

SUMMARY
Spt6 is a conserved factor that controls transcription and chromatin structure across the genome. Although Spt6 is viewed as an elongation factor, spt6 mutations in Saccharomyces cerevisiae allow elevated levels of transcripts from within coding regions, suggesting that Spt6 also controls initiation. To address the requirements for Spt6 in transcription and chromatin structure, we have combined four genome-wide approaches. Our results demonstrate that Spt6 represses transcription initiation at thousands of intragenic promoters. We characterize these intragenic promoters, and find sequence features conserved with genic promoters. Finally, we show that Spt6 also regulates transcription initiation at most genic promoters and propose a model of initiation-site competition to account for this. Together, our results demonstrate that Spt6 controls the fidelity of transcription initiation throughout the genome.
Keywords: Spt6, transcription start sites, intragenic promoters, chromatin structure
eTOC blurb
Doris et al. show that Spt6, a conserved transcription factor, is broadly required for the accuracy of transcription initiation by RNA polymerase II. In the absence of Spt6, transcription initiates at thousands of new sites, likely due to the presence of specific sequence features along with changes in chromatin structure.
Graphical Abstract
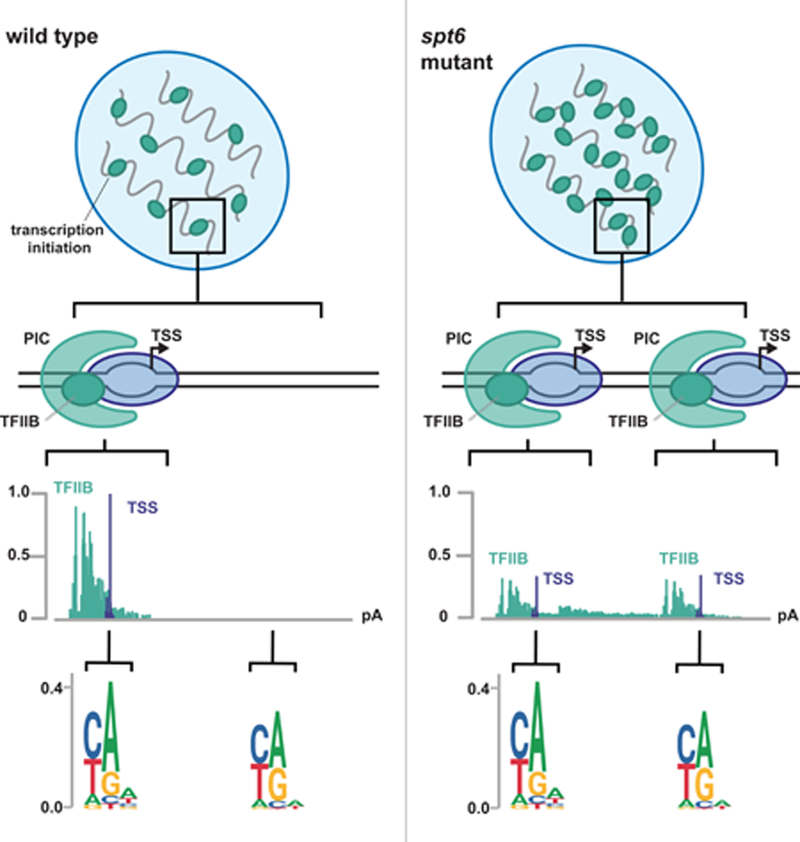
INTRODUCTION
While we once believed that transcription occurs primarily across coding regions, we now know that the transcriptional landscape is extraordinarily complicated, with transcription throughout the genome generating multiple classes of transcripts (Jensen et al., 2013). Regulation of these transcripts is exerted at several levels, including transcription initiation, elongation, termination, and RNA stability. The pervasive nature of transcription suggests that promoters are not only restricted to the 5’ ends of coding regions, but are widespread across the genome. How the cell defines and regulates initiation sites is therefore fundamental to gene expression.
Past genetic studies in yeast produced the unexpected finding that the specificity of transcription initiation is controlled in part by transcription elongation factors, including histone chaperones and modification enzymes (Cheung et al., 2008; Hennig and Fischer, 2013; Kaplan et al., 2003). One factor critical in this process is Spt6, a conserved protein that directly interacts with RNA polymerase II (RNAPII) (Sdano et al., 2017), histones (Bortvin and Winston, 1996; McCullough et al., 2015), and the essential factor Spn1/Iws1 (Diebold et al., 2010; McDonald et al., 2010). Spt6 is believed to function as an elongation factor based on its localization with elongating RNAPII (Andrulis et al., 2000; Ivanovska et al., 2011; Kaplan et al., 2000; Mayer et al., 2010) and its ability to enhance elongation in vitro (Endoh et al., 2004) and in vivo (Ardehali et al., 2009), although it has also been shown to regulate initiation (Adkins and Tyler, 2006; Ivanovska et al., 2011). During transcription, Spt6 regulates chromatin structure (Bortvin and Winston, 1996; DeGennaro et al., 2013; Ivanovska et al., 2011; Jeronimo et al., 2015; Kaplan et al., 2003; Perales et al., 2013; van Bakel et al., 2013) as well as histone modifications, including H3K36 methylation (Carrozza et al., 2005; Chu et al., 2006; Yoh et al., 2008; Youdell et al., 2008). Substantial evidence suggests that a primary function of Spt6 is as a histone chaperone, required to reassemble nucleosomes in the wake of transcription (see (Duina, 2011) for a review).
Studies in yeast have shown that Spt6 controls transcription genome-wide (Cheung et al., 2008; DeGennaro et al., 2013; Kaplan et al., 2003; Pathak et al., 2018; Uwimana et al., 2017; van Bakel et al., 2013). In spt6 mutants, the pattern of transcription dramatically changes, including altered sense transcription and increased levels of antisense transcription. Most notably, in spt6 mutants there is extensive upregulation of cryptic or intragenic transcripts that appear to initiate from within protein-coding sequences (Cheung et al., 2008; DeGennaro et al., 2013; Kaplan et al., 2003; Uwimana et al., 2017).
In this work, we address longstanding issues regarding intragenic transcription and its regulation by Spt6 in Saccharomyces cerevisiae. Previous genome-wide methods used to assay transcripts in S. cerevisiae spt6 mutants, tiled microarrays (Cheung et al., 2008) and RNA-seq (Uwimana et al., 2017), could not distinguish whether intragenic transcripts were the result of new initiation or the result of RNA processing or decay. These methods were also unable to detect intragenic transcripts from highly transcribed genes (Cheung et al., 2008; Lickwar et al., 2009). By comprehensively characterizing transcription initiation in wild-type and spt6 strains with methods that directly assay initiation, we demonstrate that intragenic transcripts result from new initiation, and that Spt6 normally represses initiation from thousands of intragenic promoters. Furthermore, we characterize the chromatin structure and sequence features of intragenic promoters, and show that intragenic promoters share some sequence characteristics with canonical promoters at the 5’ ends of genes (hereafter referred to as genic promoters). Finally, we demonstrate that, contrary to previous beliefs, Spt6 widely controls transcription initiation from genic promoters and suggest that this is due to a competition between genic and intragenic promoters. Thus, Spt6 controls the fidelity of transcription initiation across the genome.
RESULTS
Spt6 regulates transcription initiation from intragenic promoters
To overcome the limitations of previous methods used to study transcription in S. cerevisiae spt6 mutants, we adapted a transcription start site sequencing (TSS-seq) method (Arribere and Gilbert, 2013; Malabat et al., 2015) to identify the position of the RNA 5’-cap at single nucleotide resolution in wild-type and in an spt6 mutant. In the wild-type strain, TSS-seq was highly specific for reads mapping to annotated start sites, with over 70% of reads within 30 nucleotides of annotated TSSs (Pelechano et al., 2013). (Figure 1A, Figure S1A, B). As TSS-seq measures the level of 5’-ends, we found a positive correlation between RNA levels measured by TSS-seq and RNA-seq for wild-type yeast (Uwimana et al., 2017) (Figure S1C). Thus, TSS-seq determines the positions of TSSs at high resolution and quantitatively measures the levels of capped RNAs.
Figure 1.
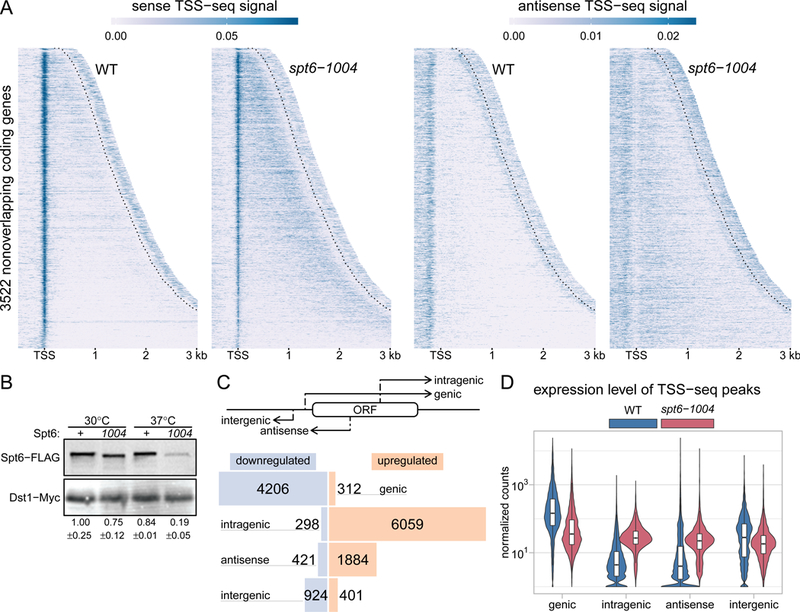
Spt6 is globally required for normal transcription initiation. (A) Heatmaps of sense and antisense TSS-seq signal in wild-type and spt6-1004 strains, over 3522 non-overlapping genes aligned by wild-type genic TSSs and sorted by length. Data are shown for each gene up to 300 nucleotides 3’ of the cleavage and polyadenylation site (CPS; indicated by the dotted line). Values are the mean of spike-in normalized coverage in non-overlapping 20 nucleotide bins, averaged over two replicates. Values above the 95th percentile are set to the 95th percentile for visualization. (B) Western blot showing levels of Spt6 protein in wild-type and spt6-1004 at 30˚C and after an 80-minute shift to 37˚C. Protein levels were quantified using anti-FLAG antibody to detect Spt6 and anti-Myc to detect Dst1 from a spike-in strain (see Methods). The numbers below the blot show the mean and standard deviation for three Westerns. (C) The diagram at the top illustrates the different classes of TSSs. The bar plot below shows the number of TSS-seq peaks differentially expressed from DESeq2 in spt6-1004 versus wild-type (see Methods), classified by genomic region. Blue bars indicate downregulated peaks and orange bars indicate upregulated peaks. (D) Violin plots showing the expression level distributions for different genomic classes of TSS-seq peaks in wild-type and spt6-1004 strains. Values are the mean of counts from two replicates, normalized using an S. pombe spike-in (see Methods).
TSS-seq analysis of the spt6-1004 mutant gave dramatically different results compared to wild type (Figure 1A, Table S1). The spt6-1004 mutation caused depletion of Spt6 to approximately 19% of wild-type levels after an 80-minute shift to the non-permissive temperature of 37°C (Figure 1B), although the cells were still viable (Kaplan et al., 2003). Under these conditions, we identified over 8,000 TSSs as significantly upregulated at least 1.5-fold in spt6-1004 compared to wild-type (Figure 1C). Approximately 6,000 of these TSSs are intragenic TSSs on the sense strand of a gene, although we also detected upregulated TSSs within annotated promoter regions, antisense intragenic (hereafter referred to as antisense), and in intergenic regions (Figure 1C). Our results show that intragenic TSSs are more common than previously known, occurring in approximately 60% of S. cerevisiae genes (Figure S1D). We note that sense strand intragenic TSSs tend to occur towards the 3’ ends of transcription units, while antisense TSSs tend to occur towards the 5’ ends (Figure 1A, S1E). We compared the set of genes we found with upregulated sense intragenic TSSs to the genes found in two previous genome-wide studies of spt6-1004 by microarrays (Cheung et al., 2008) and RNA-seq (Uwimana et al., 2017). We found considerable overlap between all three studies, though TSS-seq allowed us to identify about 1,700 additional genes with at least one intragenic TSS (Figure S1F).
We also examined the levels of different classes of transcripts as measured by TSS-seq and found that in the spt6-1004 mutant, levels for all classes became more similar to one another (Figure 1D). Notably, our results revealed that transcript levels are reduced from a majority of genic TSSs, a result that we analyze in more detail later. Taken together, our TSS-seq results demonstrate that the upregulation of thousands of capped and polyadenylated transcripts, which occurs in an spt6-1004 mutant, is due to new transcription initiation, primarily within coding regions, and that this event is more widespread than previously known.
Spt6 Controls the Localization of TFIIB
Given the dramatic changes in transcription initiation in an spt6-1004 mutant, we wanted to assay initiation using an independent approach, and to determine if intragenic promoters contain an RNAPII pre-initiation complex (PIC). Therefore, we measured genomic binding of TFIIB, a member of the RNAPII PIC, in wild-type and spt6-1004 strains. To do this, we used ChIP-nexus (He et al., 2015), a modification of ChIP-exo (Rhee and Pugh, 2012), which measures the occupancy of a chromatin-bound protein at high resolution by exonuclease digesting the DNA up to the point of crosslinking and sequencing the position of the digested ends. We found that TFIIB binding patterns as measured by ChIP-nexus are reproducible (Figure S2A) and consistent with previous TFIIB ChIP-exo results (Figures S2B, S2C, Table S2).
In the wild-type strain, TFIIB ChIP-nexus signal was primarily localized upstream of previously annotated TSSs, as expected. Using the ChIP-seq peak-calling tool (Zhang et al., 2008b), a TFIIB peak was found overlapping the window extending 200 base pairs upstream of 89% (4297/4917) of wild-type genic TSS-seq peaks. In contrast, in the spt6-1004 mutant, the pattern of TFIIB binding was vastly altered, with TFIIB infiltrating coding regions in concordance with our TSS-seq results (Figure 2A, 2B). To test whether the increase in TFIIB binding over gene bodies was caused by an increased level of TFIIB in the spt6-1004 mutant, we measured TFIIB protein levels and found that they were actually reduced to approximately 70% of wild-type levels (Figure S2D). We conclude that in the spt6-1004 mutant, a more limited pool of TFIIB protein is much more widely associated across the genome than in wild type.
Figure 2.
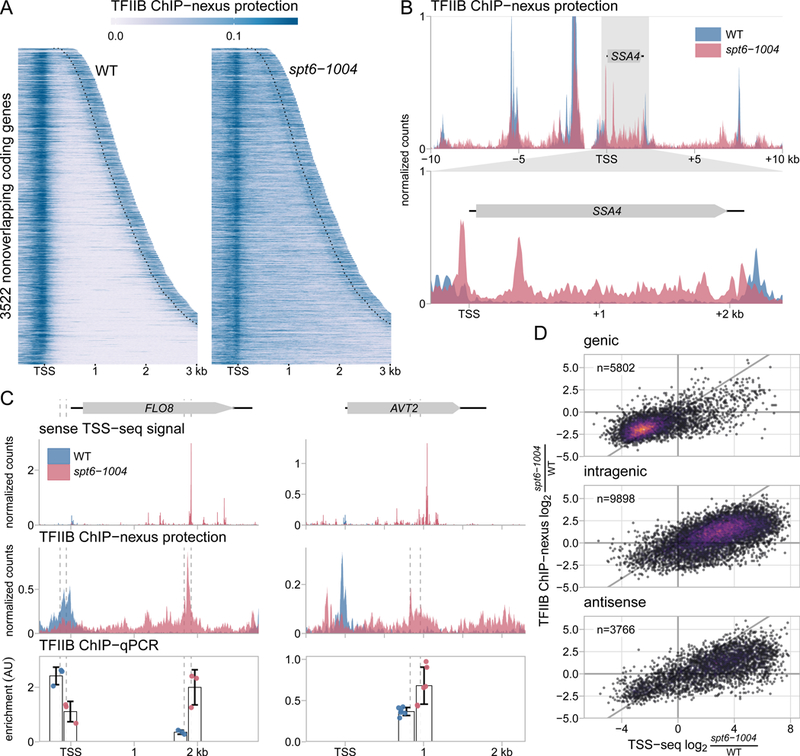
Spt6 is required for genome-wide localization of TFIIB. (A) Heatmaps of TFIIB binding as measured by ChIP-nexus in wild-type and spt6-1004 strains, over the same regions shown in Figure 1A. The values are the mean of library-size normalized coverage in 20 basepair windows, averaged over two replicates. The position of the CPS is shown by the dotted lines. Values above the 85th percentile are set to the 85th percentile for visualization. (B) The upper panel shows TFIIB binding in wild-type and spt6-1004 strains over 20 kb of chromosome II flanking the SSA4 gene, as measured by TFIIB ChIP-nexus. The lower panel shows an expanded view of TFIIB binding over the SSA4 gene. (C) TSS-seq, TFIIB ChIP-nexus, and TFIIB ChIP-qPCR measurements at the genic and intragenic promoters of the FLO8 and AVT2 genes in wild-type and spt6-1004 strains. TSS-seq counts are normalized to spike-in, ChIP-nexus values are normalized to library size, and ChIP-qPCR is normalized to amplification of a region of the S. pombe pma1+ gene used as a spike-in control. Vertical dashed lines represent the coordinates of qPCR amplicon boundaries. (D) Scatterplots of fold-change in spt6-1004 over wild-type strains, comparing TSS-seq and TFIIB ChIP-nexus. Each dot represents a TSS-seq peak paired with the window extending 200 nucleotides upstream of the TSS-seq peak summit for quantification of TFIIB ChIP-nexus signal. Fold-changes are regularized fold-change estimates from DESeq2, with size factors determined from the S. pombe spike-in (TSS-seq) or the S. cerevisiae counts (ChIP-nexus). The diagonal line is y=x.
The altered binding pattern of TFIIB in spt6-1004 (Figures 2A, 2B) made defining sites of intragenic initiation by TFIIB peak calling difficult. With the same parameters used to call peaks in the wild-type strain, MACS2 identified TFIIB peaks in spt6-1004 upstream of 85% (4050/4763) of genic TSSs, but only identified TFIIB peaks upstream of 37.0% (2240/6059) of spt6-1004 upregulated intragenic TSS-seq peaks. Two examples of these intragenic TFIIB peaks were verified by ChIP-qPCR of TFIIB (FLO8 (Figure 2C) and VAM6 (Figure S2E)). Given the spreading-like nature of TFIIB association in many places in the spt6-1004 mutant, it seemed plausible that there was an increased level of TFIIB upstream of the upregulated intragenic TSSs in spt6-1004, but that the nature of the TFIIB binding prevented a peak from being called. Two examples of this are at AVT2 (Figure 2C) and YPT52 (Figure S2E). Therefore, we dispensed with TFIIB peak-calling and simply quantified the change in TFIIB signal in spt6-1004 compared to wild type over the window 200 base pairs upstream of TSS-seq peaks. From this analysis, we found that the results from both assays were in agreement: 90.3% of genic promoters changed in the same direction by both assays while approximately 81% of sense and antisense intragenic promoters changed in the same direction (Figure 2D). We note that despite the challenge in calling intragenic TFIIB peaks, we did identify around 1500 intragenic TFIIB peaks that did not have a TSS-seq peak within 200 base pairs in either direction (Table S2). These may represent intragenic initiation events not captured by TSS-seq, either due to non-productive initiation or transcript instability. Overall, the TFIIB ChIP-nexus results support our TSS-seq results and show that Spt6 controls TFIIB localization across the genome.
Spt6 Controls Nascent Transcription on Both the Sense and Antisense Strands
As TSS-seq and TFIIB ChIP-nexus measure steady-state levels of transcripts and PICs, respectively, we also performed native elongating transcript sequencing (NET-seq) (Churchman and Weissman, 2011), which quantitatively measures the position of elongating RNAPII at single-nucleotide resolution. Although NET-seq was unable to provide information about intragenic transcription due to the overlap with genic transcription (Lickwar et al., 2009), it was able to provide other new information about the requirement for Spt6 in transcription. In wild-type cells, our NET-seq results were similar to those previously reported (Churchman and Weissman, 2011), with a high level of RNAPII over approximately the first 750 bp of the sense strand of transcription units and a lower level downstream. In contrast, in the spt6-1004 mutant, we observed reduced levels of RNAPII over the 5’ region with a relative increase downstream (Figure 3A; S3A, S3B). The reduced RNAPII density over the 5’ region provides independent evidence that genic transcription initiation is generally decreased in spt6-1004. The apparent increase in elongating RNAPII density over the 3’ regions of genes in spt6-1004 is likely caused by a combination of intragenic initiation and a slower rate of elongation (Ardehali et al., 2009; Endoh et al., 2004).
Figure 3.
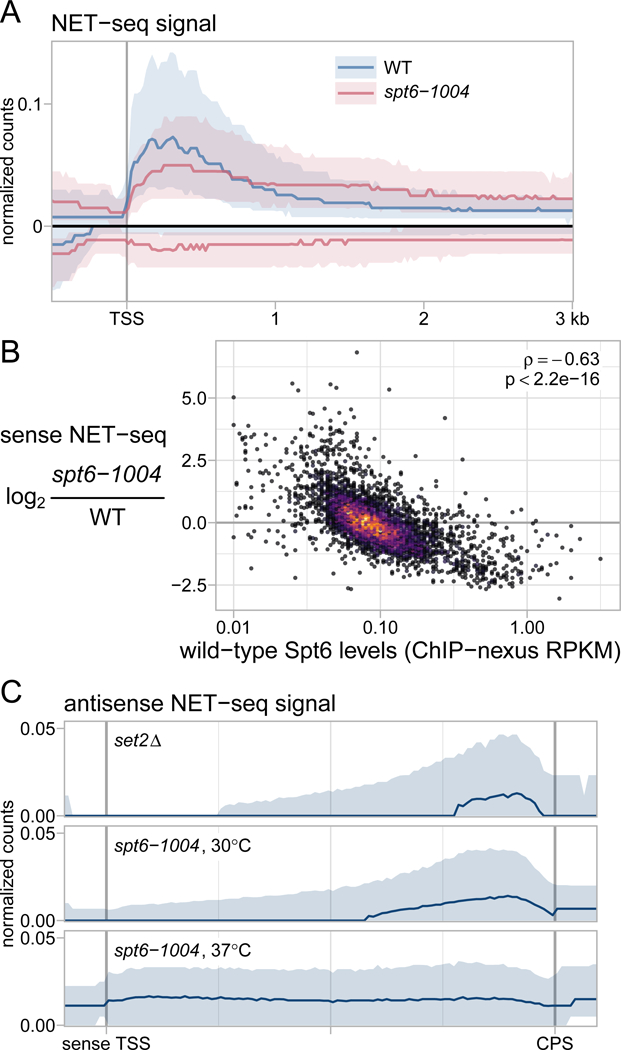
Spt6 is required for normal levels and distribution of elongating RNA polymerase II. (A) The average sense and antisense NET-seq signal in wild-type and spt6-1004 strains after a shift to 37˚C, over 3522 nonoverlapping genes. Sense and antisense signals are depicted above and below the x-axis, respectively. The solid line and shadings represent the median and inter-quartile range, which are shown in order to give an idea of how the signal varies among the thousands of genes with diverse characteristics being represented in the plot. The values are the mean of library-size normalized coverage in nonoverlapping 20 nucleotide bins, averaged over two replicates. (B) A scatterplot of NET-seq fold-change in the spt6-1004 mutant versus Spt6 occupancy in the wild-type strain as measured by Spt6 ChIP-nexus. Each dot represents NET-seq and Spt6 ChIP-nexus sense-strand signals summed over the entire length of the transcription unit. NET-seq fold-changes are regularized fold-change estimates from DESeq2. The Pearson correlation coefficient and associated p-value (Student’s t-distribution) are shown. (C) Average antisense NET-seq signal in the spt6-1004 strain at permissive (30˚C) and nonpermissive (37˚C) temperatures, compared to a set2Δ strain. The values are as in Figure 3A, with the solid line and shadings representing the median and inter-quartile range over 3522 nonoverlapping genes scaled to the same length.
NET-seq also allowed us to test whether the level of Spt6 recruited to a gene corresponds to the degree of the requirement for Spt6 in active transcription. To do this, we performed ChIP-nexus of Spt6 in wild-type cells and compared that to the change in NET-seq signal in the spt6-1004 mutant. From this analysis, we discovered a correlation between these two measurements: the genes with the greatest level of Spt6 in wild-type were those whose active sense-strand transcription was decreased the most in the spt6-1004 mutant (Figure 3B). As there is a very strong correlation between the chromatin association of Spt6 and RNAPII (Figure S3C; (DeGennaro et al., 2013; Ivanovska et al., 2011; Mayer et al., 2010; Perales et al., 2013)), this shows that highly transcribed genes are those that are most dependent upon Spt6, in agreement with a recent study (Pathak et al., 2018). These results support our TSS-seq and TFIIB ChIP-nexus results which suggested that transcription initiation from genic promoters is decreased in an spt6-1004 mutant (Figures 1D, 2D), and further suggest that the degree of decrease correlates to the level of active transcription.
Our NET-seq results also revealed new information regarding Spt6 and antisense transcription. First, while our TSS-seq results suggested that most new antisense initiation in the spt6-1004 mutant occurs towards the 5’ end of transcription units (Figure 1A), our NET-seq results showed antisense transcription to be elevated more broadly over transcription units (Figure 3A, S3B). This difference may result from antisense initiation from intergenic regions downstream of most genes (seen to right of the CPS line in Figure 1A; (Murray et al., 2012)). Second, as previous studies have demonstrated that spt6-1004 mutants are defective for Set2-dependent H3K36 methylation (Carrozza et al., 2005; Chu et al., 2006; Youdell et al., 2008), and that set2Δ mutants also have elevated antisense transcription (Kim et al., 2016; Li et al., 2007; McDaniel et al., 2017; Venkatesh et al., 2016), we compared our NET-seq results for spt6-1004 to previous NET-seq results for set2Δ (Churchman and Weissman, 2011). We included analysis of an spt6-1004 mutant grown at 30˚C, when Spt6 protein is still present, and after a shift to 37˚C, when Spt6 protein is depleted. There is no detectable H3K36 methylation in the spt6-1004 mutant at either temperature (Chu et al., 2006; Youdell et al., 2008). Our results (Figure 3C) show that spt6-1004 grown at 30˚C has a similar effect on antisense transcription as set2Δ. However, after a shift to 37˚C, the spt6-1004 mutant has more widespread derepression of antisense transcription than seen in set2Δ. These results suggest that the antisense effect in spt6-1004 at 30˚C is primarily due to loss of H3K36 methylation, while the effect after a shift to 37°C is due to additional spt6-1004 specific effects, possibly due to changes in chromatin structure.
Spt6 is Required for Normal Nucleosome Occupancy and Positioning
Several studies have shown that Spt6 is required for normal chromatin structure in S. cerevisiae (Bortvin and Winston, 1996; Ivanovska et al., 2011; Jeronimo et al., 2015; Kaplan et al., 2003; Perales et al., 2013; van Bakel et al., 2013). However, to correlate our TSS-seq results with high-resolution and quantitative analysis of chromatin structure, we performed MNase-seq to re-examine the requirement for Spt6 in chromatin structure. Our MNase-seq results from wild-type cells showed the expected signature over coding regions, including nucleosome-depleted regions 5’ of genes and a phased pattern of nucleosomes over gene bodies (Figure 4A, S4A). In contrast, the pattern of nucleosome signal is drastically altered in the spt6-1004 mutant, as previously observed (DeGennaro et al., 2013; van Bakel et al., 2013).
Figure 4.
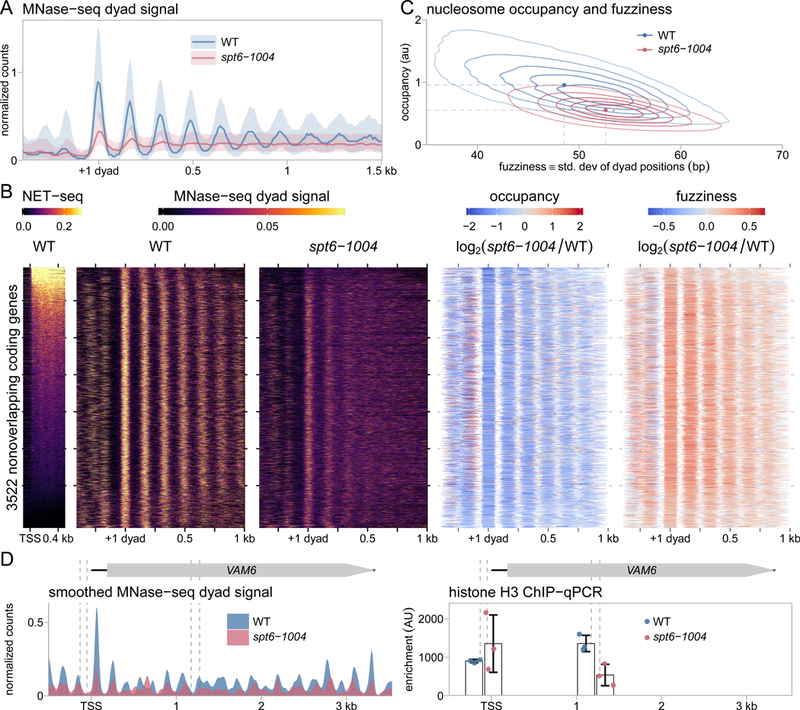
Genome-wide defects in chromatin structure in an spt6-1004 mutant. (A) Average MNase-seq dyad signal in wild-type and spt6-1004 strains, over 3522 nonoverlapping genes. The values are the mean of spike-in normalized coverage in nonoverlapping 20 nucleotide bins, averaged over two replicates (spt6-1004) or one experiment (wild-type). The solid line and shadings represent the median and inter-quartile range. (B) The leftmost panel shows the NET-seq signal in a window extending 500 nucleotides downstream of the TSS, sorted from top to bottom by the level of the signal. The second and third panels show heatmaps of the spike-in normalized MNase-seq dyad signal from wild-type and spt6-1004 strains over 3522 nonoverlapping coding genes aligned by wild-type +1 nucleosome dyad and sorted by total sense NET-seq signal. The last two panels show the spike-in normalized changes in nucleosome occupancy and fuzziness. The increased occupancy indicated just upstream of the +1 dyad is likely caused by nucleosomes occupying NDRs in the spt6-1004 mutant. (C) A contour plot showing the global distribution of nucleosome occupancy and fuzziness in wild-type and spt6-1004 strains. (D) MNase-seq and histone H3 ChIP-qPCR measurements of nucleosome signal at the VAM6 gene in wild-type and spt6-1004 strains. MNase-seq coverage is spike-in normalized dyad signal, smoothed using a Gaussian kernel with a 20 bp standard deviation, and averaged by taking the mean of two replicates (spt6-1004) or one experiment (wild-type). Histone H3 ChIP-qPCR enrichment is normalized to amplification at the S. pombe pma1+ gene as a spike-in control. Vertical dashed lines represent the coordinates of the qPCR amplicon boundaries.
Differences in nucleosome signal are caused by different features, including occupancy and fuzziness (Chen et al., 2013). To determine the contribution of these to the altered nucleosome signal observed in spt6-1004, we quantified our MNase-seq data using DANPOS2 (Chen et al., 2013). In wild type, the population of nucleosomes varied greatly in occupancy and fuzziness, with more highly occupied nucleosomes tending to be less fuzzy (more well positioned) (Figure 4B, 4C). In contrast, the distribution of nucleosomes in spt6-1004 was more homogeneous, with a global decrease in occupancy and increase in fuzziness. To verify the decreased level of nucleosome occupancy, we performed histone H3 ChIP at three genes and found a lower level in the spt6-1004 mutant compared to wild type, in agreement with previous results (Perales et al., 2013) (Figure 4D, S4C). This reduction may be caused in part by reduced expression of histone genes in spt6 mutants (our TSS-seq data; (Compagnone-Post and Osley, 1996)). In summary, Spt6 plays a major role in determining nucleosome occupancy and positioning.
Previous work showed that genes with high levels of transcription show a relative decrease in positioned nucleosome signal compared to genes with low levels of transcription (Shivaswamy et al., 2008). This trend is reflected in our wild-type MNase-seq data (Figure 4B, S4B). Furthermore, our previous work, based on the analysis of a smaller number of genes, suggested that highly transcribed genes were most prone to nucleosome loss in an spt6-1004 mutant (Ivanovska et al., 2011). However, from our new MNase-seq results, the severity of the changes in nucleosome signal in spt6-1004 with respect to occupancy and fuzziness do not depend on the transcription level (Figure 4B). We note that the weak nucleosome patterning observed in spt6-1004 at highly transcribed genes compared to moderately transcribed genes is expected given that nucleosomes are already more disordered at highly transcribed genes in wild type (Figure 4B, S4B). These results suggest that Spt6 controls chromatin structure genome-wide independently of the level of transcription.
Intragenic Promoters Have Some Sequence Characteristics of Canonical Promoters
Our TSS-seq analysis identified over 6,000 sense-strand intragenic TSSs that are derepressed in an spt6-1004 mutant. To compare these promoters to canonical promoters at the 5’ ends of genes, we examined their chromatin structure and DNA sequence. Using the wild-type and spt6-1004 MNase-seq data flanking the intragenic TSSs, we found that intragenic TSSs fell into two clusters that differed primarily by the phasing of the nucleosome array relative to the intragenic TSS (Figure 5A; Methods). In wild-type chromatin, the intragenic TSSs in both clusters tended to occur at the border between regions of nucleosome enrichment and depletion, (Figure 5A), although nucleosome positioning around these TSSs is modest compared to the positioning adjacent to canonical promoters. This is likely due to the preference of sense-strand intragenic TSSs to occur towards the 3’ ends of transcription units, where nucleosome fuzziness increases (Mavrich et al., 2008). As expected, the average nucleosome signal around both clusters of intragenic TSSs is decreased in the spt6-1004 mutant. In spite of the differences between the chromatin structure of the two clusters in wild-type strains, their expression levels in an spt6-1004 mutant are in similar (Figure 5B).
Figure 5.

Chromatin structure and sequence features of intragenic promoters. (A) The average MNase-seq dyad signal and GC percentage for two clusters of intragenic TSSs that are upregulated in an spt6-1004 mutant, as well as all genic TSSs detected in wild type or spt6-1004. The clusters were determined from the MNase-seq signal flanking the TSS (see Methods). (B) Violin plots showing the distributions of TSS-seq signal for the two clusters of intragenic TSSs that are upregulated in an spt6-1004 mutant, and the distributions of their TFIIB ChIP-nexus signal in the window extending 200 nucleotides upstream of the TSS-seq peak. Counts are size factor normalized using the S. pombe spike-in (TSS-seq) or S. cerevisiae counts (TFIIB ChIP-nexus). (C) Sequence logos of the information content of TSS-seq reads overlapping genic and intragenic peaks in spt6-1004 cells. (D) Scaled density of the TATA box upstream of TSSs. For each category, a Gaussian kernel density estimate of the positions of exact matches to the motif TATAWAWR is multiplied by the total number of TATA occurrences in the category and divided by the total number of regions in the category. (E) Volcano plot of motif enrichment and depletion upstream of intragenic TSSs upregulated in spt6-1004. Odds ratios and false discovery rate are determined by Fisher’s exact test, comparing to random locations in the genome. Factors may appear more than once if they have multiple motifs in the databases that were searched.
Given that intragenic TSSs occur at specific sites, it seemed plausible that the alterations in chromatin structure are necessary, but not sufficient for an intragenic promoter. Therefore, we looked at the DNA sequence around the intragenic TSSs. First, as AT-rich sequences are unfavorable for nucleosomes and are often found in promoters (Iyer and Struhl, 1995; Kaplan et al., 2009; Tillo and Hughes, 2009; Zhang et al., 2009), we examined the GC content of the DNA sequence flanking intragenic TSSs and found a decrease in GC content just upstream of the TSSs in both clusters, albeit more modest than at genic promoters (Figure 5A). Second, we aligned the intragenic TSS-seq reads and discovered a sequence motif almost identical to the consensus initiation sequence, (A(Arich)5NPyA(A/T)NN(Arich)6) previously observed for genic S. cerevisiae TSSs (Malabat et al., 2015; Zhang and Dietrich, 2005) (Figure 5C). Third, we searched for TATA elements with perfect matches to the consensus sequence TATAWAWR (Basehoar et al., 2004). We found this consensus sequence at 10.7% of the regions upstream of spt6-1004 sense-strand intragenic TSSs compared to 23.7% for all genic TSSs and 8.8% over random sites in the genome (Figure 5D). The intragenic promoters with a consensus TATA had modestly greater expression than those without. When we analyzed the top 1000 most upregulated intragenic TSSs (out of 6059), the percentage with TATA elements increased to 15.4%. In summary, intragenic promoters are enriched for classes of sequence elements found at many genic promoters.
Finally, we quantified the enrichment or depletion of sequence-specific transcription factor binding site motifs upstream of intragenic TSSs and found many members of both classes (Figure 5E). The most enriched motifs, a subset of those found upstream of genic promoters (Figure S5), are for transcription factors that are activated by cellular stresses (for example, Rpn4, Pdr1/3, and Mot3), some of which may reflect the temperature shift used to deplete Spt6. This supports a previous observation that some intragenic promoters can be induced by stress (Cheung et al., 2008; McKnight et al., 2014; Tamarkin-Ben-Harush et al., 2017). We also observed a significant depletion for multiple motifs, including those for Abf1 and Reb1, two factors required for NDRs at many genic promoters (Badis et al., 2008; Kaplan et al., 2009; Lee et al., 2007; Tsankov et al., 2010; Yarragudi et al., 2007). The depletion for these motifs highlights the lack of a typical NDR for intragenic promoters.
A General Requirement for Spt6 in Genic Promoter Function
Our TSS-seq data revealed the unexpected finding that Spt6 is required for normal expression levels from most genic promoters. Out of 5,274 genes, 3,857 (73.1%) were downregulated in the spt6-1004 mutant, 284 (5.4%) were upregulated, and 1,133 (21.5%) were not significantly changed. Furthermore, the TFIIB ChIP-nexus signal also decreased for most genic promoters (Figure 2D), suggesting that the changes in the spt6-1004 mutant are caused by changes in initiation, rather than at a post-initiation step. We verified the change over the genic promoter of two genes by ChIP-qPCR of TFIIB (Figure 6A). Thus, Spt6 plays a global role in the expression of genic promoters.
To see whether promoter chromatin architecture might contribute to the differential regulation of genes by Spt6, we examined our MNase-seq data for the genic TSSs downregulated, upregulated, and not significantly changed in spt6-1004. Interestingly, each group has a distinct nucleosome profile (Figure 6B). Genes that are downregulated in spt6-1004 and therefore require Spt6 for normal initiation have the wild-type profile of an NDR upstream of a strong +1 nucleosome peak. In the spt6-1004 mutant, the MNase profile of these genes reflects the changes expected from the metagene MNase profile in Figure 4A, with a slightly shallower NDR and reduced +1 nucleosome occupancy (Figure 6B). In contrast, genes that are upregulated in spt6-1004 and are therefore normally repressed by Spt6 have, on average, neither a detectable NDR nor a +1 nucleosome peak in either wild-type or spt6-1004. Finally, genes not significantly affected in spt6-1004 have a third nucleosome pattern, between the other two classes of genes. Thus, the three classes of genes differentially regulated by Spt6 have distinct chromatin architectures over their promoters.
Figure 6.
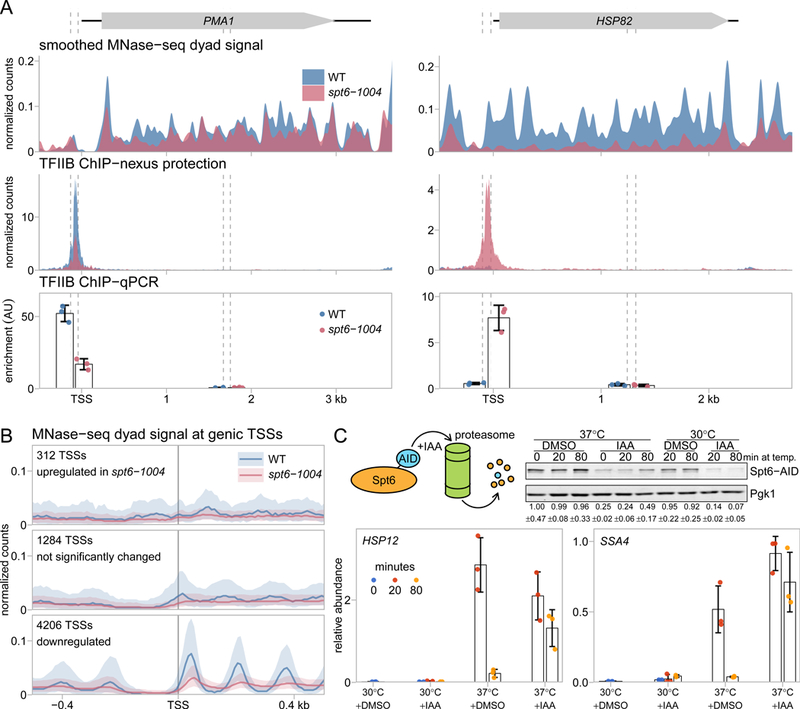
Spt6 function is necessary to control genic transcription. (A) MNase-seq, TFIIB ChIP-nexus, and TFIIB ChIP-qPCR measurements at the PMA1 and HSP82 genes in wild-type and spt6-1004 strains, plotted as in 2B and 4D. For the ChIP-qPCR analysis, the mean and standard deviation are plotted for three experiments. (B) The average MNase-seq dyad signal at genic TSSs in wild-type and spt6-1004 strains, grouped by the differential expression status of the TSS. The solid line and shading represent the median and inter-quartile range. (C) RT-qPCR analysis of HSP12 and SSA4 RNA levels, testing the effects of temperature shift and Spt6 depletion. The top left panel shows a diagram of auxin-dependent degradation system used to deplete Spt6 and the top right panel shows a Western measuring the level of Spt6 protein, with and without depletion. The bottom panels show the RNA levels for HSP12 and SSA4 at times after a temperature shift from 30˚C to 37˚C. In these experiments, either DMSO or IAA were added 30 minutes before the zero time point. Plotted are the mean and standard deviation for three experiments, normalized to SNR190 RNA.
Our analysis shows that the group of genes strongly repressed by Spt6 includes several that are normally induced by heat shock. To understand how Spt6 regulates this class of gene, we tested whether the induction of two genes, SSA4 (Werner-Washburne et al., 1987) and HSP12 (Praekelt and Meacock, 1990), required only the depletion of Spt6 or whether their induction also required the temperature shift used to deplete Spt6 in the spt6-1004 mutant. To separate the effects of Spt6 depletion and temperature shift, we used an auxin-inducible degron system (Nishimura et al., 2009) to deplete Spt6, allowing us to independently vary Spt6 depletion and temperature shift. Measurement of RNA levels by RT-qPCR (Figure 6C) showed that both genes were induced only after a shift to 37˚C, independently of whether Spt6 was depleted (see 20-minute time point). However, at 80 minutes after the shift to 37˚C, when adaptation to heat shock normally occurs, RNA levels were still high when Spt6 was depleted. These results show that Spt6 is required for the repression of some heat shock-induced genes during adaptation after the temperature shift, consistent with previously described roles for Spt6 (Adkins and Tyler, 2006) and the histone chaperone Spt16 (Jensen et al., 2008; Rowley et al., 1991).
DISCUSSION
In this work, we have integrated multiple quantitative genomic approaches to study the conserved transcriptional regulator Spt6 in S. cerevisiae, leading to new insights into Spt6 function and into the potential for expression of alternative transcripts. Our results have shown, for the first time on a genomic scale, that the thousands of intragenic and antisense transcripts produced in an spt6 mutant are due to new transcription initiation from RNAPII promoters. In addition, we identified sequence motifs at intragenic promoters that are also found at canonical promoters, indicating that promoter-like sites exist broadly within genes and are normally maintained in a repressed state by Spt6. Furthermore, we showed that Spt6 plays a genome-wide role in the regulation of initiation from genic promoters. Together, these results demonstrate that Spt6 plays a critical role in determining the specificity of transcription initiation in vivo.
The mechanism by which Spt6 normally represses thousands of intragenic promoters is uncertain. One study showed that Spt6 depletion allows ectopic localization of histone Htz1, suggesting that Spt6 represses intragenic promoters by excluding Htz1 (Jeronimo et al., 2015). However, our analysis suggests that intragenic promoters are not significantly enriched for the ectopic Htz1 locations previously found (data not shown). As Spt6 is also required for the recruitment of other proteins to transcribed chromatin, including the histone chaperone Spt2 (Chen et al., 2015; Nourani et al., 2006), as well as for histone H3 K36 methylation (Carrozza et al., 2005; Chu et al., 2006; Youdell et al., 2008), there are likely many aspects of Spt6 function that contribute to the repression of intragenic promoters.
As Spt6 is primarily associated with transcribed regions (DeGennaro et al., 2013; Ivanovska et al., 2011; Mayer et al., 2010) and it enhances the rate of elongation (Ardehali et al., 2009; Endoh et al., 2004), it was unexpected to discover that it regulates initiation from genic promoters. We suggest that Spt6 regulates these promoters indirectly, by controlling the total number of active promoters. In a wild-type yeast cell growing in rich medium, there are ~5,000 expressed promoters and ~4,000–5,000 copies of most PIC proteins, including TFIIB (Ho et al., 2018). In contrast, in an spt6-1004 mutant, there is a large increase in the number of active promoters, driving over 13,000 TSSs. Given the decreased level of TFIIB in an spt6-1004 mutant (~70% of wild-type levels), we suggest that the three-fold increase in the number of TSSs results in a competition for a limited supply of PIC components, resulting in decreased expression from genic promoters. In support of this, our results show that in wild type there is a large difference in average expression levels between different classes of TSSs, while in the spt6-1004 mutant, the differences in the expression levels between the classes are diminished (Figure 1D), as if, in the mutant, all promoters have an approximately equal opportunity to recruit PICs.
Past studies of spt6-1004 suggested that intragenic transcripts may encode functional information that is used in certain conditions (Cheung et al., 2008). In addition to yeast, intragenic transcription occurs in mammalian cells in a widespread fashion under certain conditions (Carvalho et al., 2013; Muratani et al., 2014). Furthermore, intragenic transcripts can encode N-terminally truncated proteins that have distinct functions compared to their full-length counterparts. Examples include oncogenes (Wiesner et al., 2015), stress response genes (Tamarkin-Ben-Harush et al., 2017), and p53 family genes (Wilhelm et al., 2010). For two of the yeast genes that encode functional intragenic transcripts, ASE1 (McKnight et al., 2014) and KAR4 (Gammie et al., 1999), we also observed intragenic initiation in spt6-1004. However, not all intragenic promoters are active in spt6-1004. For example, a recent study showed that Gcn4 activates transcription from many intragenic sites (Rawal et al., 2018) and most of those are not activated in an spt6-1004 mutant. In addition to encoding N-terminally truncated proteins, intragenic promoters can play other types of regulatory roles, such as interference with normal gene expression (Kim et al., 2017; Xie et al., 2011). The continued analysis of intragenic transcription will likely lead to new insights into the flexibility of genomes in encoding functional information.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Correspondence and requests for materials should be addressed to Fred Winston (winston@genetics.med.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains used in this study are listed in Table S3. All strains were constructed by standard procedures, using either yeast transformation or crosses. All oligonucleotides used for PCR are listed in Table S4. The spt6-1004 temperature-sensitive mutant and wild-type strains were grown as previously described (Cheung et al., 2008): cells were grown in YPD at 30°C to a concentration of approximately 1 × 107 cells/ml (OD600=0.6), at which point an equal volume of YPD medium pre-warmed to 44°C was added, and the cultures were shifted to 37°C for an additional 80 minutes.
METHOD DETAILS
Transcription start site sequencing
Yeast strains FY2180 and FY2181 were grown in 100 ml cultures at 30°C and shifted to 37°C as described above. After determining the cell concentration using a hemocytometer, S. pombe cells (strain 972) were added to each culture at a level of 10%, to be used for spike-in normalization. Total RNA was isolated as previously described (Ausubel, 1991). Poly(A)-enriched RNA was isolated from 300 ∝g of total RNA with 300 ∝l of Dynabeads oligo(dT)25 (Invitrogen), using the manufacturer’s instructions and eluted in water. Prior to each subsequent step of library construction, RNA samples were heat denatured at 80°C for two minutes and rapidly cooled on ice, followed by addition of 40 U of RNasin (Promega). Between each enzymatic reaction, samples were purified using an RNA binding column (Zymo Research). Ten to fifteen ∝g of poly(A) RNA was dephosphorylated with 30 units of calf intestinal phosphatase (CIP; NEB) for one hour at 37°C. CIP was removed from the reaction by heat inactivation followed by phenol extraction, and traces of phenol were removed using the above-mentioned RNA column. The m7GpppN cap was then cleaved from the RNA with 12.5 units of CapClip (CELLSCRIPT) for one hour at 37°C and the decapped RNA, containing a 5’ monophosphate, was ligated to 25 pmoles of a DNA/RNA chimeric linker (oSMDAP4; Table S4) containing a randomized RNA linker sequence of six nucleotides at the 3’ end and a 5’ biotin moiety in a 10 µl reaction with 20 units of T4 RNA ligase 1 (NEB) and 2 mM ATP. Ligation products were column purified as before and eluted into fragmentation buffer (Ingolia et al., 2009) calibrated to enrich for 90–120 nucleotide oligomers. Fragmented RNA was then size selected and purified from a 10% acrylamide urea gel (Invitrogen). PNK removal of the 3’ phosphate group and 3’-end ligation of the RNA to a random linker pool (Mayer et al., 2015) was done as previously described (Couvillion and Churchman, 2017), except after ligation the biotinylated RNA was affinity purified with 10 ∝l of Dynabeads M-270 streptavidin (Invitrogen) using the manufacturers instructions. Bead-bound RNA was eluted into 50 ∝l of elution buffer (0.1% SDS, 10 mM Tris 7.5) at 90°C for 5 minutes, and reverse transcribed with 3 pmoles of RT primer (oSMDRT2; Table S4) by heating for 5 min at 65°C, with 200 units SSIII Reverse Transcriptase (Invitrogen) at 48°C for 45 minutes. The cDNA was gel purified as above, and PCR amplified for 10–14 cycles using previously described indexing and sequencing primers for Illumina sequencing (Couvillion and Churchman, 2017).
ChIP-qPCR and ChIP-nexus
For TFIIB studies, yeast strains FY3126 and FY3127 were grown in YPD at 30°C and then shifted to 37°C as described above. The cultures were cooled to 25°C using pre-chilled medium at 4°C before cross-linking in 1% formaldehyde while shaking at 25°C for 30 minutes, followed by quenching in 125 mM glycine at 25°C for 10 minut es. For Spt6 and Rpb1 ChIP-nexus, strain FY3128 was grown without the temperature shift. Chromatin was extracted using standard methods (DeGennaro et al., 2013) and sheared in a QSONICA sonicating water bath. For ChIP-qPCR spike-in normalization, each S. cerevisiae chromatin sample was mixed with 50% S. pombe chromatin (strain FWP561) by mass for TFIIB ChIP and 30% by mass for histone H3 ChIP. Chromatin precipitations were performed overnight at 4°C with 4 ∝g of anti-H3 (ab1791; Abcam) per 300 ∝g of chromatin or 20 ∝l of Pan Mouse IgG Dynabeads (Invitrogen) per 500 ∝g of chromatin. Real-time qPCR was performed as previously described (DeGennaro et al., 2013) using primer pairs listed in Table S4.
Each ChIP-nexus library used 2.5–3 mg of S. cerevisiae chromatin containing 5% S. pombe chromatin added by mass (strains FWP561 or FWP489) for downstream spike-in normalization between samples. To generate sequencing libraries for TFIIB and Spt6 bearing TAP tags, chromatin was affinity purified using 100 ∝l Pan Mouse IgG Dynabeads (Invitrogen). For RNAPII (Rbp1) libraries, chromatin was immunoprecipitated with 40 ∝g of 8WG16 antibody (BioLegend) that was pre-bound to 100 ∝l of ProteinG Dynabeads (Invitrogen). Library constructions for Illumina sequencing were performed essentially as previously described (He et al., 2015), except buffers were optimized for yeast: Buffer A (10 mM TE, 0.1% Triton X), Buffer B (50 mM HEPES.KOH pH 7.4, 140 mM NaCl, 1 mM EDTA, 1% Triton-X, 0.1% sodium deoxycholate), Buffer C (Buffer B with 250 mM NaCl), Buffer D (10 mM Tris pH 7.5, 250 mM LiCl ,10 mM EDTA, 0.5% IGEPAL CA-360, 0.1% sodium deoxycholate).
MNase-seq
MNase digestion was performed as previously described (Rando, 2010) with some modifications, using strains FY87 and FY3125. Cultures of 500 ml were grown in YPD at 30˚C, then shifted to 37˚C as described above. At a density of approximately 1 × 107 cells/ml (OD600 = 0.5), cells were crosslinked using 2% formaldehyde for 30 minutes and then treated for 10 minutes with 125 mM glycine before collecting an equal number of cells for each strain. The cells were resuspended in 40 ml of sorbitol buffer (1 M sorbitol, 50 mM Tris pH 7.4, 10 mM β-mercaptoethanol) and incubated for 30 minutes at 30°C with 10 mg of zymolase 100T (US Biological) per gram of cells. Spheroplasting efficiency was assessed by microscopy and was more than 95% of total cells. The spheroplasts were collected and resuspended in NP buffer (1 M sorbitol, 50 mM sodium chloride, 10 mM Tris pH 7.4, 5 mM magnesium chloride, 1 mM calcium chloride, 0.075% NP-40, 1 mM β-mercaptoethanol, 500 µM spermidine). Micrococcal nuclease (MNase; Sigma) was dissolved in Ex50 buffer (10 mM Hepes pH 7.6, 50 mM sodium chloride, 1.5 mM magnesium chloride, 0.5 mM EGTA, 10% glycerol, 1mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride) prepared to produce 500 units per 840 µl stock as recommended by the manufacturer. The spheroplasts were divided into aliquots and incubated for 20 minutes at 37°C with increasing amounts of MNase, ranging f rom 2 to 15 µl of the stock. Digestion was stopped by addition of stop buffer (5% SDS, 100 mM EDTA), samples were subjected to proteinase K digestion and reverse-crosslinking at 65°C overnight, followed by DNA purification. The efficiency of MNase digestion was quantified using DNA fragment size analysis (Agilent Bioanalyzer) to establish an MNase titration curve for each strain. The MNase concentrations which yielded approximately 80% mononucleosomal DNA were selected for the library construction. The samples were mixed with the MNase-digested spike-in DNA from S. pombe (strain 972) based on the original cell count (100 ng of spike-in DNA per MNase digested DNA from 7 × 108 S. cerevisiae cells). Mononucleosomal DNA was purified using size-selected gel extraction. The sequencing libraries were constructed as described before (DeGennaro et al., 2013).
NET-seq
NET-seq was performed on strains grown at both 30°C and 37°C. Strains FY2912 and FY2913 were grown at 30°C, the cultures were split and hal f was shifted to 37°C as described above. NET-seq was performed as previously described (Churchman and Weissman, 2011).
Western blotting
To measure FLAG-Spt6 and TFIIB-TAP protein levels, strains FY3126 and FY3127 were grown with the 37°C temperature shift as described above. Prior to pelleting the cells, strain FY2354 expressing DST1-MYC was added to each culture at 50% concentration by cell number used for spike-in normalization. Cell extracts were made by bead beating in LB-140 buffer (50 mM HEPES.KOH pH 7.4 140 mM NaCl 1 mM EDTA 1% TritonX-100 0.1% NaDeoxycholate0.1% SDS) along with protease inhibitors (1mM phenylmethylsulfonyl fluoride, 2 µg/mL leupeptin, 2 µg/mL pepstatin, 0.4 mM dithiothreitol), and SDS-PAGE gels were loaded by mass. For protein detection, primary antibodies used were anti-FLAG diluted 1:5000 (clone M2; SIGMA), anti-Protein A diluted 1:1500 (clone SPA-27; SIGMA), anti-cMyc diluted 1:1000 (clone A-14 Santa Cruz), anti-PGK1 diluted 1:20000 (clone 22C5D8; Invitrogen) and anti-V5 diluted 1:2000 (clone R960–25; Invitrogen). Secondary detection used anti-mouse and anti-rabbit IR-dye-coupled antibodies from Li-Cor Biosciences. Protein bands were detected using the Li-Cor Aerius and intensities were quantified by measuring their integrated density with Adobe Photoshop Extended version 19.1.4.
Auxin induced degradation
Yeast strain FY3122 was grown in YPD at 30°C to a concentration of approximately 1 × 107 cells/ml (OD600=0.6), at which point cells were treated with 25 ∝M 3-Indoleacetic acid (IAA; SIGMA) or DMSO for 30 minutes prior to the zero timepoint or shifting to 37°C as described above. Samples were taken for Western (see above) and RT-qPCR analysis at the indicated timepoints described in the text. RT-qPCR was done as previously described (DeGennaro et al., 2013). Primer pairs for SSA4 and HSP82 genes were as previously published (Anandhakumar et al., 2016) and listed in Table S4.
Data management
All data analyses were managed using the Snakemake workflow management system (Koster and Rahmann, 2012), and are available at github.com/winston-lab.
TSS-seq library processing
Removal of adapter sequences and random hexamer sequences from the 3’ end of the read and 3’ quality trimming were performed using cutadapt (Martin, 2017). The random hexamer molecular barcode on the 5’ end of the read was then removed and processed using a custom Python script (Mayer et al., 2015). Reads were aligned to the combined S. cerevisiae and S. pombe reference genomes using Tophat2 without a reference transcriptome (Kim et al., 2013), and uniquely mapping reads were selected using SAMtools (Li et al., 2009). Reads mapping to the same location as another read with the same molecular barcode were identified as PCR duplicates and removed using a custom Python script (Mayer et al., 2015). Coverage of the 5’-most base, corresponding to the TSS, was extracted using bedtools genomecov (Quinlan and Hall, 2010) and normalized to the total number of reads uniquely mapping to the S. pombe genome. Quality statistics of raw, cleaned, non-aligning, and uniquely aligning non-duplicate reads were assessed using FastQC (Andrews, 2014).
TSS-seq peak calling
TSS-seq data for a single TSS tends to occur as a group of highly-correlated signals over a window of nucleotides, rather than at a single nucleotide. Therefore, for identification of TSSs and quantification for analyses such as differential expression, it is necessary to perform peak-calling. TSS-seq peak calling was performed using a 1-D watershed segmentation algorithm, followed by filtering for reproducibility by the Irreproducible Discovery Rate (IDR) method (Boleu et al., 2015; Li et al., 2011). First, a smoothed version of the TSS-seq coverage was generated for each sample using adaptive two-stage kernel density estimation with a discretized Gaussian kernel (pilot bandwidth = 10 nt, bandwidth = 10 nt, α = 0.2). The adaptive kernel adjusts the kernel bandwidth to be smaller in regions of high signal density and larger in regions of lower signal density (Silverman, 1986), allowing the smoother to better accommodate both ‘sharp’ TSSs where the signal is distributed over a relatively small window as well as ‘broad’ TSSs where the signal is more dispersed. Following smoothing, an initial set of peaks is formed by assigning all nonzero signal in the original, unsmoothed coverage to the nearest local maximum of the smoothed coverage, and taking the minimum and maximum genomic coordinate of the original coverage as the peak boundaries for each local maximum of the smoothed coverage. Peaks are then trimmed to the smallest genomic window that includes 95% of the original coverage, and the probability of the peak being generated by noise is estimated by a Poisson model where h, the expected coverage, is the maximum of the expected coverage over the chromosome and the expected coverage in the 2000 nt window upstream of the peak (as for the ChIP-seq peak caller MACS (Zhang et al., 2008b)). Finally, peaks are ranked by their significance under the Poisson model, and a final list of peaks for each condition is generated using the IDR method (IDR = 0.1) (Boleu et al., 2015; Li et al., 2011).
TSS-seq differential expression analysis
For TSS-seq differential expression, TSS-seq peak-calling was performed as described above for both S. cerevisiae and the S. pombe spike-ins. The read counts for each peak in each condition were used as the input to differential expression analysis by DESeq2 (Love et al., 2014), with the alternative hypothesis |log2(fold − change)| > 1.5 and a false discovery rate of 0.1. To normalize by spike-in, the size factors of the S. pombe spike-in counts were used as the size factors for S. cerevisiae, although we note that due to the median of ratios normalization method used in DESeq2, the major TSS-seq results of this work are still observed when the S. cerevisiae size factors are used.
ChIP-nexus library processing
Filtering for reads containing the constant region of the adapter on the 5’ end of the read, 3’ adapter removal and 3’ quality trimming were performed using cutadapt (Martin, 2017). The random pentamer molecular barcode on the 5’ end of the read was then removed and processed using a modified custom Python script (Mayer et al., 2015). Reads were aligned to the combined S. cerevisiae and S. pombe genomes using Bowtie2 (Langmead and Salzberg, 2012), and uniquely mapping reads were selected using SAMtools (Li et al., 2009). Reads mapping to the same location as another read with the same molecular barcode were identified as PCR duplicates and removed using a custom Python script (Mayer et al., 2015). Coverage of the 5’-most base, corresponding to the point of crosslinking, was extracted using bedtools genomecov (Quinlan and Hall, 2010). The median fragment size estimated by MACS2 (Zhang et al., 2008b) over all samples was used to generate coverage of factor protection and fragment midpoints, by extending reads to the fragment size, or by shifting reads by half the fragment size, respectively. Coverage was normalized to the total number of reads uniquely mapping to S. cerevisiae. Quality statistics of raw, cleaned, non-aligning, and uniquely aligning non-duplicate reads were assessed using FastQC (Andrews, 2014).
TFIIB ChIP-nexus peak-calling
TFIIB ChIP-nexus peak calling was performed using MACS2 (Zhang et al., 2008a), using 160 bp for the model-building bandwidth, 1000bp as the size of the large local region used to model expected counts, and the default false discovery rate of 0.05. Reads mapping to the same base were kept since PCR duplicates were filtered out using the molecular barcode. MACS2 was chosen over several ChIP-nexus and ChIP-exo specific peak calling tools because the specialized tools tended to split each TFIIB peak into multiple subpeaks, likely due to the multiple crosslinking points of TFIIB to the DNA (Rhee and Pugh, 2012).
Reannotation of S. cerevisiae TSSs using TSS-seq data
TSS-seq coverage from two replicates of a wild-type S. cerevisiae strain grown at 30°C in YPD (data not shown) was averaged and used to adjust the 5’ ends of an annotation file of major transcript isoforms based on TIF-seq data (Pelechano et al., 2013). The 5’ end of the original annotation was changed to the position of maximum TSS-seq signal in a window 250nt in each direction if the TSS-seq signal at that position was greater than the 95th percentile of all non-zero TSS-seq signal.
Classification of TSS-seq and TFIIB ChIP-nexus peaks into genomic categories
TSS-seq and TFIIB ChIP-nexus peaks were assigned to genomic categories based on their position relative to the transcript annotation described above and an annotation of all verified open reading frames (ORF) and blocked reading frames in S. cerevisiae (Crooks et al., 2004; Engel et al., 2014). First, genic regions were defined as follows: If a gene was present in both the transcript and ORF annotations, the genic region was defined as the interval (annotated TSS – 30 nucleotide, start codon]. If a gene was present in the transcript annotation but not the ORF annotation, the genic region was defined as the interval (annotated TSS-30nt, annotated TSS+30nt]. If a gene was present only in the ORF annotation, the genic region was defined as the interval (start codon-30nt, start codon]. For the purposes of peak classification, regions were considered overlapping if they had at least one base of overlap. Peaks were classified as genic if they overlapped a genic region on the same (TSS-seq) or either (TFIIB ChIP-nexus) strand. Peaks were classified as intragenic if they were not classified as a genic peak, and additionally overlapped an open or closed reading frame on the same (TSS) or either (TFIIB ChIP-nexus) strand. TSS-seq peaks were classified as antisense if they overlapped a transcript on the opposite strand. TSS-seq and TFIIB ChIP-nexus peaks were classified as intergenic if they did not overlap a transcript, reading frame, or genic region on either strand.
TSS information content
TSS-seq alignments were pooled for all replicates in a condition, and the DNA sequence flanking the position of every read overlapping TSS-seq peaks of a particular genomic category was extracted using SAMtools (Li et al., 2009) and bedtools (Quinlan and Hall, 2010). The information content of the sequences was quantified with WebLogo (Crooks et al., 2004), with the zeroth-order Markov model of the S. cerevisiae genomic sequence as the background composition. Sequence logos were plotted with helper functions from ggseqlogo (Wagih, 2017).
TFIIB ChIP-nexus differential binding analysis
For TFIIB ChIP-nexus differential binding analysis, TFIIB peaks were called as described above. A non-redundant list of peaks called in any condition was generated using bedtools, and the counts of fragment midpoints for each peak in each condition were used as the input to differential binding analysis by DESeq2 (Love et al., 2014), with the alternative hypothesis |log2(fold − change)| > 2 and a false discovery rate of 0.1. For estimation of changes in TFIIB binding upstream of TSS-seq peaks, TFIIB fragment midpoint counts were used as the input to differential binding analysis by DESeq2, using S. cerevisiae counts for size factors.
NET-seq library processing
Removal of adapter sequences from the 3’ end of the read and 3’ quality trimming were performed using cutadapt (Martin, 2017). Reads were aligned to the S. cerevisiae genome using Tophat2 without a reference transcriptome (Kim et al., 2013), and uniquely mapping reads were selected using SAMtools (Li et al., 2009). Coverage of the 5’-most base of the read, corresponding to the 3’-most base of the nascent RNA and the active site of elongating RNA polymerase, was extracted using bedtools genomecov (Quinlan and Hall, 2010) and normalized to the total number of uniquely mapped reads. Quality statistics of raw, cleaned, non-aligning, and uniquely aligning reads were assessed using FastQC (Andrews, 2014).
MNase-seq library processing
Paired-end reads were demultiplexed using fastq-multx (Aronesty, 2013), allowing one mismatch to the barcode. Filtering for the barcode on read 2 and 3’ quality trimming were performed with cutadapt (Martin, 2017). Reads were aligned to the combined S. cerevisiae and S. pombe genome using Bowtie 1 (Langmead et al., 2009), and correctly paired reads selected using SAMtools (Li et al., 2009). Coverage of nucleosome protection and nucleosome dyads were extracted using bedtools (Quinlan and Hall, 2010) and custom shell scripts to get the entire fragment or the midpoint of the fragment, respectively. Smoothed nucleosome dyad coverage was generated by smoothing dyad coverage with a Gaussian kernel of 20 bp bandwidth. Coverage was normalized to the total number of correctly paired S. pombe fragments. Quality statistics of raw, cleaned, non-aligning, and correctly pairing reads were assessed using FastQC (Andrews, 2014).
MNase-seq quantification
Quantifications of nucleosome occupancy, fuzziness, and position shifts were calculated using DANPOS2 (Chen et al., 2013) with the total counts in mutant libraries scaled by the mean observed spike-in percentage in the mutant libraries over the mean observed spike-in percentage in the wild-type libraries for spike-in normalization.
Clustering of MNase-seq signal at spt6-1004 intragenic TSSs
Spike-in normalized MNase-seq dyad signal in the window 150bp to either side of the summit of the 6059 intragenic TSS-seq peaks upregulated in spt6-1004 over wild-type was binned by taking the mean signal in non-overlapping 5bp bins, and then averaged by taking the mean of two replicates (spt6-1004) or one experiment (wild-type). The wild-type and spt6-1004 data were used as equally weighted 6059×60 input layers to a super-organizing map (SOM)(Wehrens and Buydens, 2007) trained using the input data to assign similar MNase-seq observations in 60-dimensional input space to similar nodes in a 2-dimensional (6×8) rectangular grid. The 48 ‘code vectors’ representing the typical MNase-seq pattern for each node were then clustered by agglomerative hierarchical clustering using sum of squares distance and Ward linkage. The resulting dendrogram was cut to produce the two clusters of MNase-seq signal shown in Figure 5. The choice to cut the dendrogram to produce two clusters was made because clusters created from deeper cuts tended to have nucleosome phasing patterns similar to the original two clusters. We note that the two clusters are stable under repeated training of the SOM with different random seeds. By chance, some random seeds will result in a third cluster which joins after the two major clusters have joined in the hierarchical clustering. However, this cluster is usually much smaller than the major clusters (<20 iTSSs) and can be grouped visually into one of the two major phasing patterns.
Intragenic TSS position bias
As TSS-seq peaks are required to not overlap genic regions in order to be classified as intragenic, the expected distribution if intragenic TSSs were randomly distributed along the length of an ORF is not uniform. Therefore, the expected random distribution of intragenic TSSs was determined by taking all position of the ORF that the TSS could have taken and still been called intragenic. The random distribution was then compared to the observed distribution of intragenic starts by binning start locations to the nearest tenth of a percentage of relative distance along the ORF, and applying a permutation test on the chi-squared test statistic.
Motif enrichment
FIMO (Grant et al., 2011) was used to search the S. cerevisiae genome for 3010 motifs from six databases (de Boer and Hughes, 2012; MacIsaac et al., 2006; Newburger and Bulyk, 2009; Pachkov et al., 2013; Teixeira et al., 2018; Zhu and Zhang, 1999). The zeroth-order Markov model of the S. cerevisiae genome sequence was used as a background model, with a p-value cutoff of 1e-5. For determining the enrichment of motif sites upstream of TSSs, the regions extending 200 base pairs upstream of TSS summits were taken and merged if they were overlapping. Motifs were considered to be present in a region if the entire motif was overlapping the region. The frequency of motif occurrences in the regions of interest was compared to the frequency of occurrences in the regions upstream of 6000 randomly chosen locations, using Fisher’s exact test.
Enrichment of TATA boxes
Enrichment of TATA boxes was tested as for the other motifs described above, except for the following differences: First, the query motif used was TATAWAWR, where the ambiguous bases are equiprobable. Second, the p-value was 6e-4, chosen because it was the threshold required for only exact matches to be returned. Third, the TATA motif was required to be on the sense strand relative to the TSS in order to be counted as a match.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical tests employed for each experiment are described in the figure legends or in the methods section. For TSS-seq, NET-seq, and all ChIP-nexus experiments, two biological replicates were sequenced for each condition. For MNase-seq, one experiment was sequenced for wild-type and two replicates were sequenced for spt6-1004.
DATA AND SOFTWARE AVAILABILITY
The raw sequencing data reported in this paper has been deposited at the NCBI Gene Expression Omnibus, accession number GSE115775. An archived version of all data analyses needed to generate the figures in this paper starting from the raw data is deposited at Zenodo: https://doi.org/10.5281/zenodo.1325930. Raw image data are available at Mendeley: http://dx.doi.org/10.17632/k5686bfpcv.2
Supplementary Material
Highlights.
Upon depletion of Spt6 thousands of intragenic promoters are activated.
Sequence features plus altered chromatin structure likely lead to this activation.
Spt6 depletion also causes decreased expression of most genic promoters.
This decrease in expression likely results from competition for initiation factors.
ACKNOWLEDGMENTS
We thank Josh Arribere and Wendy Gilbert for critical advice on adapting TSS-seq from TL-seq; Burak Alver, Peter Park, and Julia di Iulio for bioinformatics support; Kevin Harlen, Ameet Shetty, and Rajaraman Gopalakrishnan for advice and discussions; Mary Couvillion and Blake Tye for helpful comments on the manuscript; and Natalia Reim for providing yeast strain FY3122. Part of this research was conducted on the O2 High Performance Computer Cluster supported by the Research Computing Group at Harvard Medical School. This work was supported by an American Cancer Society Fellowship to S.M.D.; NIH fellowship F32GM119291 to O.V., NIH grant R01HG007173 to L.S.C., and NIH grant R01GM032967 to F.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes five figures and three tables and can be found with this article online.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adkins MW, and Tyler JK (2006). Transcriptional activators are dispensable for transcription in the absence of Spt6-mediated chromatin reassembly of promoter regions. Mol Cell 21, 405–416. [DOI] [PubMed] [Google Scholar]
- Anandhakumar J, Moustafa YW, Chowdhary S, Kainth AS, and Gross DS (2016). Evidence for Multiple Mediator Complexes in Yeast Independently Recruited by Activated Heat Shock Factor. Mol Cell Biol 36, 1943–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2014). FastQC: a quality control tool for high throughput sequence data. In Babraham Bioinformatics
- Andrulis ED, Guzman E, Doring P, Werner J, and Lis JT (2000). High-resolution localization of Drosophila Spt5 and Spt6 at heat shock genes in vivo: roles in promoter proximal pausing and transcription elongation. Genes Dev 14, 2635–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardehali MB, Yao J, Adelman K, Fuda NJ, Petesch SJ, Webb WW, and Lis JT (2009). Spt6 enhances the elongation rate of RNA polymerase II in vivo. EMBO J 28, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E (2013). Comparison of sequencing utility programs. The Open Bioinformatics Journal 7, 1–8. [Google Scholar]
- Arribere JA, and Gilbert WV (2013). Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res 23, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, and Struhl K, eds. (1991). Current Protocols in Molecular Biology (New York, Greene Publishing Associates and Wiley-Interscience; ). [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. (2008). A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, and Pugh BF (2004). Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709. [DOI] [PubMed] [Google Scholar]
- Boleu N, Kundaje A, and Bickel PJ (2015). Irreproducible discovery rate
- Bortvin A, and Winston F (1996). Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science 272, 1473–1476. [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. [DOI] [PubMed] [Google Scholar]
- Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, and de Almeida SF (2013). Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41, 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Rufiange A, Huang H, Rajashankar KR, Nourani A, and Patel DJ (2015). Structure-function studies of histone H3/H4 tetramer maintenance during transcription by chaperone Spt2. Genes Dev 29, 1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, and Winston F (2008). Chromatin-and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 6, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Sutton A, Sternglanz R, and Prelich G (2006). The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol Cell Biol 26, 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnone-Post PA, and Osley MA (1996). Mutations in the SPT4, SPT5, and SPT6 genes alter transcription of a subset of histone genes in Saccharomyces cerevisiae. Genetics 143, 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion MT, and Churchman LS (2017). Mitochondrial Ribosome (Mitoribosome) Profiling for Monitoring Mitochondrial Translation In Vivo. Curr Protoc Mol Biol 119, 4 28 21–24 28 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer CG, and Hughes TR (2012). YeTFaSCo: a database of evaluated yeast transcription factor sequence specificities. Nucleic Acids Res 40, D169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGennaro CM, Alver BH, Marguerat S, Stepanova E, Davis CP, Bahler J, Park PJ, and Winston F (2013). Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol Cell Biol 33, 4779–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold ML, Koch M, Loeliger E, Cura V, Winston F, Cavarelli J, and Romier C (2010). The structure of an Iws1/Spt6 complex reveals an interaction domain conserved in TFIIS, Elongin A and Med26. EMBO J 29, 3979–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duina AA (2011). Histone Chaperones Spt6 and FACT: Similarities and Differences in Modes of Action at Transcribed Genes. Genet Res Int 2011, 625210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M, Zhu W, Hasegawa J, Watanabe H, Kim DK, Aida M, Inukai N, Narita T, Yamada T, Furuya A, et al. (2004). Human Spt6 stimulates transcription elongation by RNA polymerase II in vitro. Mol Cell Biol 24, 3324–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, Dwight SS, Hitz BC, Karra K, Nash RS, et al. (2014). The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 4, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie AE, Stewart BG, Scott CF, and Rose MD (1999). The two forms of karyogamy transcription factor Kar4p are regulated by differential initiation of transcription, translation, and protein turnover. Mol Cell Biol 19, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Johnston J, and Zeitlinger J (2015). ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotechnol 33, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig BP, and Fischer T (2013). The great repression: chromatin and cryptic transcription. Transcription 4, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho B, Baryshnikova A, and Brown GW (2018). Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst 6, 192–205 e193. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovska I, Jacques PE, Rando OJ, Robert F, and Winston F (2011). Control of chromatin structure by spt6: different consequences in coding and regulatory regions. Mol Cell Biol 31, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, and Struhl K (1995). Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J 14, 2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MM, Christensen MS, Bonven B, and Jensen TH (2008). Requirements for chromatin reassembly during transcriptional downregulation of a heat shock gene in Saccharomyces cerevisiae. FEBS J 275, 2956–2964. [DOI] [PubMed] [Google Scholar]
- Jensen TH, Jacquier A, and Libri D (2013). Dealing with pervasive transcription. Mol Cell 52, 473–484. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Watanabe S, Kaplan CD, Peterson CL, and Robert F (2015). The Histone Chaperones FACT and Spt6 Restrict H2A.Z from Intragenic Locations. Mol Cell 58, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, and Winston F (2003). Transcription elongation factors repress transcription initiation from cryptic sites. Science 301, 1096–1099. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, Morris JR, Wu C, and Winston F (2000). Spt5 and spt6 are associated with active transcription and have characteristics of general elongation factors in D. melanogaster. Genes Dev 14, 2623–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. (2009). The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lu C, Srinivasan S, Awe S, Brehm A, and Fuller MT (2017). Blocking promiscuous activation at cryptic promoters directs cell type-specific gene expression. Science 356, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee BB, Oh YM, Zhu C, Steinmetz LM, Lee Y, Kim WK, Lee SB, Buratowski S, and Kim T (2016). Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nat Commun 7, 13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J, and Rahmann S (2012). Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, and Nislow C (2007). A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Pattenden SG, Seidel C, and Workman JL (2007). Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev 21, 1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Borwn JB, Huang H, and Bickel PJ (2011). Measuring reproducibiliyt of high-throughput experiments. The Annals of Applied Statistics 5, 1752–1779. [Google Scholar]
- Lickwar CR, Rao B, Shabalin AA, Nobel AB, Strahl BD, and Lieb JD (2009). The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS One 4, e4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, and Fraenkel E (2006). An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C, Feuerbach F, Ma L, Saveanu C, and Jacquier A (2015). Quality control of transcription start site selection by nonsense-mediated-mRNA decay. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2017). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, and Pugh BF (2008). A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, and Churchman LS (2015). Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, and Cramer P (2010). Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol 17, 1272–1278. [DOI] [PubMed] [Google Scholar]
- McCullough L, Connell Z, Petersen C, and Formosa T (2015). The Abundant Histone Chaperones Spt6 and FACT Collaborate to Assemble, Inspect, and Maintain Chromatin Structure in Saccharomyces cerevisiae. Genetics 201, 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SL, Hepperla AJ, Huang J, Dronamraju R, Adams AT, Kulkarni VG, Davis IJ, and Strahl BD (2017). H3K36 Methylation Regulates Nutrient Stress Response in Saccharomyces cerevisiae by Enforcing Transcriptional Fidelity. Cell Rep 19, 2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SM, Close D, Xin H, Formosa T, and Hill CP (2010). Structure and biological importance of the Spn1-Spt6 interaction, and its regulatory role in nucleosome binding. Mol Cell 40, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight K, Liu H, and Wang Y (2014). Replicative stress induces intragenic transcription of the ASE1 gene that negatively regulates Ase1 activity. Curr Biol 24, 1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Deng N, Ooi WF, Lin SJ, Xing M, Xu C, Qamra A, Tay ST, Malik S, Wu J, et al. (2014). Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat Commun 5, 4361. [DOI] [PubMed] [Google Scholar]
- Murray SC, Serra Barros A, Brown DA, Dudek P, Ayling J, and Mellor J (2012). A pre-initiation complex at the 3’-end of genes drives antisense transcription independent of divergent sense transcription. Nucleic Acids Res 40, 2432–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, and Bulyk ML (2009). UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37, D77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Nourani A, Robert F, and Winston F (2006). Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol Cell Biol 26, 1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachkov M, Balwierz PJ, Arnold P, Ozonov E, and van Nimwegen E (2013). SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res 41, D214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak R, Singh P, Ananthakrishnan S, Adamczyk S, Schimmel O, and Govind CK (2018). Acetylation-Dependent Recruitment of the FACT Complex and Its Role in Regulating Pol II Occupancy Genome-Wide in Saccharomyces cerevisiae. Genetics. [DOI] [PMC free article] [PubMed]
- Pelechano V, Wei W, and Steinmetz LM (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales R, Erickson B, Zhang L, Kim H, Valiquett E, and Bentley D (2013). Gene promoters dictate histone occupancy within genes. EMBO J 32, 2645–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praekelt UM, and Meacock PA (1990). HSP12, a new small heat shock gene of Saccharomyces cerevisiae: analysis of structure, regulation and function. Mol Gen Genet 223, 97–106. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ (2010). Genome-wide mapping of nucleosomes in yeast. Methods Enzymol 470, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal Y, Chereji RV, Valabhoju V, Qiu H, Ocampo J, Clark DJ, and Hinnebusch AG (2018). Gcn4 Binding in Coding Regions Can Activate Internal and Canonical 5’ Promoters in Yeast. Mol Cell 70, 297–311 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, and Pugh BF (2012). ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol Chapter 21, Unit 21 24. [DOI] [PMC free article] [PubMed]
- Rowley A, Singer RA, and Johnston GC (1991). CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol Cell Biol 11, 5718–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdano MA, Fulcher JM, Palani S, Chandrasekharan MB, Parnell TJ, Whitby FG, Formosa T, and Hill CP (2017). A novel SH2 recognition mechanism recruits Spt6 to the doubly phosphorylated RNA polymerase II linker at sites of transcription. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, and Iyer VR (2008). Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol 6, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman BW (1986). Density estimation for statistics and data analysis (Chapman and Hall; ). [Google Scholar]
- Tamarkin-Ben-Harush A, Vasseur JJ, Debart F, Ulitsky I, and Dikstein R (2017). Cap-proximal nucleotides via differential eIF4E binding and alternative promoter usage mediate translational response to energy stress. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MC, Monteiro PT, Palma M, Costa C, Godinho CP, Pais P, Cavalheiro M, Antunes M, Lemos A, Pedreira T, et al. (2018). YEASTRACT: an upgraded database for the analysis of transcription regulatory networks in Saccharomyces cerevisiae. Nucleic Acids Res 46, D348–D353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo D, and Hughes TR (2009). G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 10, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov AM, Thompson DA, Socha A, Regev A, and Rando OJ (2010). The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol 8, e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwimana N, Collin P, Jeronimo C, Haibe-Kains B, and Robert F (2017). Bidirectional terminators in Saccharomyces cerevisiae prevent cryptic transcription from invading neighboring genes. Nucleic Acids Res 45, 6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, and Nislow C (2013). A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9, e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Li H, Gogol MM, and Workman JL (2016). Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat Commun 7, 13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O (2017). ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647. [DOI] [PubMed] [Google Scholar]
- Wehrens R, and Buydens LMC (2007). Self-and super-organizing maps in R: the kohonen package. In Journal of Statistical Software, pp. 1–19.21494410
- Werner-Washburne M, Stone DE, and Craig EA (1987). Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol Cell Biol 7, 2568–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner T, Lee W, Obenauf AC, Ran L, Murali R, Zhang QF, Wong EW, Hu W, Scott SN, Shah RH, et al. (2015). Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 526, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, Itie-Youten A, Wakeham A, Arsenian-Henriksson M, Melino G, et al. (2010). Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 24, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, and Impey S (2011). KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J 30, 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarragudi A, Parfrey LW, and Morse RH (2007). Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res 35, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Lucas JS, and Jones KA (2008). The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev 22, 3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdell ML, Kizer KO, Kisseleva-Romanova E, Fuchs SM, Duro E, Strahl BD, and Mellor J (2008). Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol Cell Biol 28, 4915–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fletcher AG, Cheung V, Winston F, and Stargell LA (2008a). Spn1 regulates the recruitment of Spt6 and the Swi/Snf complex during transcriptional activation by RNA polymerase II. Mol Cell Biol 28, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008b). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, and Struhl K (2009). Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16, 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Dietrich FS (2005). Mapping of transcription start sites in Saccharomyces cerevisiae using 5’ SAGE. Nucleic Acids Res 33, 2838–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, and Zhang MQ (1999). SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 15, 607–611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.