

Abstract
Mucolipidosis type IV (MLIV) is an autosomal recessive, lysosomal storage disorder causing progressively severe intellectual disability, motor and speech deficits, retinal degeneration often culminating in blindness, and systemic disease causing a shortened lifespan. MLIV results from mutations in the gene MCOLN1 encoding the transient receptor potential channel mucolipin-1. It is an ultra-rare disease and is currently known to affect just over 100 diagnosed individuals. The last decade has provided a wealth of research focused on understanding the role of the enigmatic mucolipin-1 protein in cell and brain function and how its absence causes disease. This review explores our current understanding of the mucolipin-1 protein in relation to neuropathogenesis in MLIV and describes recent findings implicating mucolipin-1's important role in mTOR (mechanistic target of rapamycin) and TFEB (transcription factor EB) signaling feedback loops as well as in the function of the greater endosomal/lysosomal system. In addition to addressing the vital role of mucolipin-1 in the brain, we also report new data on the question of whether haploinsufficiency as would be anticipated in MCOLN1 heterozygotes is associated with any evidence of neuron dysfunction or disease. Greater insights into the role of mucolipin-1 in the nervous system can be expected to shed light not only on MLIV disease but also on numerous processes governing normal brain function.
Keywords: TFEB, mTOR, heterozygote, lysosome, lysosomal disease, autophagy
Graphical abstract
Mucolipidosis type IV (MLIV) is a rare, autosomal recessive, lysosomal storage disorder caused by mutations in the endosomal/lysosomal protein, mucolipin-1. This review explores the roles of aberrations to endosomal/lysosomal trafficking, autophagy, lysosomal exocytosis, mTOR (mammalian target of rapamycin) and TFEB (transcription factor EB) signaling, and heavy metal regulation in MLIV neuropathogenesis. We also share pilot data on the question of whether Mcoln1 murine heterozygotes are associated with any evidence of neuron dysfunction or disease. The goal of this review is to provide greater insights into mucolipin-1's role in the nervous system and the multifaceted pathways contributing to MLIV disease pathogenesis.
Introduction
Lysosomal disorders and the mucolipidoses
While individually rare, when grouped together lysosomal disorders have a collective frequency of about 1 in every 7700 live births, presenting a significant health burden (Hoffmann and Mayatepek 2005). Lysosomal disorders were first described in the 1820s, yet it was not until the 20th century that the disorders were clinically differentiated and their underlying causes characterized. The discovery of lysosomes by Christian de Duve in 1955 was followed by the articulation of the lysosomal storage disease concept by H.G. Hers in 1965 (de Duve et al. 1955; Hers 1965). As a result, lysosomal disorders emerged as a class unifying a spectrum of over 50 individual diseases (Futerman and van Meer, 2004; Hers and Hoof, 1973; Platt and Walkley, 2004). Broadly described, most of these conditions result in intralysosomal accumulation of macromolecules arising from an inherited metabolic defect, hence the often used term “storage” disorders (Hers and Hoof 1973). Though tissue and organ penetrance varies across this class of diseases, the resulting lysosomal system dysfunction is often accompanied by neurological involvement (Platt and Walkley 2004). One such lysosomal disease exhibiting severe neurological manifestations is the autosomal recessive disorder, mucolipidosis type IV (Slaugenhaupt et al. 1999).
In 1974, a young male presenting with corneal clouding was first described as having a new variant of mucolipidosis, termed MLIV (Berman et al. 1974). Until this point, the class of mucolipidoses (MLs) included MLI (originally described as a sialidosis, caused by defects in neuraminidase), MLII (I-cell disease, caused by mutations in N-acetylglucosamine-1-phosphotransferase (GlcNAc phosphotransferase) leading to alterations in lysosomal enzyme targeting), and MLIII (pseudo-Hurler polydystrophy, also caused by defects in GlcNAc phosphotransferase) (Pshezhetsky et al. 1997; Bonten et al. 1996; Raas-Rothschild et al. 2000; Spranger and Wiedemann 1970; Kudo et al. 2006). The MLs are an interesting example of a class of lysosomal disorders grouped together due to similar clinical manifestations and storage pattern, yet arising from mutations in distinct genes (Spranger and Wiedemann 1970; Slaugenhaupt 2002). The 1974 case was described as a new variant of mucolipidosis given the unique combination of neurologic deficits, neonatal corneal clouding, and ultrastructural evidence of electron dense inclusion bodies in the bone marrow, fibroblasts, and liver - in the absence of skeletal abnormalities and apparent lack of changes in enzymatic activity (Berman et al. 1974). Within the year of describing this new variant, additional patients presenting with similar features were described by Isabelle Rapin and colleagues (Tellez-Nagel et al., 1976), and the name mucolipidosis type IV for this new clinical variant became established in the literature (Tellez-Nagel et al. 1976; Berman et al. 1974).
Natural history of MLIV and identification of MCOLN1
Much of what is known of MLIV's clinical progression comes from initial studies of the first MLIV patients and comprehensive analyses of clinical chart data and family interviews originating from the 1970s and 1980s (Amir et al. 1987; Berman et al. 1974; Tellez-Nagel et al. 1976; Chitayat et al. 1991). This seminal study provided the first and only brain biopsy tissues from an MLIV patient examined to date (Tellez-Nagel et al. 1976). MLIV disease results in intellectual disability, systemic pathologies such as kidney involvement and achlorhydria, motor and speech deficits, retinal degeneration, and a shortened lifespan (Amir et al. 2013; Schiffmann et al. 1998; Schiffmann et al. 2014). During the first year of life, children present with psychomotor delays and visual impairment, and storage accumulation can already be detected upon tissue biopsy (Amir et al. 1987; Tellez-Nagel et al. 1976). One of the most unique characteristics of MLIV is achlorhydria with increased plasma gastrin secretion, which has been utilized as a diagnostic factor even in infant patients (Goldin et al. 2008; Chandra et al. 2011; Schiffmann et al. 1998). Dysmyelination and reduced size of the corpus callosum have also been reported in very young MLIV patients indicating that there is likely a developmental component to the disease in addition to later neurodegeneration (Schiffmann et al. 2014; Frei et al. 1998). Affected individuals go on to exhibit developmental and motor milestone delays, and impaired fine motor movement (Amir et al. 1987). However, while some patients experience deteriorating motor function over time, others are able to develop motor skills such as walking, sitting upright, or even standing with support (Tellez-Nagel et al. 1976; Amir et al. 1987). There is currently no cure or corrective therapy for MLIV disease. Small molecule therapy using miglustat has recently been shown to delay cerebellar disease in the MLIV mouse model, but has not yet been tested in human patients (Boudewyn et al. 2017). Bone marrow transplant (BMT) has also been explored in the mouse model, which was reported to amelio transmembrane domains 5 and rate motor phenotypes (Walker and Montell 2016). However, it remains unlikely that BMT would rescue clinical phenotypes overall in humans given that mucolipin-1 is a transmembrane protein (as described below) and thus would not be amenable to secretion for cross correction of diseased neurons.
Importantly, there is a need in the MLIV community for a comprehensive natural history study to chronicle MLIV disease symptoms and identify measurable clinical biomarkers over the course of life. It has been noted for example that many patients in the second to third decade of life suffer kidney disease and kidney failure, which as a major cause of morbidity in MLIV represents one of the significant knowledge gaps in MLIV research (personal communication, Dr. Rebecca Oberman, Director, ML4 Foundation). Such a study would provide information needed to establish measurable readouts for future clinical trials of emerging therapies, and to better understand pressing questions such as kidney disease etiology.
The majority of MLIV patients who have been described to date are of Ashkenazi Jewish descent (Slaugenhaupt et al. 1999; Raas-Rothschild et al. 1999; Gideon et al. 2005). In 1999, Slaugenhaupt and colleagues utilized genome scans from 13 Ashkenazi Jewish families to map the location of the MLIV gene (Slaugenhaupt et al. 1999). Using linkage analysis, the gene was mapped to chromosome 19p13.2-13.3, and two founder haplotypes were identified in this population (Slaugenhaupt et al. 1999; Acierno et al. 2001). Approximately 95% of all MLIV patients possess one of these two founder haplotypes, referred to as the major and minor mutations, localized to the MCOLN1 gene (Bargal et al. 2000). MCOLN1 contains 14 exons and encodes the 580-amino acid, 65 kDa mucolipin-1 protein. The most common mutation in MLIV accounts for 72% of MLIV cases and results in a splice mutation at IVS3-2A→G (Bargal et al. 2000; Edelmann et al. 2002). The second most common haplotype accounting for 23% of MLIV cases is caused by a 6434 base pair (bp) deletion including the first 6 exons and the first 12 bps of exon 7. The remaining cases of MLIV (∼5%) can be traced to over 20 independent mutations (Slaugenhaupt 2002; Altarescu et al. 2002; Mirabelli-Badenier et al. 2014; Saijo et al. 2016; Bargal et al. 2000; Shiihara et al. 2018). The extent of protein dysfunction or absence has been reported to correlate with the severity of disease (Altarescu et al. 2002).
The TRP proteins and mucolipin-1 in the endosomal/lysosomal system
The mucolipin-1 protein, or TRPML1, is a member of the transient receptor potential family of proteins (Montell 2005). The Transient Receptor Potential (TRP) channel family is comprised of seven subfamilies made up of over 20 different channel proteins, including the TRPC, TRPV, TRPM, TRPN, TRPA, TRPML, and TRPP (Clapham et al. 2001; Minke 2010). TRP proteins can be found widely distributed in mammalian tissues with a range of subcellular locations and functions, but are united by a similar structure built on 6 transmembrane domains (Clapham et al. 2001). The mammalian mucolipin TRP (TRPML) family consists of mucolipin-1, 2, and 3. Mucolipin-1 is predominantly localized to the surface of late endosomes and lysosomes, and can be found in every tissue with the highest expression in brain, kidney, liver, spleen, and heart (Cheng et al. 2010). Like other TRP channels, mucolipin-1 contains 6 transmembrane domains, and harbors a serine lipase domain, a nuclear localization signal, and lysosomal targeting motifs comprised of di-leucine C-terminal motifs and two N-terminal proline rich sequences between the first and second transmembrane domains (Fig. 1) (Bach 2001; Kiselyov et al. 2005; Vergarajauregui and Puertollano 2006). The N- and C- terminal ends of mucolipin-1 extend into the cytosol (Fig. 1) (Sun et al. 2000). The N-terminal di-leucine motif is also purported to interact with clathrin adaptors AP1 and AP3, while the C-terminal di-leucine motif is predicted to interact with AP2, thus promoting plasma membrane internalization (Vergarajauregui and Puertollano 2006). The C-terminal end also possesses palmitoylation sites at cysteine residues, promoting membrane localization (Pryor et al. 2006; Vergarajauregui and Puertollano 2006; Miedel et al. 2006; Kiselyov et al. 2005; Venkatachalam et al. 2006). A loop is present between transmembrane domains 5 and 6 which serves as the channel's pore (Fig. 1) (Kiselyov et al. 2005; Zhang et al. 2017b; Chen et al. 2017). The intraluminal loop between transmembrane domains 1 and 2 also contains a serine-lipase region, a proline rich domain, and a proteolytic cleavage site at Arg200 (Fig. 1) (Kiselyov et al. 2005). This site undergoes proteolytic cleavage in a post Golgi compartment (Kiselyov et al. 2005). It is thought that following cleavage, mucolipin-1 may form multimers and even heteromultimers (Zeevi et al. 2010; Cyntia et al. 2009).
Figure 1. Structure and functional motifs of mucolipin-1.
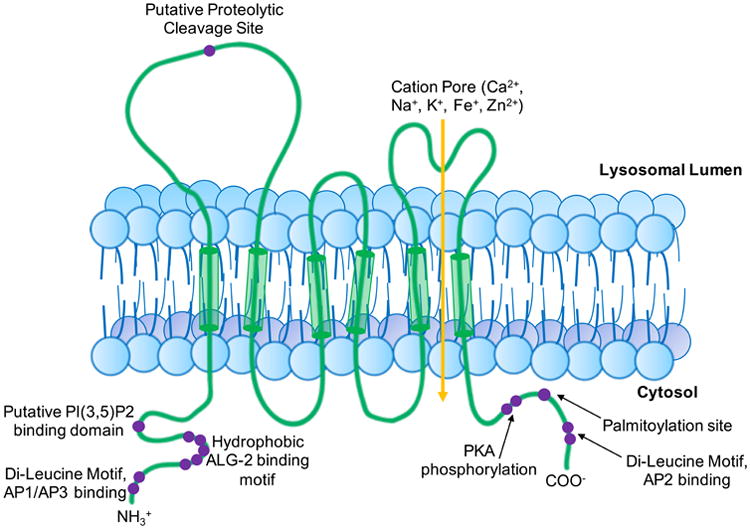
Schematic depicting mucolipin-1's structure, including 6 transmembrane domains (TMD), a proteolytic cleavage site between TMDs 1 and 2, and a pore between TMDs 5 and 6. Protein interacting motifs are noted in purple. N terminal domains contain binding sites for AP1/AP3 proteins, PI(3,5)P2, ALG-2 binding motifs, and di-leucine lysosomal targeting domains. The pore is permeable to calcium, iron, zinc, manganese, magnesium, potassium, sodium, and hydrogen ions. The C terminus contains binding sites for PKA phosphorylation, palmitoylation for membrane targeting, AP2 binding sites, and another lysosomal targeting di-leucine sequence. (Kiselyov et al. 2005; Clapham et al. 2001; Zeevi et al. 2007; Li et al. 2017; Zhang et al. 2017b; Vergarajauregui et al. 2008b; Abe and Puertollano 2011; Dong et al. 2010; Vergarajauregui et al. 2009; Vergarajauregui and Puertollano 2006; LaPlante et al. 2002)
Currently, the general consensus in the field suggests mucolipin-1 is a nonselective, inwardly rectifying (i.e., lysosome to cytosol) cation channel, with permeability to calcium, iron, sodium, potassium, manganese, magnesium, zinc, and hydrogen ions (Fig. 1) (Cheng et al. 2010; Dong et al. 2009; Soyombo et al. 2006). Though mucolipin-1 is permeable to a number of cations, the channel property that appears to be most integrally related to our current understanding of the protein's cellular function is its permeability to calcium. Mucolipin-1's role as a lysosomal calcium channel potentially places this TRP channel at the crux of autophagic regulation, trafficking regulation, and even mTOR (mammalian [or mechanistic] target of rapamycin) and TFEB (transcription factor EB) signaling - yet as described below, these roles and mucolipin-1's permeability to other cations implicate multiple pathogenic pathways in the context of MLIV disease.
Understanding neuronal dysfunction and degeneration in MLIV
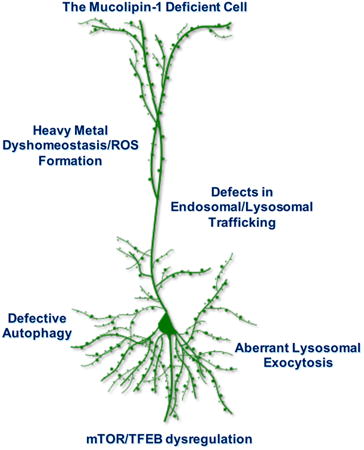
While studies in recent years have provided new insights into the many roles played by mucolipin-1 in cells, as well as a better characterization of the underlying neuropathology of MLIV disease, the distinct connections between the loss of function of this protein and the neuronal phenotypes that result remain poorly understood. Here we provide a focus on five potential neuropathogenic pathways and how they may be related to the observed abnormalities in MLIV disease – specifically, aberrations in endosomal/autophagosomal/lysosomal trafficking, alterations to autophagy, dysregulation of lysosomal exocytosis, changes in the mTORC1/TFEB signaling axis, and altered regulation of heavy metal homeostasis.
Endosomal Lysosomal Trafficking
Mucolipin-1 has long been implicated as having a role in the first pathogenic pathway: trafficking and fusion of late endosomes and lysosomes in neurons and other cell types. Early studies of MLIV model systems demonstrated the presence of enlarged late endosomes/lysosomes and accumulation of autophagosomes consistent with such trafficking defects in the absence of mucolipin-1 (Chandra et al. 2011; Venkatachalam et al. 2008; Venugopal et al. 2009). Studies more specifically dissecting the endosomal pathway indicate the absence of mucolipin-1 leads to malfunctions in retrograde lipid transport within the late endocytic pathway (Shen et al. 2012; Pryor et al. 2006; Ye et al. 2004; Miedel et al. 2008). The loss of this TRP channel's local, lysosomal calcium source has also been reported to lead to improper fission events needed to re-form lysosomes from autolysosomes and to recycle transport vesicles from the endosomal network (Miller et al. 2015; Dong et al. 2010). Defects in endosomal/lysosomal trafficking emerge as a logical candidate underlying the heterogenous lysosomal storage phenotype present in MLIV disease, particularly in regards to complex gangliosides (Micsenyi et al. 2009; Venugopal et al. 2007). Gangliosides are synthesized within the Golgi network, and rely on proper vesicular transport to be delivered to their destinations in the plasmalemma. Following residence in the plasmalemma, they are endocytosed and recycled via the lysosomal system, with precursors for subsequent ganglioside synthesis being made available by this process (Riboni and Tettamanti 1991; Ghidoni et al. 1989; Möbius et al. 1999). The described fusion and endocytic trafficking errors with the loss of mucolipin-1 may lead to a failure to efficiently catabolize gangliosides such as GM1 and GM2, with these becoming trapped within organelles as a result. Indeed, observed storage patterns in Mcoln1-/- brain tissues indicate that individual types of gangliosides (GM1, GM2 and GM3) are not always localized to the same storage compartment, a finding similar to other lysosomal diseases, and can even be found within vesicles separate from LAMP-positive late endosomes/lysosomes (Micsenyi et al. 2009). Neurons as post-mitotic cells may be uniquely sensitive to such perturbations to proper endosomal/lysosomal cycling, but whether such failure is uniquely contributing to storage in MLIV-affected neurons remains uncertain.
Interestingly, mucolipin-1's role in lipid trafficking may even be important in the pathogenic cascade of other disorders. In cultured Niemann-Pick type C (NPC) knockout CHO cell lines, mucolipin-1-mediated lysosomal calcium release is reduced and is inhibited by sphingomyelin (Shen et al. 2012; Wang et al. 2015). Further, in cultured human retinal epithelial cells (ARPE-19) and in the ABCA4 mouse model of Stargardt's retinal dystrophy, induced lysosomal accumulation via the sphingomyelinase inhibitor desipramine and lysomotopic agent chloroquine reduced mucolipin-1 mediated calcium signaling (Más Gomez et al. 2018). These studies suggest that the luminal lysosomal environment could affect mucolipin-1 function, and therefore that chronic lysosomal storage may promote additional secondary abnormalities in lysosomes. (Shen et al. 2012). As these findings may have a broad influence a variety of lysosomal disorders, efforts to understand the role of accrued storage affecting mucolipin-1 function as a further mechanism of neuropathogenesis may be informative.
Autophagy
Furthermore, along the endosomal/lysosomal pathway in the mucolipin-1-deficient cell, there is also a large body of evidence in MLIV indicating a second pathogenic pathway: defective autophagic progression and function. LC3 is a useful marker of autophagic flux, as during the formation of autophagosomes, cytosolic LC3-I is conjugated to phosphatidylethanolamine to form LC3-II, tethering LC3 to the growing autophagosome membrane (Settembre et al. 2008; Tanida et al. 2008). LC3-II turnover levels therefore can serve as an indicator of both autophagosomal maturation (by the level of LC3-II relative to LC3-I) and degradation (turnover of LC3-II within the autolysosome) (Tanida et al. 2008). Studies in both human MLIV fibroblasts as well as neuronal cultures from MLIV mice indicate that LC3-II levels are significantly higher than WT, and are not responsive to challenge with rapamycin (Curcio-Morelli et al. 2010). This finding is suggestive of a defect in the completion of autophagosome degradation during macroautophagy (hereafter referred to as autophagy), and is consistent with findings of an accumulation of autophagosomes in MLIV disease (Wong et al. 2012). It has also been shown that mitochondria accumulate in MLIV disease, which is further evidence consistent with defective autophagy (in this case, mitophagy) in this disorder (Jennings et al., 2006).
Another widely studied marker of defective autophagy is p62 aggregation - this too has been reported to occur in the MLIV mouse model and even in human MLIV cells (Micsenyi et al. 2009; Curcio-Morelli et al. 2010; Vergarajauregui et al. 2008a; Saijo et al. 2016). In order to better characterize aggregation of p62 in MLIV disease we carried out a series of confocal imaging studies on cells in the cerebral cortex, hippocampus and cerebellum of the Mcoln1-/- mouse model. We found that aggregates of p62 in the neocortex occur as large, clustered rings, as well as smaller punctae, which are particularly abundant in the cerebellum (Fig. 2). Large clusters of p62 such as those documented in the MLIV brain (and other lysosomal diseases) are not reported in WT tissues, yet smaller p62 punctae are typically observed and in fact can be enhanced in response to starvation in culture (Itakura and Mizushima 2011). Overall, the size of aggregates in MLIV brain regions we studied ranged from small (i.e., 1 micron or less in diameter), to large (reaching 12 microns in diameter), with the frequency of occurrence being relatively low in neocortex compared to the granule cell layer of the cerebellum (data not shown). The prevalence of large aggregates appeared to be less than that observed in other lysosomal diseases, e.g., in the mouse model of CLN2 disease (Micsenyi 2013). Here, p62 aggregation has been associated with lysosomal membrane permeability (LMP) rather than a defect in autophagy (Micsenyi et al., 2013). Whether LMP is a phenomenon occurring in MLIV disease is presently unknown, but leakage of cathepsin B from lysosomes can induce cell death and has been suggested as a potential apoptotic mechanism in this disease (Colletti et al., 2012; de Castro et al. 2016). Importantly, release of cathepsins from damaged lysosomes is associated with other modes of cell death as well, including caspase-independent cell death (Bröker et al. 2004). Thus, at present, whether the formation of p62 aggregates in the MLIV brain reflects a simple blockage in autophagy, or a more complex event occurring in response to LMP (as described in CLN2 disease), or involving mTORC1/TFEB dysregulation (see below), or a complex multifaceted autophagic defect, remains to be determined. Similar to how p62 aggregates may arise from a multifaceted defect in MLIV, it is possible that the heterogenous storage observed in MLIV cells arises from these multiple events growing out of the loss of mucolipin-1 as described above. These include compromises in autophagy, lipid trafficking, and even a third possible pathway of neuropathogenesis - disturbances to lysosomal exocytosis.
Figure 2. Aggregations of p62 in the Mcoln1-/- mouse brain.
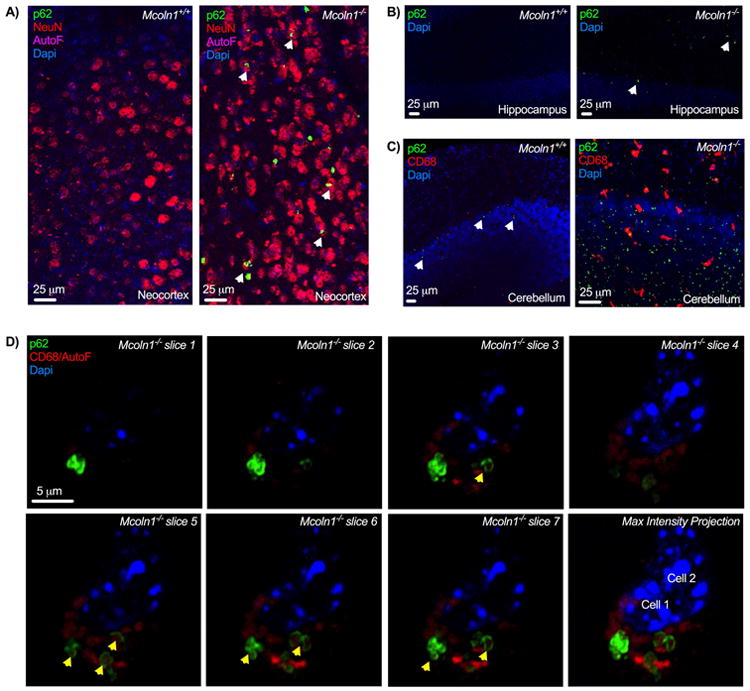
A) p62 aggregation in Mcoln1-/- mouse neocortex. p62 aggregates were not observed in WT (left panel), but aggregations positive for p62 can be detected in scattered neurons (NeuN+, p62 in neurons denoted by arrows) from all Mcoln1-/- mouse neocortical layers, shown here in cortical layer VI Mcoln1-/- (right panel). B) p62 aggregation in Mcoln1-/- hippocampus. Hippocampal sections from the CA1 region of WT (left panel) and Mcoln1-/- (right panel) mouse tissues. P62 positive structures were again not readily detected in WT hippocampus, while scattered punctate aggregates could be found in Mcoln1-/-, as indicated by white arrows. C) p62 aggregation in Mcoln1-/- mouse cerebellum. Image of WT cerebellum (left panel)) demonstrating occasional p62-positive puncta (noted by arrows), and from Mcoln1-/- mice (right panel), showing an abundant presence of small inclusions positive for p62. D) Example of typical neocortical p62+aggregate morphology. High magnification (63x) single plane images of p62 aggregates through a dorsal neocortical cell exhibiting clusters of aggregates, forming rings around diffuse, autofluorescent material. Yellow arrows denote distinguishable “rings” of p62. CD68 signal was tagged using Alexa Fluor 546 secondaries, and as autofluorescent material in MLIV fluoresces within near red wavelengths, CD68 signal overlapped with autofluorescent storage (labeled here as CD68/AutoF). Last image is a maximum intensity projection of previous slices in panel. Unless otherwise noted, all images are maximum intensity projections of acquired optical slices and collected from 6-9 month old mice. 9-11 Mcoln1-/- mice per panel were probed for p62 aggregation with matching WT counterparts.
Lysosomal exocytosis
Mucolipin-1 is well documented to play an important role in regulating lysosomal exocytosis (Medina et al. 2011; LaPlante et al. 2006; Lima et al. 2012; Park et al. 2015). It has been shown that ionomycin-induced lysosomal exocytosis in MLIV patient fibroblasts fails to deliver LAMP1 to the plasma membrane, indicative of deficient exocytosis (LaPlante et al. 2006). This finding is further supported by studies inducing gain of function mutations in mucolipin-1, which cause increased rates of lysosomal exocytosis (Dong et al. 2009). Lysosomal exocytosis through mucolipin-1 is also reported to be required for large particle phagocytosis, wherein calcium efflux through mucolipin-1 mediates the exocytotic delivery of lysosomal membrane to neighboring phagosomes (Samie et al. 2013). In contrast to these studies on defective exocytosis in the absence of mucolipin-1, a recent report examining pancreatic acinar cells and neurons from the Mcoln1 mouse model provided the surprising finding that exocytosis appeared enhanced rather than compromised (Park et al. 2015). Proteins necessary for synaptic exocytosis such as SNAP25 and VAMP2 also appeared increased in MLIV mouse cortex, cerebellum, and medulla, potentially implicating further exocytotic alterations in the MLIV brain (Park et al. 2015) Determining the exact implications of exocytotic dysfunction at the organelle level involving lysosomes and at the synaptic level in neurons clearly are important to understanding neuropathogenesis in MLIV.
mTOR, TFEB, and a complex feedback loop with mucolipin-1
The fourth neuropathogenic pathway emerging in MLIV focuses on the complex mechanisms underlying the master cell regulator mTOR and the CLEAR (coordinated lysosomal expression and regulation) network activator, TFEB, investigation on which has caused the lysosome to become the center of a wealth of research in recent years (Jaworski and Sheng 2006; Peña-Llopis et al. 2011; Troca-Marín et al. 2012; Sharma et al. 2010; Settembre et al. 2013a; Medina et al. 2011; Sardiello et al. 2009; Martina et al. 2014a). These signaling hubs form an axis sensitive to cellular nutrient states to modulate cellular resources via autophagy, and in the case of mTORC1, even to dock on the lysosomal surface itself as a signaling platform (Settembre et al. 2013b). For lysosomal diseases, this presents the possibility that the mTORC1-mediated metabolic sensing mechanism becomes compromised, potentially contributing to alterations in this, and its sister signaling pathway, TFEB, and the myriad pathways they control. The mTOR complex, comprised of mTORC1 and the more enigmatic mTORC2, plays many roles, influencing broad cellular processes that include starvation/nutrient responses, autophagy, lysosomal biogenesis and exocytosis, dendritogenesis and synaptic modulation, and lipid metabolism (Troca-Marín et al. 2012; Hay and Sonenberg 2004; Settembre et al. 2012; Urbanska et al. 2012; Sarbassov et al. 2005; Jaworski and Sheng 2006). A delicate ensemble of proteins modulates mTORC1's cellular location and activity to orchestrate these many events. In response to high amino acid availability mTORC1 translocates from the cytosol to the surface of late endosomes/lysosomes, where it is activated and initiates downstream signaling to inhibit autophagy and promote cellular growth (Sancak et al. 2010). This translocation to the lysosomal surface involves a concerted effort among numerous proteins, including Rag GTPases, the Ragulator protein complex, and p62 (Duran et al. 2011; Linares et al. 2013; Sancak et al. 2010). A partial summary of these complex interrelationships in relation to the known functions of mucolipin-1 is provided in Fig. 3.
Figure 3.
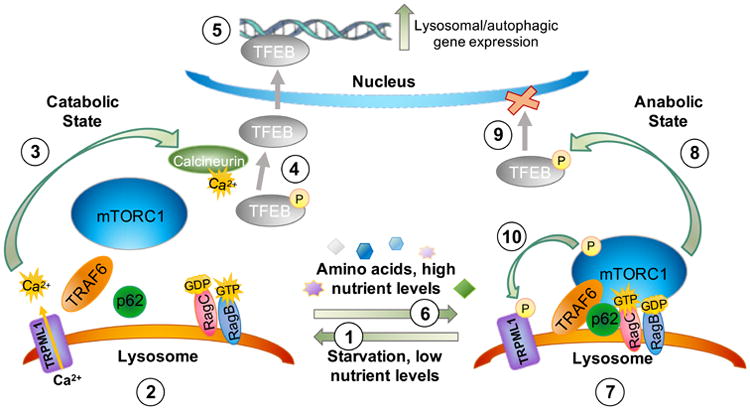
Role of mucolipin-1 (TRPML1) during mTOR and TFEB signaling. As described in the text, in response to a low nutrient state (1) mTOR dissociates from the lysosome and lysosomal calcium efflux is triggered through mucolipin-1 (2), allowing for activation of the phosphatase calcineurin (3). Calcineurin in turn dephosphorylates TFEB (4), which promotes its translocation across the nuclear envelope to activate lysosomal biogenesis and autophagy gene expression (5). In response to a high nutrient state (6), mTORC1 scaffolding proteins are recruited to the lysosomal surface and mTORC1 docks on this lysosomal platform (7). Activated mTORC1 then phosphorylates TFEB (8), which promotes its association with 14-3-3 proteins sequestering TFEB within its cytosolic location and thereby downregulates lysosomal biogenesis/autophagy gene expression (9). In response to nutrient availability, mTORC1 also phosphorylates mucolipin-1 directly (10), which reduces its channel activity and would further promote cytosolic localization of TFEB in a phosphorylated state. Mucolipin-1's ability to respond to low nutrient states by releasing calcium (see 2, above) has been suggested to be due to relief of mTOR-mediated phosphorylation of mucolipin-1. These key roles of mucolipin-1 in the nutrient sensing pathway governed by mTORC1 have been previously highlighted in other reviews (Venkatachalam et al. 2013). (Medina et al. 2015; Wang et al. 2015; Onyenwoke et al. 2015; Roczniak-Ferguson et al. 2012; Settembre et al. 2013a).
In MLIV, it has been shown that Trpml-deficient flies exhibit reductions in downstream targets of mTOR, such as phosphorylated S6 kinase, suggestive of reduced TORC1 activation (Wong et al. 2012). Interestingly, administering a high protein diet to trpml-deficient flies restores activation of mTOR and reduces mutant phenotypes such as pupal lethality and improves overall survival (Wong et al. 2012). Cell culture and fly studies have gone on to show an interaction of mucolipin-1 with AMPK (Onyenwoke et al. 2015), and demonstrate mTORC1-mediated JNK activation is diminished in trpml-deficient flies and is the underlying cause behind defects in NMJ synaptic development (Wong et al. 2015). These studies provided the first insights into potential perturbations to the mTOR signaling axis in MLIV. As flies express one homolog of mucolipin, it is possible that Drosophila trpml acts in pathways different from other mammalian systems. For example, studies from MLIV patient fibroblasts report no detectable changes in phosphorylated levels of mTOR and downstream member p70S6 kinase (Curcio-Morelli et al. 2010). Recent evidence in cell culture suggests that mTORC1 itself influences mucolipin-1 activity by phosphorylating the protein on its C-terminus, at sites Ser572 and Ser576 (Fig. 3) (Onyenwoke et al. 2015). Further, recent studies using cell culture suggest that during a prolonged starvation state, mucolipin-1 is necessary for the reactivation of mTORC1, consistent with mucolipin-1 and mTORC1 being linked via a complex negative feedback loop sensitive to cellular nutrient levels (Sun et al. 2018).
In addition to the role of mucolipin-1 during prolonged starvation in the mTORC1 pathway as described above, this TRP channel also plays a role in the TFEB-mediated response to acute low nutrient availability. In a state of low cellular resources with reduced mTORC1 activity, TFEB is not phosphorylated by mTORC1 and is further susceptible to dephosphorylation by the phosphatase calcineurin (Medina et al. 2015). As a result, TFEB is translocated to the nucleus were it would initiate lysosomal biogenesis and trigger autophagy upregulation, thus utilizing the lysosomal system to restore nutrient caches (Medina et al. 2015). Activation of calcineurin requires calcium from local lysosomal calcium stores specifically through mucolipin-1 (Fig. 3) (Medina et al. 2015; Wang et al. 2015). Similarly, TFE3, another member of the TFE transcription factor family, appears to mimic the starvation response activity of TFEB, and would therefore be an important marker to explore in MLIV given the high expression of TFE3 in brain relative to the other TFEs (Martina et al. 2014b; Martina et al. 2014a). In addition to the regulatory influence of mucolipin-1 on TFEB signaling, it has been reported in immortalized cell culture studies that mucolipin-1 activity is required for fission of new lysosomes from parental lysosomes (Miller et al. 2015; Wang et al. 2015). This is additional evidence that mucolipin-1 plays an intimate role in the regulation of lysosomal biogenesis, both in its initiation (via TFEB-network activation) and completion (via proper fission of nascent lysosomes).
Collectively, these findings are consistent with mucolipin-1 being part of an important feedback loop between mTOR and TFEB, these being connected by the recent finding of mTORC1-mediated phosphorylation of mucolipin-1 discussed above (Onyenwoke et al. 2015). When phosphorylated, mucolipin-1 is inhibited (Vergarajauregui et al. 2008b), which in turn reduces its channel activity leading to other downstream effects such as reduced activation of TFEB as a transcription factor (Onyenwoke et al. 2015). Considering these seminal findings implicating mucolipin-1's role in this fourth neuropathogenic pathway, the mTOR/TFEB signaling axis, a number of cellular consequences can be predicted in the mucolipin-1-deficient cell. For example, as TFEB regulates expression of MCOLN1 just as it does many genes coding for lysosomal proteins, this reduced transcription would be expected to lead to less mucolipin-1 expression amongst deficiencies and alterations to autophagy and lysosomal biogenesis (Palmieri et al. 2011; Sardiello et al. 2009; Medina et al. 2011) (Fig. 3, 4). Meanwhile, irregularities in the mTORC1 pathway may lead to aberrations in cell growth, structural changes to dendritic complexity and cell size, and alterations in synaptic function (Settembre et al. 2013a; Medina et al. 2015; Sardiello et al. 2009; Urbanska et al. 2012; Chen et al. 2014b; Takei and Nawa 2014; Bowling and Klann 2014). In specialized post-mitotic cells like neurons, perturbations to the mTOR/TFEB signaling axis in response to varying nutrient availability may culminate in subtle differences across cell types, and perhaps a subset of cells becoming locked in a futile metabolic cycling state (Wang et al. 2015). Such changes may also help explain neurological disease progression as these defects would be expected to progressively accumulate cellular damage over time.
Figure 4. TFEB signaling in the mucolipin-1-deficient cell.
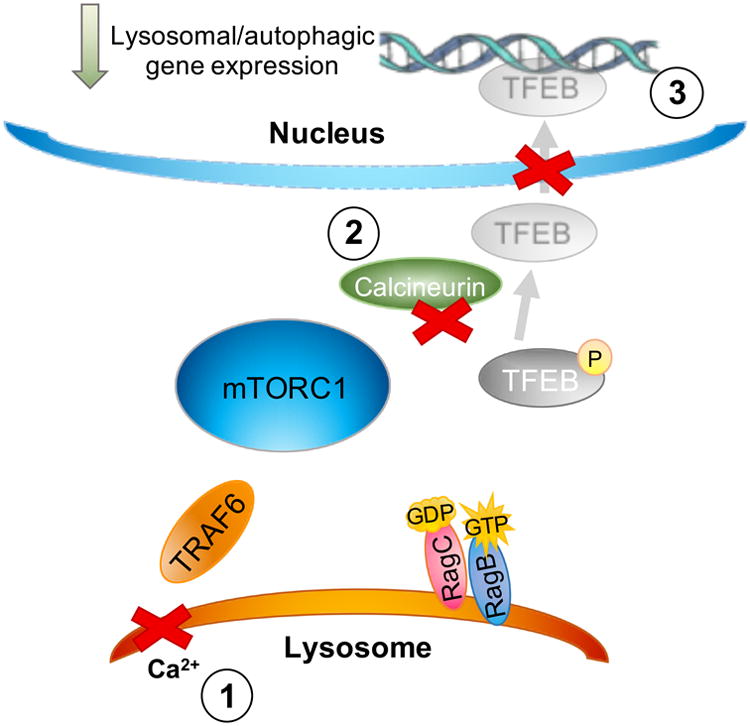
As described in the text and Fig. 3, mucolipin-1 plays an integral role in the mTORC1/TFEB nutrient response pathway – with the reported consequences of mucolipin-1's absence reviewed here. As described in the text, loss of mucolipin-1 removes the primary calcium source required to activate the phosphatase calcineurin, thus impairing TFEB dephosphorylation and its nuclear translocation (1-3). This may contribute to the mucolipin-1-deficient cell failing to properly respond to low nutrient availability via TFEB-mediated activation of the lysosomal network. (Medina et al. 2011; Medina et al. 2015; Wang et al. 2015). A full review of the role of TFEB in regulating the lysosomal network has been recently presented elsewhere (Sardiello 2016).
Heavy metal regulation and oxidative stress
Lastly, a fifth pathway of neuropathogenesis has emerged in MLIV – that of regulating lysosomal heavy metal homeostasis, particularly of iron and zinc (Dong et al. 2008; Silva et al. 2014; Eichelsdoerfer et al. 2010; Kiselyov et al. 2011). It has been shown that mucolipin-1 normally plays a role in iron handling, and its loss therefore would explain iron dysregulation noted in mucolipin-1-deficient cells and iron deficiency in patients (Grishchuk et al. 2015; Coblentz et al. 2013; Dong et al. 2008; Schiffmann et al. 1998). Notably, iron catalyzes the production of reactive oxygen species (ROS), and it is suggested that this buildup of iron in mucolipin-1-deficient cells is a contributing mechanism to degeneration and mitochondrial deterioration (Coblentz et al. 2013). Further, mucolipin-1's activity regulating lysosomal zinc levels has been shown to contribute to zinc elevation in lysosomes and in MLIV brain tissues (Silva et al. 2014; Eichelsdoerfer et al. 2010; Chen et al. 2014a; Cuajungco et al. 2014). Zinc dyshomeostasis in MLIV neurons may therefore represent another key neuropathogenic pathway given the relationship between zinc, mitochondrial toxicity, and zinc-associated accumulation of ROS (Eichelsdoerfer et al. 2010; Cuajungco and Lees 1997; Choi et al. 1988; Sensi et al. 2003; Bishop et al. 2007; Dineley et al. 2005). Iron buildup and heavy metal overload has even been suggested to play a role in the onset of axonal dystrophy (Chiueh 2001), another documented feature of Purkinje cells in MLIV disease (Micsenyi et al. 2009). In fact, iron and heavy metal dysregulation has long been associated with neurodegenerative diseases such as Alzheimer's, Huntington's, Parkinson's, and progressive palsy disease (a historical review available by Gerlach et al. 2002), and associations between metal dysregulation in the brain and neurodegenerative disease continue to be identified (Salazar et al. 2008). Enriching our understanding of heavy metal regulation in the context of MLIV may uncover additional mechanisms of cell death in specific neuronal populations.
In MLIV disease, the two cell types best documented to undergo degeneration and death are cerebellar Purkinje cells and retinal photoreceptors (Grishchuk et al. 2016; Micsenyi et al. 2009; Venugopal et al. 2007; Boudewyn et al. 2017). However, the link between loss of this protein in brain cells and subsequent neuronal dysfunction and degeneration remain enigmatic. Oxidative stress has long been implicated in cell death mechanisms in more common neurodegenerative diseases – and is an appropriate candidate for cell degeneration in MLIV that unifies many reported pathologies in this disease. As described above, the formation of ROS and mitochondrial aberrations have been well documented to occur in MLIV models, and further, it is known that mitochondria play a central role in the production of ROS (Nazıroğlu et al. 2012; Jennings et al. 2006; Coblentz et al. 2013; Coblentz et al. 2014; Venkatachalam et al. 2008). The role of ROS in neuropathogenesis therefore may represent a significant pathway in MLIV as it has recently been shown that mucolipin-1 may also act as a ROS sensor, during which these reactive moieties stimulate calcium release through mucolipin-1 and activate TFEB, promoting clearance of damaged organelles (Zhang et al. 2016). This finding revealed not only that mucolipin-1's loss can lead to mitochondrial damage and a cellular failure to degrade dysfunctional mitochondria, but is further evidence supporting mucolipin-1's link to TFEB and regulation of autophagy (Medina et al. 2015; Zhang et al. 2016; Peña and Kiselyov 2015). Much like the loss of mucolipin-1 in the TFEB-mediated starvation response, it is possible TFEB activation in response to ROS may similarly be impacted and fail to clear out damaged mitochondria producing these toxic ROS in MLIV.
Connecting mucolipin-1 to neuropathogenesis
The above-described changes – in endosomal/lysosomal trafficking, autophagy, lysosomal exocytosis dysregulation, mTORC1/TFEB signaling axis aberrations, and heavy metal dyshomeostasis with ROS formation - are all likely contributing factors to the kaleidoscopic pathologies canonical to MLIV (Fig. 5). Defects along each of these steps may help explain the heterogenous storage phenotype and organelle dysfunction observed in MLIV brain tissues (Fig. 5). Considering the mounting evidence of mucolipin-1's multipronged role in lysosomal signaling and function, the question arises as to what the consequences are for neurons and other brain cells in the partial absence of this protein, that is, would partial expression of the protein as would occur in heterozygotes lead to neuronal dysfunction?
Figure 5. A proposed complex network of disease pathogenesis and potential therapeutic targets surrounding the mucolipin-1-deficient cell.
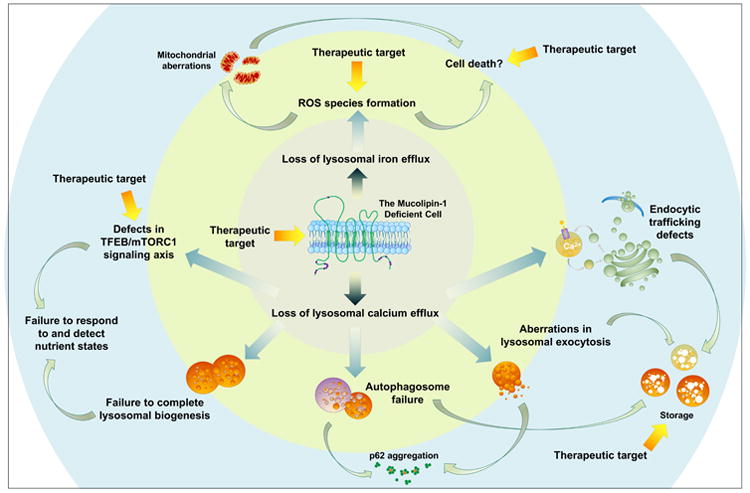
This schematic summarizes the multifaceted defects that may ensue in the absence of mucolipin-1. Loss of this cation-permeable TRP channel leads to defective lysosomal cation flux (inner circle), including loss of iron and calcium efflux (Dong et al. 2008; Dong et al. 2009). This loss subsequently leads to secondary defects (yellow middle circle). The absence of mucolipin-1-mediated calcium is followed by a failure to complete fusion of organelles within the endosomal/lysosomal network and a failure to balance the mTOR/TFEB signaling axis in response to cellular nutrient states (Medina et al. 2015; Medina et al. 2011; Curcio-Morelli et al. 2010; Miedel et al. 2008; Pryor et al. 2006). These perturbations would be expected to lead to a compromise in autophagy, aberrations in endocytic trafficking, and possibly defects in mTORC1 signaling. These secondary defects may cause further dysfunction in the mucolipin-1-deficient neuron and lead to the pathologies documented within the brain (outer blue circle), including p62 aggregation, heterogenous storage, and changes to the lipid composition of cells (Micsenyi et al. 2009; Boudewyn et al. 2017; Grishchuk et al. 2014). Such chronic changes beginning early in life could contribute to developmental abnormalities, synaptic dysfunction, and even perhaps structural changes to neurons or cell death. Further still, loss of lysosomal iron efflux through mucolipin-1 (inner circle) is reported to contribute to the formation of reactive oxygen species (ROS), which leads to mitochondrial aberrations (middle yellow circle) (Zhang et al. 2016; Colletti et al. 2012; Grishchuk et al. 2015). Many of these defective pathways would be anticipated to intersect and exacerbate one another. Defects in autophagy for example may contribute to a failure to degrade damaged lysosomes or mitochondria, the buildup of which may contribute to cell death mechanisms in MLIV through release of apoptotic-triggering cathepsins (Colletti et al. 2012). Understanding the interplay between this deeply intertwined network, including in different cell types, will be important to understanding the full picture of MLIV neuropathogenesis and help us better understand how to treat this disorder. Therapeutic targets are also highlighted with yellow arrows denoting potential intervention points, including compensation for mucolipin-1's loss (inner circle, perhaps with gene therapy), targeting reactive oxygen specials for example with antioxidants (middle circle), or targeting mTOR/TFEB signaling (perhaps with mTOR and TFEB modulators), storage (perhaps with substrate reduction therapy), or cell death (perhaps with miglustat or other therapeutics when cell death mechanisms are better identified in this disease). This does not include all potential therapeutics, and additional broader therapies such as anti-inflammatories or other small molecule-based treatments remain to be explored in the context of MLIV.
The heterozygous state of Mcoln1
In 1974, one of the first clinical studies of an MLIV patient included analysis of skin fibroblasts from the individual's healthy mother, aged 27 (Tellez-Nagel et al. 1976). This pioneering analysis revealed ultrastructural storage material identical in morphology to that seen in her son's cultures, yet occurring to a lesser degree and with lower frequency (Tellez-Nagel et al. 1976). Interestingly, no specific study has been carried out to date investigating any possible dosage effects of heterozygosity in individuals with mutations in MCOLN1. Studies of carriers of MLIV mutations probing for any potential effects of haploinsufficiency may further our understanding of the disease as well as mucolipin-1 protein function and may prove informative for families affected by MLIV. In addition to the major MLIV-causing mutations, independent mutations causing MLIV continue to be identified (Slaugenhaupt 2002; Shiihara et al. 2018; Casteels et al. 1992; Bargal et al. 2001; Saijo et al. 2016). As the array of mutations to mucolipin-1 existing in the population continue to be unveiled, understanding the potential impact of these mutations on carriers will become increasingly important. One means of doing this would be to evaluate family members of MLIV patients with known mutations for health-related issues incurred as a consequence of MCOLN1 heterozygosity. The importance of this issue is underscored by findings in other lysosomal disorders in which heterozygotes of lysosomal disease-causing mutations are documented to manifest clinical symptoms, as described below.
Lysosomal diseases stem from defects ranging from lysosomal enzyme loss to the absence of lysosomal transmembrane proteins vital to proper organelle function (Platt and Walkley 2004). Storage disorders resulting from the loss of lysosomal enzymes are not typically thought to result in clinical phenotypes within the heterozygote, as the partial expression of lysosomal hydrolases that would occur is believed sufficient to provide enzymatic activity to prevent any metabolic dysfunction (Brown et al. 1996). Yet conditions like Late Onset Tay-Sachs (LOTS), caused by reduced function of lysosomal β-hexosaminidase and characterized by late life onset of neurodegeneration, have been documented (Deik and Saunders-Pullman 2014; Neudorfer et al. 2005). A large body of evidence also has shown that individuals carrying single mutations in the GBA gene encoding the lysosomal enzyme, glucocerebrosidase, responsible for the complex, recessive disorder Gaucher disease have also been documented to exhibit clinical symptoms, such as impaired cognition and hyposmia (McNeill et al. 2012). Being a carrier of a GBA mutation has also been identified as a major risk factor for Parkinson disease and parkinsonian syndrome (Kluenemann et al. 2013; Sidransky and Lopez 2012; Goker-Alpan et al. 2004; Bras et al. 2008; Halperin et al. 2006). In light of emerging evidence of lysosomal proteins such as the NPC1 protein and now even mucolipin-1 in the neuropathogenesis of such age-related disorders as Parkinson disease, and more recently, Alzheimer disease, the relationship between impaired lysosomal function and increased risk for age-related brain disorders is becoming increasingly tightened (Kresojević et al. 2018; Zhang et al. 2017a).
In contrast to lysosomal disorders caused by absence of lysosomal enzymes, those caused by loss or malfunction of an endosomal/lysosomal transmembrane protein offer a greater possibility of a “dosage effect” in carriers. NPC disease, for example, results from mutations in either the lysosomal transmembrane protein NPC1 or its soluble protein counterpart NPC2, which together function as cholesterol transporters (Kruth et al. 1986). Canonical NPC disease results in severe and progressive neurological symptoms, cognitive decline, profound lipid storage (most notably of unesterified cholesterol), hepatosplenomegaly, and premature death (Platt and Walkley 2004; Ikonen 2004; Kruth et al. 1986). Much like for carriers of GBA mutations, adult heterozygous carriers for NPC1 mutations have exhibited multiple cases of parkinsonian syndrome indicated by resting tremor, motor difficulties, and bradykinesia (Kluenemann et al. 2013; Halperin et al. 2006; Sidransky and Lopez 2012; Josephs 2004). Further, cultured fibroblasts and blood samples from NPC-mutation carriers and healthy controls revealed that heterozygotes exhibited storage of cholesterol esters and a metabolic profile intermediate between NPC patients and healthy controls (Kruth et al. 1986; Kresojević et al. 2018; Probert et al. 2017). Foam cells, or storage-laden cells which are given their name due to their “foamy” appearance when viewed by an electron microscope, are a characteristic feature of NPC tissues and have even been noted in the bone marrow of parents and siblings of NPC patients (Harzer et al. 2014). Storage accumulation of unesterified cholesterol, vacuolization, and GM2 accumulation have also been documented by Walkley and colleagues in the feline model of NPC1 within obligate heterozygotes (Brown et al. 1996). As in aged adults, Npc1 heterozygous mice also exhibit reduced motor coordination as well as Purkinje cell loss and increased tau protein (Yu et al. 2005). Such findings clearly show that the consequences of heterozygosity can be replicated in animal models of NPC1 disease (Yu et al. 2005; Hung et al. 2016). Similar findings also can be found outside of NPC disease as well. Asymptomatic siblings and parents of individuals with juvenile Batten (CLN3) disease (which also result from the loss of a lysosomal transmembrane protein) have been documented to possess cytoplasmic inclusions within lymphocytes (Markesbery et al. 1976).
These cases – Gaucher, Niemann-Pick C1, and juvenile Batten diseases – highlight the vital role of proper lysosomal function in maintaining neuronal health and raise the possibility that heterozygosity for mutations in the global gene network of lysosomal transmembrane proteins, such as mucolipin-1, may contribute to perturbed cellular function. Understanding if MLIV mutations in heterozygous individuals cause similar metabolic defects or a predisposition to neurological symptoms during aging would be informative to MLIV carriers and potentially improve care for identified affected individuals. Further, understanding neurological health in the heterozygous state may shed light on the role of mucolipin-1 in normal brain health and maintenance.
In order to begin addressing these questions and in the absence of comprehensive data from human MLIV carriers, we turned to the Mcoln1 mouse model to evaluate the consequences of heterozygosity for mucolipin-1 expression. In other lysosomal disorders exhibiting haploinsufficiency such as NPC1 disease, animal models and even human patients often do not display overt neuronal features until later in life (Yu et al. 2005). If heterozygosity for mucolipin-1 expression impacts neuronal function, lysosomal storage-related features may similarly only appear during senescence. We predicted that if mucolipin-1 heterozygosity leads to insufficient lysosomal function, aged mice might develop a milder form of the canonical neurological features detectable in the Mcoln1 homozygous mutant, such as lysosomal storage, glial activation, behavioral deficits, or ultrastructural abnormalities. Such an investigation would also determine if the MLIV mouse is an appropriate model for assessing milder forms of the disease. As no phenotypes have thus far been observed in heterozygotes at 6-9 months of age from previously published work, this study was directed at assessing aged mice from 15-25 months old to correspond to murine “old age” (Flurkey et al., 2007). A particular focus was given to mice aged 20-25 months for exacerbated signs of aging or delayed neurological manifestations intermediate between the aged WT state and Mcoln1-/- tissues.
Methods
Mouse generation and collection
Heterozygous (Mcoln1+/-) mice were crossed to obtain littermate wild type (Mcoln1+/+), heterozygous mice (Mcoln1+/-) and mutant (Mcoln1-/-) mice genotyped per previously described methods (Venugopal et al. 2007) and included both males and females. Mcoln1−/− mice (C57/BL6 background) were generously provided by the laboratories of Drs. Susan Slaugenhaupt at Harvard Medical School and James Pickel at the National Institutes of Health. Mice were housed in standard IACUC-approved cages with corn-cob and cotton cloth-square bedding, with access to food and water ad libitum. Mouse were housed 2-5 animals per cage, with mixed genotypes housed together, and animal husbandry performed routinely to monitor the welfare of all mice. For p62 analyses, mice were collected at end-stage in the MLIV mouse model at 6-9 months of age alongside WT controls, with a total of 32 mice used (16 Mcoln1-/-, 16 WT). For aged heterozygous studies, mice were aged to 15 months through 25 months of age corresponding to murine old age (Flurkey et al., 2007). A total of 47 male and female mice were used for this study (24 heterozygotes and 23 WTs). Prior to tissue collection, mice were anesthetized using an overdose of ketamine/xylazine delivered via intraperitoneal injection to minimize pain and discomfort, and once insensate euthanized transcardially perfused with normal saline and tissues collected and frozen on liquid nitrogen, then perfused and fixed with 4% paraformaldehyde per previously described protocols (Micsenyi et al. 2009). The use of an overdose of the xylazine/ketamine mixture used in these studies is an accepted and IACUC-approved procedure for reducing pain and suffering in rodents. This study was not pre-registered. All procedures using animals were approved by the Institutional Animal Care and Use Committee (IACUC, approval numbers 20160903 and 20160913) at the Albert Einstein College of Medicine.
Behavioral analyses
Motor assessment was performed using a balance beam task as previously described (Boudewyn et al. 2017). In brief, following an equilibration period to the testing room within their home cages, training on a 3-inch wide-set flat beam was performed to promote passage across the balance beam. During testing, three round wooden beams of increasing diameter (18 mm - “narrow”, 24 mm – “medium”, 30 mm – “wide”) were elevated using tripods, with cushion-covered bins located underneath as an added safety precaution. Hides were located at the end of each beam, with a light gradient cast over the beams to stimulate beam crossing (ie, beginning of the beam was bright; end of the beam was dark). Mice were run through the narrow, medium, and then wide beams and the number of slips were counted manually. Prior to testing, mice were assigned a random number in order to blind the tester to genotype and age. Random numbers were allocated via a simple randomization method of assigning mice sequential numbers within a cage in the random order in which they were selected – ie, cage 1, mice 1 through 5 were determined at random (#1 ID being randomly assigned to first mouse picked up, #5 being last (5th) mouse picked up, cage 2, #6 ID for first mouse picked up, #11 for last mouse (5th in a cage) picked up, etc), with these randomly assigned numbers retained for any subsequent behavioral testing to preserve blinded coding. Tails were color-coded for mice housed within a cage to represent the assigned blinded number (ie, repeating patterns of red-blue-green-white-black tail codes to correspond to sequential ID numbers). No pain or discomfort was anticipated or observed during this task. Differing sample sizes for each beam were due to varying ability of aged mice to complete all three beams during testing, and only mice capable of completing a beam were included for each test. All work was performed in accordance with the Albert Einstein College of Medicine Animal Phenotyping Core Facility protocols and IACUC approved protocols.
Immunofluorescent staining and confocal imaging
For immunofluorescent staining, fixed tissues were sectioned at 35 micron thickness in coronal orientation for cerebrum and sagittal orientation for cerebellum using a Leica V100S vibratome and stained per previously established protocols (Boudewyn et al. 2017; Micsenyi et al. 2013). Primary antibody(ies) included anti-CD68 (1:500, rat monoclonal antibody, AbD Serotec MCA1957, RRID:AB_322219), anti-p62 C terminal specific (1:500, guinea pig polyclonal antibody, American Research Products 03-GP62-C, RRID:AB_1542690), anti-GFAP (1:1000, rabbit polyclonal antibody, Sigma G9269, RRID:AB_477035), and anti-NeuN (1:1000, mouse monoclonal antibody, Chemicon MAB377, RRID:AB_2298772). For those sections stained in conjunction with anti-guinea pig p62 (such as in Fig. 2), pre-adsorbed secondaries were purchased to provide minimal cross reactivity (Alexa dyes 647 and Cy3, diluted as above). 35-micron sections were mounted without antibody staining and imaged under the same conditions as stained sections to control for auto-fluorescence. Confocal microscopy was performed with a Zeiss Meta 510 Duo V2 laser scanning microscope as previously described (Boudewyn et al. 2017). Slides were blinded to genotype by being assigned a coded record number and unveiled following processing. Image analysis and processing was performed using ImageJ software and MetaMorph software (Molecular Devices).
Western blot analysis
Cerebellar tissues were homogenized and prepared for western blot analysis per the full protocol described in (Boudewyn et al. 2017). In brief, tissues were homogenized with a Dounce homogenizer in freshly made ice cold lysis buffer containing 50nM TrisHCL, 150 mM NaCl, 1% Igepal, 1% Sodium Deoxycholate, 1% Sodium Dodecyl Sulfate, 1% Triton × 100, and Complete Mini Protease Inhibitor Cocktail (Roche, 11-836-153-001), then homogenate passed through a 27-gauge needle. Following protein quantification, samples were run on an SDS-PAGE 12% Tris-Glycine gel. Blots were imaged using an Odyssey CLx imaging station equipped with Image Lite Studio Software and quantified with Image Lite Studio Software. The intensity of bands for calbindin were normalized to those of beta-actin to correct for loading differences, with antibodies including: anti-calbindin 1:1000, rabbit polyclonal antibody, Millipore AB1778 and anti-beta actin 1:3000, rabbit polyclonal antibody, Sigma A2066.
Tissue processing and imaging via transmission electron microscopy (TEM)
Tissues were prepared for electron microscopy by previously established methods (Micsenyi et al. 2009). In brief, tissues were micro-dissected into <1mm blocks of neocortical, hippocampal, and cerebellar sections. Cerebellar sections were further dissected into anterior, central, and posterior + nodular zones (Sillitoe and Joyner 2007) to allow for regional analysis. Tissue blocks were rinsed in 0.1M cacodylate buffer (Fisher Scientific 50-980-232/Electron Microscopy Sciences (EMS) #11652), fixed using Trump's fixative (Fisher Scientific 50-980-239/EMS #11750), then placed in 2% osmium (Fisher Scientific 50-332-20/EMS #19150) and dehydrated in graded ethanol steps. Tissues were then rinsed in Acetonitrile (Fisher Scientific 50-980-146/EMS #10020) and incubated in a 1:1 solution of Acetonitrile:Epon at room temperature overnight, then placed into fresh Epon solution, embedded in Epon, and cut on an ultramicrotome. Blocks were mounted on grids and stained with uranyl acetate and lead citrate, and then they were imaged using a Phillips CM10 transmission electron microscope for ultrastructural analysis. Images were processed using ImageJ and Fiji software.
Statistical Analyses
All data were compiled using GraphPad Prism Software Version 7.0a and statistically analyzed using JMP Software from SAS Version 13.0.0. To determine the statistical significance of behavioral data, ANOVA was performed. If ANOVA detected a difference, post-hoc parametric Tukey-Kramer HSD comparisons were then performed and a difference was deemed as significant where p < 0.05. Tests for normalcy and outliers on these pilot data were not performed. Sample size calculations were not performed prior to testing, and sample sizes were obtained based on literature standards (Bailey et al. 2008). Post-hoc sample calculations were performed via power analysis of pilot data. For example, for the hard balance beam, any future studies would require group samples of 45 to achieve 80.6% power to reject the null hypothesis of equal means when the population mean difference is 5.6 with a standard deviation of 11.7 for heterozygous mice, and 5.8 for WT mice with a significance level (alpha) of 0.05 using a two sided two-sample unequal-variance t-test. For statistical analyses of western blots, band measurements were analyzed using an unpaired t-test with Welch's correction. For all graphs, mean ± standard error is plotted.
Results
Upon gross assessment, end stage (6-9 months) homozygous affected (Mcoln1-/-) mice exhibit features such as bladder enlargement, hunched posture, and hindlimb clasping behavior (Venugopal et al. 2007). These characteristics were not observed at any point in heterozygous mice or WTs from 15 months of age out to 25 months of age (20-25 months). The mutant MLIV mouse model exhibits poor coordination on a balance beam that is accompanied by Purkinje cell loss as measured by calbindin D western blot analysis (Boudewyn et al. 2017). MLIV mice also exhibit widespread storage of gangliosides like GM2 in the neocortex and other parts of the CNS (Boudewyn et al. 2017; Micsenyi et al. 2009). To evaluate if our cohort of Mcoln1 heterozygotes displayed similar abnormalities, we carried out balance beam assessments, western blots for calbindin D, and stained tissues for GM2 ganglioside. Mice were tested on three balance beams of decreasing difficulty (i.e., increasing width) and the number of slips counted to assess cerebellar-mediated motor coordination (Fig. 6). No significant differences were detected when comparing heterozygote performance to that of age-matched WTs (Fig. 6A-C). Given the high variability in balance beam performance in both heterozygote and WT mice (likely due to effects of old age), as a further test of cerebellar integrity, we also examined this brain region for evidence of loss of Purkinje cells based on an analysis of calbindin-D levels. Calbindin is a calcium buffering protein highly expressed in Purkinje cells, and therefore is a useful and widely used marker of Purkinje cell loss (Celio 1990; Barski et al. 2003; Sarna et al. 2003). Calbindin density was probed via western blot but no significant decrease in levels compared to age-matched WT mice was observed (aged 20-25 months, Fig. 6D). We also examined heterozygotes for evidence of GM2 storage in neurons in cerebrum and cerebellum (which is characteristic of mutant Mcoln1 mice) (Micsenyi et al., 2009), but again no differences from WT mice were observed (data not shown). While the above studies were not able to detect any significant behavioral or gross cellular phenotypes in aged heterozygote Mcoln1 mice, it is possible that aged heterozygotes could still manifest changes at the organelle level. To test for this, ultrastructural studies were performed to probe for abnormal MLIV-related subcellular features.
Figure 6. Preliminary data on behavioral performance and calbindin density in senescent Mcoln1 heterozygous mice.
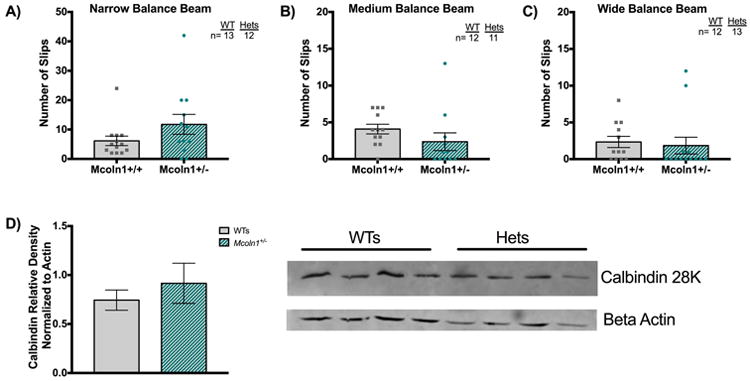
A) Average number of slips on the narrow (i.e., most difficult), medium (intermediate difficulty, B), and widest (easiest, C) beam are plotted. While heterozygotes on the hard beam appears to have slightly more slips than WTs, no significant differences were detected for this or other beams analyzed by ANOVA (A-C). N=number of mice tested on each plot. D) Calbindin density detected via western blot of heterozygous versus WT tissues (n=4 mice aged 20-25 months per genotype, average density plotted +/- SEM, p=0.4914) suggested Purkinje cell health is comparable between the two genotypes. Statistical analyses performed using unpaired t-test with Welch's correction.
Previous electron microscopy studies of patient biopsy tissues and published data in the MLIV mouse illustrate the presence of highly characteristic storage bodies – so-called compound bodies - in the MLIV brain (Tellez-Nagel et al. 1976; Micsenyi et al. 2009). The current study delineated here extends this earlier work and reveals MLIV neocortical cells with compound bodies along with multilamellar bodies congesting the cytoplasm of both neurons and support cells (representative examples in Fig. 7). Cytoplasmic structure of neurons was found to be disorganized and accompanied by enlarged mitochondria as previously reported (Micsenyi et al. 2009; Jennings et al. 2006).
Figure 7. Canonical ultrastructural features of Mcoln-/- mice compared to aged heterozygous and WT mice.
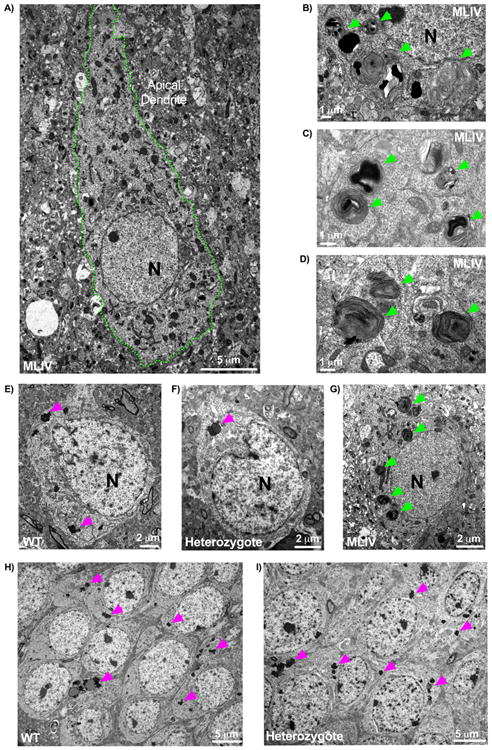
A) Characteristic pathology in Mcoln1-/- 3-month old tissues revealed by electron microscopy. Shown here is a neocortical neuron with the neuronal cytoplasm found congested with multiple compound, electron dense storage bodes as well as granulomembranous storage bodies and an enlarged apical dendrite (cell membrane outlined in green). B-C) Electron dense, compound storage bodies typically found in 3-month-old mutant mice, taken here from neocortex and highlighted by green arrows. E-G) Neocortical neurons from (E) WT, (F) heterozygote, and (G) mutant. E-F) WT and heterozygote neocortex exhibited neurons possessing lipofuscin accumulation (pink arrowheads) but otherwise devoid of pathological, compound storage bodies readily detectable in the mutant (G, green arrowheads). Neither heterozygous or WT neurons (both shown here at 18+ months) were found to exhibit abnormal structures, such as the swollen apical dendrites captured in 3-month old mutant neocortex (A). H-I) Hippocampal sections from WT (H) and heterozygous (I) mice. Similar to findings in cerebral cortex, hippocampal neurons displayed comparable features between WT (H) and heterozygotes (I) such as lipofuscin granulation storage with no evidence of pathological storage. Lipofuscin was not found to be quantitatively increased in the heterozygote in either cerebral cortex or hippocampus (data not shown). All tissues shown here are representative images from mice aged 18+ months, with the exception of the mutant at 3 months. 2 heterozygotes, 4 WT, and 4 Mcoln1-/- mice were probed for pilot EM data.
As the above-described features are readily evident in the mutant MLIV mouse even at early ages, we predicted that if partial expression of mucolipin-1 is insufficient to maintain neuronal health the heterozygote MLIV mouse would likely undergo at least an intermediate level of storage detectable as compound storage bodies. Sections of neocortex, hippocampus, and cerebellum were examined, with the latter organized into anterior, central, and posterior + nodular zones (due to the progressive lobular pathology noted in the MLIV mutant cerebellum). No compound bodies, lamellar bodies with electron dense material, or similar distinctive features were detected in heterozygote or WT mice aged 15-25 months (Fig. 7). Indeed, heterozygote tissues were not observed to display any ultrastructural characteristics differing from WT in any brain region analyzed. Lipofuscin granules could be detected readily in all regions in both WT and heterozygous mice but did not appear to occur at a higher rate in either genotype (Fig. 7). Lipofuscin was identified morphologically per established electron microscopy guidelines (Peters et al. 1991) and is consistent with lipofuscin-associated autofluorescence detected via immunofluorescent staining.
Discussion
The lack of an overt cellular or behavioral phenotype in aged Mcoln1 heterozygotes suggests either that partial expression of mucolipin-1 is sufficient for proper cellular function, or that mucolipin-1 channel function may be partially compensated for by other proteins. Mucolipin-1 is ubiquitously expressed and the only mucolipin channel believed to be expressed in the brain (Cheng et al. 2010). It is possible that CNS compensation for mucolipin-1's partial loss of function could arise from transmembrane proteins with similar pore activity, such as the so-called two pore channels (TPCs). The TPCs are endolysosomal ion channels with similar calcium release activity to mucolipin-1 (Wang et al. 2017; Grimm et al. 2012). Much like mucolipin-1, these proteins are also permeable to sodium and are similarly activated by PI(3,5)P2, suggesting the possibility that these proteins may share activation mechanisms and potentially even similar functions (Dong et al. 2010; Wang et al. 2017). It is thought that the permeability to cations and response to PI(3,5)P2 exhibited by both mucolipin-1 and TPCs may facilitate membrane fusion events by modulating these gradients, such that increases in the levels of PI(3,5)P2 promote lysosomal calcium (documented by mucolipin-1 activity) or sodium (documented by TPC activity) efflux into the cytosolic space (Wang et al. 2017; Dong et al. 2010). It is possible that TPCs located within the endosomal/lysosomal system which exhibit similar channel conductance may compensate for such activity in the face of reduced expression of mucolipin-1 in the heterozygous state. Still other channels may compensate for mucolipin-1's loss, perhaps even from the greater TRP family – TRPM (melastatin) family members for example also exhibit similar channel conductance, and some TRPM members are expressed in the brain (Fonfria et al. 2006). Alternatively, the mouse model may be limited in replicating any phenotypes that could develop in aged human populations. In the Mcoln1 mutant, the timeline of disease onset and progression differs from human disease manifestations. In human MLIV, children present early in life and experience a relatively stable adolescence, while MLIV mice are relatively stable early in life and develop overt symptoms in adulthood at 6 months of age (Amir et al. 1987; Venugopal et al. 2007). It is possible that a heterozygous timeline could similarly differ between mammalian species, or not develop in the mouse model compared to human populations. Future survey of human carriers of MCOLN1 mutations may be informative to monitor the health of individuals over time.
Conclusions and Pathogenic Crosstalk in the Absence of Mucolipin-1
In this review, we have summarized how the loss of mucolipin-1 leads to a complex, multifaceted pathogenic cascade that contributes to cellular damage culminating in neuron and brain dysfunction. Specifically, we described how primary defects in calcium and heavy metal regulation following loss of mucolipin-1 are believed to impair a host of critical processes in cells, including endosomal/lysosomal fission and fusion events, mTORC1/TFEB signaling and autophagy regulation, and the cellular response to ROS formation (Fig. 5) (Venugopal et al. 2009; Zhang et al. 2016; Curcio-Morelli et al. 2010; LaPlante et al. 2006; Coblentz et al. 2013). While these consequences of the loss of mucolipin-1 are all likely contributors to the neuropathogenesis of MLIV disease, how they interrelate and influence each other remains largely unexplored and deserving of additional studies. It is also important to recognize that while this review provides a discussion of how multifaceted defects in the absence of mucolipin-1 may relate specifically to neuronal dysfunction; it is not a full narrative of how such defects may impact systemic MLIV disease. Reviews of how mucolipin-1's loss can impact immunity and even cancer have been presented elsewhere (Grimm et al. 2018). Other important areas of research, including mucolipin-1's role in renal disease, remain largely unaddressed. Despite the broad implications of the importance of mucolipin-1 function, our studies in the aged Mcoln1 heterozygote did not reveal abnormalities indicative of loss of mucolipin-1 function, suggesting that partial expression of the protein is adequate to maintain normal function of these intricate pathways, at least in the mouse.
The field of mucolipin-1 and MLIV research to date has unveiled new avenues in our understanding of how the loss of this critical protein leads to the complex neurodevelopmental and neurodegenerative symptoms experienced by patients. Understanding the manner in which these cascades exacerbate and interplay with one another is a crucial step toward understanding not only MLIV pathogenesis but also new approaches to therapy.
Supplementary Material
Acknowledgments
The authors would like to dedicate this review to the memory and legacy of the late Dr. Isabelle Rapin, a pioneer in the field of neuro-metabolic disorders and one of the first clinicians to describe individuals affected by mucolipidosis type IV. We would also like to thank two members of the Walkley lab (Xin Huang and Bin Cui) for technical assistance, and the Rose F. Kennedy Intellectual and Developmental Disabilities core facilities. The latter include the Animal Phenotyping Core directed by Dr. Maria Gulinello and the Neural Cell Engineering and Imaging Core directed by Dr. Kostantin Dobrenis. We would also like to thank the ML4 Foundation for their support and encouragement throughout this study. This project was supported by National Institute of Health grants, R01 HD045561 and U54 HD090260. The authors have no conflict of interest to declare. Data in this paper are from a thesis to be submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine.
Abbreviations
- TFEB
transcription factor EB
- mTOR
mechanistic/mammalian target of rapamycin
- MLIV
mucolipidosis type IV
- NPC
Niemann-pick type C
- MLI
mucolipidosis type 1
- MLII
mucolipidosis type II
- MLIII
mucolipidosis type III
- CLN2
neuronal ceroid lipofuscinosis type 2
- LOTS
late-onset Tay Sachs
- GlcNAc-phosphotransferase
N-acetylglucosamine-1-phosphotransferase
- TRP
transient receptor potential
- TRPML
transient receptor potential channel mucolipin
- TRPC
“TRP” canonical family
- TRPV
“TRP” vanilloid family
- TRPM
“TRP” melastatin family
- TRPN
“TRP” NOMPC-like family
- TRPA
“TRP” ankyrin family
- TRPP
“TRP” polycystin family
- TPC
two-pore channels
- AP1-3
clathrin adaptor protein complexes 1-3
- TMD
transmembrane domain
- BMT
bone marrow transplant
- CHO
Chinese hamster ovary
- ARPE
immortalized human retinal epithelial cell line
- LC3
microtubule associated protein 1A/1B-light chain 3
- LMP
lysosomal membrane permeability
- CLEAR
coordinated lysosomal expression and regulation
- ROS
reactive oxygen species
- GBA
glucocerebrosidase
- GBA2
non-lysosomal glucocerebrosidase
- CNS
central nervous system
- EM
electron microscopy
Footnotes
Involves human subjects: If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods
ARRIVE guidelines have been followed: Yes
=> if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Conflicts of interest: None
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
Open Science Badges: No, I am not interested to achieve Open Science Badge(s) => if yes, please see Comments from the Journal for further information => if no, no information need to be included in the manuscript
References
- Abe K, Puertollano R. Role of TRP channels in the regulation of the endosomal pathway. Physiology (Bethesda) 2011;26:14–22. doi: 10.1152/physiol.00048.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acierno JS, Kennedy JC, Falardeau JL, Leyne M, Bromley MC, Colman MW, Sun M, et al. A Physical and Transcript Map of the MCOLN1 Gene Region on Human Chromosome 19p13.3–p13.2. Genomics. 2001;73:203–210. doi: 10.1006/geno.2001.6526. [DOI] [PubMed] [Google Scholar]
- Altarescu G, Sun M, Moore DF, Smith JA, Wiggs EA, Solomon BI, Patronas NJ, et al. The neurogenetics of mucolipidosis type IV. 2002 doi: 10.1212/wnl.59.3.306. [DOI] [PubMed] [Google Scholar]
- Amir N, Zlotogora J, Bach G. Mucolipidosis type IV: clinical spectrum and natural history. Pediatr Off J Am Acad Pediatr. 1987;79:956–959. [PubMed] [Google Scholar]
- Amir N, Zlotogora J, Bach G, Ziotogora J. Mucolipidosis Type IV : Clinical Spectrum and Natural History The online version of this article, along with updated information and services, is located on the World Wide Web at : Mucolipidosis Type IV : Clinical Spectrum and Natural History 2013 [Google Scholar]
- Bach G. Mucolipidosis type IV. Mol Genet Metab. 2001;73:197–203. doi: 10.1006/mgme.2001.3195. [DOI] [PubMed] [Google Scholar]
- Bailey KR, Rustay NR, Crawley JN. Behavioral Phenotyping of Transgenic and Knockout Mice: Practical Concerns and Potential Pitfalls. ILAR J. 2008;47:124–131. doi: 10.1093/ilar.47.2.124. [DOI] [PubMed] [Google Scholar]
- Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G. Identification of the gene causing mucolipidosis type IV. Nature. 2000;26:120–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- Bargal R, Avidan N, Olender T, Ben-Asher E, Zeigler M, Raas-Rothschild A, Frumkin A, et al. Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. 2001;17:397–402. doi: 10.1002/humu.1115. [DOI] [PubMed] [Google Scholar]
- Barski JJ, Hartmann J, Rose CR, Hoebeek F, Mörl K, Noll-Hussong M, Zeeuw CI De, Konnerth A, Meyer M. Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J Neurosci. 2003;23:3469–77. doi: 10.1523/JNEUROSCI.23-08-03469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman ER, Liyni N, Shapira E, Merin S, Levij IS. Congenital corneal clouding with abnormal systemic storage bodies: A new variant of mucolipidosis. J Pediatr. 1974;84:519–526. doi: 10.1016/s0022-3476(74)80671-2. [DOI] [PubMed] [Google Scholar]
- Bishop GM, Dringen R, Robinson SR. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic Biol Med. 2007;42:1222–1230. doi: 10.1016/j.freeradbiomed.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Bonten E, Spoel A, van der Fornerod M, Grosveld G, d'Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;10:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- Boudewyn LC, Sikora J, Kuchar L, Ledvinova J, Grishchuk Y, Wang SL, Dobrenis K, Walkley SU. N -butyldeoxynojirimycin delays motor deficits, cerebellar microgliosis, and Purkinje cell loss in a mouse model of mucolipidosis type IV. Neurobiol Dis. 2017 doi: 10.1016/j.nbd.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling H, Klann E. Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron. 2014;83:994–6. doi: 10.1016/j.neuron.2014.08.021. [DOI] [PubMed] [Google Scholar]
- Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson's disease: Potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FAE, Giaccone G. Cathepsin B Mediates Caspase-Independent Cell Death Induced by Microtubule Stabilizing Agents in Non-Small Cell Lung Cancer Cells. Cancer Res [CANCER Res. 2004;64:27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- Brown DE, Thrall MA, Walkley SU, Wurzelmann S, Wenger DA, Allison RW, Just CA. Metabolic abnormalities in feline Niemann-Pick type C heterozygotes. J Inherit Metab Dis. 1996;19:319–330. doi: 10.1007/BF01799262. [DOI] [PubMed] [Google Scholar]
- Casteels I, I Taylor DS, Lake BD, Spalton DJ, Bach G. Mucolipidosis type IV Presentation of a mild variant. Ophthalmic Paediatr Genet. 1992;13:4. doi: 10.3109/13816819209105168. [DOI] [PubMed] [Google Scholar]
- Castro M de, Bunt G, Wouters F. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes. Cell Death Discov. 2016;2:16012. doi: 10.1038/cddiscovery.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- Chandra M, Zhou H, Li Q, Muallem S, Hofmann SL, Soyombo AA. A Role for the Ca2+ Channel TRPML1 in Gastric Acid Secretion, Based on Analysis of Knockout Mice. Gastroenterology. 2011;140:857–867.e1. doi: 10.1053/j.gastro.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun. 2014a;5:778–790. doi: 10.1038/ncomms5681. [DOI] [PubMed] [Google Scholar]
- Chen Q, She J, Zeng W, Guo J, Xu H, Bai XC, Jiang Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature. 2017;550 doi: 10.1038/nature24035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014b;206:173–82. doi: 10.1083/jcb.201403009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Shen D, Samie M, Xu H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010;584:2013–21. doi: 10.1016/j.febslet.2009.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitayat D, Meunier C, Hodgkinson K, Silver K, Flanders M, Anderson I, Little J, Whiteman D, Carpenter S. Mucolipidosis type IV: clinical manifestations and natural history. Am J Med Genet. 1991;41:313–318. doi: 10.1002/ajmg.1320410310. [DOI] [PubMed] [Google Scholar]
- Chiueh CC. Iron overload, oxidative stress, and axonal dystrophy in brain disorders. Pediatr Neurol. 2001;25:138–47. doi: 10.1016/s0887-8994(01)00266-1. [DOI] [PubMed] [Google Scholar]
- Choi DW, Yokoyama M, Koh J. Zinc neurotoxicity in cortical cell culture. Neuroscience. 1988;24:67–79. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strubing C, Strübing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Coblentz J, Croix CSt, Kiselyov K. Loss of TRPML1 promotes production of reactive oxygen species: is oxidative damage a factor in mucolipidosis type IV? Biochem J. 2013;457:361–368. doi: 10.1042/BJ20130647. [DOI] [PubMed] [Google Scholar]
- Coblentz J, Croix CSt, Kiselyov K. Loss of TRPML1 promotes production of reactive oxygen species: is oxidative damage a factor in mucolipidosis type IV? Biochem J. 2014;457:361–368. doi: 10.1042/BJ20130647. [DOI] [PubMed] [Google Scholar]
- Colletti GA, Miedel MT, Quinn J, Andharia N, Weisz Oa, Kiselyov K. Loss of lysosomal ion channel transient receptor potential channel mucolipin-1 (TRPML1) leads to cathepsin B-dependent apoptosis. J Biol Chem. 2012;287:8082–91. doi: 10.1074/jbc.M111.285536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuajungco MP, Basilio LC, Silva J, Hart T, Tringali J, Chen CC, Biel M, Grimm C. Cellular Zinc Levels are Modulated by Trpml1-Tmem163 Interaction. Traffic. 2014;15:1247–1265. doi: 10.1111/tra.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuajungco MP, Lees GJ. Zinc Metabolism in the Brain: Relevance to Human Neurodegenerative Disorders. Neurobiol Dis. 1997;4:137–169. doi: 10.1006/nbdi.1997.0163. [DOI] [PubMed] [Google Scholar]
- Curcio-Morelli C, Charles Fa, Micsenyi MC, Cao Y, Venugopal B, Browning MF, Dobrenis K, Cotman SL, Walkley SU, Slaugenhaupt Sa. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol Dis. 2010;40:370–7. doi: 10.1016/j.nbd.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyntia C, Peng Z, Bhuvarahamurthy V, A CF, F BM, F CH, A SS. Functional multimerization of mucolipin channel proteins. J Cell Physiol. 2009;222:328–335. doi: 10.1002/jcp.21956. [DOI] [PubMed] [Google Scholar]
- Deik A, Saunders-Pullman R. Atypical presentation of late-onset Tay-Sachs disease. Muscle Nerve. 2014;49:768–771. doi: 10.1002/mus.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KE, Richards LL, Votyakova TV, Reynolds IJ. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion. 2005;5:55–65. doi: 10.1016/j.mito.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–6. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem. 2009;284:32040–52. doi: 10.1074/jbc.M109.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duve C de, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60:604–617. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Dong J, Desnick RJ, Kornreich R. Carrier Screening for Mucolipidosis Type IV in the American Ashkenazi Jewish Population. Am J Hum Genet. 2002;70:1023–1027. doi: 10.1086/339519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelsdoerfer JL, Evans JA, Slaugenhaupt SA, Cuajungco MP. Zinc Dyshomeostasis Is Linked with the Loss of Mucolipidosis IV-associated TRPML1 Ion Channel. J Biol Chem. 2010;285:34304–34308. doi: 10.1074/jbc.C110.165480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey KM, Currer J, Harrison DE. In: Chapter 20 - Mouse Models in Aging Research A2 - Fox, James G. Second. Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith ALBTTM, editors. Academic Press; Burlington: 2007. pp. 637–672. in B. R. [Google Scholar]
- Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S. Tissue Distribution Profiles of the Human TRPM Cation Channel Family. J Recept Signal Transduct. 2006;26:159–178. doi: 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- Frei KP, Patronas NJ, Crutchfield KE, Altarescu G, Schiffmann R. Mucolipidosis type IV: Characteristic MRI findings. Neurology. 1998;51:565–569. doi: 10.1212/wnl.51.2.565. [DOI] [PubMed] [Google Scholar]
- Futerman AH, Meer G van. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- Gerlach M, Ben-Shachar D, Riederer P, Youdim MBH. Altered Brain Metabolism of Iron as a Cause of Neurodegenerative Diseases? J Neurochem. 2002;63:793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Fiorilli A, Trinchera M, Venerando B, Chigorno V, Tettamanti G. Uptake, cell penetration and metabolic processing of exogenously administered GM1 ganglioside in rat brain. Neurochem Int. 1989;15:455–465. doi: 10.1016/0197-0186(89)90164-2. [DOI] [PubMed] [Google Scholar]
- Gideon B, BT WM, Ruth B, Marcia Z, Joseph E. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum Mutat. 2005;26:591. doi: 10.1002/humu.9385. [DOI] [PubMed] [Google Scholar]
- Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–940. doi: 10.1136/jmg.2004.024455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin E, Caruso RC, Benko W, Kaneski CR, Stahl S, Schiffmann R. Isolated ocular disease is associated with decreased mucolipin-1 channel conductance. Invest Ophthalmol Vis Sci. 2008;49:3134–42. doi: 10.1167/iovs.07-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Bartel K, Vollmar MA, Biel M. Endolysosomal Cation Channels and Cancer—A Link with Great Potential. 2018 doi: 10.3390/ph11010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Hassan S, Wahl-Schott C, Biel M. Role of TRPML and Two-Pore Channels in Endolysosomal Cation Homeostasis. J Pharmacol Exp Ther. 2012;342:236–244. doi: 10.1124/jpet.112.192880. [DOI] [PubMed] [Google Scholar]
- Grishchuk Y, Peña KA, Coblentz J, King VE, Humphrey DM, Wang SL, Kiselyov KI, Slaugenhaupt SA. Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV. Dis Model & Mech. 2015;8:1591–1601. doi: 10.1242/dmm.021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk Y, Sri S, Rudinskiy N, Ma W, Stember KG, Cottle MW, Sapp E, et al. Behavioral deficits, early gliosis, dysmyelination and synaptic dysfunction in a mouse model of mucolipidosis IV. Acta Neuropathol Commun. 2014;2:133. doi: 10.1186/s40478-014-0133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk Y, Stember KG, Matsunaga A, Olivares AM, Cruz NM, King VE, Humphrey DM, et al. Retinal Dystrophy and Optic Nerve Pathology in the Mouse Model of Mucolipidosis IV. Am J Pathol. 2016;186:199–209. doi: 10.1016/j.ajpath.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells, Mol Dis. 2006;36:426–428. doi: 10.1016/j.bcmd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Harzer K, Beck-Wödl S, Bauer P. Niemann-Pick Disease Type C: New Aspects in a Long Published Family – Partial Manifestations in Heterozygotes BT. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V, editors. JIMD Reports. Vol. 12. Springer International Publishing; Cham: 2014. pp. 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. 2004:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hers HG. Inborn Lysosomal Diseases. Gastroenterology. 1965;48:625–633. [PubMed] [Google Scholar]
- Hers H, Hoof F. Lysosomes and Storage Diseases. NY Acad Press. 1973;184:56–57. [Google Scholar]
- Hoffmann B, Mayatepek E. Neurological Manifestations in Lysosomal Storage Disorders - From Pathology to First Therapeutic Possibilities. Neuropediatrics. 2005;36:285–289. doi: 10.1055/s-2005-872810. [DOI] [PubMed] [Google Scholar]
- Hung YH, Walterfang M, Churilov L, Bray L, Jacobson LH, Barnham KJ, Jones NC, O'Brien TJ, Velakoulis D, Bush AI. Neurological Dysfunction in Early Maturity of a Model for Niemann–Pick C1 Carrier Status. Neurotherapeutics. 2016;13:614–622. doi: 10.1007/s13311-016-0427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. Cellular pathology of Niemann-Pick type C disease. Semin Cell Dev Biol. 2004;15:445–454. doi: 10.1016/j.semcdb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34:205–19. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- Jennings JJ, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K, Jennings JJ, Jr, et al. Mitochondrial aberrations in Mucolipidosis type IV. J Biochem. 2006;281:39041–39050. doi: 10.1074/jbc.M607982200. [DOI] [PubMed] [Google Scholar]
- Josephs KA. Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology. 2004;63:2189–2190. doi: 10.1212/01.wnl.0000145710.25588.2f. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280:43218–23. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Colletti GA, Terwilliger A, Ketchum K, Lyons CWP, Quinn J, Muallem S. TRPML: Transporters of metals in lysosomes essential for cell survival? Cell Calcium. 2011;50:288–294. doi: 10.1016/j.ceca.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for Niemann–Pick C1. J Neurol Sci. 2013;335:219–220. doi: 10.1016/j.jns.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresojević N, Dobričić V, Svetel M, Kostić V. Mutations in Niemann Pick type C gene are risk factor for Alzheimers disease. Med Hypotheses. 2018;83:559–562. doi: 10.1016/j.mehy.2014.08.025. [DOI] [PubMed] [Google Scholar]
- Kruth HS, Comly ME, Butler JD, Vanier MT, Fink JK, Wenger DA, Patel S, Pentchev PG. Type C Niemann-Pick Disease. J BtOLOClCAL Chem. 1986;261:16769–16774. [PubMed] [Google Scholar]
- Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-Cell Disease) and Mucolipidosis IIIA (Classical Pseudo-Hurler Polydystrophy) Are Caused by Mutations in the GlcNAc-Phosphotransferase α/β–Subunits Precursor Gene. Am J Hum Genet. 2006;78:451–463. doi: 10.1086/500849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlante JM, Falardeau J, Sun M, Kanazirska M, Brown EM, Slaugenhaupt SA, Vassilev PM. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 2002;532:183–187. doi: 10.1016/s0014-5793(02)03670-0. [DOI] [PubMed] [Google Scholar]
- LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, Slaugenhaupt Sa, Vassilev PM. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab. 2006;89:339–48. doi: 10.1016/j.ymgme.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang WK, Benvin NM, Zhou X, Su D, Li H, Wang S, et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat Struct & Mol Biol. 2017;24:205. doi: 10.1038/nsmb.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WC, Leuba F, Soldati T, Cosson P. Mucolipin controls lysosome exocytosis in Dictyostelium. J Cell Sci. 2012;125:2315–2322. doi: 10.1242/jcs.100362. [DOI] [PubMed] [Google Scholar]
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–96. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery W, Shield E, Egel R, Jameson H. Late-infantile neuronal ceroid-lipofuscinosis: An ultrastructural study of lymphocyte inclusions. Arch Neurol. 1976;33:630–635. doi: 10.1001/archneur.1976.00500090036007. [DOI] [PubMed] [Google Scholar]
- Martina Ja, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014a;71:2483–2497. doi: 10.1007/s00018-014-1565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina Ja, Diab HI, Lishu LL, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014b;7:ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Más Gomez N, Lu W, Lim JC, Kiselyov K, Campagno KE, Grishchuk Y, Slaugenhaupt SA, Pfeffer BA, Fliesler SJ, Mitchell CH. Robust lysosomal calcium signaling through channel TRPML1 is impaired by lysosomal lipid accumulation. FASEB J. 2018;32 doi: 10.1096/fj.201700220RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A, Hardy J, Wood NW, Schapira AHV. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord. 2012;27:526–532. doi: 10.1002/mds.24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–30. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Paola S Di, Peluso I, Armani A, Stefani D De, Venditti R, Montefusco S, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsenyi M, Dobrenis K, Stephney G, Pickel J, Vanier M, Slaugenhaupt S, Walkley S. Neuropathology of the Mcoln1 -/- knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol. 2009;68:125–135. doi: 10.1097/NEN.0b013e3181942cf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsenyi M, Sikora J, Stephney G, Dobrenis K, Walkley S. Lysosomal membrane permeability stimulates protein aggregate formation in neurons of a lysosomal disease. J Neurosci. 2013;33:10815–10827. doi: 10.1523/JNEUROSCI.0987-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, Kiselyov K. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205:1477–90. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedel MT, Weixel KM, Bruns JR, Traub LM, Weisz OA. Posttranslational Cleavage and Adaptor Protein Complex-dependent Trafficking of Mucolipin-1. J Biol Chem. 2006;281:12751–12759. doi: 10.1074/jbc.M511104200. [DOI] [PubMed] [Google Scholar]
- Miller A, Schafer J, Upchurch C, Spooner E, Huynh J, Hernandez S, McLaughlin B, Oden L, Fares H. Mucolipidosis Type IV Protein TRPML1-Dependent Lysosome Formation. Traffic. 2015;16:n/a–n/a. doi: 10.1111/tra.12249. [DOI] [PubMed] [Google Scholar]
- Minke B. The history of the Drosophila TRP channel: the birth of a new channel superfamily. J Neurogenet. 2010;24:216–233. doi: 10.3109/01677063.2010.514369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabelli-Badenier M, Severino M, Tappino B, Tortora D, Camia F, Zanaboni C, Brera F, et al. A novel homozygous MCOLN1 double mutant allele leading to TRP channel domain ablation underlies Mucolipidosis IV in an Italian Child. Metab Brain Dis. 2014:681–686. doi: 10.1007/s11011-014-9612-6. [DOI] [PubMed] [Google Scholar]
- Möbius W, Herzog V, Sandhoff K, Schwarzmann G. Gangliosides are Transported from the Plasma Membrane to Intralysosomal Membranes as Revealed by Immuno-Electron Microscopy. Biosci Rep. 1999;19:307–316. doi: 10.1023/a:1020502525572. [DOI] [PubMed] [Google Scholar]
- Montell C. The TRP Superfamily of Cation Channels. Sci STKE. 2005;2005:re3–re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- Nazıroğlu M, Dikici DM, Dursun Ş. Role of Oxidative Stress and Ca2+ Signaling on Molecular Pathways of Neuropathic Pain in Diabetes: Focus on TRP Channels. Neurochem Res. 2012;37:2065–2075. doi: 10.1007/s11064-012-0850-x. [DOI] [PubMed] [Google Scholar]
- Neudorfer O, Pastores GM, Zeng BJ, Gianutsos J, Zaroff CM, Kolodny EH. Late-onset Tay-Sachs disease: Phenotypic characterization and genotypic correlations in 21 affected patients. Genet Med. 2005;7:119. doi: 10.1097/01.gim.0000154300.84107.75. [DOI] [PubMed] [Google Scholar]
- Onyenwoke RU, Sexton JZ, Yan F, Díaz MCH, Forsberg LJ, Major MB, Brenman JE. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem J. 2015;470 doi: 10.1042/BJ20150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, Ronza A di, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- Park S, Ahuja M, Kim MS, Brailoiu GC, Jha A, Zeng M, Baydyuk M, et al. Fusion of lysosomes with secretory organelles leads to uncontrolled exocytosis in the lysosomal storage disease mucolipidosis type IV. EMBO Rep. 2015 doi: 10.15252/embr.201541542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–58. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña KA, Kiselyov K. Transition metals activate TFEB in overexpressing cells. Biochem J. 2015;470:65–76. doi: 10.1042/BJ20140645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD. The fine structure of the nervous system: The neurons and supporting cells, 3rd Edition. Ann Neurol. 1991;4:588. [Google Scholar]
- Platt F, Walkley S. Lysosomal disorders of the brain. Oxford University Press; United States: 2004. [Google Scholar]
- Probert F, Ruiz-Rodado V, Vruchte D te, Nicoli ER, Claridge TDW, Wassif CA, Farhat N, Porter FD, Platt FM, Grootveld M. NMR analysis reveals significant differences in the plasma metabolic profiles of Niemann Pick C1 patients, heterozygous carriers, and healthy controls. Sci Rep. 2017;7:6320. doi: 10.1038/s41598-017-06264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor PR, Reinmann F, Gribble FM, Luzio JP, Reimann F, Gribble FM, Luzio JP. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic. 2006;7:1388–1398. doi: 10.1111/j.1600-0854.2006.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshezhetsky AV, Richard C, Michaud L, Igdoura S, Wang S, Elsliger MA, Qu J, et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat Genet. 1997;15:316. doi: 10.1038/ng0397-316. [DOI] [PubMed] [Google Scholar]
- Raas-Rothschild A, Bargal R, DellaPergola S, Zeigler M, Bach G. Mucolipidosis type IV: the origin of the disease in the Ashkenazi Jewish population. Eur J Hum Genet. 1999;7:496. doi: 10.1038/sj.ejhg.5200277. [DOI] [PubMed] [Google Scholar]
- Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, et al. Molecular basis of variant pseudo-Hurler polydystrophy (mucolipidosis IIIC) J Clin Invest. 2000;105:673–681. doi: 10.1172/JCI5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riboni L, Tettamanti G. Rapid Internalization and Intracellular Metabolic Processing of Exogenous Ganglioside by Cerebellar Granule Cells Differentiated in Culture. J Neurochem. 1991;57:1931–1939. doi: 10.1111/j.1471-4159.1991.tb06406.x. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo H, Hayashi M, Ezoe T, Ohba C, Saitsu H, Kurata K, Matsumoto N. The first genetically confirmed Japanese patient with mucolipidosis type IV. Clin case reports. 2016;4:509–12. doi: 10.1002/ccr3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26:511–24. doi: 10.1016/j.devcel.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci. 2016;1371:3–14. doi: 10.1111/nyas.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, Ronza A di, Medina DL, Valenza M, Gennarino VA, Malta C Di, et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science (80-) 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Dwyer NK, Lubensky IA, Tsokos M, Sutliff VE, Latimer JS, Frei KP, et al. Constitutive achlorhydria in mucolipidosis type IV. Med Sci. 1998;95:1207–1212. doi: 10.1073/pnas.95.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R, Mayfield J, Swift C, Nestrasil I. Quantitative neuroimaging in mucolipidosis type IV. Mol Genet Metab. 2014;111:147–51. doi: 10.1016/j.ymgme.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH. Modulation of mitochondrial function by endogenous Zn(2+) pools. Proc Natl Acad Sci U S A. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Cegli R De, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013a;15:647–58. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, Pablo R de, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–29. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013b;14:283–96. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin SS, Erdin SS, Huynh T, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer Ca, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong X, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiihara T, Watanabe M, Moriyama K, Maruyama Y, Kikuchi A, Arai-Ichinoi N, Uematsu M, Sameshima K. Mucolipidosis IV: A milder form with novel mutations and serial MRI findings. Brain Dev. 2018;38:763–767. doi: 10.1016/j.braindev.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Joyner AL. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–77. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- Silva J, Basilio L, Hart T, Tringali J, Cuajungco M. Cellular zinc levels are influenced by the interaction between the mucolipidosis IV-associated TRPML1 protein and TMEM163 protein (996.5) FASEB J. 2014;28 [Google Scholar]
- Slaugenhaupt SA. The molecular basis of mucolipidosis type IV. Curr Mol Med. 2002;2:445–50. doi: 10.2174/1566524023362276. [DOI] [PubMed] [Google Scholar]
- Slaugenhaupt SA, Acierno JS, Helbling LA, Bove C, Goldin E, Bach G, Schiffmann R, et al. Mapping of the Mucolipidosis Type IV Gene to Chromosome 19p and Definition of Founder Haplotypes. Am J Hum Genet. 1999;65:773–778. doi: 10.1086/302549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyombo Aa, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- Spranger JW, Wiedemann HR. The genetic mucolipidoses. Humangenetik. 1970;9:113–139. doi: 10.1007/BF00278928. [DOI] [PubMed] [Google Scholar]
- Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Jr, JS A, Bove C, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- Sun X, Yang Y, Zhong XZ, Cao Q, Zhu XH, Zhu X, Dong XP. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy. 2018:1–15. doi: 10.1080/15548627.2017.1389822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. doi: 10.3389/fnmol.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Tellez-Nagel I, Rapin I, Iwamoto T, Johnson A, Norton W, Nitowsky H. Mucolipidosis IV: Clinical, Ultrastructural, Histochemical, and Chemical Studies of a Case, Including a Brain Biopsy. Arch Neurol. 1976;33:828–835. doi: 10.1001/archneur.1976.00500120032005. [DOI] [PubMed] [Google Scholar]
- Troca-Marín JA, Alves-Sampaio A, Montesinos ML. Deregulated mTOR-mediated translation in intellectual disability. Prog Neurobiol. 2012;96:268–82. doi: 10.1016/j.pneurobio.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Urbanska M, Gozdz A, Swiech LJ, Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem. 2012;287:30240–56. doi: 10.1074/jbc.M112.374405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Hofmann T, Montell C. Lysosomal Localization of TRPML3 Depends on TRPML2 and the Mucolipidosis-associated Protein TRPML1. J Biol Chem. 2006;281:17517–17527. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Long a A, Elsaesser R, Nikolaeva D, Broadie K, Montell C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–51. doi: 10.1016/j.cell.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Wong CO, Montell C. Feast or famine: Role of TRPML in preventing cellular amino acid starvation. Autophagy. 2013 doi: 10.4161/auto.22260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal B, Browning M, Curcio-Morelli C, Varro A, Michaud N, Nanthakumar N, Walkley S, Pickel J, Slaugenhaupt S. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipodosis type IV. Am J Hum Genet. 2007;81:1070–1083. doi: 10.1086/521954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, LaPlante JM, Dice JF, Slaugenhaupt SA. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219:344–353. doi: 10.1002/jcp.21676. [DOI] [PubMed] [Google Scholar]
- Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008a;17:2723–37. doi: 10.1093/hmg/ddn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S, Martina JA, Puertollano R. Identification of the Penta-EF-hand Protein ALG-2 as a Ca(2+)-dependent Interactor of Mucolipin-1. J Biol Chem. 2009;284:36357–36366. doi: 10.1074/jbc.M109.047241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S, Oberdick R, Kiselyov K, Puertollano R. Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation. Biochem J. 2008b;410410:417–425. doi: 10.1042/BJ20070713. [DOI] [PubMed] [Google Scholar]
- Vergarajauregui S, Puertollano R. Two Di-Leucine Motifs Regulate Trafficking of Mucolipin-1 to Lysosomes. Traffic. 2006;7:337–353. doi: 10.1111/j.1600-0854.2006.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci. 2015;112:201419669. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong X, Samie M, Li X, Cheng X, Goschka A, et al. TPC Proteins Are Phosphoinositide- Activated Sodium-Selective Ion Channels in Endosomes and Lysosomes. Cell. 2017;151:372–383. doi: 10.1016/j.cell.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CO, Li R, Montell C, Venkatachalam K. Drosophila TRPML is required for TORC1 activation. Curr Biol. 2012;22:1616–21. doi: 10.1016/j.cub.2012.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong COO, Palmieri M, Li J, Akhmedov D, Chao Y, Broadhead GTT, Zhu MXX, et al. Diminished MTORC1-Dependent JNK Activation Underlies the Neurodevelopmental Defects Associated with Lysosomal Dysfunction. Cell Rep. 2015;12:2009–2020. doi: 10.1016/j.celrep.2015.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye CP, Quinn SJ, Goldin E, Brown EM, Slaugenhaupt S, Vassilev PM. Functional links between mucolipin-1 and Ca2+ - dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun. 2004;322:1384–1391. doi: 10.1016/j.bbrc.2004.08.045. [DOI] [PubMed] [Google Scholar]
- Yu W, Ko M, Yanagisawa K, Michikawa M. Neurodegeneration in Heterozygous Niemann-Pick Type C1 (NPC1) Mouse: IMPLICATION OF HETEROZYGOUS NPC1 MUTATIONS BEING A RISK FOR TAUOPATHY. J Biol Chem. 2005;280:27296–27302. doi: 10.1074/jbc.M503922200. [DOI] [PubMed] [Google Scholar]
- Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta. 2007;1772:851–858. doi: 10.1016/j.bbadis.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Zeevi DA, Lev S, Frumkin A, Minke B, Bach G. Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy. J Cell Sci. 2010;123:3112–3124. doi: 10.1242/jcs.067330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fang Y, Cheng X, Lian YJ, Xu H, Zeng ZS, Zhu H. Curcumin Exerts Effects on the Pathophysiology of Alzheimer's Disease by Regulating PI(3,5)P2 and Transient Receptor Potential Mucolipin-1 Expression. 2017a doi: 10.3389/fneur.2017.00531. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang S, Li N, Zeng W, Gao N, Yang M. Cryo-EM structures of the mammalian endo-lysosomal TRPML1 channel elucidate the combined regulation mechanism. Protein Cell. 2017b;8:834–847. doi: 10.1007/s13238-017-0476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. 2016 doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.