

Abstract
A real time method to measure intracellular H2O2 would be very impactful in characterizing rapid changes that occur in physiologic and pathophysiologic states. Current methods do not provide the sensitivity, specificity and spatiotemporal resolution needed for such experiments on intact cells. We developed the use of HyPer, a genetic indicator for H2O2 that can be expressed in the cytosol (cyto-HyPer) or the mitochondria (mito-HyPer) of live cells. INS-1 cells or islets were permeabilized and the cytosolic HyPer signal was a linear function of extracellular H2O2, allowing fluorescent cyto-HyPer signals to be converted to H2O2 concentrations. Glucose increased cytosolic H2O2, an effect that was suppressed by overexpression of catalase. Large perturbations in pH can influence the HyPer signal, but inclusion of HEPES in the perfusate prevented pH changes, but did not affect glucose-induced cyto-HyPer signals suggesting this effect is largely pH-independent. Using the assay, two fundamental questions were addressed. Knockdown of SOD2, the mitochondrial form of SOD, completely suppressed glucose-induced H2O2. Further, glucose also induced mitochondrial superoxide and H2O2 production, which preceded the appearance of cytosolic H2O2. Therefore, glucose-induced H2O2 largely originated from mitochondria. Finally, glucose-induced HyPer signal was less than 1/20th of that induced by toxic levels of H2O2. Overall, the use of HyPer for real time imaging allowed resolution of acute changes in intracellular levels of H2O2 and will have great utility for islet studies involving mechanisms of H2O2 mediating signaling and oxidative stress.
INTRODUCTION
Importance of H2O2 in islets
Failure of pancreatic β cells to compensate for increased insulin resistance is a major determinant of diabetes [1, 2]. Understanding the importance of specific control steps in the regulation of insulin secretion, such as glucokinase and ATP-sensitive potassium (KATP) channels has shed light on some causes of impaired insulin secretion [3, 4]. There is evidence that H2O2 can act as a signal mediating both basal [5] and glucose-induced insulin secretion [6, 7]. Indeed islets secrete insulin in response to low levels of H2O2 [8, 9]. It has been reported that intracellular H2O2 increases in response to glucose, and the effect of glucose on H2O2 and insulin secretion rate is blocked by scavengers of H2O2 [6, 7]. Sources of H2O2 in the β cell may reside in the cytosol from the action of NADPH oxidase [10] and the mitochondria through the action of the respiratory chain [11]. On the other end of the spectra, high levels of H2O2 may lead to oxidative stress, which is harmful to the islet [12]. This action of H2O2 may be mediating the loss of function and β cell death during episodes of excess glucose and/or fatty acids [13], as well as exposure to cytokines released in response to inflammation and autoimmune attack [14]. In these studies, intracellular H2O2 was determined by conventional indicators such as DCF (2’,7’-dichlorofluorescein) [15], which are known to be non-specific and irreversible [16]. Thus, technical difficulties have precluded definitive testing of these highly significant findings. In addition, these indicators cannot distinguish between H2O2 residing in the cytosol from that in the mitochondria, critical knowledge needed for understanding the source and point of action of H2O2 signaling. What is needed is a highly specific and sensitive dye to H2O2 that can be directed at specific intracellular compartments.
Novel H2O2 indicator with high sensitivity
A recently developed genetic probe for H2O2, named HyPer, has high sensitivity, is specific to H2O2 relative to other ROS known to reside in the cells, and can be targeted to intracellular organelles [17, 18]. Previous studies have shown that the sensor expressed in islets was indeed sensitive to exogenous H2O2 and glucose [19]. However, these authors also pointed to the known pH-sensitivity of the dye that would be problematic when measuring glucose effects with the mitochondrial probe where the pH increases from 7.2 to 8 upon stimulation. Since HyPer is uniquely sensitive and specific (with respect to H2O2 and ROS) compared to other H2O2-sensitive dyes and sensors, we endeavored to use HyPer to measure H2O2 while correcting as needed for pH changes by concomitantly imaging a pH-sensitive dye with HyPer. Our data shows that for cytosolic H2O2, physiologic changes in pH (as that elicited by glucose) did not contribute to changes in HyPer signal. In addition, we found HyPer to be reproducible, dose dependent and chemically specific. The use of HyPer in islets will provide islet researchers with a powerful tool for the accurate monitoring of compartmentalized H2O2 signals to tackle key questions including the role of H2O2 in signaling and oxidative stress.
MATERIALS AND METHODS
Chemicals
Krebs-Ringer bicarbonate solution (KRB), used for all perifusion/imaging analysis, contained 98.5 mM NaCl, 4.9 mM KCl, 1.2 mM potassium phosphate, 1.2 mM magnesium sulfate, 25.9 mM sodium bicarbonate, 2.6 mM CaCl2, varying amounts of glucose (all from Sigma Aldrich, St. Louis, MO), and 20 mM HEPES (Research Organics, Cleveland, OH). Potassium cyanide (KCN) and H2O2 were both purchased from Sigma-Aldrich.
Culture of INS-1 832/13 cells
INS-1 832/13 cells (henceforth referred to as INS-1 cells) were kindly provided by Dr. Christopher Newgard and were cultured as previously described [20]. Three days before the experiment, cells were harvested by exposure to Trypsin for 5 minutes, plated on to glass coverslips coated with Matrigel (Corning, Corning, NY) and the coverslips were placed in petri dishes containing the specified adviruses in RPMI Media 1640 (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 2 mM L-Glutamine, 1 mM Pyruvate, 50 μM Beta-mercaptoethanol, 20 mM HEPES and 1% Pen/Strep.
Rat islet isolation and culture
Islets were harvested from Sprague-Dawley male rats (≈ 250 g, Charles River) anesthetized by intraperitoneal injection of sodium pentobarbital (35 mg/230 g rat). All procedures were approved by the University of Washington Institutional Animal Care and Use Committee. Islets were prepared and purified as previously described [21], and then cultured at 37°C in RPMI Media 1640 (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) for specified times with HyPer dye or other adviruses.
Viral packaging and expression of HyPer
Two forms of the H2O2 sensor were used: pHyPer-dMito vector and pHyPer-cyto vector (FP941 and FP942, Evrogen, Moscow, Russia) that localize to cytosol and mitochondria respectively [17]. Adenoviruses containing cyto-HyPer and mito-HyPer were generated by Vector Biolabs (Malvern, PA) as previous reported [22]. The H2O2 sensors were transduced in intact islets or cells during incubation in RPMI media supplemented with 10% heat-inactivated fetal bovine serum and the adenoviruses at a multiplicity of infection of 10–100 for three days at 37° C.
Permeabilization of INS-1 cells
To test the sensitivity of the HyPer sensor to known concentrations of H2O2, INS-1 cells expressing cyto-HyPer plated in 24-well dishes cells were permeabilized. Just prior to analysis, cells were washed with KRB followed by KG buffer (140 mM potassium glutamate, 5 mM ATP, 5 mM NaCl, 7 mM MgSO4, 0.4 mM EGTA, 1% BSA, and 20 mM HEPES, pH 7.4). They were then incubated for 15 min with 0.5 ml of KG buffer containing varying concentrations of streptolysin-O (Sigma-Aldrich) to permeabilized the cells. Trypan blue exclusion test demonstrated that at 3.2 units/ml streptolysin-O more than 90% of the cells were permeabilized (Fig. S1). This concentration of streptolysin-O was used for permeabilizing cells while they were in the flow system for the time indicated.
Overexpression of cytosolic and mitochondrial catalase, and knockdown of superoxide dismutase
The mitochondrial targeted catalase construct was kindly provided by Dr. Peter Rabinovitch. The adenovirus construct and the adenovirus-packaged cytosolic catalase were both generated by Vector Biolabs. Freshly isolated islets from rats or cultured INS-1 cells were infected with the above adenoviruses at a multiplicity of infection of 10–100 for 3 days. Appropriate gene expression was confirmed by Western blot or confocal imaging.
To make the SOD2 shRNA, the most efficient knockdown construct for SOD2 was screened and identified (see Supplemental Methods for details), which was then placed into the pLenti6/BLOCK-iT0222-DEST expression vector. The lentiviral vector was inserted in HEK-293T cells by calcium phosphate-mediated co-transfection with plasmids containing viral genome components. Vector preparations were concentrated 100 fold. INS-1 cells were plated at a density of 100,000 cells per 35 mm dish and, after 24 h of proliferation, were transduced with 50 µl of concentrated virus in 1 ml of INS-1 media containing 8 µg/ml polybrene (Sigma-Aldrich, St. Louis, MO) overnight. This amount was found to result in an 80% decrement in SOD2 mRNA as reflected by Quantitative PCR performed on an Mx3005P® Multiplex QPCR System (Stratagene, La Jolla, CA) with samples loaded in triplicate using ~60 ng cDNA.
Western Analysis for catalase
Protein samples were prepared from islets samples using a lysis buffer (Cell Signaling) containing protease inhibitors (Sigma). After SDS-PAGE, proteins were transferred onto nitrocellulose membranes (Bio-Rad Laboratories), and blocked in 5% nonfat milk before incubation with primary antibodies: anti‐catalase (1:1000, Athens Research and Technology) and anti-Actin (1:5000, Sigma). The secondary antibodies (1:5000, Invitrogen) were labeled with Alexa Fluor detection reagent, and images taken by Odyssey Infrared Technology (LI-COR).
Confocal imaging of HyPer indicators
To determine the intracellular localization of cyto-HyPer and mitochondrial localization of mito-HyPer, we loaded the mito-HyPer expressing islets with MitoTracker Red FM (500 nM for 20 min, M22425, Thermo-Fisher Scientific). Confocal imaging was carried out using a Leica TCP SP8 confocal microscope with a 40x, 1.3 NA oil immersion objective. Cyto-HyPer and mito-HyPer were sequentially excited with 405 and 488 nm lasers and the emissions were collected at 505–550 nm. For MitoTracker Red, 552 nm excitation was used and emission collected at >560 nm.
Single and Dual channel perifusion system for real time imaging
Real time imaging experiments were carried out while islets or INS-1 cells were perifused using a commercially available temperature controlled perifusion dish (as previously described [23]). In order to accommodate side-by-side comparison of islets with and without overexpression of catalase, the standard single port perifusion dish (Bioptechs, Butler, PA), was custom-modified so that it had two inlet and outlet ports (Fig. 1). Two rectangular sections were cut from a gasket made out of two stacked circles of Parafilm so that the voids created when the gasket was placed into the perifusion dish acted as individual perifusion chambers, each fed and drained by one of the two ports.
Figure. 1. Schematic of the dual channel fluidics/imaging chamber.
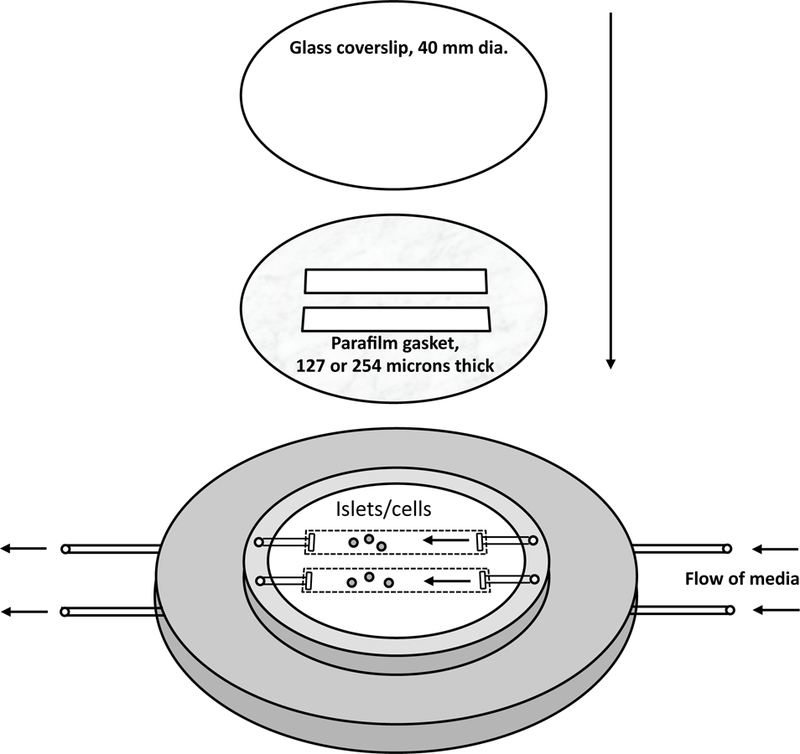
A Bioptechs FCS2, a closed system, parallel plate flow cell, was modified in order to accommodate two inflow and outflow ports. The gaskets were then cut so that the ports supplied and drained flow to and from two separated cell channels. For cells one Parafilm gasket was used, and for islets, two were stacked prior to cutting the rectangular holes. This system allowed for two perifusions to be carried out in parallel such as when comparing genetically altered islets or cells to control cells.
Real time epi-fluorescent imaging of intracellular H2O2, superoxide and pH
After the islets or cells were loaded into the perifusion chambers, the chambers were sealed and mounted on to the stage of a Nikon Eclipse TE-200 inverted microscope. KRB was pumped through the two chambers at flow rates of 130 µL/min for each using a Masterflex L/S peristaltic pump (Cole-Parmer, Vernon Hills, IL). The HyPer signals were generated by dual fluorescence excitation via a xenon arc lamp (Lambda LS-1620, Sutter Instrument Company, Novato, CA) through either a 405/30nm or a 480/40nm bandpass filter and detected at 510nm through a longpass dichroic mirror with a cutoff of lower 500nm. The images were taken using a digital camera (Photometrics Cool Snap HQ2 CCD camera, Tucson, AZ) through a 40x Super Fluor Nikon objective (DIC H/N2). At the end of each experiment, an inhibitor of the electron transport chain (KCN) was added to the media to stop the generation of H2O2 from the mitochondria, and the steady state values in the presence of KCN used for background correction. Note that KCN may also have other effects in addition to blocking cytochrome c oxidase that might prevent the signal from going all the way to baseline, but it did induce a level of signal that was lower than all other conditions that were investigated. Data was expressed ratiometrically, where the excitation intensities at 480nm excitation were divided by those obtained during excitation at 405nm. MitoSOX Red (M36008, Thermo-Fisher Scientific, Waltham, MA), a dye sensitive to superoxide that localizes to the mitochondria, and SNARF-1 (5-(and-6)-Carboxy SNARF®−1, Acetoxymethyl Ester, Acetate, C1271, Thermo-Fisher Scientific), a pH-sensitive dye that distributes in the cytosol, were visualized by excitation at 546 nm and detection at 605 nm.
Continuous measurement of oxygen consumption rate and insulin secretion rate
A flow culture system was used that concomitantly measured oxygen consumption rate while collecting outflow fractions for subsequent measurement of insulin (described previously [21, 24, 25]). OCR was calculated as the flow rate (approximately 80 µL/min) times the difference between inflow and outflow oxygen tension and measured by detecting signal from an oxygen-sensitive dye painted on the inside of the perifusion chamber [21]. Insulin in the outflow fractions was measured using an RIA kit (Linco Research Inc., St. Charles MO).
Static assessment of insulin secretion
Insulin secretion rate was determined statically with multiple conditions as previously described [26]. Briefly, islets were handpicked in parallel into a Petri dish containing KRB, 0.1% BSA and 3 mM glucose and incubated at 37 °C/5% CO2 for 60 min. Subsequently, islets were placed into wells of 96-well plates (10 islets/well) containing indicated amounts of glucose, and incubated for 60 more min. At the end of this period, supernatant was assayed for insulin using a Linco RIA kit.
Viability staining with Acridine Orange and Propidium Iodide
Islets were imaged after staining of islets with acridine orange (10 mmol/L) and propidium iodide (15 mmol/L) (AO/PI) [27].
RESULTS
Sensitivity of expressed H2O2 sensor: Response of cyto-HyPer to extracellular H2O2
To test the sensitivity and response time of the cytosolic H2O2 sensor, INS-1 cells were permeabilized with streptolysin O prior to loading them into the perifusion system. Various concentrations of H2O2 ranging from 30 nM up to 1 μM were added to the inflow at 20-minute intervals (Fig. 2A). Starting at 65 nM, the fluorescence increased in proportion to the H2O2 concentration until 600 nM was reached (Fig. 2B). At 1 μM, the signal began to saturate. The response time of the sensor was slightly delayed at low extracellular concentrations, but was within 1 minute at 600 nM. Since the concentration of H2O2 was certainly reduced by anti-oxidant machinery in the cell, this estimate of response time was an upper limit. Taken together, the relation between cyto-HyPer signal and cytosolic concentrations of H2O2 is linear between 65 and 600 nM, and has a kinetic resolution that is at most on the order of a few minutes.
Figure 2. Real time (A) and steady state (B) responses of cyto-HyPer in permeabilized INS-1 cells.

(A) INS-1 cells were imaged during perifusion with stepwise increasing amounts of H2O2 in the buffer as indicated (typical calibration curve). (B) Steady state values were calculated as the average of the final five minutes of each H2O2 concentration. A trendline was drawn through the linear region from 65 to 600 nM, where 30 nM was indistinguishable from 0 nM fluorescence
Test of H2O2 specificity by cyto-HyPer
Having characterized the sensitivity of cyto-HyPer sensor to H2O2, we next addressed whether it was specific for H2O2. To do this, we over-expressed cytosolic catalase, an enzyme that converts H2O2 to water with very high specificity, in islets and INS-1 cells. Catalase was overexpressed after 2 days of incubation with an adenovirus containing DNA for either cytosolic or mitochondrial catalase, as reflected by Western blot (Fig. 3A). Using the two-channel perifusion system, we conducted side-by-side comparisons between real time responses of cyto-HyPer to glucose in islets or INS-1 cells with and without high levels of catalase. In the absence of catalase over-expression, glucose led to an increased fluorescence ratio in both islets (Fig. 3B) and INS-1 cells (Fig. 3C). Over-expression of cytosolic catalase in islets, and mitochondrial catalase in INS-1 cells, completely inhibited the glucose-induced increases in signal, indicating that the change in fluorescence in response to glucose was largely determined by H2O2. There was, however, a slight decrease in cyto-HyPer fluorescence in response to glucose in the INS-1 cells, when mitochondrial catalase was being overexpressed, an effect that we did not investigate.
Figure 3. Effect of catalase overexpression on cyto-HyPer.
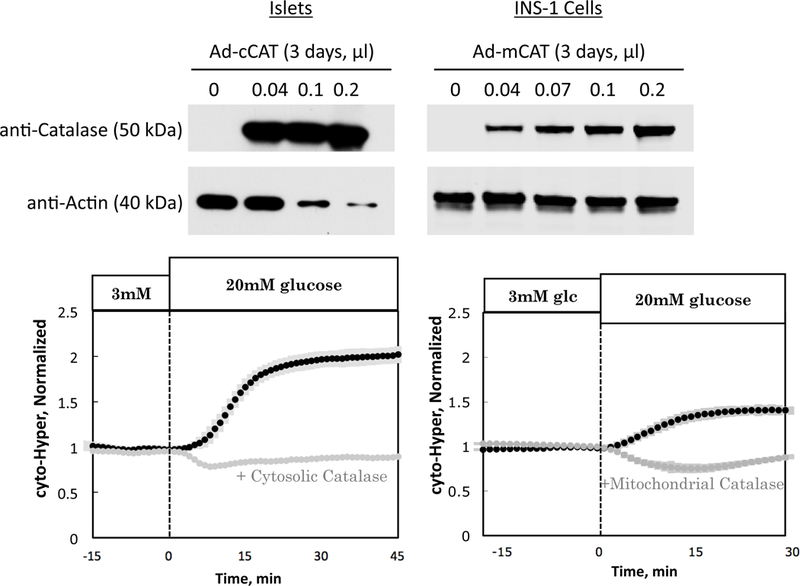
(A) Western analysis of cytosolic catalase in islets and mitochondrial catalase in INS-1 cells Glucose-induced increase in cyto-HyPer signal is suppressed in response to (B) overexpression of cytosolic catalase in rat islets (n = 4) and (C) overexpression of mitochondrial catalase in INS-1 cells (n = 4), demonstrating specificity of HyPer for H2O2.
During glucose stimulation of the islet, changes in pH do not effect H2O2 measurements
A previous study described the sensitivity of HyPer fluorescence to elevated pH when islets were exposed to high levels of base [19]. Accordingly we endeavored to test whether stabilization of changes in intracellular pH could be accomplished by use of the pH buffer HEPES, and whether pH changes in the physiologic range affect HyPer fluorescence. When perifused in standard KRB buffer (in the absence of HEPES), islet pH (as reflected by the pH sensor SNARF-1) decreased in response to 20 mM glucose, an effect which was rapidly reversed upon returning to 3 mM glucose (Fig. 4A). In contrast, when 20 mM HEPES was added to the KRB buffer, pH was no longer sensitive to changes in glucose (Fig. 4A). Therefore, at least for changes in glucose, the use of HEPES maintains intracellular pH so it could not contribute to glucose changes in cyto-HyPer. We then measured the effect of glucose-induced changes in pH on cytosolic H2O2 by comparing the response of H2O2 in the presence and absence of HEPES in the buffer. Somewhat unexpectedly, the H2O2 responses to glucose were identical whether the pH changes were occurring or were prevented by HEPES (Fig. 4B). This data is actually in concordance with the original methods paper introducing HyPer where it was shown that near physiologic pH, the cyto-HyPer signal is insensitive to pH, and it is only at high pH when the effects on HyPer are significant ([17], Supplementary Fig. 1). Thus it appears that for glucose stimulation, pH sensitivity of HyPer is not an issue, and the use of HEPES provides a method to stabilize changes in pH to test this assumption.
Figure 4. Lack of effect of glucose-induced changes in pH on cytosolic H2O2.
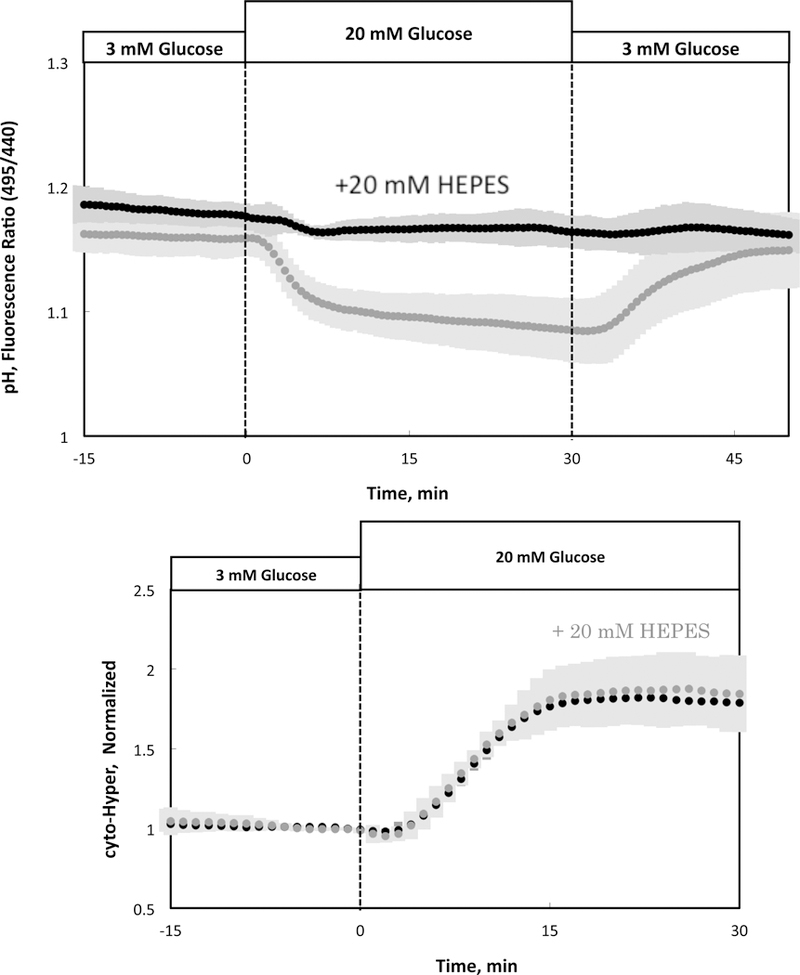
(A) Glucose-stimulated pH in the presence and absence of 20 mM HEPES in the buffer (n = 3). (B) Glucose-induced changes in cytosolic H2O2 by islets in the presence or absence of 20 mM HEPES (n = 3).
Identification of source of cytosolic H2O2 in islet cells
Previous studies have suggested that sources of H2O2 in islets are located in the cytosol, including NADPH oxidase [28], autooxidation [29], and the hexosamine pathway [30], as well as mitochondrial oxidation [31] (Fig. 5). Since in many cell types mitochondria are the major source of ROS [32], we tested whether mitochondrial superoxide is the major source of cyto-HyPer-detected H2O2 by knocking down mitochondrial superoxide dismutase (SOD2) in INS-1 cells. The SOD2 shRNA was titrated to achieve an 80% reduction in SOD2 mRNA in INS-1 cells ((0.56 vs. 2.75 ng SOD2/ng RNA in the transfected vs. control INS-1 cells)). Similar to the over-expression of catalase, knock down of SOD2 prevented the glucose-induced increase in cytosolic H2O2 (Fig. 6). This suggests that the major source of the intracellular H2O2 is from the conversion of mitochondrial superoxide to H2O2 by the action of SOD2. Attempts made to knockdown islet SOD2 were not successful for reasons that were not clear.
Figure. 5. Production and conversion of H2O2.
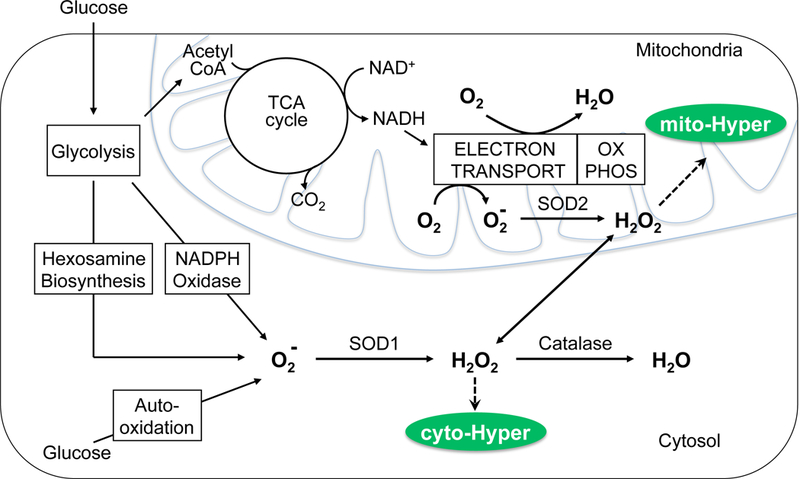
H2O2 is produced in the cytosol and mitochondria by conversion of superoxide and the action of SOD. H2O2 in the cytosol and mitochondria are detected by cyto-HyPer and mito-HyPer respectively.
Figure 6. Effect of SOD2 (mitochondrial SOD) knockdown on glucose-induced increase in H2O2.
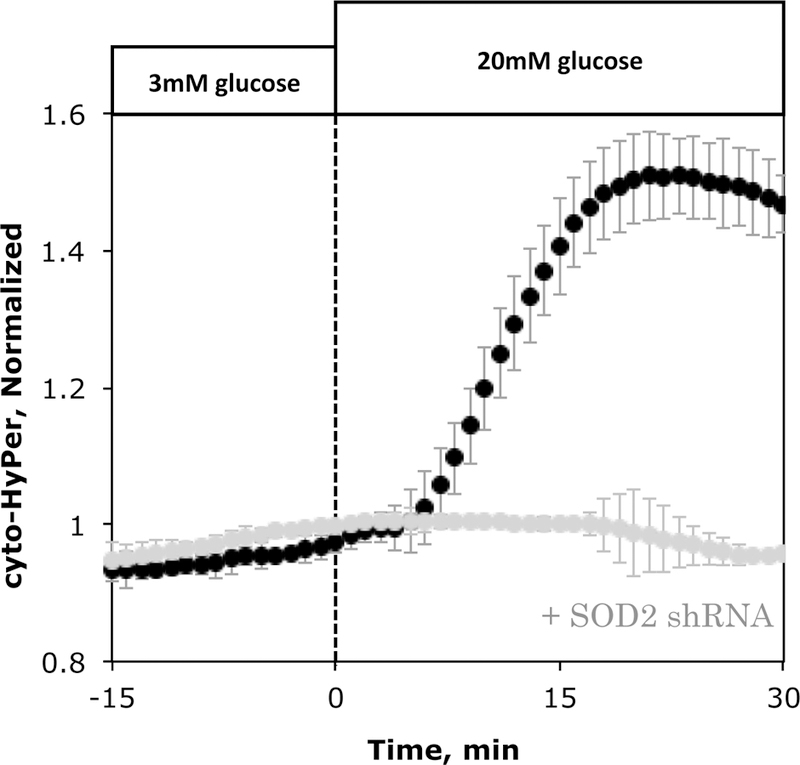
Response in H2O2 to glucose by INS-1 cells cultured for 3 days in the presence (n= 2) or absence of shRNA (n = 4) for SOD2.
Glucose responses of intracellular generation of superoxide and mitochondrial and cytosolic H2O2
To further test the source of H2O2 and temporal resolution for the sensors, the kinetics of the response of superoxide, mitochondrial H2O2 and cytosolic H2O2 were measured by the dyes MitoSOX Red, mito-HyPer and cyto-Hyper, respectively. To confirm that the mito-HyPer was specifically distributed in the mitochondria, confocal images of islets expressing mito-Hyper and cyto-Hyper were compared to islet fluorescence after loading with MitoTracker Red. The fluorescence pattern of MitoTracker Red was localized to forms consistent with mitochondrial distribution. The spatial distribution of the fluorescence for mito-HyPer and MitoTracker Red were well correlated (Fig. 7A), and in contrast to cyto-Hyper, showed a lack of signal from nuclei and cytosol. Without expression of HyPer, islets did not have significant non-specific fluorescent signal.
Figure 7. Kinetics of superoxide, mitochondrial and cytosolic H2O2 in response to glucose.
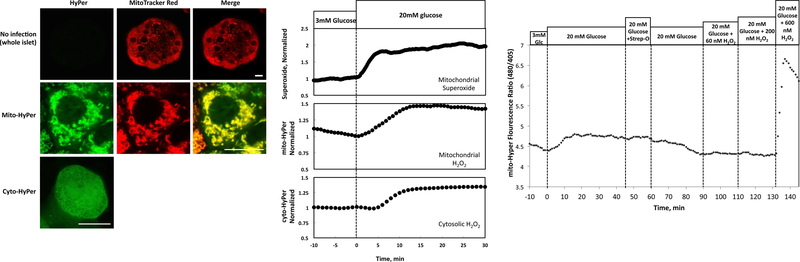
(A) Confocal imaging of islets and islet cells infected with or without mito-HyPer and cyto-HyPer. Top row of panels: images of whole islets loaded with or without MitoTracker Red. Middle row of panels: single islet cell expressing mito-HyPer and loaded with MitoTracker Red. Bottom panel: image of single islet cell expressing cyto-HyPer. White scale bar is 10 microns in length. (B) Kinetic responses of islets loaded with MitoSOX Red (a sensor for mitochondrial superoxide), or expressing mito-HyPer or cyto-HyPer (n = 4). (C) Lack of response of mito-HyPer to low levels of extracellular H2O2 in permeabilized islets (typical response).
From the previous data, intracellular H2O2 is largely being produced from mitochondrially-generated superoxide, which then traverses the mitochondrial membrane to the cytosol. Consistent with this scenario, superoxide responded to glucose prior to the increase in mitochondrial H2O2, and there was a lag in cyto-HyPer of about 5 minutes relative to mito-HyPer (Fig. 7B). As H2O2 is generally thought to be freely diffusible across membranes, we sought to confirm this finding by an alternate method. To do this, we used permeabilized INS-1 cells expressing mito-HyPer, and exposed them to increasing concentrations of H2O2, in a similar fashion to the experiment shown in Fig. 2A performed with cyto-HyPer. In contrast to cytosolic H2O2, which increased in response to 65 nM extracellular H2O2, increases in mitochondrial H2O2 was seen at 600 nM but not 200 nM extracellular H2O2. Thus, in two different experimental conditions, and when H2O2 transport was in the direction of cytosol to mitochondria, or vice versa, there was a significant barrier to equilibrium between the two compartments.
Comparison of physiologic and toxic levels of H2O2
It is a hallmark of type 2 diabetes that beta cell mass is decreased [33]. High levels of ROS generated by increased glucose metabolism have been proposed to result in damage to beta cells, providing a mechanism for how hyperglycemia could lead to decreased beta cell mass [34]. An accurate, and calibrated assay for H2O2 allows comparison of the H2O2 levels achieved by increased glucose and the amount of H2O2 needed to result in damage to the beta cell. To implement this test, we first determined the amount of exogenous H2O2 that is needed to bring about significant islet cell death. Islet membrane integrity was evaluated for periods of up to 24 hours following a 15-minute exposure to varying amounts of H2O2 (Fig. 8A). At low concentrations (below 50 μM) no dead cells were detected, whereas exposure to 100 μM resulted in some cells on the periphery of the islet to stain for propidium iodide entry (reflecting breakdown of the plasma membrane). At 500 μM H2O2, much of the islet stained red for propidium iodide within 6 hours (although the core was spared). Thus, under these conditions, it takes more than 100 μM H2O2 to damage the islets. We also performed experiments where islets were chronically exposed to H2O2 for 24 hours, and statically assessed for changes in insulin secretion rate. No changes were observed by islets exposed to 10, 30 or 100 μM H2O2, but the rate was reduced by more than 90% after exposure to 500 μM H2O2 (data not shown), a dose response similar to that obtained and using boluses and membrane integrity as an endpoint and in a previous study [28].
Figure 8. Comparison of glucose-induced H2O2 levels vs. H2O2 observed in the presence of toxic concentrations of extracellular H2O2.
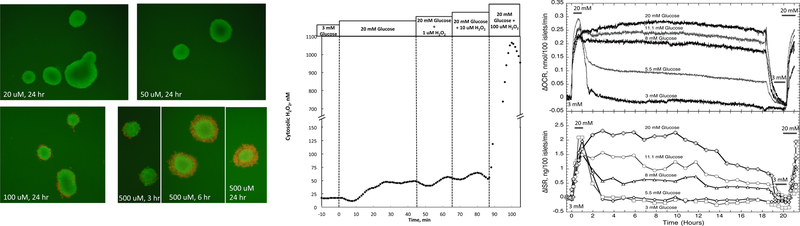
(A) Islets were incubated for 15 minutes in the presence of the indicated concentrations of H2O2 and then assessed for viability as reflected by membrane patency at the times shown. (Red cells no longer have membrane integrity.) (B) Cytosolic H2O2 in response to glucose and extracellular H2O2 (typical response). Toxic concentrations of extracellular H2O2 were accompanied by supraphysiologic cytosolic levels of H2O2. (C) Oxygen consumption rate and insulin secretion rate as a function of exposure to various concentrations of glucose in the culture media (n = 3). Prior and subsequent to the culture period, acute islet response to an increase in glucose concentration from 3 to 20 mM was assessed.
In order to evaluate the potential for glucose metabolism to produce enough H2O2 to have toxic effects, we compared the amount of glucose-generated intracellular H2O2 to that which was present when islets were exposed to toxic amounts of exogenous H2O2. To do this, fluorescent data was converted to actual concentrations of H2O2 by permeabilizing the cells at the end of the experiment and carrying out a stepwise concentration dependency as was done to generate Fig. 2A. An increase in glucose (to 20 mM) also increased cytosolic H2O2 to about 50 nM (Fig. 8B). When the exogenous H2O2 was raised to 100 μM, the minimal amount needed to induce a loss of viability, cytosolic H2O2 was increased to more than 20 times that reached at high glucose. Thus, increases in endogenously generated H2O2 by high glucose were many times lower than that needed to induce toxic effects in vitro.
To further confirm that high glucose levels do not impair metabolic or secretory function, oxygen consumption and insulin secretion by islets were measured continuously for 24 hours using a flow culture system while exposed to various concentrations of glucose in islet culture media. Oxygen consumption rates were proportional to the concentration of glucose, and did not decrease for any of the glucose levels (Fig. 8C). Insulin secretory rates were also proportional to glucose concentration, but as observed in previous studies [35], waned over time after about 10 hours following the stimulation by glucose. At the beginning and end of the experiment, the ability of the flow-cultured islets to respond acutely to an increase in glucose from 3 to 20 mM was assessed. All islet oxygen consumption and insulin secretion responses were the same, except for islets exposed to 3 mM glucose overnight whose responses were much diminished. Thus, with respect to 24 hour exposures, exposure to high glucose resulted in more metabolically and functionally robust islets, consistent with the scenario that glucose cannot alone induce toxic levels of H2O2.
DISCUSSION
H2O2 has been hypothesized to have a major role in islet function and pathology, both as a signaling molecule [6, 7] and as a mediator of beta-cell damage during oxidative stress [11, 36]. However, a major barrier to progress in testing and further investigating these biochemical concepts is the lack of a real time assay for measuring the time course and absolute concentrations of H2O2 in intact islets. A previous study demonstrated the ability of HyPer to express in islets and track H2O2 levels [19], however, pH-sensitivity was thought to be an obstacle to its general use. Accordingly, we built upon this previous study, addressed the pH-sensitivity and validated the H2O2 assay for use in islets. We then applied the assay to address two fundamental questions about the source and concentration range of intracellular H2O2.
Validation of a real time H2O2-specific assay
The validation of cyto-HyPer involved two basic tests: sensitivity and specificity. The assay response was linear between 65 and 600 nM, occurred with less than minute-to-minute resolution, and catalase, an enzyme with high specificity for H2O2, completely suppressed glucose-induced increase in H2O2. In general, the responses in INS-1 cells and whole islets were similar, but INS-1 cells were used when the adviruses would not result in desired changes in protein levels, for instance for knocking down SOD2. In addition, in the presence of HEPES, pH didn’t change in response to glucose, and the H2O2 response to glucose was the same in both the presence and absence of HEPES. Therefore, while we present a method to circumvent the potential pH-sensitivity of the H2O2 signal by buffering intracellular pH, it proved unnecessary since the small change in pH induced by glucose did not alter the HyPer signal. This was likely due to the fact that the HyPer protein sensitivity to pH below about 7.6 is greatly reduced ([17], Supplementary Fig. 1). With respect to mito-HyPer, we did not have a method for measuring the mitochondrial pH, nor do we have an approach that necessarily stabilizes it. However, with the exception of a delay in the cyto-HyPer signal relative to the mito-HyPer, the signals were in most cases qualitatively similar, making it likely that the mito-HyPer is also a valid kinetic measure of H2O2. Thus, cyto-HyPer can quantify physiologic changes of H2O2 in real time in intact whole islets, and the glucose response was specific for H2O2 and not affected by pH.
Physiologic vs. harmful levels of H2O2 in islets
As islets in the transition to diabetes are exposed to lengthy durations of hyperglycemia, it has been proposed that high metabolic flux through the electron transport chain generates harmful levels of ROS [12], which in turn could lead to loss of beta cell mass. In our study, consistent with other reports [37, 38], glucose caused an increase in both cytosolic and mitochondrial islet H2O2. However, this is the first study that quantified the intracellular concentrations of H2O2, which increased from 20 to 50 nM when islets were exposed to 3 and 20 mM glucose respectively. In contrast, the amount of H2O2 observed in the presence of toxic levels of H2O2 was more than 20 times that seen in the presence of 20 mM glucose. This is consistent with the stability of oxygen consumption observed during exposure to high glucose, as well as another study where viability and secretory function of isolated islets cultured in vitro was maintained for 7 days or more in the presence of very high levels of glucose [39]. To be sure, conditions in vivo may result in different/higher levels of intracellular H2O2, and furthermore the effects of H2O2 could be more potent in in vivo conditions. But nonetheless, it seems that the vastly different concentration dependency of toxicity vs. H2O2 levels would suggest that a mere 2.5-fold increase of H2O2 levels would be unlikely to result in H2O2-mediated damage.
Generation of H2O2 in islets mainly by conversion of mitochondrial superoxide
Due to the potential for endogenously generated H2O2 to induce damage and/or act as a physiologic signal, there has been much interest in the intracellular mechanisms mediating H2O2. Past publications have considered several biochemical pathways operating in the cytosol of the islet that lead to production of H2O2 including the actions of NADPH oxidase [28], autooxidation [29], and the hexosamine biosynthesis pathway [30]. In contrast, in regard to many cell types, the mitochondria are thought to be the main source of H2O2 [32]. The results of our studies, including the loss of glucose-stimulated H2O2 response after mitochondrial SOD knockdown, and the delay in appearance of H2O2 in the cytosolic relative mitochondrial H2O2, strongly suggest that mitochondrial superoxide is the major source of H2O2 in the islet. During this experimental investigation it was interesting to observe an unexpected asymmetry in the transfer of H2O2 between the cytosol and the mitochondria. Although it is commonly held that H2O2 is very permeable to lipid membranes and that the mitochondrial membrane would not represent a kinetic barrier, we observed a delay in response of cytosolic H2O2 in response to mitochondrially-generated H2O2, and a complete lack of increase in mitochondrial signal at low levels of cytosolic H2O2 induced by extracellular H2O2. Whether this was due to impermeability of H2O2 by the mitochondrial membrane, or the action of antioxidants around the cytosolic side of the mitochondrial membrane is not established by this study. However, the time course of H2O2 distribution into different compartments has implications for the potential for H2O2 to act as a signal.
Conclusions
We have developed an assay that measures H2O2 levels in real time, where only mild changes in pH are occurring;
Data supports predominant source of H2O2 under glucose stimulation is mitochondrial superoxide;
H2O2 is not freely permeable to either the plasma membrane or the mitochondrial membrane;
The concentrations of glucose-stimulated H2O2 in isolated islets are far below those that are acutely toxic.
Supplementary Material
ACKNOWLEDGEMENTS
The mitochondrial targeted catalase construct was kindly provided by Dr. Peter Rabinovitch. INS-1 832/13 cells were kindly provided by Dr. Christopher Newgard. We would like to thank Dr. Jean-Christophe Jonas for reading the manuscript and helpful comments.
FUNDING
This research was funded by grants from the American Diabetes Association/Gail Patrick Estate (Grant 1-15-IN-65), NIH (P30 DK-17047 (Cell Function Analysis Core), NCATS R41 TR001196), and the Washington State Life Sciences Discovery Fund (4553677).
ABBREVIATIONS USED:
- AO
acridine orange
- cyto-HyPer
pHyPer-cyto
- DCF
2', 7'-dichlorofluorescein
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- H2O2
hydrogen peroxide
- INS-1 cells
INS-1 813/32 cells
- KCN
potassium cyanide
- KRB
Krebs-Ringer bicarbonate
- mito-HyPer
pHyPer-dMito
- PI
propidium iodide
- ROS
reactive oxygen species
- SOD2
superoxide dismutase 2
REFERENCES
- 1.Prentki M and Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leahy JL (2005) Pathogenesis of type 2 diabetes mellitus. Arch Med Res 36, 197–209 [DOI] [PubMed] [Google Scholar]
- 3.Matschinsky FM (1996) Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45, 223–241 [DOI] [PubMed] [Google Scholar]
- 4.Ashcroft FM (2007) The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab 293, E880–889 [DOI] [PubMed] [Google Scholar]
- 5.Crutzen R, Shlyonsky V, Louchami K, Virreira M, Hupkens E, Boom A, Sener A, Malaisse WJ and Beauwens R (2012) Does NAD(P)H oxidase-derived H2O2 participate in hypotonicity-induced insulin release by activating VRAC in beta-cells? Pflugers Arch 463, 377–390 [DOI] [PubMed] [Google Scholar]
- 6.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE and Collins S (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56, 1783–1791 [DOI] [PubMed] [Google Scholar]
- 7.Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani AL, Ktorza A, Casteilla L and Penicaud L (2009) Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 58, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janjic D, Maechler P, Sekine N, Bartley C, Annen AS and Wolheim CB (1999) Free radical modulation of insulin release in INS-1 cells exposed to alloxan. Biochem Pharmacol 57, 639–648 [DOI] [PubMed] [Google Scholar]
- 9.Maechler P, Jornot L and Wollheim CB (1999) Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 274, 27905–27913 [DOI] [PubMed] [Google Scholar]
- 10.Syed I, Kyathanahalli CN and Kowluru A (2011) Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: role of protein prenylation. Am J Physiol Regul Integr Comp Physiol 300, R756–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, Oliveira-Emilio HC, Carpinelli AR and Curi R (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583, 9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drews G, Krippeit-Drews P and Dufer M (2010) Oxidative stress and beta-cell dysfunction. Pflugers Arch 460, 703–718 [DOI] [PubMed] [Google Scholar]
- 13.Robertson RP and Harmon JS (2006) Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med 41, 177–184 [DOI] [PubMed] [Google Scholar]
- 14.Padgett LE, Broniowska KA, Hansen PA, Corbett JA and Tse HM (2013) The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 1281, 16–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV and Wheeler MB (2010) Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 59, 448–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlsson M, Kurz T, Brunk UT, Nilsson SE and Frennesson CI (2010) What does the commonly used DCF test for oxidative stress really show? Biochem J 428, 183–190 [DOI] [PubMed] [Google Scholar]
- 17.Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV and Lukyanov S (2006) Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature methods 3, 281–286 [DOI] [PubMed] [Google Scholar]
- 18.Malinouski M, Zhou Y, Belousov VV, Hatfield DL and Gladyshev VN (2011) Hydrogen peroxide probes directed to different cellular compartments. PLoS One 6, e14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roma LP, Duprez J, Takahashi HK, Gilon P, Wiederkehr A and Jonas JC (2012) Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic beta-cells using ratiometric fluorescent proteins: confounding effects of pH with HyPer but not roGFP1. Biochem J 441, 971–978 [DOI] [PubMed] [Google Scholar]
- 20.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M and Newgard CB (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 [DOI] [PubMed] [Google Scholar]
- 21.Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J and Reems J (2004) Regulation of ATP/ADP in pancreatic islets. Diabetes 53, 401–409 [DOI] [PubMed] [Google Scholar]
- 22.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr., Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W and Tian R (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rountree AM, Reed BJ, Cummings BP, Jung SR, Stanhope KL, Graham JL, Griffen SC, Hull RL, Havel PJ and Sweet IR (2013) Loss of coupling between calcium influx, energy consumption and insulin secretion associated with development of hyperglycaemia in the UCD-T2DM rat model of type 2 diabetes. Diabetologia 56, 803–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweet IR, Cook DL, Wiseman RW, Greenbaum CJ, Lernmark A, Matsumoto S, Teague JC and Krohn KA (2002) Dynamic perifusion to maintain and assess isolated pancreatic islets. Diabetes Technol Ther 4, 67–76 [DOI] [PubMed] [Google Scholar]
- 25.Sweet IR, Khalil G, Wallen AR, Steedman M, Schenkman KA, Reems JA, Kahn SE and Callis JB (2002) Continuous measurement of oxygen consumption by pancreatic islets. Diabetes Technol Ther 4, 661–672 [DOI] [PubMed] [Google Scholar]
- 26.Niswender CM, Willis BS, Wallen A, Sweet IR, Jetton TL, Thompson BR, Wu C, Lange AJ and McKnight GS (2005) Cre recombinase-dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase A. Genesis 43, 109–119 [DOI] [PubMed] [Google Scholar]
- 27.Bank HL (1988) Rapid assessment of islet viability with acridine orange and propidium iodide. In vitro cellular & developmental biology : journal of the Tissue Culture Association 24, 266–273 [DOI] [PubMed] [Google Scholar]
- 28.Michalska M, Wolf G, Walther R and Newsholme P (2010) Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Biosci Rep 30, 445–453 [DOI] [PubMed] [Google Scholar]
- 29.Robertson RP, Harmon J, Tran PO, Tanaka Y and Takahashi H (2003) Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 52, 581–587 [DOI] [PubMed] [Google Scholar]
- 30.Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A and Weir GC (2001) Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem 276, 31099–31104 [DOI] [PubMed] [Google Scholar]
- 31.Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M and Araki E (2003) Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun 300, 216–222 [DOI] [PubMed] [Google Scholar]
- 32.Jezek P and Hlavata L (2005) Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol 37, 2478–2503 [DOI] [PubMed] [Google Scholar]
- 33.Matveyenko AV and Butler PC (2008) Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab 10 Suppl 4, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson RP (2004) Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 279, 42351–42354 [DOI] [PubMed] [Google Scholar]
- 35.Bolaffi JL, Heldt A, Lewis LD and Grodsky GM (1986) The third phase of in vitro insulin secretion. Evidence for glucose insensitivity. Diabetes 35, 370–373 [DOI] [PubMed] [Google Scholar]
- 36.Evans JL, Goldfine ID, Maddux BA and Grodsky GM (2003) Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 52, 1–8 [DOI] [PubMed] [Google Scholar]
- 37.Ihara Y, Toyokuni S, Uchida K, Odaka H, Tanaka T, Ikeda H, Hiai H, Seino Y and Yamada Y (1999) Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 48, 927–932 [DOI] [PubMed] [Google Scholar]
- 38.Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW and Philipson LH (2003) Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J Biol Chem 278, 9796–9801 [DOI] [PubMed] [Google Scholar]
- 39.Liang Y, Najafi H, Smith RM, Zimmerman EC, Magnuson MA, Tal M and Matschinsky FM (1992) Concordant glucose induction of glucokinase, glucose usage, and glucose-stimulated insulin release in pancreatic islets maintained in organ culture. Diabetes 41, 792–806 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.