

Summary
We present a perspective of our view of the application of cryo electron microscopy (cryoEM) to structure based drug design (SBDD). We discuss the basic needs and requirements for SBDD, the current state of cryoEM, and the challenges that need to be overcome for this technique to reach its full potential in facilitating the process of drug discovery.
Introduction
The importance of structural information in drug discovery has been understood since the mid-80s, and by the early 90s the first success stories started to appear (Roberts et al., 1990) (Erickson et al., 1990) (Dorsey et al., 1994). Nowadays, structure guided drug design is an integral part of the discovery platform in most pharmaceutical industries. This rapid evolution has been accelerated by the advent of proteomics and structural genomics, developments in information technology, fast-paced advances in cloning, expression, and purification of targets, development of high-throughput crystallography, and numerous advances in structure determination using nuclear magnetic resonance (NMR). Availability of faster computers and relatively inexpensive computer clusters has also played a role, increasing the speed of in silico lead identification and optimization.
Structure guided drug design is a very iterative process, that can last years before a new drug candidate is obtained, and involves many fields of research, from structural and computational chemistry to biology, pharmacology, and medicinal chemistry, just to name a few (Anderson, 2009). The very first (and necessary) step is the determination of the structure of the target of interest. Until now, the major methods utilized for structure determination were x-ray crystallography, and NMR, with homology modeling stepping in when an experimental structure could not be obtained. Crystallography and NMR combined have produced over 130,000 structures in the public domain (as of January 2018, the PDB contains 122,681 x-ray structures and 12,102 NMR structures), and possibly many thousands more in the private sector, providing a wealth of structural information covering a wide spectrum of targets, from small soluble proteins to large assemblies to membrane bound targets. Nevertheless, structural information for a large portion of targets of interest to the pharmaceutical industry is still unknown, mainly for reasons that include molecular size, complexity, flexibility, and difficulties in production and purification that make them unsuitable for the established structure determination techniques like crystallography and NMR. In addition, structures determined by x-ray crystallography very often represent just one snapshot of one specific conformation of the target protein, which does not necessarily represent the most common conformation present at a cellular level and often cannot provide detailed information about flexibility and conformational motions.
In recent years, cryo electron microscopy (cryoEM) has emerged as a complementary technique to crystallography and NMR (Frank, 2017). While EM analysis of biological specimens has been in use for ~40 years, only in the past ~5 years has the method become capable of producing near-atomic level structures suitable for application in drug discovery (for example, (Shen et al., 2016) (Guo et al., 2017a) (Shalev-Benami et al., 2017) (Guo et al., 2017b) (Hirschi et al., 2017)). CryoEM also potentially provides several advantages over the more traditional methods: access to larger and/or more complex biological systems that have been intractable to analysis by x-ray or NMR; the advantage of analyzing proteins in solution (albeit vitrified) and, perhaps most importantly, the possibility of characterizing multiple conformational or compositional states from the same sample (Roh et al., 2017) (Frank et al., 2016), hence providing insights into states of the macromolecule theoretically closer to those that are biologically relevant (Nogales and Scheres, 2015).
While there are many relevant biological questions that can be answered by lower resolution structures (for example, the nuclear pore complex structure (Aitchison and P. Rout, 2012) (von Appen and Beck, 2016); heat shock protein binding modes (Shi et al., 2013); Glutamate receptor activation and mechanism (Meyerson et al., 2014)), the process of structure guided drug design has some specific requirements (resolution being only one of them) that need to be considered when selecting a protein target. In this review we will discuss the requirements for small molecule structure guided drug discovery, what the current state of cryoEM is in this area, what it can bring to the table, and what can be done (or expected) in the future to move this technique to the forefront of drug discovery.
Small molecule structure guided drug discovery: when and why
Structural information can be used to guide a drug discovery program from the very beginning, from understanding at an atomic level the mode of action of the target in question, to providing structure based alternatives to the traditional screening procedures, to structurally validating hits identified with other techniques (Figure 1). In very early stages, a structure can provide insights into the target activation and modulation, and possible conformational flexibility derived by interactions with partner(s) (small molecules or larger substrates). At this point, even low resolution structures (worse than 3–4 Å) can be used as input for computer based approaches such as in silico screening, docking and design, which can identify and suggest preferred pharmacophores, optimal binding vectors, and essential interactions; this information can then be incorporated in a rational drug design process (Śledź and Caflisch, 2017). When accompanied by concurrent binding, activity and pharmacology assays, this approach can result in a faster discovery of initial hits, even without the need for further structural data. If a robust crystal system is in place, structure based fragment identification has also been shown to be a viable method for the identification of diverse chemical matter that can then be used as starting point for drug development (Jhoti et al., 2007).
Figure 1:
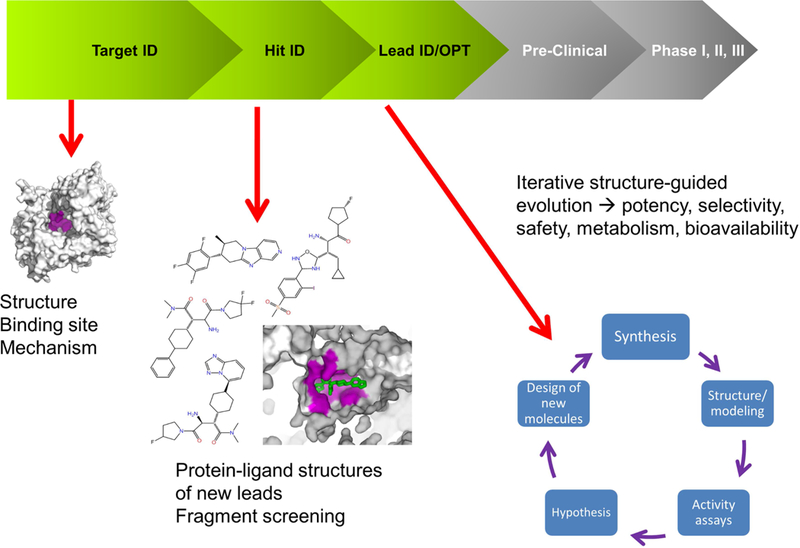
Schematic representation of the drug discovery and development process. The areas in the R&D process where structural information can be used are highlighted. In the Target ID space, one structure may be sufficient to identify the target binding site and characterize its mechanism. During the Hit ID stage, a few structures may be required to structurally characterize the lead compounds identified during screening. If a robust structural system is in place, X-ray can be used to run fragment screening campaigns. The heaviest request for structural data happens at the Lead ID/Opt stage, in which several iterations (from compound synthesis to structure/acitivity to SBDD back to synthesis) may happen in few weeks.
The subsequent steps will generally require a more intense structural effort. Structural biology groups will be required to produce structures of the select target bound to different pharmacophores (or compound classes); this may be just a handful of structures, if only one example small molecule class is deemed sufficient but there may be as many as hundreds of structures if structural support is required throughout the entire process (Prongay et al., 2007) (Stamford et al., 2012) (Scapin, 2015). Structural knowledge may be sought, for example, when apparently simple chemical changes to the small molecule cause major deviations from the expected pharmacology, or when large modifications are introduced into the small molecules to address issues such as solubility and cell penetration, or when ongoing issues such as potency and selectivity need to be addressed. This lead optimization is largely the time in which structure determination is most heavily used, albeit maybe not the period in which structural information is the most impactful. Very often it becomes a routine exercise, an assay run side by side with other assays, a fact that is helped by the increased ease of access to synchrotrons and their high level of automation in data collection and processing.
Small molecule structure guided drug discovery: requirements
There are several requirements that need to be kept in mind when discussion SBDD. The most relevant may be the resolution of the reconstructed maps. In the grand scheme of things, answers can be found at any resolution, it depends on the questions asked. For example, analyses of large conformational changes to understand the mechanism of action of a given protein can be done (and be very informative) even at resolutions of 4–8 Å or less (for example (Blees et al., 2017) (McGoldrick et al.,2018) (Lu et al., 2017)). But for a SBDD process we need to understand, explain and eventually predict changes in atoms positions, atom-atom interactions, and main chain or side chain motions as small as 0.5–1.5 Å. Accuracy in atomic positions then becomes essential. The error on atomic coordinates is resolution dependent: a typical 2.0 Å crystallographic model has a coordinate error of less than 0.2 Å (Murshudov and Dodson, 2997), but for lower resolution structures, when even assigning the correct side chain position becomes a challenge, the uncertainty becomes quite high. As a result, ideally, a resolution of 3 Å or better is necessary for structure based predictions and drug design. Structures at lower resolution can still be useful; molecular dynamics and other computational methods can be used to fit and minimize the structure and assign the most probable loop conformations and residue rotamers (Trabuco et al., 2008) (Zhao et al., 2013) (Arenz et al., 2016) (Tordai et al., 2017), as well as build de novo structures (Das and Baker, 2008) or identify the position of chemically relevant waters (Wang et al., 2011) (Jukič et al., 2017). The combination of low resolution experimental structures and modeling can become a powerful tool for SBDD, particularly if orthogonal validation methods and checks are available.
Other aspects that are often overlooked in the context of SBDD are timing and throughput. In order to initiate a successful structure guided drug development program, it is necessary to characterize and validate the selected target(s) from a three-dimensional point of view as early as possible in the program. This target validation step may be accomplished by generating just one or two structures, for example one of the target alone, and one in complex with its natural substrate or other modulators. In many cases, if the conformational differences between the various states are large enough, even medium to low resolution structures can be extremely useful in understanding the mechanism or identifying binding sites (Gao et al., 2016) (Guo et al., 2017b) (Meyerson et al., 2014). Early on in the project, structure determination can take a few months to a year, and still provide enough novel information to be useful. On the other hand, in order to be useful and make an impact in all other steps of the process, and especially during the compound optimization cycle, structural information needs to be delivered quickly, ideally within 24–48 hours from receiving the compounds. Chemistry and biology can generate and validate dozen of new compounds per week: assay results alone, when combined with molecular modeling, can easily guide the drug development process, and if the structural information is not timely, it could quickly become irrelevant.
Lastly, the conformational state(s) of the macromolecular target need to be carefully analyzed: mutations and/or truncations required for sample stability and crystallizability, the buffer(s) used for purification and storage, and crystal packing may affect the overall structure or induce local changes that can affect ligand binding (Scapin et al., 2001) (Cousido-Siah et al., 2012). This may be the area that where cryoEM could have the highest impact as a tool for structure determination, since it is a “solution based” methodology that can be applied to native proteins and provide in some cases a different view of the macromolecule (Bartesaghi et al., 2014).
Single particle cryoEM as a tool for SBDD: present state and outlook
As has now been much discussed (Frank, 2017) (Kühlbrandt, 2014) (Method of the Year, 2016), cryoEM has recently gone through a very dramatic expansion and is now being rapidly adopted by most structural biologists. This expansion was triggered when a new camera that could directly detect electrons became commercially available about 5 years ago (Li et al., 2013) (Faruqi et al., 2003), providing the final technical advance that allowed near-atomic resolution structures to be reconstructed from images of single particles embedded in vitreous ice. For very well behaved samples (e.g. apoferritin, 20S proteasome, aldolase) it is now possible to reach a resolution below 3A using a few hours of data collection and processing (Figure 2). Unfortunately most samples are not as well behaved. There are still some major challenges that need to be addressed to improve the chances of a successful outcome when approaching a structural analysis using cryoEM.
Figure 2.
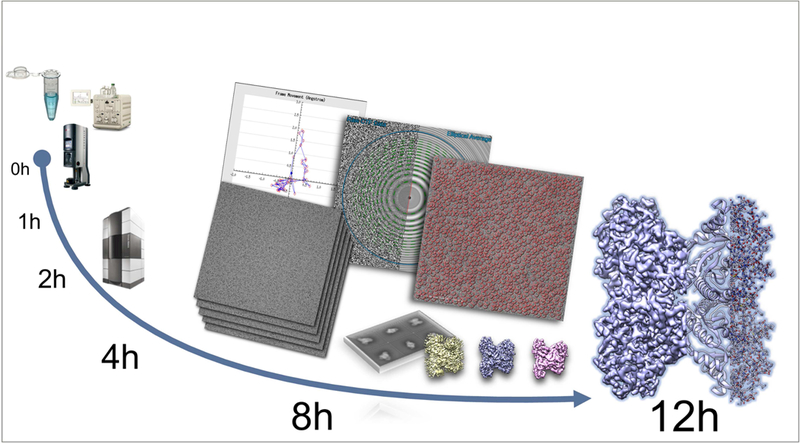
Timeline for CryoEM structure determination. The figure shows the timeline from data collection to a 2.5Å structure for a well behaved aldolase sample (unpublished results, Ed Eng et al.. NYSBC). Steps from data collection to particle picking are almost all fully automated and require minimal user input (Suloway, et al., 2005) (Lander, et al., 2009). Reconstruction was carried out with Cryosparc (Punjani, et al., 2017).
Sample Preparation
The first hurdle that needs to be overcome is preparing specimens suitable for imaging in a transmission electron microscope, preferably in a reproducible way so that conditions identified for one target-ligand complex can be readily used for all subsequent complexes of the same target. Ideally these specimens will have the single particles embedded in a layer of vitreous ice just a little bit thicker than the diameter of the particles, and the particles should be distributed isotopically so as not to adopt a preferred orientation. The current method used for cryoEM specimen preparation is to apply a small aliquot (3uL) of sample onto an EM grid (typically copper or gold mesh covered by a thin fenestrated substrate of either carbon or gold), remove most of the sample by applying filter paper directly to one or both sides of the grid, and then immediately plunge the grid into a cryogen (typically ethane or propane). This process is not well controlled at the nanoscale level and the layer of vitreous ice that results is often of quite variable thickness (Figure 3) with the consequence that time must be spent searching out areas of the grid with ice of suitable thickness, reducing the throughput at the microscope. A much more critical problem however is that when the thin layer of liquid is formed just prior to vitrification, the particles have ample opportunity to collide with and potentially interact with the air-water interface. This may result in the particles adopting preferred orientations (Glaeser and Han, 2017) (Tan et al., 2017) and in the worst case, denaturing either partially or completely. To address this problem we have been developing a new process for preparing vitrified grids that uses a piezo dispensing tip to deliver 50 pL droplets to a self-wicking grid as it flies past the tip on its way to the liquid ethane (Jain et al., 2012) (Razinkov et al., 2016) (Dandey et al., 2018) (Wei et al., 2018). This method provides more control of the ice thickness (Figure 3) and particle distribution and we have used it to successfully freeze dozens of different samples several of which have resulted in sub 3Å structures. A further benefit of this new method is that the sample spends less time in the thin liquid layer prior to vitrification and as a result the deleterious effects of the air-water interface are reduced. We have shown that if we reduce the spot to plunge time to ˜100ms (compared to 1s when using traditional methods) we can substantially reduce preferred orientation at least in some samples (paper in preparation). The system, which we call Spotiton, is in the process of being commercially developed and the hoped for outcome is a fully automated and very reproducible process, factors which will likely be important in a commercial drug development pipeline.
Figure 3.

Sample preparation. The figure shows a comparison between a grid vitrified using a standard plunger (top) and a grid prepared using SpotItOn (Jain, et al., 2012) (Razinkov, et al., 2016), bottom. In the grid vitrified with the standard plunger the ice distribution is highly variable across the grid, and identification of suitable areas for data collection can be a lengthy process. In the SpotItOn grid, one single narrow strip of very uniform ice is visible and almost every square in the stripe is available for data collection.
Data Collection
A further challenge in using cryoEM for SBDD is that the resolution achieved for many structures falls short of what is desirable. Since 2014 (the year of the “resolution revolution” (Kühlbrandt, 2014)) the number of EM structures at a resolution of 4 Å or better has been steadily increasing, and as of January 2018 there are 672 entries in the Electron Microscopy Database (EMDB) for single particles that meet the “4 Å or better” criteria (Figure 4). Of these, only 75 are better than 3 Å and only 12 reach the resolution of 2.5 Å or better; many of these are of “test cases” used to test the advances in the technology and to push the resolution limits (for example (Kimanius et al., 2016) (Fan et al., 2017) (Herzik et al., 2017) (Feng et al., 2017) (Danev et al., 2017)). Among the sub-3 Å structures, five contain small molecule compounds and could be view as a starting point for SBDD (Shalev-Benami et al., 2017) (Merk et al., 2016) (Gao et al., 2016) (Banerjee et al., 2016) (Bartesaghi et al., 2015) (Wong et al., 2017). The limits in resolution are often inherent to the sample itself, arising from heterogeneity, flexibility, preferred orientation, denaturation, aggregation, etc., and can sometimes be somewhat overcome by increasing the number of particles contributing to the map: this approach though requires more time spent at the microscope, with data collections spanning days to weeks. As timing and fast throughput are essential to allow for structural information to make an impact in a drug discovery process, in our opinion it will be critical to improve the efficiency of data collection at the microscope by an order of magnitude for cryoEM to be competitive in the later stages of SBDD. These improvements may come from a variety of technical advances, including new cameras with higher speeds and larger pixel arrays, better stages that are more stable and can be used to rapidly move from one target to the next with no delay due to thermal instabilities or hysteresis, and better automation software at every stage of data collection (from sample loading and exchanging, to calibration, target queuing and image acquisition). This can limit the need for slow steps and guide the optimization of data collection protocols, similarly to what is done with synchrotron automated data collections (Winter and McAuley, 2011).
Figure 4:
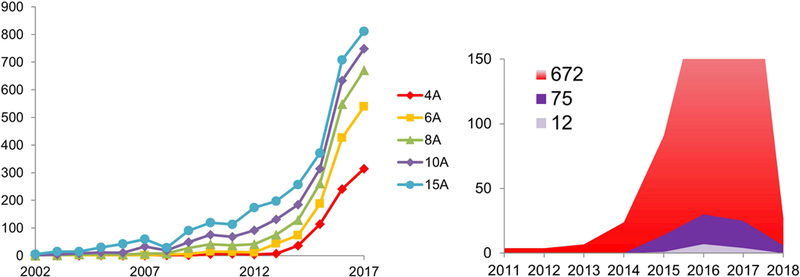
Analysis of EM maps deposited in the EMDB. Left: trend of released maps achieving given resolution levels (as of December 2017). Right: as of December 2017, 645 maps at a resolution of 4 Å or better were deposited in the EMDB (red shaded area). These included 69 maps better than 3 Å and 12 at a resolution greater than 2.5 Å. During January 2018, 27 maps better than 4 Å and 6 better than 3.0 Å were added to the EMDB.
Data Processing, structure determination and structure validation
With a speedup in data collection, the workflow bottleneck will be moved to the processing phase. While most of the best used software packages are now semi automated (e.g. EMAN (Tang et al., 2007), Relion (Scheres, 2012), CryoSparc (Punjani et al., 2017), cisTEM (Grant et al., 2018), Scipion (de la Rosa-Trevín et al., 2016)) there are none so far that can take in images and produce a 3D map without the help of human intervention. Most of the intervention is required for selecting particles from the individual images and in assessing the degree of heterogeneity in 2D classes and 3D maps. While most particle picking is done automatically, it normally initially requires some input form the user in terms of optimizing parameters or providing templates. Many users also sort out “good” from “bad” particles by doing several rounds of 2D alignment and classification and rejecting classes that appear to be junk or otherwise unsuited for further processing. The 3D reconstruction also requires user input in terms of deciding how many 3D classes to determine and which can be carried forward to refinement. It is our belief that many of these steps that currently require human input will be replaced in the near future with deep learning methods. For example several “general” particle pickers are under development, see for example (Wang et al., 2016a), and it seems feasible that similar methods may be developed to accept or reject 2D and 3D classes. Once this is achieved, then a truly fully automated pipeline can be developed that can provide feedback during data collection and drive decisions as to the optimal number of images that should be acquired for any given sample (which may vary from few to many thousands).
For SBDD applications however the maps are not the end-point, but rather the experimental set onto which the pursued atomic structure needs to be built, with enough precision and accuracy that allow the model to be used in all subsequent steps of drug design. Automated model building, refinement and validation then become as important as obtaining good maps in the first place. Ideally, the resulting map (and 3D model(s), when available) can be input to a density-to-structure pipeline that would automatically carry out structure solution, ligand fitting and model refinement. In crystallography, software packages such as Phenix.refine (Afonine et al., 2012), ARP/wARP (Lamzin et al., 2012) and GlobalPhasing PipeDream (Sharff et al., 2011) are designed to link and automate (in different ways and to a different extent, depending on the package) a structure solution stage, structure refinement step(s) and automated ligand fitting. While these packages are still mostly designed for use with x-ray diffraction data, modifications have been implemented to allow working with EM maps (for example CCPEM (Wood et al., 2015) (Burnley et al., 2017), Phenix CryoFit (Kirmizialtin et al., 2015) or Rosetta (Wang et al., 2016b)). Next steps should be linking these EM-specific variations into a single pipeline, which would ideally require minimal user input, and output a final model ready to be used for further drug design. The last step of the process (validation of the resulting maps and coordinates) should not be underestimated: the quality of the model will affect the quality of the results obtained using that model as a starting point for any of the computational approaches (virtual screening, docking, ligand design) used in the SBDD process. The need for validations processes and standards for both maps and coordinates is well understood within the community: in 2010 the Electron Microscopy Validation Task Force (EM VTF) met at Rutgers University to consider possible validation standards for 3D maps and models produced by electron microscopy reconstruction methods, and produced a series of recommendations summarized in (Henderson et al., 2012). Since then, two map validation servers and a 3DEM validation pipeline, which produces an EM validation report similar to the validation reports produced by wwPDB for structures from X-ray crystallography, have been introduced and are available through the wwPDB and the EMDB (Patwardhan and Lawson, 2016). Inclusion of the validation report when submitting manuscripts for review should become a standard procedure.
Industrial interest and some potentially applicable results
A clear confirmation of the transition of cryoEM to a mainstream structural biology tool, and of its potential for facilitating SBDD, comes from the fact that many pharmaceutical and biotech companies have embraced this new technique, and are quickly setting up new internal facilities and/or collaborations with existing academic laboratories or companies that specialize in the technique (Peplow, 2017). A few companies historically focused on providing crystallography services are also moving into the field, and now offer cryoEM as a tool for structure determination. Two of the largest suppliers of crystallography tools are now offering a variety of cryoEM supplies, bringing both expertise and innovation to the field (see table 1 for a summary).
Table 1:
Commercial options for cryoEM imaging and supplies
| Name | Type | |
|---|---|---|
| Cambridge Pharmaceutical cryoEM consortium | Consortium | https://www2.mrc-lmb.cam.ac.uk/cambridge-pharmaceutical-cryo-em-consortium/ |
| NanoImaging Services | CRO | https://www.nanoimagingservices.com/ |
| TetraGenetics | CRO | http://www.tetragenetics.com/drug-discovery-technology/structure-based-design/ |
| Novalix | CRO | http://www.novalix-pharma.com/ |
| Creative Biostructure | CRO | https://www.creative-biostructure.com/cryo-em-services 4.htm |
| MiTeGen | Supplier | https://www.mitegen.com/product-category/cryo-em/ |
| Molecular Dimensions | Supplier | https://www.moleculardimensions.com/products/c503-Cryo-Electron-Microscopy |
| SubAngstrom | Supplier | www.subangstrom.com |
Scapin et al., present their perspective of the possible application of cryo-electron microscopy (cryoEM) to structure-based drug design (SBDD) approach. They discuss SBDD requirements, cryoEM current state, and the challenges that need to be overcome for this technique to reach its full potential in facilitating the process of drug discovery.
Most papers of possible interest for drug development reveal previously unknown structures of difficult targets that have the potential to facilitate further drug design, or of interesting target-drug complexes. For example, Baretić and coworkers (Baretić et al., 2017) report the structure of human ataxia telangiectasia mutated (ATM) protein, a large kinase involved in DNA repair and cancer: the protein is shown in two conformations (open and closed) and provides the first glimpse into the kinase structure; Fitzpatrick et al (Fitzpatrick et al., 2017) report the first structure of Tau filaments from Alzheimer’s patients’ brain, identifying a stretch of amino acids that could be the template for aggregation, and suggesting that targeting this peptide may provide a new way for the design of specific inhibitors of Tau aggregation; the structure of TRPV1 in lipid nanodiscs (Gao et al., 2016) allows for the first visualization of the interactions of lipids with transmembrane helices, and the identification of the binding site for two known drugs, capsazepine and resiniferatoxin. Scapin et al very recently reported the first structure of insulin bound to its receptor (Scapin et al., 2018)), a very well established target for the treatment of diabetes. Other similar examples have been summarized elsewhere (Subramaniam et al., 2016) (Merk et al., 2016).
There still are very few reports of cryoEM driven SBDD (optimization of a drug based on structural information). A single exception is the paper by Wong and coworkers (Wong et al., 2017), in which they identified the antimalarial mefloquine (MFQ) binding mode to Plasmodium falciparum 80s ribosome, and used the information to develop MFQ derivatives with improved potency (Figure 5). Nevertheless, the outlook is promising: the field is growing steadily, and both hardware and software are still improving. If sample preparation can become more automated, and we can get one order of magnitude improvement in the efficiency of data collection and analysis, cryo-EM could become a very useful technique for visualizing protein-ligand complexes and accelerate the process of drug discovery.
Figure 5:
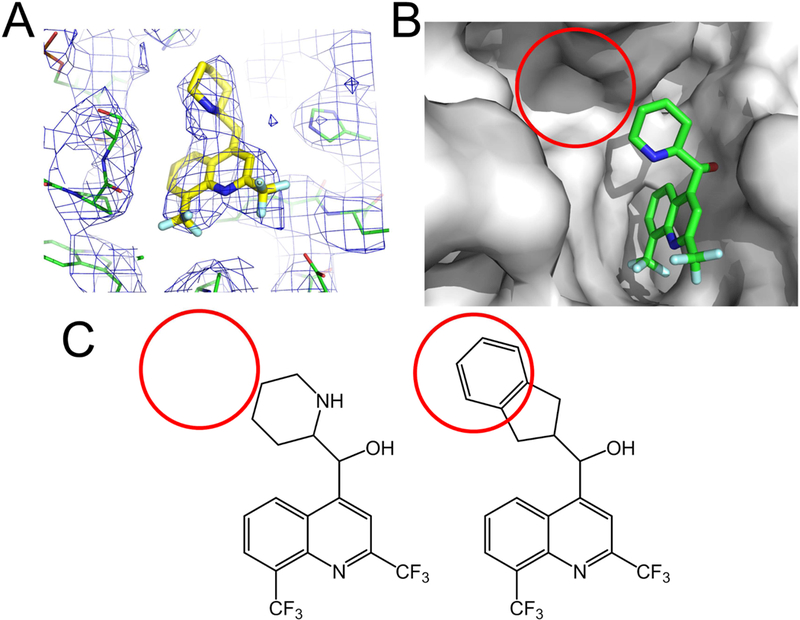
Using cryoEM structures to guide structure based drug design. A) Density for the antimalaria mefloquine as visualized in the 3.2 Å cryoEM map (Panel A and B were generated with PyMol, from map EMD-8576 and PDB 5UMD). B) The structure revealed an empty pocket within the ligand binding site (red circle). C) Structure guided design of modified mefloquine, with the piperidine replaced by a larger substituents, resulted in derivatives with a ~2-fold fold potency enhancement towards the parasite (Wong, et al., 2017).
Acknowledgements
We thank Edward Eng, Laura Kim and other members of the Simons Electron Microscopy Center at the New York Structural Biology Center for the Aldolase data and figure. Some of the work presented here was conducted at the National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the NIH National Institute of General Medical Sciences (GM103310) and the Simons Foundation (349247).
REFERENCES
- Afonine P, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams P (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr, Volume 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison J and P.Rout M (2012). The Yeast Nuclear Pore Complex and Transport Through It. Genetics, 190(3), 855–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, A (2009). The process of structre based drug design. Chemistry & Biology, Volume 10, 787–797. [DOI] [PubMed] [Google Scholar]
- Arenz S, Bock LV, Graf G, Innis CA, Beckmann R, Grubmϋller H, Vaiana AC, Wilson DN (2016). A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat Commun, 7, 12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. (2016). 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science, Volume 351, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Pollard HK, Fisher DI, Johnson CM, Santhanam B, Truman CM, Kouba T, Fersht AR, Phillips C, and Williams RL (2017). Structures of closed and open conformations of dimeric human ATM. Sci Adv, 3, e1700933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Matthies D, Banerjee S, Merk A, and Subramaniam S (2014). Structure of β- galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy. Proc Natl Acad Sci U S A, 111, 11709–11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, and Subramaniam S (2015). 2.2 A resolution cryo-EM structure of beta-galactosidase in complex with a cell-permeant inhibitor. Science, Volume 348, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, Moeller A, and Tampe R (2017). Structure of the human MHC-I peptide-loading complex. Nature, 551, 525–528. [DOI] [PubMed] [Google Scholar]
- Burnley TCM, Palmer C, Winn M (2017). Recent developments in the CCP-EM software suite. Acta Cryst, D73, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousido-Siah A,Petrova T, Hazemann I, Mitschler A, Ruiz FX, Howard E, Ginell S, Atmanene C, Van Dorsselaer A, Sanglier-Cianferani S, et al. (2012). Crystal packing modifies ligand binding affinity: the case of aldose reductase. Proteins, 80, 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandey V Wei, H, Zhang, Z, Tan, YZ, Acharya, P, Eng, ET, Rice, WJ, Kahn, PA, Potter, CS, Carragher, B (2018). Spotiton: New Features and Applications. J Struct Biol. 202, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Tegunov D and Baumeister W (2017). Using the Volta phase plate with defocus for cryo-EM single particle analysis. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R and Baker D (2008). Macromolecular modeling with rosetta. Annu Rev Biochem, 77, 363–382. [DOI] [PubMed] [Google Scholar]
- de la Rosa-Trevin J, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, G0mez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, e. al. (2016). Scipion web tools: Easy to use cryo-EM image processing over the web. J Struct Biol, 195, 93–99. [DOI] [PubMed] [Google Scholar]
- Dorsey B, Levin RB, McDaniel SL, Vacca JP, Guare JP, Darke PL, Zugay JA, Emini EA, and Schleif WA (1994). L-735,524: The Design of a Potent and Orally Bioavailable HIV Protease Inhibitor. J. Med. Chem 37, 3443–3451. [DOI] [PubMed] [Google Scholar]
- Erickson J, Neidhart DJ, VanDrie J, Kempf DJ, Wang XC, Norbeck DW, Plattner JJ, Rittenhouse JW, Turon M, et al. (1990). Design, activity, and 2.8 A crystal structure of a C2 symmetric inhibitor complexed to HIV-1 protease. Science, 249, 527–533. [DOI] [PubMed] [Google Scholar]
- Fan X, Zhao L, Liu C, Zhang JC, Fan K, Yan X, Peng HL, Lei J, and Wang HW (2017). Near- Atomic Resolution Structure Determination in Over-Focus with Volta Phase Plate by Cs-Corrected Cryo- EM. Structure, 25, 1623–1630. [DOI] [PubMed] [Google Scholar]
- Faruqi A, Cattermole DM, Henderson R, Mikulec B, and Raeburn C (2003). Evaluation of a hybrid pixel detector for electron microscopy. Ultramicroscopy, 94, 263–276. [DOI] [PubMed] [Google Scholar]
- Feng X, Fu Z, Kaledhonkar S, Jia Y, Shah B, Jin A, Liu Z, Sun M, Chen B, Grassucci RA, et al. (2017). A Fast and Effective Microfluidic Spraying-Plunging Method for High-Resolution Single-Particle Cryo-EM. Structure, 25, 663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, et al. (2016). Cryo-EM Analysis of the Conformational Landscape of Human P- glycoprotein (ABCB1) During its Catalytic Cycle. Mol Pharmacol, 90, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J (2017). Advances in the field of single-particle cryo-electron microscopy over the last decade. Nature Protocols, 12, 209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D and Cheng Y (2016). TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature, 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser R and Han B (2017). Opinion: hazards faced by macromolecules when confined to thin aqueous films. Biophys Rep, 3, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife, 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, She J, Zeng W, Chen Q, Bai XC, and Jiang Y (2017a). Structures of the calcium-activated, non-selective cation channel TRPM4. Nature, 552, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, et al. (2017b). Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell, 171, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure, 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzik M, Wu M and Lander G (2017). Achieving better-than-3- angstrom resolution by single-particle cryo-EM at 200 keV. Nat. Methods, 14, 1075–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi M, Herzik MA, Wie J, Suo Y, Borschel WF, Ren D, G.C. Lander, and Lee SY (2017). Cryo- electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature, 550, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain T, Sheehan P, Crum J, Carragher B, and Potter CS (2012). Spotiton: a prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J Struct Biol, 179, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhoti H, Cleasby A, Verdonk M and Williams G (2007). Fragment-based screening using X-ray crystallography and NMR spectroscopy. Curr Opin Chem Biol, 11, 485–489. [DOI] [PubMed] [Google Scholar]
- Jukič M, Konc J, Gobec S and Janežič D (2017). Identification of Conserved Water Sites in Protein Structures for Drug Design. J Chem Inf Model, 57, 3094–3103. [DOI] [PubMed] [Google Scholar]
- Kimanius D, Forsberg B, Scheres S and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W (2014). The resolution revolution. Science, 343, 1443–1444. [DOI] [PubMed] [Google Scholar]
- Lamzin V, Perrakis A and Wilson K (2012). ARP/wARP - automated model building and refinement In: Arnold E, Himmel D & Rossmann M, eds. International Tables for Crystallography. Volume F: Crystallography of biological macromolecules. Dordrecht: Kluwer Academic Publishers, 525–528. [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. (2009). Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol, 166, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, and Cheng Y (2013). Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods, 10, 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wu J, Dong Y, Chen S, Sun S, Ma YB, Ouyang Q, Finley D, Kirschner MW, and Mao Y (2017). Conformational Landscape of the p28-Bound Human Proteasome Regulatory Particle. Mol Cell, 67, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizialtin S, Loerke J, Behrmann E, Spahn CMT, and Sanbonmatsu KY (2015). Using Molecular Simulation to Model High-Resolution Cryo-EM Reconstructions. Methods Enzymol, 558, 497–514. [DOI] [PubMed] [Google Scholar]
- McGoldrick LL, Singh AK, Saotome K, Yelshanskaya MV, Twomey EC, Grassucci RA, and Sobolevsky AI (2018). Opening of the human epithelial calcium channel TRPV6. Nature 553, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, and Subramaniam S (2016). Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell, 165, 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Method of the Year, (2016). Nature Methods, 13(1). [DOI] [PubMed] [Google Scholar]
- Meyerson J, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, and Subramaniam S (2014). Structural mechanism of glutamate receptor activation and desensitization. Nature, 514, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G and Dodson E (1997). Simplified error estimation a la Cruickshank in macromolecular crystallography. CCP4 Newsletter, January. [Google Scholar]
- Nogales E and Scheres S (2015). Cryo-EM: A Unique Tool for the Visualization of Macromolecular Complexity. Mol Cell, 58, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan A and Lawson C (2016). Databases and Archiving for CryoEM. Methods Enzymol, 579, 393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peplow M (2017). Cryo-electron microscopy makes waves in pharma labs. Nature Reviews Drug Discovery 16, 815–817. [DOI] [PubMed] [Google Scholar]
- Prongay A, Guo Z, Yao N, Pichardo J, Fischmann T, Strickland C, Myers J Jr, Weber PC, Beyer,
- B.M., Ingram R, et al. (2007). Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1- (cyclobutylmethyl)-2,3-dioxopropyl]-3- [2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl- 1-oxobutyl]- 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. J Med Chem, 50, 2310–2318. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein J, Fleet D and Brubaker M (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods, 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Razinkov I, Dandey V, Wei H, Zhang Z, Melnekoff D, Rice WJ, Wigge C, Potter CS, and Carragher B (2016). A new method for vitrifying samples for cryoEM. J Struct Biol, 195, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, N.,Martin, J.A., Kinchington, D., Broadhurst, A.V., Craig, J.C., Duncan, I.B., Galpin, S.A., Handa,
- B K, Kay J, Kröhn A, et al. (1990). Rational design of peptide-based HIV proteinase inhibitors. Science, 248, 358–361. [DOI] [PubMed] [Google Scholar]
- Roh S, Hryc CF, Jeong HH, Fei X, Jakana J, Lorimer GH, and Chiu W (2017). Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc Natl Acad Sci U S A, 114, 8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin G (2015). Structural Chemistry and Molecular Modeling in the Design of DPP4 Inhibitors In: Multifaceted Roles of Crystallography in Modern Drug Discovery. Dordrecht: Springer. [Google Scholar]
- Scapin G,Dandey VP, Zhang Z, Prosise W, Hruza A, Kelly T, Mayhood T, Strickland C, Potter CS , and Carragher B (2018). Structure of the Insulin Receptor-Insulin complex by single particle cryoEM analysis. Nature, 556, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin G, Patel S, Patel V, Kennedy B, and Asante-Appiah E (2001). The structure of apo protein- tyrosine phosphatase 1B C215S mutant: more than just an S --> O change. Protein Sci, 10, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S (2012). A Bayesian View on Cryo-EM Structure Determination. J Mol Biol, 415, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev-Benami M, Zhang Y, Rozenberg H, Nobe Y, Taoka M, Matzov D, Zimmerman E, Bashan A, Isobe T, Jaffe CL, Yonath A, and Skiniotis G (2017). Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat Commun, 8, 1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharff A, Keller P, Vonrhein C, Smart O, Womack T, Flensburg C, Paciorek C and Bricogne G (2011). Pipedream, version 1.2.1, Global Phasing Ltd, Cambridge, United Kingdom [Google Scholar]
- Shen P, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE, and Cao E (2016). The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell, 167, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J Koteiche HA, McDonald ET, Fox TL, Stewart PL, and McHaourab HS (2013). Cryoelectron microscopy analysis of small heat shock protein 16.5 (Hsp16.5) complexes with T4 lysozyme reveals the structural basis of multimode binding. J Biol Chem, 288, 4819–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śledź P and Caflisch A (2017). Protein structure-based drug design: from docking to molecular dynamics. Curr Opin Struct Biol, 48, 93–102. [DOI] [PubMed] [Google Scholar]
- Stamford A, Scott JD, Li SW, Babu S, Tadesse D, Hunter R, Wu Y, Misiaszek J, Cumming JN, Gilbert EJ, et al. (2012). Discovery of an Orally Available, Brain Penetrant BACE1 Inhibitor that Affords Robust CNS Aβ Reduction. ACS Med Chem Lett, 3, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Earl LA, Falconieri V, Milne JL, and Egelman EH (2016). Resolution advances in cryo-EM enable application to drug discovery. Curr Opin Struct Biol, 41, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated Molecular Microscopy: The New Leginon System. J Struct Biol, 151, 41–50. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol, 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Tan Y, Baldwin PR, Davis JH, Williamson JR, Potter CS, Carragher B, and Lyumkis D (2017). Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods, 14, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordai H, Leveles I and Hegedus T (2017). Molecular dynamics of the cryo-EM CFTR structure. Biochem Biophys Res Comm, 491, 986–993. [DOI] [PubMed] [Google Scholar]
- Trabuco G, Villa E, Mitra K, Frank J, and Schulten K (2008). Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure, 16, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Appen A and Beck M (2016). Structure Determination of the Nuclear Pore Complex with ThreeDimensional Cryo electron Microscopy. J Mol Biol, 428 2001–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Gong H, Liu G, Li M, Yan C, Xia T, Li X, and Zeng J (2016). DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM. J Struct Biol, 195, 325–336. [DOI] [PubMed] [Google Scholar]
- Wang L, Berne B and Friesner R, 2011. Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci USA, 108, 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Song Y, Barad BA, Cheng Y, Fraser JS, and DiMaio F (2016). Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife, 5, e17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Dandey VP, Zhang Z, Raczkowski A, Rice WJ, Carragher B, and Potter CS (2018). Optimizing “Self-Wicking” Nanowire Grids. J Struct Biol, 202, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G and McAuley K (2011). Automated data collection for macromolecular crystallography. Methods, 55, 81–93. [DOI] [PubMed] [Google Scholar]
- Wong W, Bai XC, Sleebs BE, Triglia T, Brown A, Thompson JK, Jackson KE, Hanssen E, Marapana DS, Fernandez IS, et al. (2017). Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat Microbiol, 2, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C, Burnley T, Patwardhan A, Scheres S, Topf M, Roseman A, and Winn M (2015). Collaborative Computational Project for Electron cryo-Microscopy. Acta Cryst, D71, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, and Zhang P (2013). Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature, 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]