

Summary
ADP-ribosylation of proteins can profoundly impact their function and serves as an effective mechanism by which bacterial toxins impair eukaryotic cell processes. Here we report the discovery that bacteria also employ ADP-ribosylating toxins against each other during interspecies competition. We demonstrate that one such toxin from Serratia proteamaculans interrupts the division of competing cells by modifying the essential bacterial tubulin-like protein, FtsZ, adjacent to its protomer interface, blocking its capacity to polymerize. The structure of the toxin in complex with its immunity determinant revealed two distinct modes of inhibition: active site occlusion and enzymatic removal of ADP-ribose modifications. We show that each is sufficient to support toxin immunity; however, the latter additionally provides unprecedented broad protection against non-cognate ADP-ribosylating effectors. Our findings reveal how an interbacterial arms race has produced a unique solution for safeguarding the integrity of bacterial cell division machinery against inactivating post-translational modifications.
In Brief
A widely conserved toxin for interbacterial competition targets proteins for ADP-ribosylation, while antitoxins fight back using dual mechanisms of active site occlusion and enzymatic reversal of the modification, conferring broad immunity to diverse toxins.
Graphical Abstract
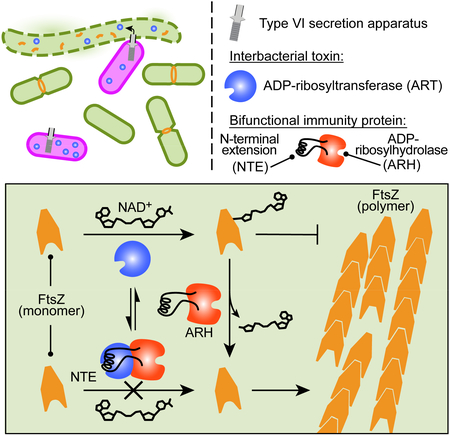
Introduction
Microbial communities are of fundamental importance to virtually all natural ecosystems. Although the term “communities” implies cooperation, it is now appreciated that antagonistic behavior directed against closely interacting microbes serves as a key driver of composition and resilience within these consortia (Foster and Bell, 2012). Reflecting this central role of antagonism, pathways dedicated to interspecies antibacterial toxin delivery occur broadly throughout both Gram-positive and -negative bacteria (Jones et al., 2017; Russell et al., 2014; Whitney et al., 2017). These include the Esx pathway, prevalent in the phyla Firmicutes and Actinobacteria, and the type VI secretion system (T6SS), which is widespread in the phyla Proteobacteria and Bacteroidetes. Pathogenic bacteria may use these pathways to facilitate invasion of or dominance within polymicrobial infections, whereas for commensal organisms they can dictate compatibility within consortia (Sana et al., 2016; Speare et al., 2018; Verster et al., 2017).
Contact-dependent interbacterial competition is generally mediated by secreted toxic effector proteins. Complex export machinery enables these effectors to access target molecules that reside within the cell envelope of competitor cells (Russell et al., 2011; Whitney et al., 2017). The T6SS and Esx pathways deliver effectors indiscriminately; therefore, bacteria harboring effectors transported by these systems require a means of inhibiting self-intoxication. To date, protection against all characterized effectors of these systems has been shown to derive from the production of specific immunity determinants that bind cognate toxins and inhibit their enzymatic function (Alcoforado Diniz et al., 2015). Cognate effector and immunity proteins, herein referred to as E–I pairs, are without known exception encoded adjacently.
Identification and characterization of toxic effectors from a growing number of species has revealed that these proteins exhibit tremendous sequence and biochemical diversity. For instance, large superfamilies of toxins that share little homology beyond conserved catalytic residues target amide or glycosidic bonds in the bacterial cell wall, DNA, membrane phospholipids and the cellular metabolite NAD+ (Alcoforado Diniz et al., 2015; Whitney et al., 2015). By targeting highly conserved, essential primary metabolites, interbacterial effectors allow bacteria to compete effectively against a wide phylogenetic cross-section of organisms. Multiple toxins targeting a broad array of cellular structures are often employed simultaneously by a single delivery pathway, which promotes bacterial competitiveness via synergistic toxigenic activities and widens the environmental conditions under which at least one effector is efficacious (LaCourse et al., 2018). Despite a high degree of toxin diversity, genes encoding effector proteins of interbacterial antagonistic systems are often readily recognizable in bacterial genomes. These genes are typically characterized by 5’ regions encoding conserved effector targeting domains and by polymorphic toxin modules encoded at their 3’ ends. Targeting domains linked to T6SS effectors include PAAR, RHS, and VgrG, whereas LXG is the sole domain so far implicated in the recognition of antibacterial substrates by the Esx pathway (Cianfanelli et al., 2016; Whitney et al., 2017).
Interestingly, there is substantial overlap in the modes of action of bacterial toxins that target eukaryotic host cells and those that target other bacteria. For instance, structurally related NADases of Pseudomonas aeruginosa and Mycobacterium tuberculosis are employed to neutralize competing Gram-negative bacterial cells and to induce necrotic cell death in human macrophages, respectively (Sun et al., 2015; Whitney et al., 2015). In some cases, the same toxin may be capable of targeting cells of both domains, as described for T6SS-delivered phospholipases (Bleves, 2016). These results point toward the important role that interbacterial antagonism can play in shaping the toxin repertoire and emergence of bacterial pathogens. Here, we describe an interbacterial toxin that inactivates the critical cell division protein, FtsZ, via ADP-ribosyltransferase (ART) activity. This toxin is the first characterized member of a large family of related ART toxins, with phylogenetically broadly distributed representatives bearing hallmarks of interbacterial effectors exported by the T6SS and the Esx pathway. Proteins with this activity, such as cholera and diphtheria toxins, are known to be widely utilized by pathogens (Simon et al., 2014); however, proteins with this activity have not previously been demonstrated to participate in interbacterial antagonism. Finally, structural analyses reveal that protection against ART toxins is conferred by bifunctional immunity determinants, which concomitantly utilize active site occlusion to inactivate cognate effector proteins and a promiscuous ADP-ribosylhydrolase (ARH) activity to reverse ART-catalyzed modifications more broadly.
Results
Predicted ART and ARH proteins are associated with interbacterial toxin delivery systems
During the course of our efforts to define mechanisms of interbacterial competition, we found an uncharacterized protein domain common to predicted effectors of diverse contact-dependent antagonism pathways. In Gram-negative bacteria, this domain is found in proteins also containing PAAR, RHS, or both of these domains, indicative of intercellular delivery via the T6SS pathway (Figure 1A and Table S1) (Cianfanelli et al., 2016). In Gram-positive species, proteins with this domain additionally contain LXG domains, indicative of transit through the Esx pathway (Whitney et al., 2017). In all cases, the uncharacterized domain resides at the C-terminus of the protein – the stereotyped position in each of these systems for the toxin domain of their substrates. Subsequent sequence analyses showed that the domain possesses a constellation of conserved amino acids characteristic of RSE family ART enzymes (Figure 1B) (Cohen and Chang, 2018). Bacterial members of this family include eukaryotic cell-targeting toxins; however, members of this family are not known to act between bacterial cells.
Figure 1.

Proteins sharing conserved motifs with ART and ARH proteins are associated with interbacterial toxin delivery pathways. (A) and (C) Scaled depiction of representative genomic loci from the indicated species (Serratia proteamaculans, Pseudomonas caryophylli, Burkholderia mutltivora, Listeria monocytogenes) encoding predicted ART (yellow) and ARH (blue) domain-containing proteins. Regions encoding N-terminal domains of T6SS (PAAR, RHS) and Esx system (LXG) substrates shaded in grey. Black bars designate location of bases encoding the noted residues conserved in ART and ARH proteins; residues targeted for mutagenesis in this study depicted in red. (B) and (D) Sequence logos generated from alignments of ART (B) and ARH (D) family proteins associated with bacterial contact dependent antagonism pathways (locus tag numbers for genes encoding sequences used provided in Table S1). Sequences from characterized ART (CTX toxin) and ARH (DraG) shown below for reference. See also Table S1.
ADP-ribosylation can be a reversible modification, and enzymes belonging to the ARH family are known to catalyze ADP-ribose removal (Cohen and Chang, 2018). Interestingly, we noted that the genes immediately downstream of predicted antibacterial RSE family genes encode ARH family proteins (Figure 1C and Table S1). Immunity determinants of interbacterial toxins are typically encoded in this position, suggesting that the role of these proteins may be to provide protection from ART-based intoxication via hydrolysis of ADP-ribose adducts. The active site of ARH proteins contains metal ions coordinated by a network of conserved, predominately acidic, residues (Berthold et al., 2009). These amino acids are conserved in the ARH family proteins we identified, further suggesting an enzymatic mechanism of immunity (Figure 1D). Taken together with prior bioinformatic analyses (Aravind et al., 2015), these observations led us to hypothesize that toxins with ART activity are widespread mediators of bacterial competition.
A predicted ART from Serratia proteamaculans mediates T6S-dependent interbacterial intoxication
To investigate the potential for ART enzymes to mediate interbacterial antagonism, we initially focused on a predicted ART toxin of Serratia proteamaculans. This protein, which we termed Tre1 (type VI secretion ADP-ribosyltransferase effector 1), contains an N-terminal PAAR domain, a configuration consistent with known T6SS effector proteins (Figure 1A). Relative to control E. coli cultures, those expressing Tre1 or its c-terminal toxin domain (Tre1tox) displayed a significant loss in colony forming units (Figure 2A). Time-lapse microscopy revealed that this loss in viability was associated with both cellular elongation and lysis (Figures 2B and S1A, Videos S1 and S2). To determine whether the toxicity of Tre1 derives from its predicted ART activity, we expressed a Tre1tox protein in which the catalytic glutamic acid within the RSE motif is substituted with glutamine (Tre1tox E415Q) (Tsuge et al., 2003). Despite expression levels matching that of Tre1tox, this predicted catalytically inactive protein did not reduce E. coli viability, nor did it promote cell elongation and lysis to levels approaching the wild-type (Figures 2A, 2C, S1A and S1B, Videos S1 and S2). Co-expression of Tre1tox with the predicted ARH family protein encoded downstream, which we subsequently refer to as Tri1, restored the viability of E. coli to that of the control (Figures 2A and S1A). Together, these data demonstrate that Tre1 and Tri1 are an E–I pair, and they are consistent with the toxicity of Tre1 deriving from its ART-like domain.
Figure 2.

A predicted ART domain-containing substrate of the T6SS is an antibacterial toxin that promotes competitiveness of S. proteamaculans. (A) Viable E. coli cells recovered from plating cultures carrying plasmids expressing the indicated proteins on inducing media (c.f.u. = colony forming units). (B) and (C) Representative micrographs of E. coli cells expressing Tre1tox (B) or Tre1tox E415Q (C). Frames were acquired 200 min after induction of protein expression. Scale bar = 2 μm. (D) Relative competitiveness of the indicated donor (“D”) and recipient (“R”) strains of S. proteamaculans grown in co-culture on a solid surface for 6 h. Competitive index was determined by comparing final and initial c.f.u. ratios of the two strains. (E) Interspecies competition experiments between the indicated S. proteamaculans donor strains grown in co-culture with the noted E. coli recipients, quantified as in D. Data in A, D and E are represented as means ± standard deviation (SD). Asterisks in D and E indicate statistically significant differences between competitive indices of a given donor strain toward the indicated recipients (p<0.05). For experiments shown in A, D and E, n ≥ 3. See also Figure S1 and Videos S1 and S2.
We next sought to determine whether Tre1 functions as an interbacterial T6SS toxin. To this end, we used allelic exchange to generate a strain of S. proteamaculans bearing a deletion of the tre1 tri1 loci (Figure S1C). Growth competition assays performed under contact-promoting conditions revealed that this strain exhibits a fitness defect when incubated with wild-type, but not with S. proteamaculans lacking a functional T6SS (ΔicmF) (Figure 2D). To determine the contribution of the putative catalytic activity of Tre1 to this phenotype, we generated a S. proteamaculans strain expressing the E415Q allele from the native tri1 chromosomal locus (tre1E415Q). Tre1E415Q was produced at the same level as the native protein, yet unlike wild-type S. proteamaculans, the strain producing this variant displayed equal fitness with Δtre1 Δtri1 in co-culture (Figures 2D and S1D). These data show that Tre1 and Tri1 can function as a T6S-dependent E–I pair between cells of S. proteamaculans.
In other bacteria, T6S-exported toxins can be delivered to competing Gram-negative organisms, and thus enhance the fitness of the producing organism during co-culture with other species. Indeed, we found that inactivation of the S. proteamaculans T6SS significantly diminished fitness in co-culture with E. coli (Figure 2E). We next measured the contribution of the toxic activity of Tre1 to this phenotype. The S. proteamaculans tre1E415Q strain was employed in this experiment, since removing a PAAR domain-containing effector can lead to a generalized structural perturbation of the secretory apparatus. Our results showed that Tre1 inactivation yields a strain with fitness intermediate to that of wild-type and ΔicmF strains, consistent with Tre1 constituting one component of a multi-part effector payload delivered by this S. proteamaculans T6S pathway (Figure 2E). Ectopic expression of Tri1 in E. coli abrogated the tre1-dependent fitness advantage of wild type, further confirming the contribution of this toxin to antagonism of E. coli by S. proteamaculans (Figure 2E).
Structure of the Tre1–Tri1 complex reveals two distinct immunity mechanisms
Bioinformatic analysis of Tri1 suggested that, unlike previously characterized immunity determinants of toxins delivered between bacteria, it could provide protection from intoxication via an enzymatic mechanism. However, we found that Tri1 stably interacts with Tre1, a feature shared with immunity proteins that function by inhibiting the active site of their cognate toxin (Figure S2A and S2B). To gain insight into both the mechanism of Tre1-induced toxicity and of Tri1-mediated immunity, we determined a 2.3 Å X-ray crystal structure of Tre1tox in complex with Tri1 (Figure 3A). Phases were obtained experimentally using selenomethionine-substituted protein and molecular replacement with the resulting model was used to solve the structure of the native crystal in a second space group (Table S3). In our structure, the exclusively α-helical Tri1 and mixed α,β-Tre1 share an extensive interaction surface (2,604 Å2) consisting of two distinct regions: the plane formed between the two globular domains of the proteins (1,178Å2) and an interface formed by a 65 amino acid N-terminal extension (NTE) of Tri1 (1,550 Å2). This striking structural feature of Tri1 extends away from the core of the immunity protein, reaches into the active site of the toxin, and at its distal end returns to pack into the Tre1tox–Tri1 globular domain interface (Figure 3A and 3B).
Figure 3.

Tri1 provides immunity to intoxication by Tre1 through two structurally distinct mechanisms. (A) Ribbon diagram representation of the X-ray crystal structure of Tre1tox (yellow) in complex with Tri1 (blue). Mg2+ ion and coordinating residues in the Tri1 active site shown in red. (B) Space filling depiction of Tre1 showing occlusion of active site residues R356, S381, and E415 (red) by the N-terminal extension (NTE) of Tri1 (blue). (C) and (D) Structural alignments of Tre1 (C) and Tri1 (D) with previously characterized ART and ARH proteins, respectively (grey). Region of Tri1 comprising the NTE is indicated. (E) Diagram of regions shared between Tri1 proteins and previously characterized ARH proteins (blue; NTE found only predicted ARH domain-containing immunity proteins). Gaps present in sequence alignments not shown. Conservation at each position (15 aa window) is shown above for all NTE-containing ARH domain proteins. (F) Proportion of the NTE segment consisting of the indicated secondary structure elements. For Tri1-Sp (dark grey), percentages determined from current structural analysis (PDB: 6DRE); for other Tri1 homologs (light grey), percentages represent average values from prediction analyses performed using GeneSilico Metaserver (n = 22 representative sequences). (G) Magnification of the active site of Tre1tox in complex with Tri1, showing electrostatic and hydrogen bond interactions between Arg32 of Tri1 and the catalytic glutamic acid (Glu415) of Tre1tox. (H) E. coli cells recovered following induction of heterologously expressed Tre1 and the indicated Tri1 alleles or empty vector control. Left panel (dark grey), expression of both proteins controlled by the pBAD promoter and induced by 0.2% arabinose; right panel (light grey), Tre1 expression controlled by the T7 promoter and induced by IPTG (0.01 mM) and Tri1 controlled by pBAD and induced by arabinose (indicated concentrations). Data are presented as means ± SD. Asterisks indicate significant differences in viability between populations expressing the indicated Tri1 variant proteins and the empty vector control, when induced with 0.1% arabinose (p<0.05, n ≥ 3). See also Figure S2 and Table S3.
The structure revealed that Tre1 shares a high degree of similarly to the catalytic domains of RSE-type ART enzymes (Figure 3C) (Cohen and Chang, 2018). Among this large and diverse group of proteins found in both bacteria and eukaryotes, Tre1 is most similar to a subset of those which ADP-ribosylate arginine residues, iota toxin from C. perfringens (2.1 Å r.m.s.d., 148 residues) (Tsuge et al., 2003). Like these proteins and ARTs more broadly, the active site of Tre1 is formed at the interface of two sub-domains, an N-terminal α-helical domain and a C-terminal domain that includes the β-sheet core of the protein. Examination of the predicted NAD+-binding site of Tre1 revealed conserved placement of the canonical R–S–E catalytic triad and surrounding motifs (Figure 3C).
The structure of Tri1 supported our bioinformatics predictions that place the protein in the ARH family. Indeed, the single tertiary structural distinction from DraG, a bacterial ARH protein involved in the regulation of nitrogen fixation, is that DraG lacks the NTE of Tri1 (Figure 3D and E). DraG and related ARH proteins possess a dinuclear Mn2+ or Mg2+ active site in which the metal ions are coordinated by a network of conserved, predominately acidic, residues (Berthold et al., 2009). These residues and their overall topology is conserved between DraG and Tri1; however, we identified only one Mg2+ in the Tri1 active site (Figure 3D and E).
All T6SS immunity proteins characterized to date protect against intoxication by a single mechanism – occlusion of the active site of cognate effectors. The structure of Tri1 suggested that this protein may employ two mechanisms of immunity against Tre1: active site occlusion via its NTE and removal of the Tre1 product via its apparent ARH active site. To test this hypothesis, we evaluated the ability of Tri1 variants defective in these mechanisms individually or in combination to protect against Tre1-based intoxication. Removal of the NTE yielded insoluble protein, thus we scanned contacts between the active site of Tre1 and α2 within the Tri1 NTE to identify candidate substitutions that could specifically disrupt the interaction. We noted that the guanidinium group of Arg32 of Tri1 engages in electrostatic and bidentate hydrogen bond interactions with the catalytic glutamic acid of the toxin (Glu415) (Figure 3G). Based on these observations, we speculated that substitution of this residue with glutamate (Tri1R32E) would disrupt the Tri1 NTE–Tre1 active site interaction. Inactivation of the putative ARH active site of Tri1 was achieved by substituting aspartate 161 with asparagine (Tri1D161N); an analogous substitution of this key catalytic residue within DraG reduces the activity of that enzyme to undetectable levels (Berthold et al., 2009).
Heterologous expression in E. coli was employed to evaluate the ability of the Tri1 variants to protect cells from intoxication by Tre1tox. Despite expression levels similar to the native protein, Tri1R32E D161N failed to protect cells against Tre1tox (Figures 3H, left and S2C). Under the same conditions, Tri1 variants bearing substitutions to the ARH active site or NTE individually inhibited Tre1tox-induced toxicity to an extent indistinguishable from the wild-type. To more systematically interrogate the capacity of the two Tre1 inhibition modes of Tri1 to act independently, we developed an expression system in which levels of Tre1tox and Tri1 could be modulated by separate inducer molecules. Using this system, we identified expression regimes in which the contribution of both postulated Tri1 immunity mechanisms could be discerned (Figure 3H, right). In summary, these data indicate that Tri1 can provide immunity against Tre1 via two structurally distinct mechanisms.
Our structural and genetic results highlight the functional significance of the Tri1 NTE. This is in agreement with our finding that many predicted ART-inhibiting immunity factors, particularly those found in Gram-negative bacteria, have a short (30–60 amino acids), nonconserved region N-terminal to their ARH domain (Table S1 and Figures 1C and 3E). Although these N-terminal segments share little primary sequence conservation, structure prediction suggests that they adopt a fold consisting almost entirely of α-helical and random coil secondary elements, matching the structure observed in the NTE of Tri1-Sp (Figure 3F). Based on these data, we postulate that the dual mechanism of ART inhibition by Tri1-Sp is a widely conserved feature of ARH-family immunity proteins.
Tre1 modifies a functionally essential site of FtsZ under physiological conditions
Our data to this point supported a model in which Tre1 functions in interbacterial antagonism via the ADP-ribosylation of proteins within target cells. To define candidate substrates of the toxin, we subjected cellular proteomes derived from E. coli expressing Tre1tox or Tre1tox E415Q to mass spectrometry. This analysis identified nine ADP-ribosylated (ADPr) peptides deriving from seven proteins in Tre1tox samples; no ADPr peptides were detected in control E. coli lysates containing Tre1tox E415Q (Figures 4A and S3A–I). Our search considered all previously identified amino acid targets of ART enzymes (Cys, Gln, Arg, Asn, Thr, Glu, Asp, Lys) (Cohen and Chang, 2018); however, we detected exclusively ADP-ribose–Arg as the product of Tre1tox. This finding is consistent with the amino acid targeting specificity of the RSE family ART enzymes most structurally related to Tre1(Fu et al., 2007). Under the over-expression conditions employed in this assay, the targets of Tre1 include essential proteins involved in cell division (FtsZ), translation (EF-Tu), RNA metabolism (RNase E) and lipoprotein transport (LolD). Additionally, non-essential proteins that nonetheless contribute to critical cellular processes including central metabolism (SucB), translation fidelity (ribosomal protein L9), and the global regulation of transcription (Fis) were also modified by the toxin.
Figure 4.

Tre1 ADP-ribosylates FtsZ, disrupts Z ring formation and inhibits cell division. (A) Sequence of E. coli peptides ADP-ribosylated due to heterologous expression of Tre1tox-Sp. Red, modified residues; shading, peptides also modified by Tre1tox-Pp. (B) Tandem mass spectrum of the indicated peptide from E. coli FtsZ enriched by immunoprecipitation from cells in competition co-culture with Tre1-expressing S. proteamaculans. Fragmentation ions (b, blue; y, red) with resolved spectra and the site of ADP ribosylation (bold) are indicated. (C) Proportion of E. coli cells measuring greater than 2X the median cell length at the time of mixing (light grey), or after 6 hours co-culture on a solid medium (dark grey) with S. proteamaculans producing the indicated Tre1 proteins. Asterisk denotes statistically significant difference in E. coli cell size between the populations after competition (p<0.05, n > 1000, see also Figure S3L). (D and E) Time-lapse fluorescence and phase contrast microscopy sequences of E. coli cells expressing FtsZ-FP and carrying plasmids for the inducible expression of Tre1 (D) or Tre1tox E415Q (E). Cells were cultivated in a microfluidic chamber to enable the introduction of inducer. All cells in time-lapse series were defined by image segmentation with custom analysis software and Z ring dynamics of a representative cell (outlined in solid red, daughters outlined in dashed red) are provided (lower single cell image series) (Stylianidou et al., 2016). (F) Ratio of cell area (red) and average number of FtsZ-FP foci in E. coli cells expressing Tre1tox (black) or Tre1tox E415Q (blue) over the indicated time period. Tre1tox or Tre1tox E415Q induction, dashed line; 95% confidence intervals, grey shading. See also Video S3 and Figure S3.
ART over-expression could lead to non-specific protein modification and thus obscure the identity of physiological target(s). The following lines of evidence suggested that among those proteins identified by our mass spectrometric analyses, FtsZ, an essential cell division factor, is a physiologically relevant target of Tre1. First, we conducted a similar mass spectrometric analysis on lysates derived from E. coli expressing a homolog of S. proteamaculans Tre1 (Tre1-Sp) from Pseudomonas putida GB-1 (Tre1-Pp) (45% identical over 217 amino acids in the toxin domain). This revealed FtsZ as one of four candidate Tre1-Pp targets in common with those of Tre1-Sp (Figures 4A and S3J). Second, cells experiencing Tre1-based intoxication morphologically phenocopy those with dysfunctional cell division machinery, including dramatic elongation apparently caused by a failure to septate (Figure 2B and Video S1). Third, the amino acid position of FtsZ modified by both Tre1-Sp and Tre1-Pp, Arg174, is conserved across the Enterobacteriaceae, and it was previously identified as a critical functional site in E. coli FtsZ that is non-permissive to substitution (Koppelman et al., 2004). Therefore, modification of Arg174 by Tre1 could afford S. proteamaculans a fitness advantage against competitors from related species likely to inhabit an overlapping niche.
If FtsZ were a physiologic target of Tre1, we reasoned that the protein should be modified by the toxin within recipient cells during interbacterial competition. The tryptic peptide bearing Arg174 of FtsZ is identical between S. proteamaculans and E. coli. Therefore, to facilitate specific enrichment of the protein from recipient cells, we generated an E. coli strain in which native ftsZ is substituted with an allele coding for a functional hexahistidine-tagged variant of the protein (FtsZ-His6) (Oliva et al., 2003). We subjected this strain to conditions promoting contact with S. proteamaculans and subsequently employed stringent denaturing conditions to isolate E. coli FtsZ-His6 from intoxicated cells. Analysis of the resulting sample by mass spectrometry provided clear evidence of ADP-ribosylation of FtsZ at Arg174 during interbacterial competition (Figure 4B). Moreover, modification of this site was not observed in a parallel experiment employing S. proteamaculans tre1E415Q.
Next we sought to link our biochemical evidence of FtsZ ADP-ribosylation to the phenotypic outcome of intoxication by intercellularly transferred Tre1. We were unable to obtain viable E. coli expressing singly an ftsZ allele coding for an Arg174 variant of the protein, thus precluding several straightforward means of establishing this link. Instead, we exploited the distinctive morphological consequence of FtsZ inactivation – cell elongation – as a phenotypic proxy for its targeting by Tre1 during interbacterial competition. Following contact with wild-type S. proteamaculans, we observed a significant increase in the frequency of highly elongated E. coli recipient cells relative to starting populations (Figures 4C, S3K and S3L). A population of E. coli contacting S. proteamaculans tre1E415Q also became elongated, though these were observed at significantly lower frequency. We speculate that the latter is due to the action of other T6S toxins of S. proteamaculans (Figure 2E). In total, these data indicate that FtsZ is a physiologically relevant target of S. proteamaculans Tre1. Our data further suggest that ADP ribosylation of FtsZ by Tre1 disrupts cell division, leading to cell elongation.
Tre1-catalyzed ADP-ribosylation of FtsZ inhibits Z ring formation
In exponentially growing cells, FtsZ localizes predominantly to the mid-cell. There, in a GTP-dependent fashion, dynamic filaments of the protein assemble to form a characteristic ring, referred to as the Z ring, which templates the cell division machinery, including peptidoglycan biosynthetic enzymes (Erickson et al., 2010). As a first step toward determining how FtsZ function is affected by ADP-ribosylation, we used time-lapse fluorescence microscopy (TLFM) to visualize Z ring dynamics in Tre1-intoxicated cells. For these studies, we employed cells coding for a functional FtsZ variant containing mNeonGreen inserted at a permissive site in place of the wild-type ftsZ allele (FtsZ–FP) (Moore et al., 2017). Importantly, in the three-dimensional structure of FtsZ, the site of mNeonGreen insertion is distal to Arg174, the residue targeted by Tre1. Prior to induction of Tre1 and throughout a 60 min course of Tre1E415Q induction, FtsZ–FP was pervasively observed as an apparent ring at cell septa (Figures 4D–F and Video S3). However, within 10 minutes following induction of the active toxin, the fraction of cells with clearly formed Z rings decreased appreciably. By 60 min post-induction, intact Z rings were rarely observed and, instead, FtsZ–FP appeared distributed in a collection of puncta throughout the cell. These changes in FtsZ dynamics corresponded closely in time with inhibited cell division, as quantified by cell area (Figure 4F).
Our TLFM data suggested that the activity of Tre1 disrupts the capacity of FtsZ to form or maintain Z rings. These findings within cells motivated us to further dissect the mechanism of Tre1-mediated FtsZ inactivation using in vitro approaches. We began by measuring the capacity of Tre1tox to act on purified FtsZ. Our assays demonstrated that Tre1tox ADP ribosylates FtsZ with approximately 16-fold greater efficiency than a control protein, bovine serum albumin (BSA) (Figure 5A). Notably, this finding is consistent with FtsZ acting as a preferred, but not exclusive substrate, of Tre1 in vivo (Figure 4A).
Figure 5.

Tre1 inhibits FtsZ polymerization in vitro. (A) Autoradiograph of SDS-PAGE-resolved products of purified BSA or FtsZ incubated with the noted Tre1tox variants and [adenylate-32P]-NAD+. (B) Polymerization of FtsZ pre-incubated with the indicated concentrations of Tre1tox and Tre1tox E415Q, as measured by 90° angle light scattering. GTP was added at T0 (dashed line) to initiate the polymerization reaction. (C, D) Negative stain electron microscopy analysis of FtsZ polymers formed following 5 min incubation with the indicated concentrations of Tre1tox and a reduced concentration of crowding reagent compared to that employed for experiments depicted in B. Average filament lengths, (C), representative electron micrographs, (D). Black arrows highlight individual polymers. (E) Impact of Tre1tox addition (dashed line) on pre-formed FtsZ polymers, measured as in B. (F) Modeled structure of an E. coli FtsZ filament containing 3 monomers (shades of grey) with position ADP-ribosylated by Tre1 highlighted (R174, blue). Top, R174 in stick representation; middle, R174-ADPr in molecular surface representation; bottom, R174-ADPr in alternative rotomer that induces steric clashes between protomers. The FtsZ monomer structure was generated with MODELLER based on FtsZ from Staphylococcus aureus (PDB 5MN4) (Wagstaff et al., 2017) and the polymer was generated based on crystallographic symmetry. (G) Effect of adding FtsZ–ADPr at the indicated ratios to polymerization reactions of unmodified FtsZ. FtsZ-ADPr subunits were generated by incubating with an equimolar concentration of NAD+ to avoid excess ADP-ribosylation. Total FtsZ concentration (FtsZ + FtsZ-ADPr) was kept at 12.5 μM in all assays, and polymerization was quantified as in B.
In the presence of a molecular crowding agent and GTP, purified FtsZ initiates filament formation that can be monitored in real-time using 90° angle light scattering (Mukherjee and Lutkenhaus, 1999). Using this assay, we found that pre-incubating FtsZ with 10 nM Tre1tox in the presence of NAD+ for 5 min prior to the addition of GTP abrogates filament formation (Figure 5B). Detectable Tre1tox-mediated effects on FtsZ polymerization were observed at toxin concentrations as low as 0.5 nM, whereas no significant inhibition was observed with 10 nM Tre1tox E415Q. Filament length measurements derived from negative stain electron micrographs of these samples supported the light scattering results (Figures 5C and 5D).
The finding that Tre1 inhibits FtsZ filament formation does not rule out the possibility that Tre1 also disassembles or destabilizes existing filaments. A dual mechanism such as this could allow the toxin to act with greater efficiency in vivo. However, when we treated pre-formed FtsZ filaments with Tre1tox in the presence of NAD+ at concentrations exceeding that likely obtainable in vivo, we did not observe changes in light scattering indicative of filament perturbation (Figure 5E). Taken together with our TLFM findings, these in vitro data strongly suggest that Tre1 inhibits cell division by interfering with the capacity of FtsZ to polymerize.
The amino acid position of FtsZ that is the preferred site of modification by Tre1, Arg174, resides on a loop located proximal to the protomer interface in the filamentous form of the protein (Figure 5F, top). This observation led us to hypothesize that Tre1 inhibits FtsZ polymerization by allosterically interfering with inter-subunit interactions. As a first test of the feasibility of this mechanism, we performed molecular modeling using in silico-generated FtsZ Arg174–ADP ribose (Figure 5F, middle). These studies clearly indicated the capacity of Arg174–ADP ribose to block several intermolecular interactions of likely importance to the formation of FtsZ filaments (Figure 5F, bottom). To further interrogate the hypothesis, we generated Tre1-modified FtsZ subunits and added these at varying stoichiometries to FtsZ polymerization reactions. In preparing ADPr FtsZ subunits, excess ADP-ribosylation was avoided by limiting NAD+ to a concentration equimolar with that of FtsZ. Light scattering analyses showed that ADPr FtsZ subunits poison polymerization of the unmodified protein (Figure 5G). Although it is difficult to directly correlate light scattering values to filament length, the dose-dependent effect of modified subunits on the maximal light scattering value observed is consistent with their capacity to bind and terminate growing filaments. In total, our in vitro and in vivo data support a model for Tre1-based cellular intoxication in which the effector generates ADPr FtsZ subunits that poison polymerization and thus inhibit Z ring formation and cell division.
Promiscuous ARH domains confer broad protection against ART toxins
With FtsZ defined as a substrate of Tre1, we sought to determine whether, as predicted by our structural and genetic analyses, Tri1 is able to act reciprocally on the protein. Indeed, we found that the wild-type immunity protein, but not Tri1D161N, catalyzes the removal ADP-ribose adducts from Tre1-treated FtsZ (Figure 6A). Interestingly, in these assays we noted that Tri1 hydrolyzes ADP-ribose moieties from FtsZ and BSA with similar efficiency. This promiscuity of Tri1 raised the possibility that immunity proteins containing ARH domains may have the capacity to hydrolyze ADP-ribose modifications broadly and thereby provide protection against non-cognate ART toxins. This contrasts with previously characterized immunity determinants, which function exclusively through sequence-specific interactions with toxin active sites and thus inactivate only cognate and closely related toxins (Alcoforado Diniz et al., 2015).
Figure 6.

ARH domain-containing immunity proteins provide protection from non-cognate ART toxins. (A) Autoradiograph of SDS-resolved products resulting from incubating a mixture of FtsZ and BSA (1:16 ratio) with Tre1tox and [adenylate-32P]-NAD+ to facilitate ADPr, followed by treatment with the indicated Tri1 proteins. (B and D) Viable E. coli recovered from populations expressing Tre1tox-Sp and the indicated ARH-domain proteins. (C) Competitiveness of the indicated donor (D) strains of S. proteamaculans toward wild-type or a tri1 mutant of P. putida GB-1, after 6 h coculture on a solid medium. (E) Competitiveness of indicated donor strains of S. proteamaculans toward the noted recipient strains of P. putida B6–2 as grown in C. (F) Competitiveness of S. proteamaculans toward E. coli heterologously expressing a vector control, Tri1 from S. proteamaculans or Tri1 from P. putida B6–2, grown as in C. Data in B-F represent means ± SD. Asterisks in B-F indicate statistically significant differences between the indicated mean values (p<0.05, n = 3). See also Figure S4.
Although the Tre1 homologs from S. proteamaculans and P. putida GB-1 modify a partially overlapping set of targets, the effectors diverge significantly (45% identity within the toxin domain). This corresponds to relatively low conservation of toxin-interacting residues in the NTE domains of the respective Tri1 homologs of the organisms (24% identity, amino acids 30–60) compared to that of the ARH domains of these proteins (60% identity, amino acids 65-C-term). In line with these observations, we failed to detect an interaction between Tri1-Pp and Tre1tox-Sp by co-immunoprecipitation (Figure S4A). This lack of binding provided an opportunity to determine whether the hydrolase activity of ART immunity proteins is sufficient to confer protection against non-cognate ART toxins. We found that Tri1-Pp affords E. coli protection against Tre1-Sp-based intoxication to a level comparable to that of the cognate immunity protein, Tri1-Sp (Figure 6B). DraG, an ARH protein involved in regulating nitrogen fixation, did not provide protection from Tre1-Sp, indicating that broad substrate targeting is not a property shared by all ARH domain proteins (Figures 6B and S4B). The hydrolase activity of Tri1-Pp also accounted for the full extent of Tri1-Pp-mediated protection from Tre1 delivered by S. proteamaculans during interbacterial competition (Figure 6C). Notably, inactivation of the hydrolase domain of Tri1-Pp did not reduce P. putida fitness in the absence of intercellular Tre1-Sp delivery, suggesting that the ADP-ribose hydrolytic activity of this Tri1 homolog is dispensable for inhibition of its cognate effector.
Our findings did not rule out the possibility that the ARH activity of Tri1-Pp is specific and that its capacity to neutralize Tre1-Sp derives from the substrate overlap between this protein and its own cognate effector, Tre1-Pp (e.g. FtsZ). We therefore tested whether an ARH domain immunity protein from a distantly related species, the Gram-positive organism Listeria monocytogenes (Eri1; Esx secretion ADP-ribosyltransferase immunity 1), could also confer protection against Tre1-Sp. Remarkably, Eri1, but not a predicted catalytically inactive variant (Eri1D69N), significantly increased the viability of E. coli experiencing intoxication by Tre1tox-Sp (Figures 6D and S4C). FtsZ proteins from Listeria and other Firmicutes lack Arg174, and the proteomes of these organisms are generally highly divergent, strongly suggesting that target(s) of Ere1 (cognate effector of Eri1) – likely delivered to other Gram-positive species by the Esx secretion pathway – differ from that of Tre1-Sp and Tre1-Pp. In total, these results support the hypothesis that the broad substrate specificity of ARH domains within immunity proteins enables them to provide protection from intoxication by diverse ART toxins.
If ARH domains are sufficient to confer broad protection against ART toxins, we reasoned that proteins containing these domains could provide a fitness benefit independent of a cognate effector. Position-specific iterative searching revealed that ARH domain proteins are broadly distributed across diverse bacterial taxa, and are largely found within strains that do not encode recognizable ART toxins (Figure S5 and Table S2). With the exception of DraG and the immunity determinants we study herein (Tri1-Sp, Tri-Pp and Eri1), to our knowledge these proteins have not been characterized. To narrow our search for ARH domain proteins likely to provide immunity versus function in regulatory or housekeeping roles, we focused on those most homologous to Tri1 or Eri1 (>30% amino acid identity). In addition to genes belonging to E–I pairs, this search identified 95 predicted ARH immunity genes without adjacent toxin loci (Table S2). These isolated ARH domain immunity genes and those in apparent E–I pairs cluster together phylogenetically within three distinct paraphyletic clades, indicating the evolutionary relatedness of immunity genes found in both contexts and that ARH immunity function likely evolved independently on at least three occasions (Figure S5).
One organism we found to encode an isolated tri1 is P. putida B6–2. We inactivated this gene and assessed competitiveness of the strain with S. proteamaculans. P. putida B6–2 Δtri1 displayed reduced fitness in competition with wild-type S. proteamaculans, but not a strain expressing catalytically inactive Tre1 (Figure 6E). Heterologous expression of Tri1 from P. putida B6–2 also increased the competitiveness of E. coli grown in co-culture with S. proteamaculans to a similar degree as expression of Tri1-Sp (Figure 6F). In total, our results indicate the capacity for promiscuous ARH proteins – isolated or operating in conjunction with a cognate effector – to provide defense against a diversity of ART toxins utilized by competing bacteria.
Discussion
Proteins with ART activity are widely utilized by bacterial pathogens, and were among the earliest toxins identified. Indeed, diphtheria toxin was the first bacterial enzyme shown to inactivate a host protein via covalent modification (Honjo et al., 1969). The results we report herein suggest that ARTs are also widespread mediators of interbacterial antagonism. Given that encounters between bacteria vastly predate the evolution of eukaryotic cells, it is conceivable, or even likely that host-targeting ARTs evolved from ancestral proteins with antibacterial function. Our findings indicate that similar proteins can be targeted by both types of ART toxins; we show that the antibacterial ART of S. proteamaculans, Tre1, targets the bacterial cytoskeletal protein and tubulin homolog, FtsZ, while the structurally-related iota toxin of C. perfringens modifies actin, a major component of the eukaryotic cytoskeleton (Aktories and Wegner, 1992). Interestingly, both toxins specifically act on monomeric substrates and prevent polymerization.
Our bioinformatic analysis of polymorphic toxins with predicted ART activity led to the identification of a diverse collection of proteins for which the targets remain to be identified. Substrates modified by secreted antibacterial ARTs may encompass arginine or other residues on a variety of proteins, or they may potentially extend to nucleic acid or small molecule targets (Aravind et al., 2015). Precedence for the antibacterial activity of DNA-targeting ARTs was recently established through the characterization of a toxin/antitoxin (TA) module found in Mycobacterium tuberculosis (Jankevicius et al., 2016). Additional physiologically relevant targets of Tre1 may also exist. In heterologous expression experiments, we found that Tre1-Sp and Tre1-Pp modify small sets (n = 7) of proteins, whose constituents overlap to an extent that appears non-random (4/7). In addition to FtsZ, two essential proteins, EF-Tu and RNase E, were targeted by both toxins. Simultaneous inactivation of multiple essential proteins could have the benefit of thwarting or delaying the emergence of resistance. Given the non-native levels achieved by heterologous expression, the modification of these proteins by Tre1-Sp and Tre1-Pp may instead reflect their high abundance and arginine accessibility.
We found that Tri1 protects S. proteamaculans from self-intoxication by two distinct mechanisms: the removal of ADP-ribose moieties on Tre1-targeted proteins and toxin active site occlusion. The globular domain of Tri1 closely resembles DraG, an ADP-ribosylhydrolase with a housekeeping function unconnected to interbacterial competition. Supporting this relatedness, our phylogenetic analyses suggest that tri1-type immunity genes likely arose through duplication of housekeeping ARH genes, and that these events occurred independently on multiple occasions. Interestingly, inhibition of the Tre1 active site is mediated by the NTE of Tri1, which is not found in DraG. Selection for increased efficacy in blocking an incipient ART toxin could have led to ARH–ART protein interaction and evolution of the toxin active site-occluding NTE found in Tri1 and homologs. However, this trajectory raises the question of why Tri1 and related proteins retain functional ARH domains. Our results demonstrate that the promiscuous ADP-ribosylhydrolase activity exhibited by these proteins enables them to confer a fitness benefit during competition with species that produce divergent, non-cognate ART toxins. We find that genes encoding ARH domain proteins with NTEs are more common in bacterial genomes than corresponding E–I pairs, suggesting that the fitness benefit of the ARH domain is significant even when cognate toxins are lost. Interestingly, the antitoxin responsible for conferring protection from the recently described DNA-targeting ART toxin of M. tuberculosis, DarG, also exhibits both ARH and cognate toxin-binding activities (Jankevicius et al., 2016). While the contribution of the interaction between DarT and DarG towards the antitoxin function of DarG remains to be determined, the similarities with the E–I pair interaction described here suggest the potential of parallel evolution.
Numerous independent experimental results in this study support the conclusion that bacterial intoxication by Tre1 results – at least in part – from ADP-ribosylation of the central bacterial cell division protein FtsZ. Perhaps most compellingly, we show that physiological levels of Tre1 potently inhibit FtsZ polymerization in vitro and we demonstrate that FtsZ within a recipient cell is ADP-ribosylated in a manner dependent on interbacterial delivery of functional Tre1 during competition with S. proteamaculans. FtsZ is an attractive target for protein-modifying toxins, as it is highly conserved across bacteria, and interference with its function leads to rapid growth arrest and loss of viability (Dai and Lutkenhaus, 1991; Vaughan et al., 2004). While Tre1 is, to our knowledge, the first toxin to be identified that covalently post-translationally modifies FtsZ, several other inhibitors of its function have been characterized. House-keeping FtsZ-inhibitory proteins provide critical spatiotemporal regulation of cell division (Ortiz et al., 2016). Interestingly, the toxin component of the E. coli toxin/antitoxin module CbtA/CbtB was recently shown to disrupt Z ring formation through interaction with the FtsZ H6/H7 loop (Heller et al., 2017). However, the physiological role of this activity remains to be determined. E. coli phages λ and T7 also each encode FtsZ-interacting proteins that inhibit cell division during propagation of phage particles (Haeusser et al., 2014; Kiro et al., 2013).
Our finding that Tre1 catalyzes site-specific modification of FtsZ in vivo raises the possibility that this toxin could be applied to dissecting the mechanisms of bacterial cell division. While FtsZ plays a well characterized role in septum formation and cytokinesis, aspects of the mechanism by which this occurs remain to be clarified. For instance, two recent studies have demonstrated that FtsZ filaments exhibit treadmilling behavior during Z ring formation, rather than forming a continuous, static structure as previously thought (Bisson-Filho et al., 2017; Yang et al., 2017). Treadmilling, or the preferential addition of new subunits at one end of a polymer coupled with loss at the opposite end requires a means of establishing filament polarity; how this is achieved for FtsZ filaments remains unclear (Krupka and Margolin, 2018). The ability of Tre1 to block FtsZ polymerization by modification of a residue located on one half of the monomer interaction interface could prove useful in testing different models for the mechanism by which polarity is produced (Wagstaff et al., 2017).
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph Mougous (mougous@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture
Bacterial strains under investigation in this study were derived from S. proteamaculans 568, P. putida GB-1 and P. putida B6–2 (Abebe-Akele et al., 2015; Tang et al., 2011; Wu et al., 2011). Cultivation of these organisms was performed using Luria-Bertani media (LB) at 30°C. For S. proteamaculans, the medium was supplemented with 50 μg ml−1 kanamycin where necessary, and counter-selection for allelic exchange was performed on nutrient agar plates supplemented with 15% (w/v) sucrose. For P. putida, media were supplemented with 25 μg ml−1 chloramphenicol, 25 μg ml−1 irgasan, and 30 μg ml−1 gentamicin as needed, and counter-selection for allelic exchange was performed on LB plates supplemented with 15% (w/v) sucrose.
Escherichia coli strains used in this study included DH5α and EC100 λ pir for plasmid maintenance and toxicity assays, S17–1 λ pir for conjugal transfer of plasmids into S. proteamaculans and E. coli, SM10 for conjugal transfer of plasmids into P. putida, C600 for competition experiments with S. proteamaculans, and BL21 (DE3) for purification of Tre1, Tri1, and FtsZ. E. coli strains were grown on LB supplemented with 150 μg ml−1 carbenicillin, 50 μg ml−1 kanamycin, or 25 μg ml−1 chloramphenicol as needed.
METHOD DETAILS
Bioinformatic analysis of ART and ARH domain proteins
The protein sequence of Tre1-Sp was submitted to the EMBL-EBI HMMMER server Jackhmmer (https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer) for iterative searches against the UniprotKB database. All identified bacterial proteins were then analyzed by conserved domain searching to determine if they contained N-terminal domains associated with T6SS or Esx pathway substrates, including PAAR, RHS, VgrG, or LXG domains (Cianfanelli et al., 2016; Whitney et al., 2017). Additionally, the genetic loci encoding each protein was examined for nearby (within 10kb) genes encoding predicted secretion system structural elements. For ARH domain proteins, the DraG sequence from Rhodopseudomonas palustris was used to search against the same databases as described above. Four iterations were run, and the results were filtered to include only bacteria. To perform phylogenetic analyses, these sequences were downloaded and aligned with Clustal Omega 1.2.4 using the default settings. The resulting multi-alignment was then trimmed using trimAl 1.2 (Capella-Gutierrez et al., 2009), so that only columns with less than 60% gaps were retained. Maximum likelihood phylogenetic inference was then performed using FastTree 2.1.9 (https://bioweb.pasteur.fr/packages/pack@FastTree@2.1.9). ARH-domain immunity proteins were distinguished from those with other functions on the basis of 30% amino acid identity shared with one of the immunity proteins characterized in this study (Tri1 or Eri1) and genetic association with predicted ART toxins and T6SS or Esx pathway related genes. Additionally, orphan Tri1 proteins were distinguished from housekeeping ARH proteins by the presence of an NTE (30–60 amino acids in length). Sequence alignments and logos were generated by Geneious software (Biomatters).
Plasmid construction
All primers used in plasmid construction and generation of mutant strains are listed in Table S4. Plasmids used for heterologous expression in E. coli in this study were pETDuet-1, pBAD18, and pBAD33. For pBAD33 E. coli expression constructs, genes were cloned with a fusion to either 3′ VSV–G-linker (tri1-Sp and tri1-Pp) or 5′ VSV–G-linker (eri1 and draG) with SacI and XbaI restriction sites introduced in the cloning primers. For pBAD18 expression constructs, tre1-Sp, tre1tox-SP (266–444), and tre1tox-Pp (244–496) were cloned into the SacI and XbaI restriction sites. For the Tre1 and Tri1 purification constructs, the tre1tox and tri1 genes were amplified and cloned into MCS-1 (BamH1 and NotI sites) and MCS-2 (NdeI and XhoI sites) of pETDuet-1, respectively, generating an N-terminal hexahistidine fusion to Tre1tox. For the purification of Tri1 variants and homologs, full length tri1 was cloned into MCS1 of pETDuet-1 using the BamHI and NotI restriction sites. The resulting plasmids incorporate N terminal hexahistidine tags into the Tri1 protein variants.
Site-directed mutagenesis was used to generate the desired catalytic substitution mutants in toxin and immunity proteins from pBAD18, pBAD33, and pET-Duet-1 plasmid(s). Primers containing the desired mutations were used in combination with a second primer facing in the opposite orientation to generate a copy of the expression construct plasmid containing the point mutations incorporated into the gene coding sequence. Dpn1 digestion was then used to remove template plasmid. The remaining amplicon was transformed into DH5α and selected by growth on plates containing carbenicillin to select for pBAD18 and pET-Duet-1 or chloramphenicol for pBAD33.
In-frame deletion constructs were generated utilizing the suicide vectors pRE118 for S. proteamaculans and E. coli, and pEXG2 for P. putida (Edwards and Donachie, 1993; Rietsch et al., 2005). For the production of deletion constructs, 750 bp regions flanking the deletion were amplified, joined using splicing by overlap extension (SOE) PCR, and subsequently cloned into the appropriate vector. To generate the tre1E415Q-SP mutation construct, 750 bp regions flanking the mutation with an additional overlapping extension consisting of the desired mutation were amplified and joined using SOE PCR and subsequently cloned into pRE118. Integration of constitutive fluorescent reporters at the attTn7 site of S. proteamaculans was achieved by co-transformation of pUC18R6K-mini-Tn7T-Km-gfp and the helper plasmid pTNS3 (Choi et al., 2008; Choi and Schweizer, 2006).
Generation of mutant strains of S. proteamaculans, P. putida, and E. coli
To generate deletions and point mutations of S. proteamaculans strains, mutation constructions in pRE118 were transformed into E. coli S17–1 λ pir. E. coli S17–1 λ pir donor strains carrying the mutation constructs and S. proteamaculans recipient strains to be mutated were grown overnight on LB plates containing antibiotics as appropriate, then scraped together to create a 1:1 mixture of each donor–recipient pair that was spread on an LB agar plate and incubated at 30°C for 6 hours to facilitate plasmid transfer via conjugation. Cell mixtures were then scraped into PBS and plated on minimal medium agar plates (100 mM HEPES-KOH pH = 7.0, 0.1% (NH4)2SO4, 40 mM MgSO4, 0.2% (w/v) sucrose, 0.5 mM KH2PO4, 50 μg ml−1 kanamycin, 1.5% agar) to select for S. proteamaculans containing the mutagenesis construct inserted into the chromosome. S. proteamaculans merodiploid strains were then grown overnight in non-selective LB medium at 30°C, followed by counter selection by plating on nutrient agar plates supplemented with 15% (w/v) sucrose. Kanamycin sensitive, sucrose resistant colonies were screened for allelic replacement by colony PCR and mutations were confirmed by Sanger sequencing of PCR products.
For P. putida, mutation constructs in pEXG2 were transformed into E. coli SM10. Conjugation to enable plasmid transfer was performed as described above for S. proteamaculans, then plated on LB agar containing gentamicin and irgasan to select for P. putida with integrated plasmid. Merodiploids were grown overnight in liquid LB at 30°C, then plated on LB agar containing 15% (w/v) sucrose for counter selection. Gentamycin sensitive, sucrose resistant colonies were screened for allelic replacement by colony PCR and mutations were confirmed by Sanger sequencing of PCR products.
For expression of GFP in S. proteamaculans, pUC18R6K-mini-Tn7T-Km-gfp and the helper plasmid pTNS3 were co-transformed into S. proteamaculans strains by electroporation (Choi et al., 2008; Choi and Schweizer, 2006). After 1 h outgrowth in nonselective medium, transformations were plated on LB with kanamycin to select for mini-Tn7 integrants.
To obtain an E. coli strain in which the native version of ftsZ was replaced with ftsZ-his6, we employed the strain JKD7–1/pKD3, in which chromosomal ftsZ is inactivated and a functional copy of FtsZ is expressed from a temperature sensitive plasmid (Dai and Lutkenhaus, 1991). This strain was transformed with pJSB2-ftsZ-6His and grown overnight at 30°C on LB plates amended with carbenicillin, chloramphenicol, and 0.2% (w/v) glucose. Colonies from these plates were then streaked onto LB agar containing chloramphenicol and arabinose and incubated at 42°C to cure the strain of the temperature sensitive plasmid expressing native FtsZ, and retain pJSB2-ftsZ-6His as the sole source of functional FtsZ (Dai and Lutkenhaus, 1991; Redick et al., 2005).
Tre1 toxicity assays
To assess the toxicity of Tre1 variants and protection conferred by expression of Tri1 variants, log phase cultures of E. coli DH5α or BL21 (DE3) harboring toxin and immunity genes in pBAD18, pBAD33, and/or pET-Duet-1 were grown under non-inducing conditions, then 10-fold serial diluted. Each dilution was plated onto LB agar plates containing carbenicillin to select for pBAD18 and pET-Duet-1 or chloramphenicol for pBAD33 and appropriate inducer molecules. Unless otherwise noted, expression of both toxin and immunity proteins from pBAD18 and pBAD33 was induced by the addition of 0.2% arabinose. Toxin production from pET-Duet-1 was induced by the addition of 0.01 mM IPTG.
Bacterial competition assays
For each competition experiment, donor and recipient strains were first grown for ~18 hrs in liquid LB medium. Liquid cultures were spun for 1 min at 17,900 × g to pellet cells, culture supernatant was removed, cells were washed once with fresh LB medium, spun again, and finally resuspended in LB. Cell suspensions were then normalized to OD600 = 2.0. Mixtures of donor and recipient strains were then established at 1:1 (S. proteamaculans vs. E. coli) or 10:1 (intraspecies competitions and S. proteamaculans vs. P. putida) v/v ratios. Starting ratios of donor and recipient strains were established by performing 10-fold serial dilutions and plating on appropriate selective media (see below). Competitions were initiated by spotting 3 × 5 ul of each mixture on nitrocellulose filters placed on 3% (w/v) agar LB plates. When the recipient strain employed was E. coli, these plates additionally contained 0.2% L-arabinose to induce heterologous expression of Tri1 (or an empty vector control). Competitions were incubated for 6 h (quantification by plating) or 18 h (analysis by microscopy) at 30°C. Cells were then harvested by scraping individual spots from excised sections of the nitrocellulose filter into LB medium. Suspensions were serially diluted and plated on selective media for quantification of CFUs or were spotted on agarose pads placed on microscopes slides for visualization by phase contrast and fluorescence microscopy.
For intraspecies competition experiments, donor strains of S. proteamaculans were marked by chromosomal insertion of Km-gfp at attTn7, while recipient strains were unmarked; donor populations were quantified by enumerating CFU obtained on LB with kanamycin, and recipient populations quantified by subtracted this number from the total CFU enumerated on non-selective LB plates. For competitions between S. proteamaculans and E. coli, E. coli C600 containing the RP4 plasmid was used as the recipient strain background, and the donor strain was marked with kanamycin resistance as in the intraspecies competition experiments; donor and recipient populations were distinguished by plating on LB containing carbenicllin or kanamycin, respectively. For competitions between S. proteamaculans and P. putida, the kanamycin resistant donor strain was again employed, and the recipient strain was distinguished by its intrinsic resistance to chloramphenicol. Competitive indices for each experiment were determined by dividing the final donor to recipient ratio by the initial donor to recipient ratio.
Protein expression level analyses
To analyze the expression of all Tre1 and Tri1 variants, E. coli strains expressing the proteins from pBAD18 and pBAD33 were grown in LB medium supplemented with 0.2% arabinose at 37 °C for 3 hours and harvested at an OD600 of 1.0. For E. coli expressing toxins and the variants, Tri1-Sp was co-expressed to prevent cytotoxicity. To analyze the expression of Tre1 and Tre1E415Q from the native locus in S. proteamaculans, cells were grown in LB medium at 30°C for 18 hours and harvested at OD600 = 3.5. For each quantification assay, cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 5.0% (v/v) glycerol, 0.1% (v/v) Triton X100, 1 mM EDTA, and 1 mM DTT) and then disrupted by sonication (10 pulses, 10 seconds each) and the solution clarified by centrifugation at 20,000 × g for 30 minutes. A sample of supernatant was then mixed 1:1 with 2X SDS-PAGE sample loading buffer. Samples in SDS-loading buffer were boiled at 95 °C for 5 min and loaded at equal volumes to resolve using SDS-PAGE, then transferred to nitrocellulose membranes. Membranes were blocked in TBST (10 mM Tris-HCl pH 7.5, 150 mM NaCl2, and 0.1% w/v Tween-20) with 5% (w/v) nonfat milk for 30 min at room temperature, followed by incubation with primary antibodies (anti-Tre1 or anti-VSV-G) diluted in TBST for 1 hour at room temperature. Blots were then washed by TBST, followed by incubation with secondary antibody (Goat anti-Rabbit HRP conjugated) diluted in TBST for 30 minutes at room temperature. Finally, blots were washed by TBST again and were developed using Radiance HRP substrate (Azure Biosystems) and visualized using the Azure Biosystems c600.
Protein purification
To prevent cytotoxicity during overexpression in E. coli, all Tre1 constructs were coexpressed with Tri1 from pET-Duet-1 in E. coli BL21 (DE3). Stationary phase cultures of expression strains were used to inoculate 1 L of LB broth cultures containing carbenicillin, which were then grown to mid-log phase (OD600 = 0.4 – 0.7) in a shaking incubator at 37°C. Protein expression was induced by the addition of 0.1 mM IPTG followed by incubation at 30°C for 5 h. Cells were harvested by centrifugation at 7,000 × g for 30 minutes, followed by resuspension in 30 mL of lysis buffer (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 5 mM imidazole, 1 mM DTT, 5% glycerol, and lysozyme). Resuspended cells were incubated on ice for 30 minutes then disrupted by sonication (10 pulses, 10 seconds each) and cellular debris was removed by centrifugation at 40,000 g for 30 minutes. His-tagged Tre1tox and associated protein was purified from supernatant by gravity flow through a 2 ml Ni-NTA agarose column. Bound proteins were then eluted using a linear imidazole gradient to a final concentration of 400 mM. The purity of each protein sample was assessed by SDS-PAGE followed by Coomassie Brilliant Blue staining. Where noted, protein samples were further purified by fast protein liquid chromotagraphy (FPLC) using gel filtration on a Superdex 200 column (GE Healthcare). Fractions with high purity were used in biochemical assays. Cells expressing selenomethionine-incorporated Tre1tox–Tri1 complex were grown in SelenoMethionine Medium Complete (Molecular Dimensions) using the expression conditions described above. Cell lysis, Ni-NTA affinity purification, and gel filtration were also performed as described above.
For experiments requiring free Tre1tox, a denaturation and refolding strategy was employed to separate His-tagged Tre1tox from Tri1. First, proteins were diluted 1:25 into denaturation buffer (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 5 mM imidazole, and 8 M urea). Next, denatured proteins were run over a column containing 1 mL Ni-NTA agarose to selectively bind His-tagged Tre1tox and removed dissociated Tri1. On-column refolding was achieved by washing the column with renaturation buffer (50 mM Tris-HCl pH = 7.5, 500 mM NaCl) followed by elution of refolded protein using 50mM Tris-HCl pH = 7.5, 500 mM NaCl, 300 mM imidazole. Protein samples were subsequently dialyzed against 20 mM Tris-HCl pH = 7.5, 150 mM NaCl, 5% (v/v) glycerol and stored in −80°C.
To purify untagged Tre1tox, Tobacco Etch Virus (TEV) protease cleavage sequence (ENLYFQG) was inserted in pETDuet-1-tre1tox+tri1 between the hexahistidine tag coding sequence and the tre1 gene. The same denaturation and refolding strategy described above was employed to separate His-tagged TEV-Tre1tox from Tri1. His-tagged TEV-Tre1tox was then incubated with 1 mg/mL His-tagged TEV protease overnight, followed by passing through 1 mL of Ni-NTA agarose to remove TEV protease, hexahistidine peptide, and uncut His-tagged TEVTre1tox. Flow-through fractions with high purity of untagged Tre1tox were pooled and used in binding assays.
For purification, FtsZ was overproduced in E. coli strain BL21 (DE3) transformed with the pET-11 expression vector containing the coding region of the gene. Cultures were grown in LB medium at 37°C to OD600 = 1.0 and induced by adding 0.1 mM IPTG. After a 3-hour induction, cells were precipitated and resuspended in lysis buffer containing 50 mM Tris-HCl pH = 8.0, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1 mM PMSF, and 0.1 mg/mL lysozyme. Resuspended cells were incubated on ice for 30 minutes then disrupted by sonication (10 pulses, 10 seconds each). Cellular debris was removed by centrifugation at 40,000 g for 30 minutes. The unwanted proteins in the supernatant were precipitated with 20% ammonium sulfate. The ammonium sulfate precipitate was discarded, and the supernatant was recovered and precipitated by raising the ammonium sulfate to 30%. FtsZ was then precipitated by centrifugation at 40,000 g for 30 minutes and resuspended in buffer consisting of 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, and 5 mM MgCl2. The pellet was dialyzed against 50 mM Tris–HCl pH 8.0, 0.1 mM EDTA and 5% glycerol, aliquoted and stored frozen at–80°C.
Crystallization, x-ray data collection, and structure determination
For crystallization of native Tre1tox-Tri1 complex and selenomethionine-incorporated Tre1tox-Tri1 complex, Ni-NTA purified protein was concentrated to 5 mg/mL by spin filtration (30 kDa cutoff, Millipore) and screened against commercially available crystallization screens (Microlytic). Diffraction quality crystals were obtained by sitting drop vapor diffusion at room temperature in a solution containing 0.1 M Na2HPO4: Citric Acid pH 4.2, 1.6 / 0.4 M NaH2PO4 / K2HPO4. Native Tre1tox-Tri1 complex crystals displayed the symmetry of space group P212121 (a = 87.2 Å, b = 151.0 Å, c = 196.9 Å, α = β = γ= 90°), with four dimers in the asymmetric unit. Prior to data collection, crystals were cryoprotected in crystallization buffer consisting of 10 mM Tris-HCl pH = 7.5, and 250 mM NaCl, 30% (v/v) PEG3350, and 1 mM TCEP.
Crystals of the selenomethionine Tre1tox-Tri1 complex were obtained at 5 mg/mL in identical crystallization buffer, mixed 1:1 with identical crystallization solution as the native complex. Selenomethionine Tre1tox-Tri1 complex crystals displayed the symmetry of space group C2 (a = 136.3 Å, b = 510 Å, c = 94.9 Å, α = 90°, β = 123.7°, γ= 90°), with one dimer in the asymmetric unit. Prior to data collection, crystals were cryoprotected in crystallization solution as the native complex.
Native data were obtained at 1.000 Å wavelength and 100 K temperature at the BL502 beamline (ALS, Lawrence Berkeley National Laboratory). A complete data set was obtained from one crystal. Initial molecular replacement approaches were not sufficient for solution. Highly redundant anomalous (MAD) data were obtained at 0.9795 Å (inflection), 0.9794 Å (peak), and 0.9770 Å (high) wavelengths from a single selenomethionine crystal. Data were processed using HKL2000 (http://www.hkl-xray.com/).
Heavy atom searching using penix.autosol identified 12 possible sites, and refinement yielded an estimated Bayes correlation coefficient of 41.8 to 2.5 Å resolution. After density modification, the estimated Bayes correlation coefficient increased to 42.2. Approximately 70% of the selenomethionine model was constructed automatically, and the remaining portion was built manually. The current model (Table S2) contains one Tre1tox-Tri1 dimer. A native Tre1tox-Tri1 solution was obtained by molecular replacement with the resulting model.
Refinement was carried out against peak anomalous data with Bijvoet pairs kept separate, and against native data using phenix.refine (https://www.phenix-online.org/documentation/reference/refinement.html) interspersed with manual model revisions using the program Coot (Emsley and Cowtan, 2004) and consisted of conjugate-gradient minimization and calculation of individual atomic displacement and translation/libration/screw parameters (Painter and Merritt, 2006). Noncrystrallographic symmetry (NCS) torsion restraints were used in refinement of the native structure. Coordinates and structure factors are deposited in the RCSB Protein Data Bank (6DRE and 6DRH).
Secondary structure prediction
Secondary structure protein predictions for the NTE in Tri1 homologs was carried out using GeneSilico Metaserver (https://genesilico.pl/meta2). To select the NTE region for analysis, the sequences of Tri1 homologs were aligned with characterized ADP ribosylhydrolases in the Protein Data Bank. The amino acid sequences upstream of the first α-helix shared among all ARH domains were selected as NTE. The percentage of secondary structure elements (α-helix, β-strand, random coil) are the average of representative predictors.
ADP-ribosyltransferase and ADP-ribosylhydrolase enzymatic assays
ADP-ribosylation reactions were performed at 25°C for 30 minutes in a buffer consisting of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2 and ~ 5000 Bq/reaction [adenylate-32P]-NAD+ (PerkinElmer). Purified BSA and FtsZ were added at 2 – 32 μM, and Tre1tox was added at 10 nM as indicated. The reactions were terminated by the addition of 2X SDS loading dye and separated by SDS-PAGE, followed by soaking in gel fixing solution (10% glacial acetic acid, 25% isopropanol) for 30 minutes. The fixed gel was placed on a sheet of Whatman 3 MM filter paper, covered with plastic wrap and dried at 80°C for 1 hr under a vacuum using a conventional gel dryer (Bio-Rad). The gel was then exposed to a phosphor-imaging screen at room temperature overnight and incorporation of radiolabeled NAD+ was detected by a Typhoon FLA 9000 biomolecular imager (GE Healthcare).
ADP-ribosylhydrolase substrates were obtained by incubation of 32 μM BSA and 2 μM FtsZ with 10 nM of Tre1tox at 25°C for 30 minutes in the same buffer as ADP-ribosylation reaction. The indicated concentrations of Tri1 and the Tri1 variant proteins were then added into the solutions. The reactions were incubated at 25°C for another 30 minutes and were analyzed as for ADP-ribosylation assays described above.
Proteomic analysis of ADP-ribosylated substrates
Overnight cultures of E. coli DH5α harboring pBAD18_tre1tox or pBAD_tre1tox E415Q were diluted in LB media to an OD600 = 0.05. After incubation at 37 °C for 2.5 hours, protein expression was induced by adding 0.2 % (w/v) arabinose for 30 minutes and cells were harvested and resuspended in lysis buffer containing 100 mM ammonium bicarbonate, 8 M urea and 0.1% (w/v) RapiGest™ SF surfactant (Waters). The cell suspensions were then sonicated six times with 10 second pulse lengths followed by 30 min centrifugation at 20,000 g to remove cellular debris. 200 μg of each sample was then reduced with 5 mM TCEP for 1 hour at 37°C followed by alkylation using 10 mM iodoacetamide for 30 minutes in the dark at 25°C. Alkylation reactions were quenched using 12 mM N-acetyl-cysteine and subsequently diluted with 100 mM ammonium bicarbonate to reduce the urea concentration to 1.5 M. Samples were then treated with 10 μg of sequencing grade trypsin (Promega) for 16 hours at 37°C. The RapiGest™ SF surfactant was then precipitated by the addition of concentrated HCl (to pH 2–3) followed by incubation at 37°C for 15 minutes. Precipitated detergent was removed by centrifugation at full speed for 10 minutes. Samples were then diluted with 100% acetonitrile and 10% (w/v) trifluoroacetic acid (TFA) to a final concentration of 5% (v/v) and 0.1% (w/v) and applied to MacroSpin™ C18 columns (The Nest Group, Inc.) that had been charged with two washes of 100% acetonitrile followed by one wash with ddH2O. Bound tryptic peptides were then washed twice in 5% (v/v) acetonitrile, 0.1% (w/v) TFA before elution with 80% (v/v) acetonitrile and 0.1% (v/v) formic acid.
For samples collected after interbacterial competition, His-tagged FtsZ of E. coli was enriched by applying cell lysate (prepared and clarified as described above) to a Ni-NTA agarose column. Bound FtsZ was eluted in elution buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, and 400 mM imidazole) and precipitated at −20°C overnight by 10% TCA and 80% acetone. Precipitated protein was then washed by cold acetone and resuspended in lysis buffer containing 100 mM ammonium bicarbonate and 8 M urea and prepared for mass spectrometry analysis as described above for whole cell lysates.
Peptides were analyzed by LC-MS/MS using a Dionex UltiMate 3000 Rapid Separation nanoLC and a Q Exactive™ HF Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific Inc, San Jose, CA). Approximately 1 μg of peptide samples was loaded onto the trap column, which was 150 μm × 3 cm in-house packed with 3 um C18 beads. The analytical column was a 75 μm × 10.5 cm PicoChip column packed with 3um C18 beads (New Objective, Inc. Woburn, MA). The flow rate was kept at 300nL/minute. Solvent A was 0.1% FA in water and Solvent B was 0.1% FA in ACN. The peptide was separated on a 120-minute analytical gradient from 5% ACN/0.1% FA to 40% ACN/0.1% FA. The mass spectrometer was operated in data-dependent mode. The source voltage was 2.10 kV and the capillary temperature was 320 degrees C. MS 1 scans were acquired from 300–2000m/z at 60,000 resolving power and automatic gain control (AGC) set to 3×106. The top 15 most abundant precursor ions in each MS 1 scan were selected for fragmentation. Precursors were selected with an isolation width of 2 Da and fragmented by Higher-energy collisional dissociation (HCD) at 30% normalized collision energy in the HCD cell. Previously selected ions were dynamically excluded from re-selection for 20 seconds. The MS 2 AGC was set to 1×105.
Proteins were identified from the tandem mass spectra extracted by Xcalibur version 4.0. MS/MS spectra were searched against the Uniprot E. coli DH5α strain database using Mascot search engine (Matrix Science, London, UK; version 2.5.1). The MS 1 precursor mass tolerance was set to 10 ppm and the MS 2 tolerance was set to 0.05 Da. A 1% false discovery rate cutoff was applied at the peptide level. Only proteins with a minimum of two unique peptides above the cutoff were considered for further study. The search result was visualized by Scaffold (version 4.8.3. Proteome Software, INC., Portland, OR).
90° angle light scattering assay
90° angle light scattering experiments were performed as described previously (Mukherjee and Lutkenhaus, 1999). Purified FtsZ was added to a final concentration of 12.5 μM and pre-incubated with the indicated concentration of Tre1tox in a buffer consisting of 50 mM HEPES-KOH pH = 7.5, 300 mM K-acetate, 5 mM Mg-acetate, 1 mM NAD+, and 20% (w/v) Ficoll PM70. The sample was equilibrated at 30°C for 5 minutes and followed by the addition of 1 mM GTP to begin polymerization. 90° light scattering was then measured every 2 seconds for 15 min in a fluorescence spectrophotometer at a wavelength of 350 nm (both emission and absorbance).
For measuring impact of Tre1tox to the pre-formed FtsZ polymers, purified FtsZ was polymerized at 30°C as described above without pre-incubation with Tre1tox and NAD+. The indication concentration of Tre1tox and 1 mM NAD+ were then added to the polymerized protein. Polymerization was quantified as described above for a total of 25 minutes.
To measure the effect of ADP-ribosylated FtsZ on polymerization of unmodified FtsZ, ADP-ribosylated FtsZ subunits were generated by incubation of 12.5 μM of purified FtsZ with 12.5 μM of NAD+ and 10 nM Tre1tox at 25°C for 30 minutes. The ADP-ribosylation reaction was terminated by the addition of 100 nM Tri1 immunity protein with catalytic mutation, D161N. The ADP-ribosylated FtsZ subunits were added at the indicated ratios to unmodified FtsZ and incubated for 5 minutes, followed by the addition of 1 mM GTP to begin polymerization. Polymerization was measured as described above.
Negative Stain Electron Microscopy
To visualize the effect of toxin on preformed polymers, purified FtsZ (12.5 μM) was incubated with 0, 0.1, or 10 nM of Tre1tox in the assembly buffer (50 mM HEPES-KOH pH = 7.5, 300 mM K-acetate, 5 mM Mg-acetate, 9 % (w/v) ficoll PM70) at 20 °C for 30 min. FtsZ was then polymerized with addition of 1 mM GTP. After 30 min, the solution containing polymerized FtsZ was diluted 3 times with the assembly buffer. The diluted polymeric mixtures were adsorbed onto glow-discharged carbon-coated copper mesh grids for 60 seconds, washed once with assembly buffer and three times with deionized water, then stained with 2% uranyl formate for 30 seconds, and allowed to air dry. Grids were imaged using the FEI Tecnai Spirit 120 kV electron microscope equipped with a Gatan Ultrascan 4000 CCD Camera. The images were low-pass filtered to 20 Å to improve contrast. Polymers lengths in blinded frames from each sample were measured manually using the microscope scale bar.
Time-lapse fluorescence and phase contrast microscopy
Imaging was performed on a Nikon Eclipse Ti-E wide-field epi-fluorescence microscope, equipped with a sCMOS camera (NEO, Andor Technology) and arc lamp illumination for fluorescence imaging. We image through 60× 1.4 NA oil-immersion objective, and maintain a constant focal plane using a Nikon Perfect Focus system. The microscope is controlled by NIS-Elements. An environmental chamber encloses the microscope to maintain the temperature at 30°C during imaging.
Cells expressing Tre1 or Tre1 E415Q were prepared for microscopy according to the following protocol: (i) Cells were grown in LB medium at 37°C overnight. (ii) Overnight cultures were back-diluted to an OD600 of approximately 0.05 and incubated for 90 min to OD600 ~ 0.4. An μL each of E. coli cells expression Tre1tox or Tre1tox E415Q were spotted on a thin LB agarose (0.2–0.5%) pad on the surface of a microscope slide. To visualize cell morphology, we acquired phase contrast images for each sample at 5 min intervals.
For experiments where FtsZ-FP was visualized by fluorescence microscopy, cells were imaged using a CellASIC ONIX (Model EV262) microfluidic perfusion system paired with bacterial microfluidic plates (B04 plates from Merck). E. coli cells encoding FtsZ::mNeonGreen and carrying pBAD18 Tre1tox or Tre1tox E415Q were introduced into parallel chambers on the same microfluidic plate. Prior to induction, the LB medium within the chambers was constantly exchanged using flow pressure of 1 psi. 0.2 % of arabinose was introduced into the medium 10 minutes after the start of image acquisition. To facilitate rapid solution switching, the LB-arabinose flow pressure is initially set to 8 psi for 1 minute, and subsequently lowered back to 1 psi for the remainder of the experiment. A series of phase-contrast (to determine cell boundaries) and fluorescence images (to visualize FtsZ-mNeonGreen localization) of each sample were acquired with frames every minute. For fluorescence imaging, arc lamp illumination was used with a Nikon GFP filter cube to select the appropriate wavelengths of excitation and emission light. Exposure time for fluorescence images was 300 ms.
For imaging samples before and after competition, mixtures of S. proteamaculans attTn7::Km-gfp and E. coli were spotted on a PBS with 2% agarose pad placed on a microscope slide. Still images of the cells were acquired before and after 18 hours competition, including a phase-contrast image (to visualize cell morphology) and fluorescence image (to distinguish S. proteamaculans, which expressed GFP, from unlabeled E. coli) for each field of view.
Image analysis
Alignment
The first step in our image processing method is to remove drift in the xy plane by aligning both the phase and fluorescence images against the first image in the data set. The corrected alignment was retained for further analysis. To produce phase-contrast movies, the aligned frames were stacked and output in a movie format using build-in MATLAB functions.
Segmentation and Linking
Cell boundaries are computationally determined based on the phase-contrast image at each time point using the Wiggins Lab’s freely-available software, superSegger (http://mtshasta.phys.washington.edu/website/ssodownload.php). For time-lapse images, the software links cells frame-to-frame. Cell masks were defined to include all pixels interior to the cell boundary. These masks are retained for further computation of cell properties. In particular, cell area is taken directly as the number of pixels in the cell mask multiplied by the pixel area.
Focus detection
Foci were detected in fluorescence images as described previously (Mangiameli et al., 2017). Briefly, sub-regions around each intensity maximum were generated using a watershed process on the conjugate image. To exclude any intensity maxima that lie outside the cell boundaries, we consider only sub-regions that overlap with cell masks. Within these overlap regions, we precisely locate foci by placing a circular 3-pixel-radius disk at the maximum-intensity pixel. The intensity profile within this circular disk was modeled as a Gaussian distribution:
where G0 is the background intensity, GG is peak intensity, and b describes the focus width. The focus position is parameterized by .
When quantifying the number of foci per cell, we avoid false detection events by defining the focus score σ as follows:
Where IA is the integrated background subtracted intensity within the three-pixel-radius region (with area A), and δI is the per-pixel the standard deviation interior to the cell mask, but exterior to any circular intensity regions. The focus score is a measure of the signal-to-noise ratio of a focus, and we reject any foci scoring less than 3. Foci scoring below this cutoff were qualitatively consistent with stochastic fluctuations in intensity upon visual inspection of the images. In our quantification of FtsZ-mNeon green foci, we count all foci scoring above the cutoff within each cell mask. We note that an assembled Z ring appears as two punctate foci at the same long-axis position when the cell is viewed side-on.
Analysis of competition data
Snapshot images were first processed using superSegger (http://mtshasta.phys.washington.edu/website/ssodownload.php) to quantify and store cell properties of interest, particularly cell area and total fluorescence. S. proteamaculans were expressing cytoplasmic GFP and could thus be separated from E. coli based on high signal in the fluorescence image. The area distribution of E. coli cells was quantified before and after the competition, and the fraction of the E. coli population having areas greater than twice the median was recorded.
FtsZ structural model
A structural homology model of E. coli FtsZ was created using the MPI Bioinformatics Toolkit and sequence alignment with HHpred (https://toolkit.tuebingen.mpg.de/). The model generated with MODELLER (https://salilab.org/modeller/) was based upon a crystal structure of Staphylococcus aureus FtsZ in the open conformation (PDB 5MN4) (Wagstaff et al., 2017). A trimeric FtsZ polymer was generated based on crystallographic symmetry. An ADP-ribosyl group was manually modelled at E. coli FtsZ residue R174, using two different common arginine side chain rotamers. Model energy minimization and image rendering were performed with Chimera (https://www.cgl.ucsf.edu/chimera/).
Tre1tox-Tri1 interaction assay
Purified N-terminally His-tagged Tri1-Sp and Tri1-Pp_GB-1 were incubated with 100 μL of Ni-NTA agarose beads for 1 hour at 4°C in a binding buffer (50 mM Tris-HCl pH = 7.5, 300 mM NaCl, 5 mM MgCl2, 5% glycerol, 0.1 % Triton X100, 5 mM imidazole, 1 mM DTT). Beads were then washed with excess binding buffer to remove unbound Tri1 proteins. Purified untagged Tre1tox-Sp was then incubated with Tri1 containing Ni-NTA agarose beads for another 1 hour at 4°C, followed by a second wash step with excess binding buffer. Beads were then mixed 1:1 with 2X SDS-loading buffer and boiled at 95 °C for 5 minutes. Bound proteins were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance in E. coli viability assays and bacterial competition experiments was assessed by unpaired t-tests between relevant samples. Details of statistical significance is provided in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The coordinates and structure factors for the Tre1tox-Tri1 complex are deposited in the RCSB Protein Data Bank under accession numbers 6DRE and 6DRH.
Supplementary Material
Figure S1. Related to Figure 2. Induction of Tre1tox expression leads to death of E. coli cells. (A) Viable E. coli cells recovered following induction of expression of the indicated proteins. Expression was induced at T = 3 hours. Data represent means ± SD, n = 3. (B) Anti-Tre1 western blot indicating levels of the indicated proteins produced by E. coli cells after 3–5 h of induction (until OD600 = 1) from inducible expression plasmids. Wild-type Tri1-Sp was coexpressed to prevent cytotoxicity caused by expression of Tre1 and variants. Control band is a non-specific target of the Tre1 antibody, used to indicate equal loading. (C) Schematic depicting the tre1 (yellow) tri1 (blue) locus of wild-type S. proteamaculans and the strain bearing an in-frame deletion of the two genes. Numbers indicate last nucleotide remaining on either side of the deletion. (D) Anti-Tre1 western blot indicating levels of the protein variants shown produced from the native locus in S. proteamaculans grown in liquid LB for 18 h.
Figure S2. Related to Figure 3. Tri1 copurifies with Tre1tox and point mutations in the Tri1 active site and NTE do not affect protein stability. (A) Fast protein liquid chromatograph trace indicating absorbance intensity of the indicated fraction volumes collected during gel filtration purification on a Superdex 200 column of Tre1tox and Tri1. (B) Coomassie stained SDS-PAGE analysis of the indicated fractions eluted from the FPLC purification of Tre1tox-Tri1. (C) Anti-VSV-G western blot analysis of the indicated Tri1 variants carrying a C-terminal VSV-G tag, after 3–5 hrs of expression in E. coli (until OD600 = 1).
Figure S3. Related to Figure 4. The activity of Tre1 leads to ADP-ribosylation of several proteins and cell elongation in E. coli. (A-I) Tandem mass spectra for ADPr proteins detected in E. coli cell lysate following heterologous expression of Tre1tox-Sp for 30 min. Proteins from which peptides derive and modified residues noted at the top of each panel. Fragmentation ions with resolved spectra indicated in blue (5' direction) or red (3' direction). (J) Sequence of peptides found to be ADPr in E. coli after 30 min expression of Tre1tox-Pp. Modified arginine residues, red. (K) Fluorescence and phase contrast micrographs of samples collected from cocultures of S. proteamaculans donor strain expressing GFP and the indicated Tre1 variants (yellow), and recipient E. coli cells (blue, pseudo-colored from phase contrast image) at the start of coculture (top panels) or after 18 hours coculture on an agar surface (bottom panels). (L) Fraction of E. coli cells of the indicated lengths before and after competition with S. proteamaculans expressing wild-type or point mutant Tre1. Lengths representing overall median and 2X median cell size are indicated for reference (dashed lines). Numbers of cells measured before and after competition with each S. proteamaculans donor noted at bottom.
Figure S4. Related to Figure 6. Tri1 proteins provide protection from non-cognate Tre1 proteins through ARH activity, and not direct binding. (A) Coomassie stained SDS-PAGE analysis of Tre1tox-Sp pulled down by nickel affinity purification of the indicated His6-Tri1 variants. Proteins levels prior to column loading shown at 20% concentration at left. (B and C) Western blot analysis of VSV-G-tagged versions of the indicated ARH domain proteins produced after 3–5 hr of induction (until OD600 = 1) from expression plasmids in E. coli. The control band is a non-specific target of the VSV-G antibody, used to indicate equal loading.
Figure S5. Related to Figure 6. ARH domain proteins predicted to function as immunity determinants form three distinct clades. (A) Maximum-likelihood phylogeny of ARH-domain of a subset of proteins identified through position-specific iterative searching with DraG from Rhodopseudomonas palustris as a seed. Clades including immunity proteins characterized in this study, green; DraG-containing clade, blue. Branches corresponding to proteins characterized in this study are indicated by arrows. Clades consisting of sequences derived primarily from one phylum also labeled. (B) Sub-phylogeny of Tri1-Sp and Tri1-Pp related proteins derived from the tree in (A), indicating Shimodaira-Hasegawa test values from 1,000 resamples. A related ARH domain protein from Bradyrhizobium erythrophlei predicted to serve a housekeeping function serves as an outgroup.
Video S1. Related to Figure 2. Time lapse phase contrast microscopy series of E. coli expressing Tre1tox E415Q (left) or Tre1tox (right).
Video S2. Related to Figure 2. Time lapse phase contrast microscopy series of E. coli containing an empty vector (left) or expressing Tre1tox (right).
Video S3. Related to Figure 4. Time lapse fluorescence microscopy series of E. coli expressing FtsZ-mNeonGreen and Tre1tox E415Q (left) or Tre1tox (right).
Highlights.
Protein-targeting ADP-ribosyltransferases (ARTs) act as interbacterial toxins Immunity proteins protect from ARTs by toxin binding and ADP-ribose (ADPr) cleavage
A type VI secretion effector from Serratia acts via ADPr addition to FtsZ FtsZ–ADPr blocks polymerization and cell division, leading to cell death
Acknowledgements
We thank Harold Erickson, Joe Lutkenhaus, and Caroline Harwood for providing reagents, Rachel Klevit and Samuel Miller for sharing equipment, Maria Janowska for technical assistance, Fernando Moreno-Herrero for helpful discussion, and Simon Dove for critical reading of the manuscript. We acknowledge support from the NIH to the Northwestern Proteomics Core Facility (CA060553 and GM108569), the University of Washington Proteomics Resource (UWPR95794), DV (R01GM120553 and HHSN272201700059C) and JDM (AI080609). The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02–05CH11231. DV is a Pew Biomedical Scholar and JDM is an HHMI Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests.
References
- Abebe-Akele F, Tisa LS, Cooper VS, Hatcher PJ, Abebe E, and Thomas WK (2015). Genome sequence and comparative analysis of a putative entomopathogenic Serratia isolated from Caenorhabditis briggsae. BMC genomics 16, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktories K, and Wegner A (1992). Mechanisms of the cytopathic action of actin-ADP-ribosylating toxins. Mol Microbiol 6, 2905–2908. [DOI] [PubMed] [Google Scholar]
- Alcoforado Diniz J, Liu YC, and Coulthurst SJ (2015). Molecular weaponry: diverse effectors delivered by the Type VI secretion system. Cellular microbiology 17, 1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Zhang D, de Souza RF, Anand S, and Iyer LM (2015). The natural history of ADP-ribosyltransferases and the ADP-ribosylation system. Current topics in microbiology and immunology 384, 3–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold CL, Wang H, Nordlund S, and Hogbom M (2009). Mechanism of ADP-ribosylation removal revealed by the structure and ligand complexes of the dimanganese mono-ADP-ribosylhydrolase DraG. Proc Natl Acad Sci U S A 106, 14247–14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson-Filho AW, Hsu YP, Squyres GR, Kuru E, Wu F, Jukes C, Sun Y, Dekker C, Holden S, VanNieuwenhze MS, et al. (2017). Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleves S (2016). Game of Trans-Kingdom Effectors. Trends Microbiol 24, 773–774. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, and Gabaldon T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics (Oxford, England) 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, and Schweizer HP (2008). Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Applied and environmental microbiology 74, 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, and Schweizer HP (2006). mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nature protocols 1, 153–161. [DOI] [PubMed] [Google Scholar]
- Cianfanelli FR, Monlezun L, and Coulthurst SJ (2016). Aim, Load, Fire: The Type VI Secretion System, a Bacterial Nanoweapon. Trends Microbiol 24, 51–62. [DOI] [PubMed] [Google Scholar]
- Cohen MS, and Chang P (2018). Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol 14, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, and Lutkenhaus J (1991). ftsZ is an essential cell division gene in Escherichia coli. J Bacteriol 173, 3500–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DH, and Donachie DW (1993). Construction of a triple deletion mutant of penicillin-binding proteins 4, 5 and 6 in Escherichia coli In Bacterial Growth and Lysis: Metabolism and Structure of the Bacterial Sacculus, de Pedro MA, Holtje JV, and Loffelhardt W, eds. (London: Plenum Press; ). [Google Scholar]
- Erickson HP, Anderson DE, and Osawa M (2010). FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74, 504–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KR, and Bell T (2012). Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol 22, 1845–1850. [DOI] [PubMed] [Google Scholar]
- Fu ZQ, Guo M, Jeong BR, Tian F, Elthon TE, Cerny RL, Staiger D, and Alfano JR (2007). A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature 447, 284–288. [DOI] [PubMed] [Google Scholar]
- Haeusser DP, Hoashi M, Weaver A, Brown N, Pan J, Sawitzke JA, Thomason LC, Court DL, and Margolin W (2014). The Kil peptide of bacteriophage lambda blocks Escherichia coli cytokinesis via ZipA-dependent inhibition of FtsZ assembly. PLoS genetics 10, e1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller DM, Tavag M, and Hochschild A (2017). CbtA toxin of Escherichia coli inhibits cell division and cell elongation via direct and independent interactions with FtsZ and MreB. PLoS genetics 13, e1007007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T, Nishizuka Y, and Hayaishi O (1969). Adenosine diphosphoribosylation of aminoacyl transferase II by diphtheria toxin. Cold Spring Harb Symp Quant Biol 34, 603–608. [DOI] [PubMed] [Google Scholar]
- Jankevicius G, Ariza A, Ahel M, and Ahel I (2016). The Toxin-Antitoxin System DarTG Catalyzes Reversible ADP-Ribosylation of DNA. Mol Cell 64, 1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Low DA, and Hayes CS (2017). Can’t you hear me knocking: contact-dependent competition and cooperation in bacteria. Emerg Top Life Sci 1, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiro R, Molshanski-Mor S, Yosef I, Milam SL, Erickson HP, and Qimron U (2013). Gene product 0.4 increases bacteriophage T7 competitiveness by inhibiting host cell division. Proc Natl Acad Sci U S A 110, 19549–19554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelman CM, Aarsman ME, Postmus J, Pas E, Muijsers AO, Scheffers DJ, Nanninga N, and den Blaauwen T (2004). R174 of Escherichia coli FtsZ is involved in membrane interaction and protofilament bundling, and is essential for cell division. Mol Microbiol 51, 645–657. [DOI] [PubMed] [Google Scholar]
- Krupka M, and Margolin W (2018). Unite to divide: Oligomerization of tubulin and actin homologs regulates initiation of bacterial cell division. F1000Res 7, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCourse KD, Peterson SB, Kulasekara HD, Radey MC, Kim J, and Mougous JD (2018). Conditional toxicity and synergy drive diversity among antibacterial effectors. Nat Microbiol 3, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiameli SM, Merrikh CN, Wiggins PA, and Merrikh H (2017). Transcription leads to pervasive replisome instability in bacteria. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DA, Whatley ZN, Joshi CP, Osawa M, and Erickson HP (2017). Probing for Binding Regions of the FtsZ Protein Surface through Site-Directed Insertions: Discovery of Fully Functional FtsZ-Fluorescent Proteins. J Bacteriol 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, and Lutkenhaus J (1999). Analysis of FtsZ assembly by light scattering and determination of the role of divalent metal cations. J Bacteriol 181, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva MA, Huecas S, Palacios JM, Martin-Benito J, Valpuesta JM, and Andreu JM (2003). Assembly of archaeal cell division protein FtsZ and a GTPase-inactive mutant into double-stranded filaments. J Biol Chem 278, 33562–33570. [DOI] [PubMed] [Google Scholar]
- Ortiz C, Natale P, Cueto L, and Vicente M (2016). The keepers of the ring: regulators of FtsZ assembly. FEMS Microbiol Rev 40, 57–67. [DOI] [PubMed] [Google Scholar]
- Redick SD, Stricker J, Briscoe G, and Erickson HP (2005). Mutants of FtsZ targeting the protofilament interface: effects on cell division and GTPase activity. J Bacteriol 187, 2727–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietsch A, Vallet-Gely I, Dove SL, and Mekalanos JJ (2005). ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102, 8006–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, and Mougous JD (2011). Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell host & microbe 16, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sana TG, Flaugnatti N, Lugo KA, Lam LH, Jacobson A, Baylot V, Durand E, Journet L, Cascales E, and Monack DM (2016). Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc Natl Acad Sci U S A 113, E5044–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon NC, Aktories K, and Barbieri JT (2014). Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol 12, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speare L, Cecere AG, Guckes KR, Smith S, Wollenberg MS, Mandel MJ, Miyashiro T, and Septer AN (2018). Bacterial symbionts use a type VI secretion system to eliminate competitors in their natural host. Proc Natl Acad Sci U S A 115, E8528–E8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianidou S, Brennan C, Nissen SB, Kuwada NJ, and Wiggins PA (2016). SuperSegger: robust image segmentation, analysis and lineage tracking of bacterial cells. Mol Microbiol 102, 690–700. [DOI] [PubMed] [Google Scholar]
- Sun J, Siroy A, Lokareddy RK, Speer A, Doornbos KS, Cingolani G, and Niederweis M (2015). The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nature structural & molecular biology 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Yu H, Li Q, Wang X, Gai Z, Yin G, Su F, Tao F, Ma C, and Xu P (2011). Genome sequence of Pseudomonas putida strain B6–2, a superdegrader of polycyclic aromatic hydrocarbons and dioxin-like compounds. J Bacteriol 193, 6789–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge H, Nagahama M, Nishimura H, Hisatsune J, Sakaguchi Y, Itogawa Y, Katunuma N, and Sakurai J (2003). Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J Mol Biol 325, 471–483. [DOI] [PubMed] [Google Scholar]
- Vaughan S, Wickstead B, Gull K, and Addinall SG (2004). Molecular evolution of FtsZ protein sequences encoded within the genomes of archaea, bacteria, and eukaryota. J Mol Evol 58, 19–29. [DOI] [PubMed] [Google Scholar]
- Verster AJ, Ross BD, Radey MC, Bao Y, Goodman AL, Mougous JD, and Borenstein E (2017). The Landscape of Type VI Secretion across Human Gut Microbiomes Reveals Its Role in Community Composition. Cell host & microbe 22, 411–419 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff JM, Tsim M, Oliva MA, Garcia-Sanchez A, Kureisaite-Ciziene D, Andreu JM, and Lowe J (2017). A Polymerization-Associated Structural Switch in FtsZ That Enables Treadmilling of Model Filaments. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JC, Peterson SB, Kim J, Pazos M, Verster AJ, Radey MC, Kulasekara HD, Ching MQ, Bullen NP, Bryant D, et al. (2017). A broadly distributed toxin family mediates contact-dependent antagonism between gram-positive bacteria. Elife 6, e26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JC, Quentin D, Sawai S, LeRoux M, Harding BN, Ledvina HE, Tran BQ, Robinson H, Goo YA, Goodlett DR, et al. (2015). An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Monchy S, Taghavi S, Zhu W, Ramos J, and van der Lelie D (2011). Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol Rev 35, 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, and Xiao J (2017). GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figure 2. Induction of Tre1tox expression leads to death of E. coli cells. (A) Viable E. coli cells recovered following induction of expression of the indicated proteins. Expression was induced at T = 3 hours. Data represent means ± SD, n = 3. (B) Anti-Tre1 western blot indicating levels of the indicated proteins produced by E. coli cells after 3–5 h of induction (until OD600 = 1) from inducible expression plasmids. Wild-type Tri1-Sp was coexpressed to prevent cytotoxicity caused by expression of Tre1 and variants. Control band is a non-specific target of the Tre1 antibody, used to indicate equal loading. (C) Schematic depicting the tre1 (yellow) tri1 (blue) locus of wild-type S. proteamaculans and the strain bearing an in-frame deletion of the two genes. Numbers indicate last nucleotide remaining on either side of the deletion. (D) Anti-Tre1 western blot indicating levels of the protein variants shown produced from the native locus in S. proteamaculans grown in liquid LB for 18 h.
Figure S2. Related to Figure 3. Tri1 copurifies with Tre1tox and point mutations in the Tri1 active site and NTE do not affect protein stability. (A) Fast protein liquid chromatograph trace indicating absorbance intensity of the indicated fraction volumes collected during gel filtration purification on a Superdex 200 column of Tre1tox and Tri1. (B) Coomassie stained SDS-PAGE analysis of the indicated fractions eluted from the FPLC purification of Tre1tox-Tri1. (C) Anti-VSV-G western blot analysis of the indicated Tri1 variants carrying a C-terminal VSV-G tag, after 3–5 hrs of expression in E. coli (until OD600 = 1).
Figure S3. Related to Figure 4. The activity of Tre1 leads to ADP-ribosylation of several proteins and cell elongation in E. coli. (A-I) Tandem mass spectra for ADPr proteins detected in E. coli cell lysate following heterologous expression of Tre1tox-Sp for 30 min. Proteins from which peptides derive and modified residues noted at the top of each panel. Fragmentation ions with resolved spectra indicated in blue (5' direction) or red (3' direction). (J) Sequence of peptides found to be ADPr in E. coli after 30 min expression of Tre1tox-Pp. Modified arginine residues, red. (K) Fluorescence and phase contrast micrographs of samples collected from cocultures of S. proteamaculans donor strain expressing GFP and the indicated Tre1 variants (yellow), and recipient E. coli cells (blue, pseudo-colored from phase contrast image) at the start of coculture (top panels) or after 18 hours coculture on an agar surface (bottom panels). (L) Fraction of E. coli cells of the indicated lengths before and after competition with S. proteamaculans expressing wild-type or point mutant Tre1. Lengths representing overall median and 2X median cell size are indicated for reference (dashed lines). Numbers of cells measured before and after competition with each S. proteamaculans donor noted at bottom.
Figure S4. Related to Figure 6. Tri1 proteins provide protection from non-cognate Tre1 proteins through ARH activity, and not direct binding. (A) Coomassie stained SDS-PAGE analysis of Tre1tox-Sp pulled down by nickel affinity purification of the indicated His6-Tri1 variants. Proteins levels prior to column loading shown at 20% concentration at left. (B and C) Western blot analysis of VSV-G-tagged versions of the indicated ARH domain proteins produced after 3–5 hr of induction (until OD600 = 1) from expression plasmids in E. coli. The control band is a non-specific target of the VSV-G antibody, used to indicate equal loading.
Figure S5. Related to Figure 6. ARH domain proteins predicted to function as immunity determinants form three distinct clades. (A) Maximum-likelihood phylogeny of ARH-domain of a subset of proteins identified through position-specific iterative searching with DraG from Rhodopseudomonas palustris as a seed. Clades including immunity proteins characterized in this study, green; DraG-containing clade, blue. Branches corresponding to proteins characterized in this study are indicated by arrows. Clades consisting of sequences derived primarily from one phylum also labeled. (B) Sub-phylogeny of Tri1-Sp and Tri1-Pp related proteins derived from the tree in (A), indicating Shimodaira-Hasegawa test values from 1,000 resamples. A related ARH domain protein from Bradyrhizobium erythrophlei predicted to serve a housekeeping function serves as an outgroup.
Video S1. Related to Figure 2. Time lapse phase contrast microscopy series of E. coli expressing Tre1tox E415Q (left) or Tre1tox (right).
Video S2. Related to Figure 2. Time lapse phase contrast microscopy series of E. coli containing an empty vector (left) or expressing Tre1tox (right).
Video S3. Related to Figure 4. Time lapse fluorescence microscopy series of E. coli expressing FtsZ-mNeonGreen and Tre1tox E415Q (left) or Tre1tox (right).