suggests that TLR4-induced ATP release and autocrine stimulation of P2 receptors enhance mitochondrial activity in LPS-stimulated cells (Fig. 2C; Suppl. Video 3, Supplemental Digital Content 9). Similar to U937 macrophages, inhibitors of mitochondrial ATP synthesis impaired ATP release from LPS-stimulated human monocytes (Fig. 3, A and B). None of the mitochondrial inhibitors tested affected cell viability for up to 24 h (Suppl. Fig. 3, Supplemental Digital Content 4). Taken together, our findings show that mitochondria have a central role in the activation of monocytes and macrophages by LPS and that an initial burst of ATP release in response to TLR4 stimulation promotes an autocrine feed-forward signaling mechanism that upregulates mitochondrial activity.
Figure 3.
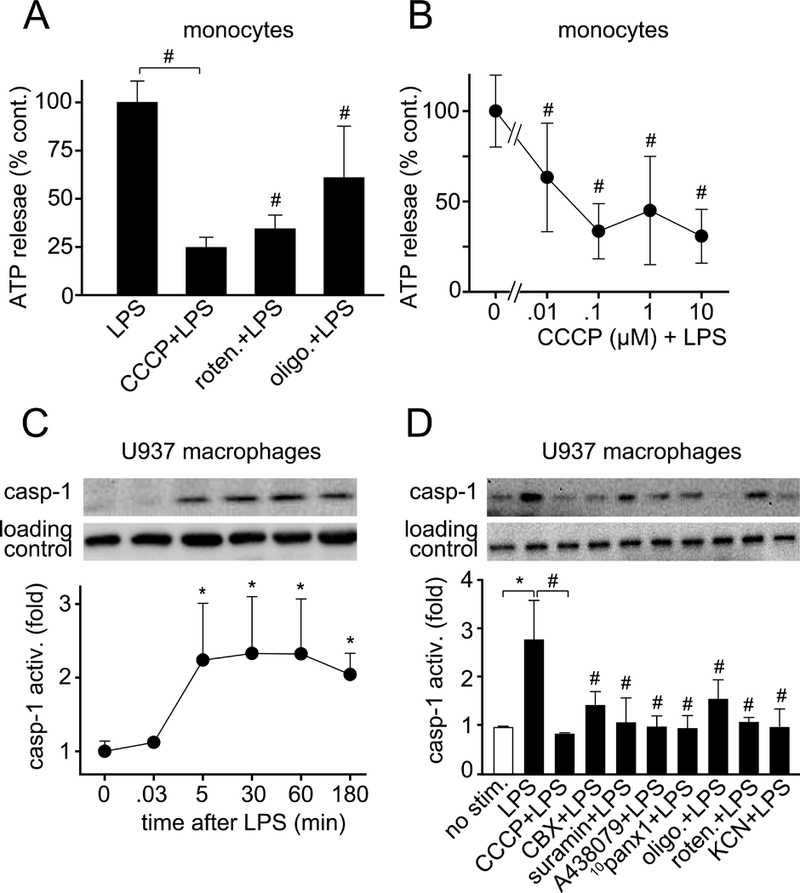
Mitochondrial ATP and autocrine feedback through purinergic receptors are required for LPS-induced caspase-1 activation. A-B, Primary human monocytes were pretreated for 5 min with the mitochondrial inhibitors CCCP (10 μM), rotenone (10 μM), or oligomycin (10 μM; A) or with the indicated concentrations of CCCP (B). Then cells were stimulated with LPS (10 ng/ml) for 5 s and ATP concentrations in the supernatants were determined as described in Fig. 1. Data shown are means ± SD of n=6 (A) or n=3 (B) independent experiments; #P<0.05 compared to LPS-stimulated controls. C, U937 macrophages were stimulated with LPS (1 μg/ml) for the indicated times. Then caspase-1 activation levels were assessed using immunoblotting and antibodies that recognize activated caspase-1 (p20 subunit). D, U937 macrophages were pretreated or not with the mitochondrial inhibitors CCCP (10 μM), rotenone (10 μM), oligomycin (10 μM) or KCN (500 μM), the panx1 channel blockers CBX (100 μM) or 10panx1 (100 μM), or the P2 receptor inhibitors suramin (pan-P2 receptors; 100 μM) or A438079 (P2X7 receptors; 10 μM) for 15 min. Then cells were stimulated with LPS (1 μg/ml) for 2 h and caspase-1 activity was measured. Total p38 MAPK was assessed as loading control. Data shown are mean values ± SD of n=3 separate experiments; *P<0.05 vs. unstimulated negative controls; #P<0.05 vs. LPS-stimulated positive control.
Purinergic signaling is required for LPS-induced caspase-1 activation and IL-1β secretion
Initial purinergic signaling generates ATP needed for TLR signaling. In addition, it likely also provides the ATP to support subsequent cell activation processes such as NLRP3 inflammasome activation and IL-1β production. IL-1β secretion requires activation of caspase-1 and the formation of an active p20 caspase-1 fragment (24, 25). LPS induced rapid and prolonged caspase-1 activation in U937 macrophages (Fig. 3C). Caspase-1 activation was blocked by inhibitors of mitochondrial ATP synthesis (CCCP, oligomycin, rotenone, or KCN) or panx1-induced ATP release (CBX, 10panx1), and by non-selective P2 (suramin) and selective P2X7 (A438079) receptor antagonists (Fig. 3, C and D). Similarly, blocking mitochondria, ATP release, or P2 receptors dose-dependently impaired LPS-induced IL-1β secretion from primary human monocytes (Fig. 4). In summary, our results show that blocking ATP release or P2 receptor signaling inhibits early events occurring within seconds of LPS stimulation, such as p38 MAPK activation, as well as later events such as transcription of pro-IL-1β and caspapse-1 activation, which precede IL-1β release (Fig. 5A). These findings demonstrate that TLR4-induced mitochondrial ATP synthesis and autocrine purinergic signaling are required for the initial activation of monocytes and macrophages and for subsequent functional responses that lead to IL-1β production via inflammasome-dependent or independent pathways.
Figure 4.
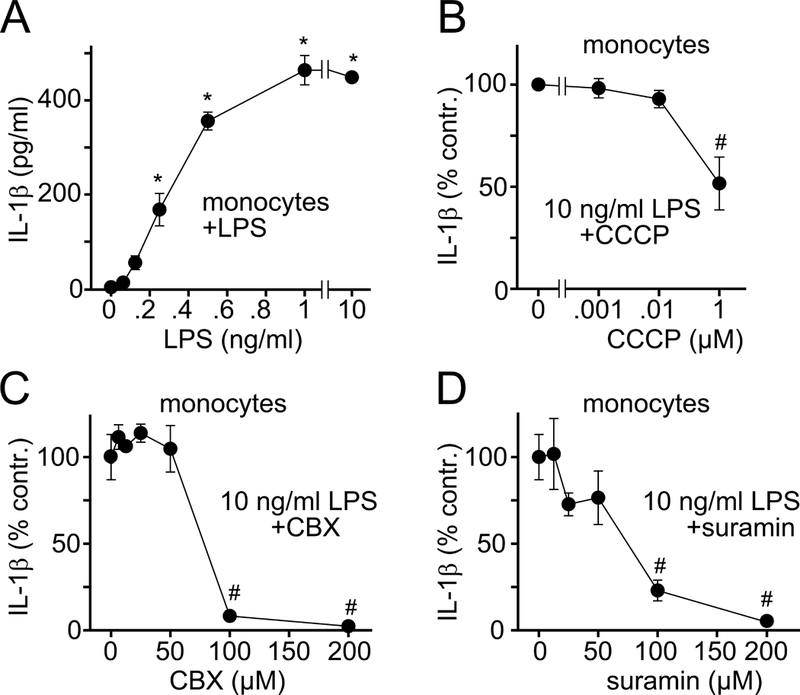
Mitochondrial ATP synthesis, ATP release, and P2 receptor stimulation are required for LPS-induced IL-1β secretion. A-D, Primary human monocytes were stimulated with LPS and IL-1β concentrations in the supernatants were measured after 24 h. B-D, Monocytes were pretreated for 5 min with CCCP, CBX, or suramin, stimulated with LPS (10 ng/ml), and IL-1β secretion was determined after 24 h. Data are means ± SD, n=3; *P<0.05 vs. unstimulated controls; #P<0.05 vs. LPS-stimulated controls.
Figure 5.
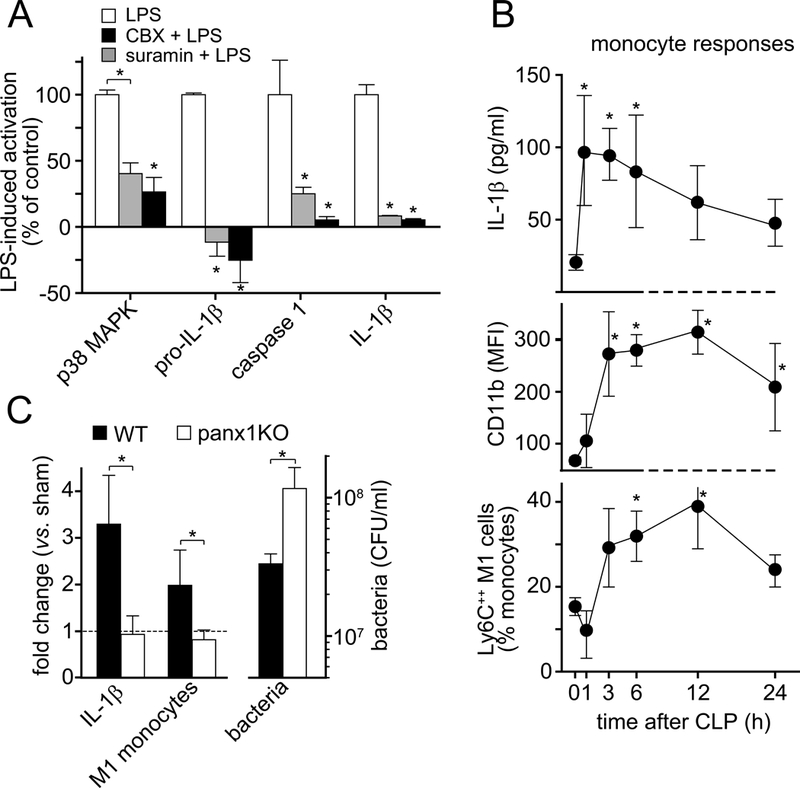
Panx1 channels are required for monocyte activation and IL-1β secretion in a mouse sepsis model. A, Differentiated U937 macrophages were stimulated for 30 s (p38 MAPK), 2 h (pro-IL-1β, caspase-1), and human monocytes (IL-1β) were stimulated for 24 h with LPS (10 ng/ml) in the presence or absence of CBX (100 μM) or suramin (200 μM) and cell activation was measured by immunoblotting or ELISA (IL-1β). Data are means ± SD, n=3; *P<0.05 vs. LPS-stimulated controls. B, C57BL/6 wild-type (WT) mice were subjected to sepsis using cecal ligation and puncture (CLP) and plasma IL-1β levels as well as CD11b and Ly6C expression by peripheral blood monocytes were measured by ELISA and flow cytometry, respectively, at the time points indicated. C, WT mice or panx1 knockout (KO) mice were subjected to CLP or sham surgery and IL-1β plasma levels and the increase in inflammatory Ly6C++ M1 monocytes were determined 4 h after CLP and compared to sham animals (left). WT or panx1 KO mice were injected i.p. with 1×108 live E. coli and bacteria in the peritoneal cavity were counted 2 h later (right). Data are expressed as mean ± SEM of n=5 animals per group; *P<0.05; MFI, mean fluorescence intensity; CFU, colony forming units.
Purinergic signaling is required for monocyte activation in sepsis and antimicrobial host defense
LPS increases IL-1β production in response to infection and sepsis (26). We subjected WT and panx1 KO mice to CLP as previously described, resulting in severe bacteremia (19). CLP triggered IL-1β production in 1 hour and caused polarization of monocytes to an inflammatory M1 phenotype characterized by increased CD11b and Ly6C expression (Fig. 5A; Suppl. Fig. 4, Supplemental Digital Content 5) (27). Panx1 KO mice failed to produce IL-1β and to activate monocytes in response to CLP and were less capable than WT mice of coping with bacteria following E. coli injection (Fig. 5B) and CLP-induced bacteremia (19). These findings demonstrate that monocytes depend on panx1-induced ATP release to recognize danger cues such as LPS and to orchestrate the necessary inflammatory response that is required to protect the host from infections.
DISCUSSION
Our findings indicate that cellular ATP release is essential for the detection of LPS and other danger molecules by monocytes and macrophages. Mitochondria fuel the initial panx1/ATP/P2 receptor signaling pathways triggered by TLR agonists as well as subsequent signaling steps that induce IL-1β secretion from stimulated cells (Suppl. Fig. 5, Supplemental Digital Content 6). NLRP3 inflammasome activation and IL-1β secretion are known to involve extracellular ATP, panx1, and P2X7 receptors (10, 28). However, the purinergic signaling events upstream of inflammasome activation have not been previously defined (7).
Here we show that TLR4 stimulation triggers rapid ATP release, which extends previous reports that ATP release contributes to inflammasome activation (12, 13, 29–31). Previous studies focused on ATP release as a trigger of inflammasome activation. In particular, external ATP at millimolar concentrations was shown to induce inflammasome activation by inducing P2X7 receptors to form large pores and causing mitochondrial disruption and pyroptosis (7). By contrast, little is known about ATP release as an upstream signaling event that facilitates cell activation. Here we show, to our knowledge for the first time, that mitochondria rapidly generate the ATP that is released and that a panx1/ATP/P2Y2 receptor signaling axis is required for TLR4 signal amplification. We propose that monocytes and macrophages use this initial panx1/ATP/P2Y2 trigger mechanism to detect danger signals and to initiate NLRP3 inflammasome priming by generating pro-IL-1β and other building blocks that are needed for the assembly of the NLRP3 inflammasome complex. Activation of NLRP3 involves a second purinergic signaling mechanism via the better-known panx1/ATP/P2X7 receptor axis. Monocytes seem to need both purinergic signaling mechanisms to detect microbial dangers, to produce IL-1β, and to cope with invading microorganisms (Suppl. Fig. 5, Supplemental Digital Content 6).
Our previous work has shown that similar purinergic signaling mechanisms regulate the functions of T cells and neutrophils (18, 32–34). Taken together, our findings demonstrate that mitochondria and autocrine purinergic signaling mechanisms regulate diverse aspects of the innate and adaptive immune responses to infections. A role for mitochondria in inflammasome activation has been previously recognized, but the underlying mechanisms have remained unclear (35–37). Our current findings indicate that one of the roles of mitochondria is to generate ATP to initiate the purinergic signaling mechanism by which cells detect TLR agonists and respond with NLRP3 inflammasome activation, if needed. This interpretation is supported by a recent report showing that mitochondrial function is required for NLRP3 inflammasome activation by extracellular ATP (38).
While NLRP3 is perhaps the best characterized inflammasome, other canonical and noncanonical inflammasome pathways involving caspase-11 have been recently described (9, 10, 39, 40). Future studies will be needed to assess whether autocrine purinergic signaling mechanisms are involved in these signaling pathways and how they contribute to host defense and inflammation in critical care patients.
TLR4 signaling is an important mechanism by which host cells detect the presence of LPS and other indicators of bacterial infections. Our results suggest that mitochondria and multiple P2 receptor subtypes are involved in the host immune responses to bacterial infection. LPS was shown to promote the polarization of monocytes towards an inflammatory phenotype that relies mainly on aerobic glycolysis (41). On the other hand, mitochondria require oxygen for ATP production, which suggests that hypoxic conditions as well as changes in the metabolic environment that impair TLR signaling and IL-1β production could impair the ability of monocytes and macrophages to defend the host from infections. This possibility is supported by our findings that panx1 KO mice failed to produce IL-1β and to effectively protect the host from bacterial infection. We conclude that the purinergic signaling mechanisms identified in our study are potential novel targets to modulate inflammation and host responses in critical care patients.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Sophie Bromberger and Yong Shen for their valuable support in revising the manuscript. We thank Drs. Yasutaka Kurishita and Itaru Hamachi (Kyoto University, Japan) for providing the fluorescent ATP probe 2–2Zn.
Footnotes
Conflicts of Interest and Source of Funding: The authors have disclosed that they do not have any conflicts of interest. This work was funded in part by grants from the National Institutes of Health, GM-51477, GM-60475, GM-116162, AI-080582, and T32 GM-103702.
Copyright form disclosure: Drs. Lee, Zhang, and Junger received support for article research from the National Institutes of Health. Dr. Li received support for article research from National Natural Science Foundation of China No.81701564. The remaining authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Auron PE, Webb AC, Rosenwasser LJ, et al. : Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A 1984; 81:7907–7911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinon F, Mayor A, Tschopp J: The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27:229–265 [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S: The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 2010; 11:373–384 [DOI] [PubMed] [Google Scholar]
- 4.Tan Y, Zanoni I, Cullen TW, et al. : Mechanisms of toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 2015; 43:909–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zanoni I, Ostuni R, Marek LR, et al. : CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell 2011; 147:868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosadini CV, Kagan JC: Early innate immune responses to bacterial LPS. Curr Opin Immunol 2017; 44:14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y, Hara H, Núñez G: Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016; 41:1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaidt MM, Ebert TS, Chauhan D, et al. : Human monocytes engage an alternative inflammasome pathway. Immunity 2016; 44:833–846 [DOI] [PubMed] [Google Scholar]
- 9.Kayagaki N, Warming S, Lamkanfi M, et al. : Non-canonical inflammasome activation targets caspase-11. Nature 2011; 479:117–121 [DOI] [PubMed] [Google Scholar]
- 10.Yang D, He Y, Muñoz-Planillo R, et al. : Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 2015; 43:923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Netea MG, Nold-Petry CA, Nold MF, et al. : Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009; 113:2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrari D, Chiozzi P, Falzoni S, et al. : Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med 1997; 185:579–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccini A, Carta S, Tassi S, et al. : ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 2008; 105:8067–8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parzych K, Zetterqvist AV, Wright WR, et al. : Differential role of pannexin-1/ATP/P2X7 axis in IL-1β release by human monocytes. FASEB J 2017; 31:2439–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Netea MG, van de Veerdonk FL, van der Meer JW, et al. : Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol 2015; 33:49–77 [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Yang J, Gao W, et al. : Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014; 513:237–241 [DOI] [PubMed] [Google Scholar]
- 17.Kurishita Y, Kohira T, Ojida A, et al. : Organelle-localizable fluorescent chemosensors for site-specific multicolor imaging of nucleoside polyphosphate dynamics in living cells. J Am Chem Soc 2012; 134:18779–18789 [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Yao Y, Sumi Y, et al. : Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signa. 2010; 3:ra45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Kondo Y, Bao Y, et al. : Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med 2017; 45:e97–e104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldassare JJ, Bi Y, Bellone CJ: The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J Immunol 1999; 162:5367–5373 [PubMed] [Google Scholar]
- 21.Han J, Lee JD, Bibbs L, et al. : A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994; 265:808–811 [DOI] [PubMed] [Google Scholar]
- 22.McCormack JG, Halestrap AP, Denton RM: Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 1990; 70:391–425 [DOI] [PubMed] [Google Scholar]
- 23.Glancy B, Balaban RS: Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012; 51:2959–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutterwala FS, Haasken S, Cassel SL: Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 2014; 1319:82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathur A, Hayward JA, Man SM: Molecular mechanisms of inflammasome signaling. J Leukoc Biol 2018; 103:233–257 [DOI] [PubMed] [Google Scholar]
- 26.Opal SM: The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol 2007; 297:365–377 [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Zhang L, Yu C, et al. : Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2014; 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426 [DOI] [PubMed] [Google Scholar]
- 29.Riteau N, Baron L, Villeret B, et al. : ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012; 3:e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gombault A, Baron L, Couillin I: ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol 2012; 3:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen HB, Briggs KT, Marino JP, et al. : TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood 2013; 122:1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Corriden R, Inoue Y, et al. : ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006; 314:1792–1795 [DOI] [PubMed] [Google Scholar]
- 33.Yip L, Woehrle T, Corriden R, et al. : Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 2009; 23:1685–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Junger WG: Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11:201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou R, Yazdi AS, Menu P, et al. : A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221–225 [DOI] [PubMed] [Google Scholar]
- 36.Gurung P, Lukens JR, Kanneganti TD: Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med 2015; 21:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tur J, Vico T, Lloberas J, et al. : Macrophages and mitochondria: a critical interplay between metabolism, signaling, and the functional activity. Adv Immunol 2017; 133:1–36 [DOI] [PubMed] [Google Scholar]
- 38.Sadatomi D, Nakashioya K, Mamiya S, et al. : Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J Biochem 2017; 161:503–512 [DOI] [PubMed] [Google Scholar]
- 39.Schroder K, Tschopp J: The inflammasomes. Cell 2010. 19;140:821–832 [DOI] [PubMed] [Google Scholar]
- 40.Lamkanfi M, Dixit VM: Mechanisms and functions of inflammasomes. Cell 2014; 157:1013–1022 [DOI] [PubMed] [Google Scholar]
- 41.Lachmandas E, Boutens L, Ratter JM, et al. : Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol 2016; 2:16246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.