

Abstract
Purpose
Radiation is used extensively to treat localized cancer, but improved understanding of its effects on the immune system have increased interest in its potential systemic (abscopal) effects, particularly in combination with checkpoint inhibitors such as anti-PD1. The majority of patients either do not respond or develop resistance to monotherapy over time. Here, we investigated the efficacy of OX40 (CD134) stimulation as an alternative immunotherapeutic approach in combination with radiotherapy (XRT) in a murine model of anti-PD1-resistant lung tumors.
Experimental Design
We established a bilateral tumor model in 129Sv/Ev mice using an anti-PD1 resistant lung tumor cell line. Primary tumors were treated with intratumoral injection of an OX40 agonist antibody, given as adjuvant therapy after XRT (36 Gy in three 12-Gy fractions), while secondary tumors were left untreated to investigate abscopal outcomes.
Results
The combination of XRT followed by OX40 stimulation effectively inhibited local and systemic antitumor growth, limited lung metastases, and improved survival rates. This treatment regimen augmented CD4+ and CD8+ T cell expansion. XRT induced the expression of OX40 on T cells in tumors and spleens and increased the percentages of splenic CD103+ dendritic cells.
Conclusion
Our data extends the benefits of radiation to systemic disease control, especially when combined with anti-OX40 agonist to promote immunologically mediated abscopal effects. Moreover, this study provides a rational treatment approach and sequence to overcome anti-PD1 resistant poorly immunogenic tumors.
Keywords: PD1 resistance, radiotherapy, immunotherapy, OX40, cancer
Introduction
Lung cancer is the leading cause of cancer death worldwide. The non-small cell subtype is the most common, with about 50% of non-small cell cases being adenocarcinomas (1). Radiation therapy (XRT) is an important component of treatment for metastatic lung cancer, but the long-term response rates after the current standard treatment of XRT with chemotherapy remain very low (2), leading to the search for more effective treatment strategies.
One such strategy, combining XRT with immunotherapy, is the subject of intense interest and investigation. The rationale for such combinations is that XRT has known effects on the immune system (both stimulatory and suppressive) and the stimulatory effects of XRT, particularly on T cells, may enhance antitumor immunity in both the local tumor microenvironment and systemically. To date, the best-studied forms of immunotherapy for this purpose have been immune checkpoint inhibitors such as antibodies to PD1/PDL1 and CTLA-4 (3,4). Although checkpoint inhibitors have shown promise for both local and systemic disease control in several preclinical and clinical studies (5), the reality is that few patients respond to such therapy, and even those who initially respond can later develop treatment resistance (6).
In an attempt to dissect the mechanisms underlying the development of resistance to checkpoint inhibitors, we recently generated a syngeneic mouse model of lung adenocarcinoma that does not respond to programmed cell death protein 1 (PD1) checkpoint blockade (7). In the current study, we used this model to test the effects of an alternative form of immunotherapy, OX40 costimulation in combination with XRT, on anti-PD1-resistant tumors. OX40 (CD134) is a type I membrane glycoprotein in the tumor necrosis factor (TNF) receptor superfamily (8). The OX40 receptor is a costimulatory molecule expressed mostly on activated effector T cells and naive regulatory T cells (Tregs). The ligand for OX40 (OX40L, CD252) is expressed on activated professional antigen-presenting cells such as dendritic cells (DCs), macrophages, and B cells (9,10). OX40 signaling on CD4+ T cells activates the NF-κB pathway and upregulates anti-apoptotic molecules such as the Bcl-2 family, which participate in T cell expansion, activation, memory generation, and cytokine production, culminating in an antitumor effect (11,12). OX40 expression on murine T cells is induced between 24 to 96 hours after antigen recognition. Murine Tregs also express OX40, which can inhibit effector T cells through the secretion of cytokines such as transforming growth factor-beta (TGF-β) and interleukins 10 and 13 (10,11,13).
For the current study, we hypothesized that XRT would induce local and systemic (abscopal) antitumor responses when combined with an agonist antibody to OX40 (α-OX40), which would enhance the infiltration of tumor-specific T cells and further induce OX40 expression in the tumor microenvironment. Moreover, because radiation-induced OX40L expression can expand costimulatory signaling through OX40 on T cells (14,15), this may further augment the antitumor effects observed both locally and systemically. The upregulation of this costimulatory signaling evades other radiation-induced regulatory immune responses, such as suppression by myeloid-derived suppressor cells and Tregs, while upregulating effector CD4+ T cells and CD103+ antigen-presenting DCs (16). Herein, we explored these mechanisms in our anti-PD1 resistant tumor model and detected significant upregulation of OX40 induced by XRT, which provided the rationale of adding α-OX40 agonist in a sequential fashion to generate robust abscopal responses.
Materials and Methods
Mice
The female 129Sv/Ev mice used in this study were 8 to 12 weeks of age. The strain was originally purchased from Taconic and bred in-house. All mice were housed at the Experimental Radiation Oncology mouse colony facility at The University of Texas MD Anderson Cancer Center according to Animal Care IACUC guidelines.
Tumor cell lines
The 344SQ tumor cell line is a murine metastatic lung cancer line from which our anti-PD1-resistant cell line was generated and derived as previously described (7). The cell lines were cultured in complete medium (RPMI-1640 supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated fetal bovine serum) and incubated at 37°C in 5% CO2.
Tumor establishment and treatments
Primary tumors were established by subcutaneous injection of 0.5 × 106 anti-PD1-resistant 344SQ cells into the right hind legs of 129Sv/Ev syngeneic mice. For the abscopal experiments, secondary tumors were established by subcutaneous injection of 0.1 × 106 anti-PD1-resistant 344SQ cells into the left hind legs of mice 4 days after injection of the primary tumors. Tumors were measured with calipers three times per week and recorded as tumor volume (in mm3) = width2 × length / 2. Mice were euthanized when tumors became ulcerated or reached a maximum size of 1500 mm3. For local irradiation of primary tumors, a Cesium source was used where mice were placed on a jig, and their entire body shielded except for the leg area to receive the designated radiation dose.
To identify the optimal radiation doses for combination XRT and α-OX40 treatment, tumor-bearing mice were injected intratumorally with α-OX40 mAb (100 μg/injection) or rat IgG isotype control (100 μg/injection) on days 7, 11, 15, 19, 23 after tumor inoculation. Radiation (XRT) was administered starting on day 12 to a total dose of 24 Gy in two 12-Gy fractions, 15 Gy in five 3-Gy fractions, 15 Gy in three 5-Gy fractions, or 36 Gy in three 12-Gy fractions. Fractions were given once per day and tumor volumes were monitored for 35 days. To optimize the timing/sequencing of α-OX40 with XRT, tumor-bearing mice were treated as follows: (a) IgG control (200 μg/injection); (b) XRT alone; (c) α-OX40 alone (3 doses 200 μg/injection), (d) α-OX40 before XRT (3 induction doses at 200 μg/injection); (e) α-OX40 during XRT (3 concurrent doses); or (f) α-OX40 after XRT (3 adjuvant doses). Tumor growth was monitored for 25 days. Finally, to explore the systemic (abscopal) effects of XRT + adjuvant α-OX40 treatment, primary tumors were intratumorally injected with α-OX40 (200 μg/injection) at 48 hours after XRT (36 Gy total in three 12-Gy fractions), and mice were monitored for over 50 days. Mice in control group were treated intratumorally with IgG sham in all experiments. Mice were euthanized as they expired and lungs were harvested for metastases counts after staining with Bouin’s fixative solution (Polysciences Inc., cat. #16045-1).
Monoclonal antibodies
The α-OX40 mAb (Clone OX-86; cat. #BE0031) and control rat IgG2a mAb (Clone 2A3; cat. #BE0089) were provided by GlaxoSmithKline. For flow cytometry purposes, fluorochrome-conjugated anti-CD3 (#100353), anti-CD4 (#100406), anti-CD8 (#100734), anti-CD45 (#103126), anti-CD11b (#101226), anti-CD11c (#117310), anti-CD103 (#121426), anti-F4/80 (#123108), anti-Ly6C (#128018), anti-Ly6G (#127616), anti-OX40 (#119414), and anti-OX40L (#108805) antibodies were purchased from BioLegend.
Tissue processing and flow cytometry
Tissue harvesting and processing were conducted as previously described (7). In short, to obtain single-cell suspensions, tumor tissues were digested with 250 μg/ml of Liberase (Roche) and incubated for 45 minutes at 37°C with shaking. Fetal bovine serum was added and samples were filtered followed by Histopaque 1077 gradient isolation of TILs. Cells were then blocked with anti-CD16/CD32 before being stained for flow cytometry. Samples were analyzed with an LSR II flow cytometer and FlowJo software.
Statistical analysis
All statistical analyses were done with Graph Pad Prism 6 software. Student’s t-tests were used to compare differences between individual groups. Mouse survival rates were analyzed by using the Kaplan–Meier method and compared with log-rank tests. A two-way ANOVA was conducted to compare tumor growth curves between groups. Statistical significance was accepted at P value ≤ 0.05.
Results
Optimizing the XRT dose in combination with anti-OX40
To identify the optimal dose of XRT to be used in combination with α-OX40 immunotherapy, we used a preclinical model of lung adenocarcinoma that does not respond to anti-PD1 treatment (7) and tested several doses and fractionation schedules. To simulate several scenarios applied in the clinic, we tested three schedules: 15 Gy in three 5-Gy fractions (lower stereotactic dose); 15 Gy in five 3-Gy fractions (conventional dose); and 24 Gy in two 12-Gy fractions (higher stereotactic dose). Of these three schedules, 24 Gy in two 12-Gy fractions produced the greatest suppression of primary tumor growth. The control (IgG) and α-OX40-only conditions (five 100-μg intratumoral injections spaced 4 days apart) had no apparent effect on tumor growth, but the combination of XRT and concurrent intratumoral α-OX40 induced an additive effect on the primary (irradiated) tumor in 344SQ anti-PD1-resistant tumor-bearing mice (Fig. 1A, 1B, and 1C). Keeping in mind the possibility of minor systemic dissemination, intratumoral administration of α-OX40 was preferred to generate tumor-specific immune effects and avoid any potential systemic toxicity. This combination treatment also significantly improved survival rates (Fig. 1D) and reduced the number of lung metastases (Fig. 1E) in this mouse model. From this work, we established that the stereotactic-equivalent high dose radiation was the optimal dose to carry forward in our experiments.
Figure 1.
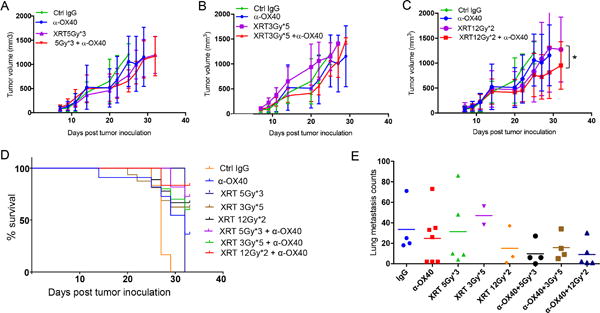
Higher radiation doses in combination with intratumoral α-OX40 control the growth of local (primary) tumors, improve survival rates, and produce fewer lung metastases. A-C, Mice (8 per group) were injected s.c. with 0.5 x 106 344SQ anti-PD1-resistant tumor cells into the right hind leg and given intratumoral injections of α-OX40 antibody (100 μg/injection) or rat IgG isotype control (100 μg/injection) on days 7, 11, 15, 19, and 23 after tumor inoculation. On day 7, the right hind legs of the mice were irradiated with one of the following schedules: A, three fractions of 5 Gy each; B, five fractions of 3 Gy each; or C, two fractions of 12 Gy each. 12Gyx2 + α-OX40 was superior to 5Gyx3 + α-OX40 group (two-way ANOVA, P<0.0001); 12Gyx2 was inferior to 12Gyx2 + α-OX40 group (two-way ANOVA, P=0.0453). D, Proportions of anti-PD1-resistant tumor-bearing mice surviving after the indicated treatments. E, lung metastasis counts enumerated at experimental endpoint (day 32) for different treatment groups as shown.
Optimizing the sequence of α-OX40 treatment and XRT
Next, to establish the optimal sequencing for combining XRT with α-OX40 in terms of suppressing the growth of primary tumors and the appearance of lung metastases, we tested two XRT schedules (three 12-Gy fractions for a total of 36 Gy, Fig. 2), which we previously found to overcome anti-PD1 resistance) (7); and (two 12-Gy fractions for a total of 24 Gy, Supplementary Fig. S1), which was the most effective in the previous experiment. The control group was given three 200-μg intratumoral injections of IgG; the OX40-only group, three 200-μg injections of α-OX40; and the XRT-only group, either 36 Gy (12 Gy × 3) or 24 Gy (12 Gy × 2) with IgG injections. The combination-group sequences for the 36-Gy XRT condition (Fig. 2) were induction α-OX40 followed by XRT; concurrent α-OX40 and XRT; and XRT followed by adjuvant α-OX40. The combination-group sequences for the 24-Gy XRT condition (Supplementary Fig. S1) were induction α-OX40 followed by XRT, and XRT followed by adjuvant α-OX40. A comparison of primary (irradiated) tumor growth indicated that the 36-Gy schedule was more effective than the 24-Gy schedule; moreover, the adjuvant combination (in which XRT was given first followed by α-OX40) was the most effective at suppressing the growth of these anti-PD1-resistant tumors in mice. Although the 24-Gy XRT dose seemed to suppress lung metastases relative to the control or α-OX40-only groups (Supplementary Fig. S1B), this effect was statistically significant only when the 36-Gy dose was used (Fig. 2B).
Figure 2.
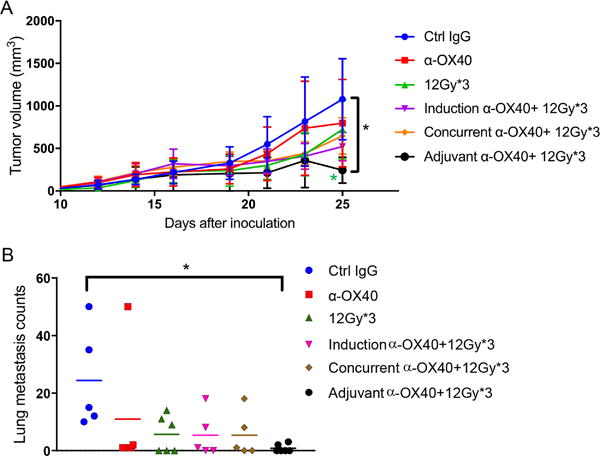
High-dose XRT followed by adjuvant α-OX40 most effectively suppressed local tumor growth and reduced lung metastases. A, Mice injected in the right hind legs with anti-PD1-resistant 344SQ tumors (n = 6 per group) were treated with intratumoral α-OX40 (three 200-μg injections) before XRT (induction); during XRT (concurrent); or after XRT (adjuvant). Tumor growth volumes were plotted. 12Gyx3 + α-OX40 was superior to 12Gyx2 + α-OX40 (Fig. 1C) (two-way ANOVA, P=0.0265); 12Gyx3 vs. 12Gyx3 + adjuvant α-OX40 (P= 0.0114 for the last point on day 25); α-OX40 vs. 12Gyx3 + adjuvant α-OX40 (two-way ANOVA, P=0.0005). B, Lungs from mice in 2A were harvested, stained with Bouin’s fixative solution, and numbers of metastases counted. *P ≤ 0.05.
Treatment with α-OX40 after radiation promotes abscopal effects in anti-PD1-resistant model
After identifying the optimal doses and sequence for XRT and α-OX40 in the previous experiments, we evaluated the abscopal effect of this treatment in the anti-PD1-resistant tumor-bearing mice. We found that five 200-μg injections of α-OX40 into the primary tumors, either alone or in combination with XRT (12 Gy × 3), demonstrated in vivo activity against 344SQ anti-PD1-resistant tumors, with both the primary (Fig. 3A) and secondary tumors (Fig. 3B) showing significant growth retardation. Further, among the combination-treated mice, survival rates up to 60 days were best for the XRT and adjuvant α-OX40 cohort (Fig. 3C). Compared to the control group, lung metastases were also diminished in the XRT plus α-OX40 group (Fig. 3D), despite the extra overall tumor burden from secondary tumors in the abscopal model.
Figure 3.
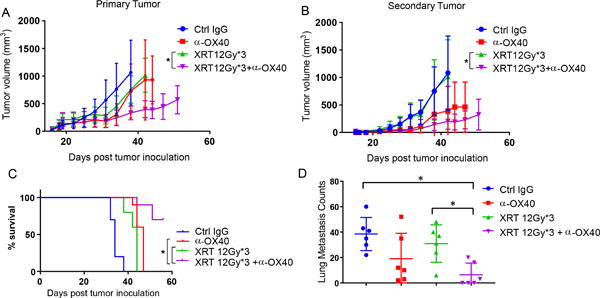
Radiation + α-OX40 promotes abscopal effects. Anti-PD1 resistant tumor cells were inoculated into 129Sv/Ev mice (n = 10 per group) bilaterally (right hind leg and left hind leg) 4 days apart. Primary tumors were irradiated 11 days after their inoculation whereas second tumors were left untreated. Intratumoral α-OX40 (total of 5 doses, 200 μg each) was administered to the primary tumor starting 2 days after last dose of XRT. A, Primary tumor measurements recorded upon XRT + α-OX40 treatment along with control groups as indicated. XRT vs. XRT + α-OX40 (two-way ANOVA, P< 0.0001). B, Secondary tumor measurements recorded as in A. XRT vs. XRT + α-OX40 (two-way ANOVA, P< 0.0001). C, Percent survivals of mice from A were recorded and graphed. XRT vs. XRT + α-OX40 (log rank test, P=0.0001); α-OX40 vs. XRT + α-OX40 (log rank test, P=0.0002). D, Lung metastases were enumerated upon sacrificing mice in their subsequent groups. XRT vs. XRT + α-OX40 (t-test, P=0.006).
Combination therapy enhances T cell infiltration into irradiated and abscopal tumors
In general, the presence of higher numbers of effector tumor-infiltrating lymphocytes (TILs) in the tumor is associated with better prognosis (17–19); indeed, antitumor immune responses depend on the proportions and functions of the cells in the tumor microenvironment (19). Thus we isolated and compared percentages of TILs obtained on day 29 from the primary and secondary (abscopal) tumors in our treatment groups. In the primary tumors, percentages of CD4+ T cells seemed to be increased (Fig. 4A and 4B) but did not reach significance. On the other hand, CD8+ T cells were upregulated significantly after combination therapy relative to tumors in the control or α-OX40-alone groups (Fig. 4A and 4C). In the secondary (unirradiated) tumors, both CD4+ and CD8+ T cells increased non-significantly after combination adjuvant α-OX40 therapy as compared to control or α-OX40-alone groups (Fig. 4D, 4E, and 4F).
Figure 4.
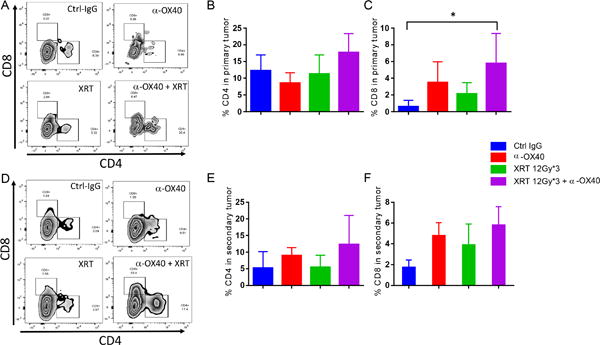
Radiation + α-OX40 upregulated CD4+ and CD8+ tumor-infiltrating T-cells in primary (irradiated) and second (non-irradiated) tumors. In the abscopal model, primary tumors A, B, C, (injected in the right hind leg and later irradiated) and secondary tumors D, E, F, (injected in the left leg and not irradiated) were harvested, processed, and analyzed by flow cytometry on day 29 for CD4+ and CD8+ T-cells after gating on CD45+ cells. *P ≤ 0.05 is considered significant.
Radiation induces the expression of OX40 receptor on CD4+ T cells
To further explore the mechanism(s) by which adjuvant α-OX40 treatment enhances tumor control with XRT, we analyzed the expression of OX40 on CD4+ and CD8+ T cells at 48 hours and 7 days after XRT (12 Gy × 3). In the 48 h analysis set, we analyzed CD4+ and CD8+ cells only in splenocytes, since immune cells are depleted at the irradiated site this soon after ablative XRT; immune cells usually start reappearing within the irradiated tumors 5-7 days later (20). Therefore we analyzed OX40 expression from splenocytes and TILs on day 7. At 48 h after XRT, percentages of OX40+ CD4 T cells (Fig. 5A) and the mean fluorescence intensity (MFI) of the OX40 receptor expression (Fig. 5B) were significantly elevated, but not as much in CD8 T cells (Supplementary Fig. S2A). Interestingly, the proportion of migratory CD103+ dendritic cells was also upregulated in the splenocytes at 48 h (Fig. 5C). By day 7, OX40 expression was slightly increased in splenic CD4+ T cells (Fig. 5D) and significantly upregulated in CD4+ T cells from TILs (Fig. 5E). Percentage of splenic CD103+ dendritic cells had equilibrated in the irradiated and non-irradiated groups by 7 days (Fig. 5F). These results suggest that there is an initial priming phase that involves CD103+ DCs in the spleen, followed by trafficking of immune cells to the tumor. Finally, there was a significant increase in the percentage of splenic neutrophils expressing OX40L 48 h after XRT treatment (Supplementary Fig. S2B). This could further contribute to OX40 signaling and T cell costimulation.
Figure 5.
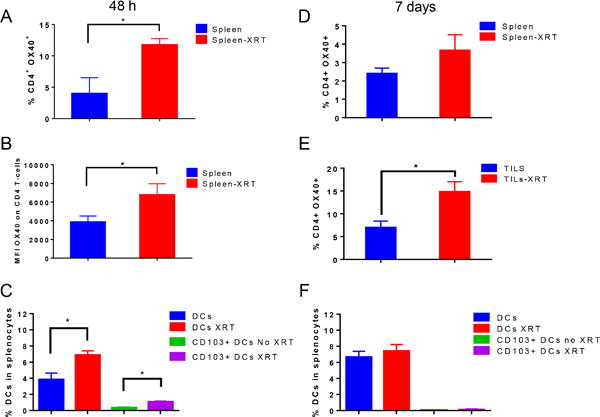
XRT induced the expression of OX40 and upregulated antigen-presenting dendritic cells. A, B, C, anti-PD1 resistant tumor-bearing mice were treated with XRT 12 Gy x 3 (n=3-5 mice per group) and splencoytes were analyzed by flow cytometry 48 h after the last dose of XRT. A, Percentages of CD4+ OX40+ T-cells B, mean fluorescence intensity (MFI) values of OX40 expression on CD4+ T-cells. C, Percentages of Dendritic cells (DCs) and CD103+ subset of DCs were reported. D, E, F, splenocytes and tumor-infiltrating lymphocytes (TILs) were analyzed by flow cytometry 7 days after XRT as in A, B, C. For OX40 expression, cells were first gated on CD45+, then on CD3+, followed by CD4+ T-cells. To analyze the dendritic cell percentages, cells were gated on CD45+, then CD3−CD11c+, or CD3−CD11c+ CD103+. *P ≤ 0.05 is considered significant.
Discussion
Target therapies based on blockade of immunosuppressive checkpoints receptors such as programmed death 1 (PD1), its ligand (PDL-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) have changed the management and treatment of cancer. Unfortunately, primary and developed resistance to these therapies remain to be a significant hurdle. Finding new alternative strategies that can reinstall and enhance therapeutic responses to immune checkpoint inhibitors are needed. Our results demonstrate that combining radiotherapy with an OX-40 agonist can improve tumor responses in an anti-PD1 resistant lung cancer model.
The view of radiotherapy as a curative, tumor cytotoxic tool is evolving, and here we showed that stereotactic-like high-dose XRT (36 Gy given in three 12-Gy fractions) triggered adaptive immune responses that mediated tumor regression. Specifically, we established that intratumoral injection of an OX40 agonist antibody, given after XRT in an adjuvant setting, effectively inhibited the growth of primary tumors, promoted systemic abscopal anti-tumor effects on secondary tumors and improved survival rates by around 60-days in the anti-PD1 resistant murine model. On the other hand, concurrent therapy with XRT + α-OX40 did not limit the tumor growth (Fig. 2a). XRT with adjuvant α-OX40 treatment also significantly reduced lung metastasis. This could be attributed to the observed increase in CD8+ T cell expansion and effector differentiation in the primary tumor.
Mechanistically, XRT prompted the expression of OX40 in CD4+ T cells in splenocytes and TILs 48 hours and 7 days after XRT, respectively. This was associated with an increase in CD103+ dendritic cells in the spleen (Fig. 5). These results provide the rationale that administering α-OX40 agonist after XRT and within a certain time frame is important for optimal outcomes. Our data showed that OX40 receptor levels went up by day 2 and were downregulated by day 7. Hence we started OX40 treatment by the second day after XRT. Previous studies highlighted the role of inhibiting histone deacetylases on the expression of TNF family members (21). Radiation as an epigenetic modulator may allow the OX40 and OX40L promoters to be acetylated and methylated, leading to a consequent increase in protein levels (15). Earlier reports have shown that OX40 stimulation followed by XRT may promote local control of tumors in Lewis Lung carcinoma (20,22). This was not the case in our anti-PD1 resistant lung cancer model. Our findings from the current study showed that adjuvant treatment has a better outcome in regard to tumor growth control and survival when compared with induction or concurrent therapies (Fig 2). Recent studies have also emphasized the importance of α-OX40 treatment timing and sequencing with other therapeutic agents (23,24). For example, in a colon cancer study, the authors demonstrated, similar to our results, that anti-OX40 given 1 day after XRT (20 Gy) had better therapeutic outcomes than anti-OX40 given 7 days prior to XRT (23). In another study highlighting the importance of sequencing, anti-OX40 given with anti-PD1 in a concurrent setting hindered the positive anti-tumor effects of anti-OX40 monotherapy (24) and induced T cell apoptosis in a TC-1 tumor model (25).
Radiation therapy has several immunologic advantages that can help restore the immune cycle (26). We previously demonstrated that radiation increased MHC-I expression driven by interferon production in tumors within 48 hours, leading to increased CD8+ T cell infiltration, priming, and activation (7). Here, we showed that stereotactic doses of radiation bridge innate and adaptive immune systems by increasing the CD103+ subset of dendritic cells (DCs) and the percentage of CD4+ T cells expressing the OX40 receptor. CD103+ DCs are identified by their expression of Batf3 transcription factor and the production of IL-12 cytokine, which favors Th1 responses (27,28) and exert an important role in priming CD8+ effector cells through cross-presentation. In our study, CD103+ DCs were upregulated significantly at 48 h after XRT but not at 7 days later (Fig 5), suggesting that XRT may initially release tumor-associated antigens that are processed and presented by these DCs, which in turn signal CD4+ and CD8+ T cells to activate and migrate to the tumor site 5 to 7 days later to elicit their anti-tumor functions.
Our study has significant translational value because 80% of non-small cell lung cancer patients are refractory to PD1 therapy, and 30% of those that initially respond develop resistance within 1 year of treatment (29). XRT with adjuvant anti-OX40 treatment provides a promising therapeutic alternative to these treatment-refractory patients.
Our study provides the basis and rationale for future studies combining XRT and anti-OX40 with other biologics to obliterate lung tumors. One such candidate is anti-CTLA-4 with the ability to deplete Tregs. In this way, an OX40 agonist would act solely on T-effectors and not on Tregs (30) further improving radiation-mediated abscopal benefits. Another potential candidate for combination therapy is an antibody that blocks PDL1. PDL1 is expressed on tumor cells as well as on myeloid-derived suppressor cells; its interaction with PD1 on T cells can result in T cell exhaustion and anergy. Therefore, a PDL1-depleting antibody may have a dual role in targeting both tumor cells and myeloid-derived suppressor cells (31). Collectively, our findings reveal the immunotherapeutic benefits of OX40 stimulation in an adjuvant setting after radiation to control both local and systemic tumors, offering a promising approach for treating anti-PD1-refractory patients.
Supplementary Material
Figure 6.
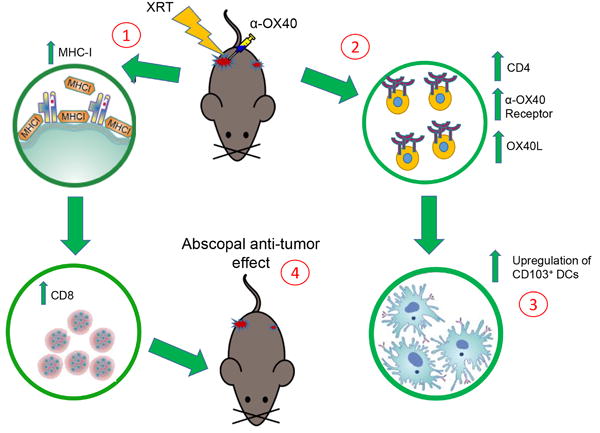
Schematic diagram summarizing the effects of XRT followed by adjuvant α-OX40. 1, XRT hits tumor cells releasing antigens and upregulating MHC-I molecules that further drive CD8+ T-cells. 2, XRT upregulates OX40 receptor on the surface of CD4+ T-cells that further augments the function of cytotoxic T cells. XRT also increases neutrophil percentages expressing the OX40L. 3, XRT upregulates systemic CD103+ dendritic cells (DCs) that cross-present antigens to CD8+ T-cells. 4, XRT in combination with α-OX40 to primary tumor promotes abscopal effects, controlling growth of second (non-irradiated) tumors.
Statement of Translational Relevance.
Immunotherapeutics have demonstrated potential to cure metastatic cancer. However, the majority of patients are resistant to monotherapy. Consequently, combinatorial strategies are under heavy investigation. Building evidence has highlighted potential for radiation and immunotherapy to reduce resistance rates and improve systemic responses. While checkpoint blockade agents such as anti-CTLA4 and anti-PD1 are well characterized, newer immunotherapeutic agents such as OX40 agonists are less understood. Currently, there are 10 clinical trials investigating the use of OX40 with other therapies. Prior to this study, the timing of treatments had not yet been elucidated. Herein we demonstrate that Radiotherapy limits tumor growth in part by prompting the release of tumor antigens, which stimulates T cells to express the OX40 receptor, where the antibody can further engage and promote systemic antitumor effects. By revealing mechanisms by which Radiation and OX40 therapies synergize, our study provides valuable insight into the optimal timing of treatment.
Acknowledgments
This work was supported by GlaxoSmithKline; the Mabuchi Research Fund; the Cancer Center Support (Core) Grant CA016672 to The University of Texas MD Anderson Cancer Center from the National Cancer Center, National Institutes of Health; Merck (LKR129022); an MD Anderson Knowledge Gap Award; a Doctors Cancer Foundation Grant; the Lung Cancer Research Foundation; the Family of M. Adnan Hamed; the Susan and Peter Goodwin Foundation; the Orr Family Foundation to MD Anderson Cancer Center’s Thoracic Radiation Oncology program; and the Wiegand Foundation. The authors thank Christine F. Wogan, MS, ELS, of MD Anderson’s Division of Radiation Oncology, for editorial contributions.
Footnotes
Author Contributions
SN, HBB, MAC and JWW designed the experiments and SN and HBB wrote the manuscript. SN performed and interpreted the experiments with HBB and MSC. SN and HBB collected research data with AIY and AL. HBB, SN, and JES analyzed the data. SN, HBB, JES, MAC, MSC, AC, JYC, QNN, DRG, AD, JVH and TRC contributed to editing the manuscript and graphical design. JWW, PH, and HLJ provided technical support and contributed to organizing progress reports. JWW, MAC, PH, NY, HJ, HBB and SN supervised the project and experimental plan.
Conflicts of interest: The authors have declared that no conflicts of interest exist.
References
- 1.Zhai L, Lauing KL, Chang AL, Dey M, Qian J, Cheng Y, et al. The role of IDO in brain tumor immunotherapy. Journal of neuro-oncology. 2014 doi: 10.1007/s11060-014-1687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart DJ. Tumor and host factors that may limit efficacy of chemotherapy in non-small cell and small cell lung cancer. Crit Rev Oncol Hematol. 2010;75(3):173–234. doi: 10.1016/j.critrevonc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seyedin SN, Schoenhals JE, Lee DA, Cortez MA, Wang X, Niknam S, et al. Strategies for combining immunotherapy with radiation for anticancer therapy. Immunotherapy. 2015 doi: 10.2217/imt.15.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishihara D, Pop L, Takeshima T, Iyengar P, Hannan R. Rationale and evidence to combine radiation therapy and immunotherapy for cancer treatment. Cancer Immunol Immunother. 2016 doi: 10.1007/s00262-016-1914-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahvi DA, Meyers JV, Tatar AJ, Contreras A, Suresh M, Leverson GE, et al. Ctla-4 blockade plus adoptive T-cell transfer promotes optimal melanoma immunity in mice. Journal of immunotherapy. 2015;38(2):54–61. doi: 10.1097/CJI.0000000000000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Molecular oncology. 2014;8(6):1132–9. doi: 10.1016/j.molonc.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zhang F, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokouchi H, Yamazaki K, Chamoto K, Kikuchi E, Shinagawa N, Oizumi S, et al. Anti-OX40 monoclonal antibody therapy in combination with radiotherapy results in therapeutic antitumor immunity to murine lung cancer. Cancer science. 2008;99(2):361–7. doi: 10.1111/j.1349-7006.2007.00664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan PY, Zang Y, Weber K, Meseck ML, Chen SH. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Molecular therapy : the journal of the American Society of Gene Therapy. 2002;6(4):528–36. doi: 10.1006/mthe.2002.0699. [DOI] [PubMed] [Google Scholar]
- 10.Linch SN, McNamara MJ, Redmond WL. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front Oncol. 2015;5:34. doi: 10.3389/fonc.2015.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. Journal of immunology. 2008;180(11):7240–8. doi: 10.4049/jimmunol.180.11.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73(24):7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological reviews. 2009;229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein MB, Garnett CT, Zhang H, Velcich A, Wattenberg MM, Gameiro SR, et al. Radiation-induced modulation of costimulatory and coinhibitory T-cell signaling molecules on human prostate carcinoma cells promotes productive antitumor immune interactions. Cancer biotherapy & radiopharmaceuticals. 2014;29(4):153–61. doi: 10.1089/cbr.2013.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumari A, Cacan E, Greer SF, Garnett-Benson C. Turning T cells on: epigenetically enhanced expression of effector T-cell costimulatory molecules on irradiated human tumor cells. Journal for immunotherapy of cancer. 2013;1:17. doi: 10.1186/2051-1426-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Cao X. Intratumoral dendritic cells in the anti-tumor immune response. Cellular & molecular immunology. 2015;12(4):387–90. doi: 10.1038/cmi.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mella M, Kauppila JH, Karihtala P, Lehenkari P, Jukkola-Vuorinen A, Soini Y, et al. Tumor infiltrating CD8 T lymphocyte count is independent of tumor TLR9 status in treatment naive triple negative breast cancer and renal cell carcinoma. Oncoimmunology. 2015;4(6):e1002726. doi: 10.1080/2162402X.2014.1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mihm MC, Jr, Mule JJ. Reflections on the Histopathology of Tumor-Infiltrating Lymphocytes in Melanoma and the Host Immune Response. Cancer immunology research. 2015;3(8):827–35. doi: 10.1158/2326-6066.CIR-15-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. Journal of immunotherapy. 2010;33(8):798–809. doi: 10.1097/CJI.0b013e3181ee7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutheesophon K, Nishimura N, Kobayashi Y, Furukawa Y, Kawano M, Itoh K, et al. Involvement of the tumor necrosis factor (TNF)/TNF receptor system in leukemic cell apoptosis induced by histone deacetylase inhibitor depsipeptide (FK228) Journal of cellular physiology. 2005;203(2):387–97. doi: 10.1002/jcp.20235. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien MA, Power DG, Clover AJ, Bird B, Soden DM, Forde PF. Local tumour ablative therapies: opportunities for maximising immune engagement and activation. Biochimica et biophysica acta. 2014;1846(2):510–23. doi: 10.1016/j.bbcan.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PloS one. 2016;11(6):e0157164. doi: 10.1371/journal.pone.0157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(20):6165–77. doi: 10.1158/1078-0432.CCR-16-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer immunology research. 2017;5(9):755–66. doi: 10.1158/2326-6066.CIR-17-0292. [DOI] [PubMed] [Google Scholar]
- 26.Weichselbaum RR, Liang H, Deng L, Fu YX. Radiotherapy and immunotherapy: a beneficial liaison? Nature reviews Clinical oncology. 2017;14(6):365–79. doi: 10.1038/nrclinonc.2016.211. [DOI] [PubMed] [Google Scholar]
- 27.Break TJ, Hoffman KW, Swamydas M, Lee CC, Lim JK, Lionakis MS. Batf3-dependent CD103(+) dendritic cell accumulation is dispensable for mucosal and systemic antifungal host defense. Virulence. 2016;7(7):826–35. doi: 10.1080/21505594.2016.1186324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Lopez M, Iborra S, Conde-Garrosa R, Sancho D. Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. European journal of immunology. 2015;45(1):119–29. doi: 10.1002/eji.201444651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clinical & translational immunology. 2016;5(4):e70. doi: 10.1038/cti.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. The Journal of clinical investigation. 2014;124(2):687–95. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.