

Abstract
Background and Purpose
Atopic dermatitis (AD) is a multifactorial skin condition with complex interactions of innate and adaptive immune responses. There are several existing therapies for AD, including topical glucocorticosteroids, emollients, phototherapies, calcineurin inhibitors and immunosuppressants, such as cyclosporine A. Although these therapies reduce inflammation, they also cause serious side effects. Therefore, it is necessary to develop new therapeutic approaches for AD treatment without side effects. There are several studies on natural materials or toxins, such as herbs, ginseng extract and snake venom, for AD treatment. However, treatment of AD with bee venom and its major component, melittin has rarely been studied.
Experimental Approach
Effects of bee venom and melittin were studied in a model of AD in vivo induced by 1‐chloro‐2,4‐dinitrobenzene (DNCB) in female Balb/c mice and in cultures of human keratinocytes, stimulated by TNF‐α/IFN‐γ. The potential pharmacological effects of bee venom and melittin on these in vivo and in vitro AD‐like skin disease models were studied.
Key Results
Bee venom and melittin exhibited potent anti‐atopic activities, shown by decreased AD‐like skin lesions, induced by DNCB in mice. In vitro studies using TNF‐α/IFN‐γ‐stimulated human keratinocytes showed that bee venom and melittin inhibited the increased expression of chemokines, such as CCL17 and CCL22, and pro‐inflammatory cytokines, including IL‐1β, IL‐6 and IFN‐γ, through the blockade of the NF‐κB and STAT signalling pathways.
Conclusions and Implications
Our results suggest that bee venom and melittin would be suitable for epicutaneous application, as topical administration is often appropriate for the treatment of AD.
Abbreviations
- AD
atopic dermatitis
- CCK
cell counting kit
- DNCB
1‐chloro‐2,4‐dinitrobenzene
- EMSA
electrophoretic mobility shift assay
- RBC
red blood cell
- STAT
signal transducer and activator of transcription
- Th cell
helper T cell
- TSLP
thymic stromal lymphopoietin
Introduction
Atopic dermatitis (AD), commonly known as atopic eczema, is a multifactorial skin disease, with complex interactions of innate and adaptive immune responses based on a genetic, pharmacological and psychological predisposition (Boguniewicz and Leung, 2011). AD, which is one of the most common chronic inflammatory skin diseases, is characterized by an impaired epidermal barrier, eczematous lesions, pruritus, dry skin, an abnormal immune response and an IgE‐mediated allergy to various exogenous antigens (Lee and Lee, 2014). The pathogenesis of AD involves a complex inflammatory process (Tan and Corren, 2011). Two competing hypotheses are generally presented regarding the pathogenesis of AD. The first proposes a primary immune functional disorder, resulting in IgE sensitization and a subsequent epithelial‐barrier obstruction (Czarnowicki et al., 2017). The second suggests that a disturbance of the skin barrier, either caused by an intrinsic genetic deficiency in epidermal skin barrier formation or as a result of an environmental transition, leads to atopic disease (Brandt and Sivaprasad, 2011). While debate around these theories remains, it is obvious that a number of genetic and environmental factors contribute to skin barrier dysfunction and immune dysregulation in AD (Blakely et al., 2016).
The first hypothesis of AD pathogenesis invokes an imbalance between helper T (Th) cells 1 and 2 by the presence of a specific IgE response in association with Th2 immune responses (Choi and Kim, 2013). In healthy individuals, balance exists among important subsets of helper T cells (e.g. Th1, Th2, Th17 and Th22). The pathogenesis of acute AD is related to Th2‐dominant inflammation, characterized by the dermal infiltration of Th cells, macrophages and eosinophils, increase in allergen‐specific IgE, mast cell activation and Th2 cytokine production (Mizutani et al., 2015). Th2 cells secrete the cytokines http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4996 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4997, that up‐regulate antibody formation via B cells, eosinophils, mast cells and other pathways (Kidd, 2003). IL‐4 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4980 have established roles in B cell differentiation and class switching, thus providing a link to characteristic elevations of serum IgE levels in AD, as well as eosinophil and mast cell infiltration into the skin (Chan et al., 2013). The pathogenesis of chronic AD is related to a predominance of Th1 cells and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4968, and chronic AD skin lesions undergo tissue remodelling caused by chronic inflammation (Coondoo, 2012). There are also increased numbers of mast cells but virtually no accumulation of neutrophils. Th1 cells secrete the cytokine IFN‐γ and activate inflammatory pathways mainly via macrophage activation (Kidd, 2003).
The second hypothesis, epidermal barrier dysfunction, involves dry, itchy skin, which is exacerbated by a mechanical injury inflicted by scratching. This enables the entry of antigens via the skin to result in the production of inflammatory cytokines (Oyoshi et al., 2009). A number of the barrier genes are localized on chromosome 1 in a cluster epidermal differentiation complex and include those for filaggrin, loricrin and involucrin. During the transition from stratum granulosum to stratum corneum, profilaggrin is cleaved into multiple filaggrin monomers that provide physical strength by aggregating the keratin bundles (Kalinin et al., 2002). Loss‐of‐function mutations in the filaggrin gene have been identified as the main genetic predisposing factor for AD and many other allergic diseases, including asthma (Irvine et al., 2011). According to the findings of the filaggrin‐deficient mice experiments, skin barrier function depends on the normal differentiation of keratinocytes (Kawasaki et al., 2012). Thus, keratinocytes, as the major group of epidermal cells, play an important role in the pathogenesis of inflammatory skin diseases, such as AD (Jung et al., 2012).
Many commercial formulations for the treatment of AD are available in gels, creams, lotions or ointments but show limited effectiveness (Shah et al., 2012). Furthermore, there are several modes of therapy for AD, such as topical glucocorticosteroids, emollients, phototherapies, calcineurin inhibitors and immunosuppressants, such as cyclosporine A (Misery, 2011; Berke et al., 2012). These therapies reduce inflammation, but they also cause a range of serious side effects. For example, topical steroid therapy is the most effective remedy for AD, but the continual repetitive application of steroids leads to side effects that include skin thinning and atrophy (Del Rosso and Friedlander, 2005). Therefore, the development of new therapeutic approaches without side effects is necessary for improving AD treatment. There are several studies on natural materials or toxins, such as herbs, ginseng extract and snake venom, for AD treatment (Sohn et al., 2011; Park et al., 2016). A recent study investigated the effects of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=275 from bee venom on AD‐like skin lesions in mice, induced by house dust mite extract (Jung et al., 2017). However, a beneficial effect of bee venom on AD has not been clearly recognized.
Purified bee venom is a natural toxin produced by honeybees (Apis mellifera) and has been extensively used as a traditional medicine for various diseases (Son et al., 2007). Bee venom contains various peptides, including melittin, apamin, adolapin and mast cell degranulating peptide along with enzymes, biological amines and non‐peptide components (Lariviere and Melzack, 1996). Melittin, the major component (50% of dry weight) of bee venom, is a small linear peptide composed of 26 amino acids. Several studies have examined the biological and pharmacological activities of bee venom and melittin, confirming that they possess radioprotective, anti‐inflammatory, antibacterial, antiviral and anti‐cancer activities (Raghuraman and Chattopadhyay, 2007; Lee and Bae, 2016). In addition, they have pain‐relief effects, anti‐rheumatoid arthritis effects and aid immune modulatory activity (An et al., 2016). A recent study investigated the effects of melittin on Propionibacterium acnes‐induced inflammatory responses in vitro and in vivo (Lee et al., 2014). However, the precise mechanism by which bee venom and melittin are thought to act in AD is not fully understood. Based on the various physiological activities of bee venom and melittin, they should be effective in AD. The excellent anti‐inflammatory effects of bee venom and melittin is expected to maximize their effects in an inflammatory skin condition such as AD.
Therefore, the present study was designed to determine the effect of bee venom and melittin in an AD‐like model. Consequently, this study investigated the potential pharmacological effects of bee venom and its major component, melittin, on two models of AD, that induced by 1‐chloro‐2,4‐dinitrobenzene (DNCB) in vivo and that induced by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074/IFN‐γ in human keratinocyte cultures.
Methods
Animals
All animal care and experimental procedures were carried out strictly in accordance with the criteria of the Institutional Animal Care and Use Committee of Catholic University of Daegu and were approved by the Institutional Animal Care and Use Committee of Catholic University of Daegu (Daegu, Korea; Approval number DCIAFCR‐151007‐9‐Y and DCIAFCR‐160428‐Y). The animals were treated humanely, and all efforts were made to minimize the animals' suffering and the numbers used. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Six‐week‐old female Balb/c mice (18–23 g; Samtoko, Gyeonggi‐do, Korea) were individually housed in polycarbonate animal cages and maintained under a constant temperature (22 ± 2°C) and humidity (55%). The mice had free access to water and food and were kept in 12 h light–dark cycles. After 1 week of acclimatization, the Balb/c mice were randomly divided into nine groups (n = 5 per group) as follows: (1) an untreated group (normal control, NC); (2) a DNCB (1‐chloro‐2,4‐dinitrobenzene; Sigma, St. Louis, MO, USA)‐sensitized and challenged group (DNCB); (3) a DNCB‐sensitized and challenged group with 0.1 g placebo treated group (Pla); (4–6) DNCB‐sensitized and challenged groups with 100, 200 and 500 μg of bee venom, mixed with placebo (the groups were named BV100, BV200 and BV500); (7–9) DNCB‐sensitized and challenged groups with 100, 200 and 500 μg of melittin, mixed with placebo (Mel100, Mel200 and Mel500). The doses of bee venom and melittin used here were based on previous studies (Lee et al., 2014). The bee venom, melittin or placebo were applied topically to the shaved dorsal skin.
Cell cultures
The human keratinocyte HaCaT cell line (Cell Lines Service GmbH, Eppelheim, Germany) was cultured in high glucose DMEM supplemented with 10% (v/v) FBS and antibiotics (100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin) at 37°C in a humidified 5% CO2 incubator. HaCaT (5.0 × 105 cells·mL−1) cells were seeded in the complete medium. Twenty‐four hours later, the medium was changed to serum‐free medium with bee venom (1, 10 and 100 ng·mL−1; Sigma) and melittin (0.1, 0.5 and 1 μg·mL−1; Enzo, Plymouth, PA, USA). After 30 min, the cells were stimulated with 10 ng·mL−1 TNF‐α/IFN‐γ for 9 h. PBS (pH 7.4), FBS, DMEM, penicillin and streptomycin were purchased from Gibco (Grand Island, NY, USA). Recombinant human TNF‐α and IFN‐γ were purchased from R&D Systems (Minneapolis, MN, USA).
Mouse model of AD
The Balb/c mice were anaesthetized by isoflurane inhalation (Ifran; HANA Pharm, Seoul, Korea) by using RC2 Rodent Circuit Controller (VETEQUIP, CA, USA). During anaesthesia, dorsal skin was shaved with an electric clipper and hair removal creams. Twenty‐four hours later, the mice were sensitized with 200 μL of 1% DNCB in an acetone : olive oil (3:1) solution applied topically to the shaved dorsal skin, twice over one week. Mice were then challenged with 200 μL of 0.4% DNCB similarly applied, twice a week for 5 weeks (Chan et al., 2013; Choi et al., 2013). Two weeks after the first induction of skin inflammation, bee venom or melittin (100, 200 and 500 μg mixed with a placebo) was applied to the dorsal skin five times per week for 4 weeks. The placebo used is a pharmaceutically produced gel with no biological activity, a mixture of carbomer, polyethylene glycol 400, trolamine, and purified water (Dongsung Bio Pharm., Seoul, Korea). The animal model of atopic dermatitis in this paper was based on the ‘Guideline for the efficacy of herbal medicinal herb medicine‐Atopic dermatitis’ of South Korea Ministry of Food and Drug Safety. The experimental protocol used in the present study is depicted schematically in Figure 1A. At the end of each treatment period, the mice were killed by CO2 asphyxiation and blood samples were collected by cardiac puncture. Blood samples were allowed to clot for 1 h at room temperature before centrifuging for 20 min at 10770× g. The serum thus obtained from the blood samples was stored at −70°C for further analysis. Immediately after blood collection, the dorsal skin was excised for further analysis. The experimenters were blinded to the treatments given to the animals and to the histological analyses and data analyses.
Figure 1.
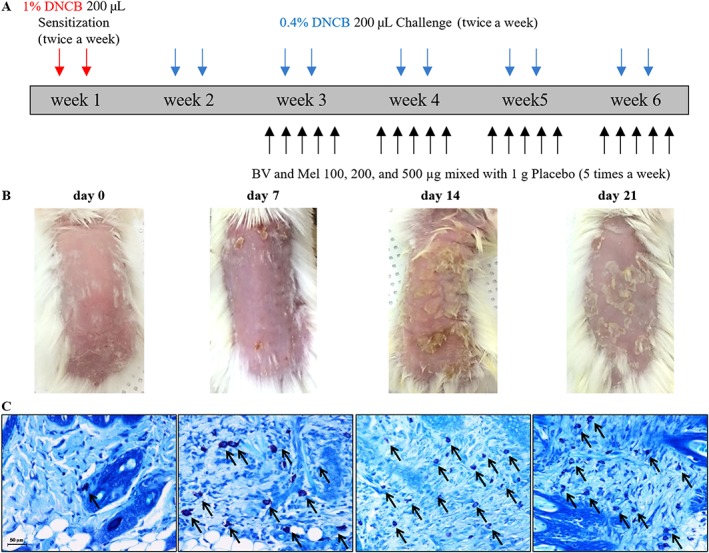
DNCB‐induced AD‐like symptoms in Balb/c mice. (A) Scheme of the experimental procedure. (B) Typical clinical features and (C) corresponding histopathological analysis of mouse dorsal skin are shown. The typical mast cell infiltrations are indicated by arrows. Scale bar = 50 μm.
Histological analysis
All skin tissue specimens were fixed in 10% formalin for 24 h at room temperature. After fixation, sections, cut perpendicular to the anterior–posterior axis of the dorsal skin, were dehydrated in graded ethanol, cleared in xylene and embedded in paraffin. The sections (4 μm) were mounted on glass slides, rehydrated to distilled water and stained with haematoxylin and eosin (H&E). As part of the histological assessment, all slides were examined under a slide scanner (3DHISTECH Panoramic MIDI, Budapest, Hungary). The thickness of the epidermis and dermis was analysed with iSolution DT software (IMT i‐Solution, Burnaby, BC, Canada).
Mast cell infiltration and degranulation
After mice were killed, the DNCB‐treated site (1 × 1 cm) was excised, fixed in formalin for 24 h and embedded in paraffin. Deparaffinized skin sections were stained with Giemsa for histological analysis. To measure the mast cell infiltration and degranulation, each slide was counted from all fields per section at 400× magnification. As part of the histological assessment, all slides were examined under a slide scanner. The granulation index per single mast cell was calculated by dividing the number of degranulated mast cells by the total number of mast cells.
ELISA
Levels of IgE (Bethyl Laboratories, Montgomery, TX, USA), IL‐4, IFN‐γ and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5083 R&D Systems) in mouse serum samples were measured using‐ELISA kits according to the manufacturer's instructions. The absorbance at 450 nm was measured with an ELISA reader (BMG Labtech, Ortenaukreis, Germany).
Preparation of splenocytes
The spleen was aseptically removed from each Balb/c mouse and a single‐cell suspension was prepared by forcing the spleen through a 71 μm stainless steel mesh. Red blood cells (RBCs) were lysed using RBC lysis buffer (Qiagen, Hilden, Germany), and the lysate was centrifuged at 1500× g for 10 min at 4°C. Splenocytes were suspended in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) medium supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin, and spleen cells (5.0 × 105 cells per well) were seeded in plates for each group and incubated for 72 h. Supernatants were harvested and measured for IgE using ELISA kits.
Preparation of total and nuclear protein from HaCaT cells
For total protein assays, cells were lysed in lysis reagent (Cell Lytic™ M, Sigma‐Aldrich, St. Louis, MO, USA), according to the instruction manual. Nuclear and cytosolic protein were collected using NE‐PER nuclear and cytoplasmic extraction reagent kits (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. The protein concentration of all samples was measured with the Bradford assay (Bio‐Rad Laboratories, Hercules, CA, USA), from the optical density at 595 nm, using a spectrophotometer. .
Western blot analysis
The protein samples were separated on precast gradient polyacrylamide gels (Bolt™ 4–12% Bis‐Tris Plus Gels, Thermo Fisher Scientific) and transferred to nitrocellulose membranes (GE Healthcare Life Science, Marlborough, MA, USA) by using Bolt Mini Blot Module and Mini Gel Tank (Thermo Fisher Scientific), according to the manufacturer's recommendations. The membrane was blocked in 5% BSA. The blocked membrane was probed with a primary antibody and HRP‐conjugated secondary antibody. Following a repeat of the wash step, the membrane was kept in enhanced chemiluminescence detection reagents (Thermo Fisher Scientific) for 1 min. Signal intensity was measured with an image analyser (ChemiDoc™ XRS+ system, Bio‐Rad Laboratories). The primary antibodies used were anti‐IL‐1β (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 (Abcam, Cambridge, MA, USA), anti‐IFN‐γ (Abcam), anti‐GAPDH, anti‐ JAK2, anti‐phospho‐JAK2, anti‐STAT 1 and 3, anti‐phospho‐STAT1 and 3, anti‐IKKα, anti‐phospho‐IKKα, anti‐IκBα, anti‐phospho‐IκBα, anti‐NF‐κB and anti‐phospho‐NF‐κB p65 were purchased from Cell Signaling Technology (Beverly, MA, USA). In addition, anti‐Lamin B from Invitrogen was used.
Quantitative real‐time PCR
Total mRNA was extracted from the dorsal skin and HaCaT cells by the TRIzol reagent (Thermo Fisher Scientific), according to the manufacturer's recommendations. Reverse transcription reaction was performed with AccuPower RT Premix and Oligo dT18 (Bioneer, Daejeon, Korea), according to manufacturer's instructions. Real‐time PCR was performed in a LightCycler nano System (Roche Applied Science, Mannheim, Germany) using a LightCycler DNA Master SYBR Green I (Roche Applied Science). PCR mixtures contained 100 ng of cDNA and 0.5 μM each of forward and reverse primers. The samples were denatured at 95°C for 10 min, followed by 45 cycles of annealing and extension at 95°C for 20 s, 60°C for 20 s and 72°C for 20 s. Expression values were normalized to GAPDH. Quantitative real‐time PCR products were further confirmed by melting curve analysis. The primer sequences were as follows: IFN‐γ, forward, 5′‐TGG CAT AGA TGT GGA AGA AAA GAG‐3′, and reverse, 5′‐TGC AGG ATT TTC ATG TCA CCA T‐3′; IL‐4, forward, 5′‐ACAGGAGAAGGGACGCCAT‐3′, and reverse, 5′‐GAAGCCCTACAGACGAGCTCA‐3′; http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=797, forward, 5′‐AGG GAC CAC ACA GAG AC‐3′, and reverse, 5′‐CTC GAG CTG CGT GGA TGT GC‐3′; http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=798, forward, 5′‐ATG GCT CGC CTA CAG ACT GCA CTC‐3′, and reverse, 5′‐CAC GGC AGC AGA CGC TGT CTT CCA‐3′; GAPDH, forward, 5′‐GGA GCC AAA AGG GTC ATC AT‐3′, and reverse, 5′‐GTG ATG GCA TGG ACT GTG GT‐3′.
Immunohistochemical staining
Paraffin‐embedded tissue sections (4 μm thick) were deparaffinized with xylene, dehydrated in gradually diminishing concentrations of ethanol and treated with 3% hydrogen peroxide in methanol for 10 min to block endogenous peroxidase activity. The tissue sections were immersed in 10 mM sodium citrate buffer (pH 6.0) for 5 min at 95°C. The last step was repeated using a 10 mM sodium citrate solution (pH 6.0). The sections stayed in the same solution while cooling for 20 min, and they were rinsed in PBS. The sections were then incubated with a primary antibody (1:100 dilution) for 1 h at 37°C. The primary antibodies were as follows: anti‐TSLP, anti‐CD3 and anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2995 (Abcam). The signal was visualized using an Envision System (DAKO, CA, USA) for 30 min at 37°C. 3,3′‐diaminobenzidine tetrahydrochloride (DAB) was used as the coloring reagent, and haematoxylin was used as the counter‐stain. The slides were examined with a slide scanner, Pannoramic MIDI, and analysed with iSolution DT software.
Immunofluorescence staining and confocal microscopy
The paraffin‐embedded skin tissue sections were deparaffinized with xylene and dehydrated in gradually decreasing concentrations of ethanol. The tissue sections were then placed in a blocking serum (5% BSA in PBS) at room temperature for 1 h. A primary antibody (1:500 dilution) was incubated at room temperature for 2 h, and a secondary antibody incubation (1:200 dilution) was performed at room temperature for 1 h. The antibodies included filaggrin (Abcam) and a goat anti‐rabbit secondary antibody conjugated with Alexa Fluor 488 (Thermo, MA, USA). Sections were then counterstained with Hoechst 33342. The slides were mounted using a VECTASHIELD Mounting Medium (VECTOR Laboratories, Burlingame, CA, USA). Stained slides were viewed under a confocal microscope system (Nikon A1 microscope equipped with a digital camera, Nikon, Tokyo, Japan).
HaCaT cells were seeded at a density of 2.0 × 105 cells per well in 2‐well chamber slide (Eppendorf, Hamburg, Germany). After 24 h, the medium was changed to a serum‐free medium containing the indicated concentration of bee venom (1, 10 and 100 ng·mL−1) or melittin (0.1, 0.5 and 1 μg·mL−1). After 2 h, the cells were co‐treated with 10 ng·mL−1 TNF‐α/IFN‐γ for 1 h. The treated cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. Fixed cells were treated with 0.1% Triton X‐100 in PBS for 2 min to permeabilize. Following permeabilization, the cells were blocked in PBS containing 5% BSA at room temperature for 1 h. After blocking, the cells were incubated with diluted primary antibody overnight at 4°C, and the reaction with secondary antibody was performed at room temperature for 2 h. The nuclei were stained with Hoechst 33342 solution for 10 min at 37°C. Slides were mounted using fluorescence mounting medium (DAKO). Specimens were examined and photographed using a confocal microscope system. The antibodies used are as follows: anti‐phospho‐STAT1, anti‐phospho‐STAT3, anti‐F‐actin and anti‐mouse‐biotinylated, and anti‐rabbit‐biotinylated secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 555.
Cell viability assays
The viability of HaCaT cells was assessed using cell counting kit (CCK)‐8 assays (Dojindo, Kumamoto, Japan). The HaCaT cells were seeded in 96‐well plate at 5.0 × 104 cells·mL−1 and allowed to attach for 24 h. The medium was replaced with serum‐free media. Cells were treated with serum‐free media containing different concentrations of bee venom (1, 10 and 100 ng·mL−1) and melittin (0.1, 0.5 and 1 μg·mL−1) for 9, 12 and 24 h. After experimental treatment, 10 μL of WST‐8 solution (2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium, monosodium salt) was added to each well, and the HaCaT cells were incubated for an additional 2–4 h at 37°C. The absorbance values were measured at 450 nm using a microplate reader.
Electrophoretic mobility shift assay (EMSA)
The Lightshift® Chemiluminescent EMSA Kit (Thermo) was used for the EMSA assay, according to the manufacturer's instructions. The image analyser (Chemidoc XRS+ system) was used to detect the chemiluminescence of biotin‐labelled DNA. The NF‐κB (5′‐AGT TGA GGG GAC TTT CCC AGG C‐3′) oligonucleotide probe was end‐labelled with DIG‐ddUTP.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). All data are presented as means ± SEM. Statistical significance was tested using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA). Group means were compared by one‐way ANOVA with Tukey's multiple comparison test. Tukey's tests were run only when F achieved P < 0.05 and there was no significant variance inhomogeneity. Differences with P < 0.05 were considered significant.
Materials
Bee venom was supplied by Chung Jin Biotech Co. (Ansan, Korea) and melittin by Enzo Life Sciences (Farmingdale, NY, USA)
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
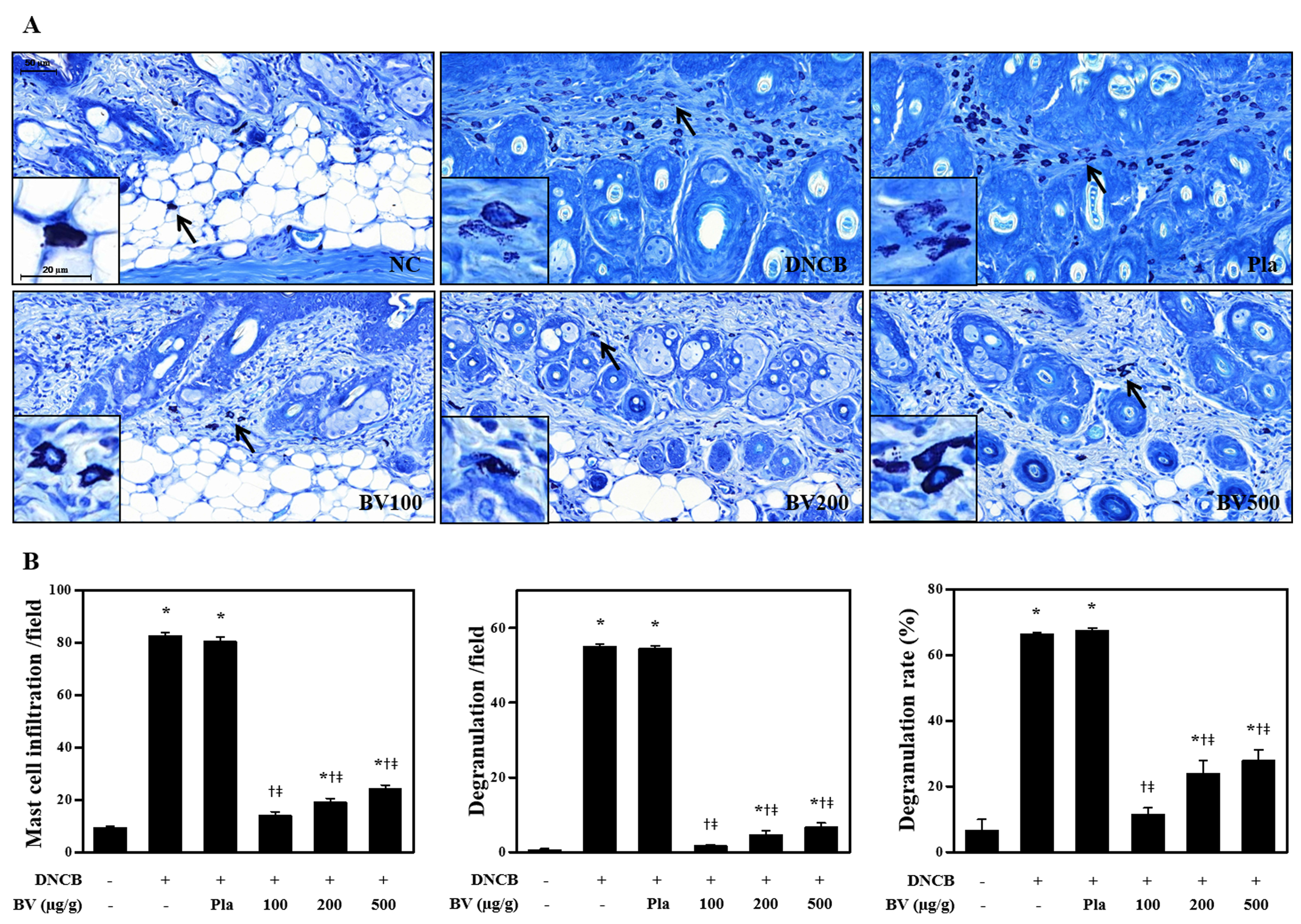
DNCB is capable of inducing AD‐like skin lesions in a mouse model
DNCB is known to induce AD‐like skin lesions. When compared with mice at day 0, corresponding to the induction period of skin inflammation, the DNCB‐treated mice showed significantly increased physical signs of AD, such as pruritus, oedema, erythema, scarring and excoriation, during a 3 week period (Figure 1B). Histopathological analysis confirmed that DNCB induced mast cell accumulation and degranulation (shown as arrows in the figure) in the samples of dorsal skin (Figure 1C).
Bee venom and melittin alleviated DNCB‐induced inflammation and mast cells in AD‐like skin lesions in an animal model
AD‐like skin lesions are characterized by epidermal and dermal thickening, the dermal infiltration of CD4+ T cells and the local expression of Th2 cytokines (Fröhlich et al., 2009). Therefore, we investigated the effects of bee venom (results shown in Supporting Information) and of melittin (results shown in Figures) on DNCB‐induced AD‐like symptoms and responses using H&E staining. Bee venom or melittin treatment significantly decreased the dorsal skin thickness, compared with that of the DNCB group (Figure 2B, Supporting Information Figure S1B). Histological analysis confirmed that bee venom and melittin inhibited pathological changes including the infiltration of inflammatory cells in skin lesions (Figure 2A, Supporting Information Figure S1A). Mast cells have been linked to the pathophysiology of allergic disorders, including AD (Galli and Tsai, 2012). According to the evaluation of mast cell infiltration and degranulation using Giemsa staining, the mast cell infiltration and degranulation in skin lesions was increased in the DNCB group, while the topical application of bee venom or melittin resulted in a decrease of infiltration and degranulation of mast cells (Figure 3, Supporting Information Figure S2).
Figure 2.
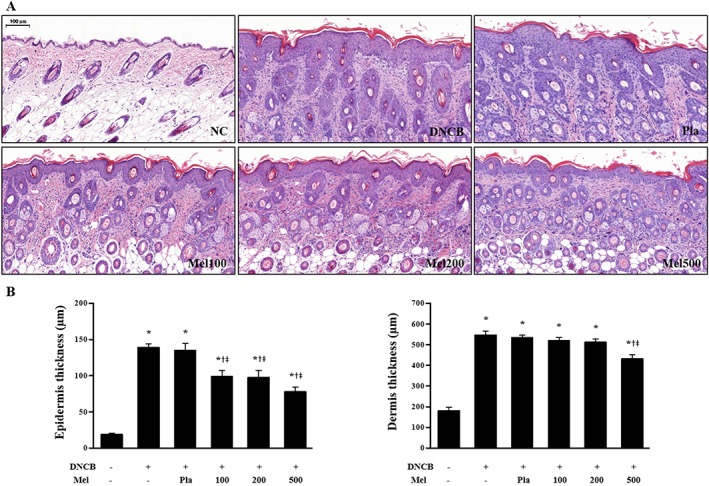
Effects of melittin on DNCB‐induced changes in (A) epidermal hyperplasia and a heavy dermal cell infiltration and (B) epidermal thickness and dermal thickness (n = 5). Scale bar = 100 μm. *P < 0.05 versus NC; + P < 0.05 versus DNCB; ǂ P < 0.05 versus Pla; NC, normal control; DNCB, DNCB‐sensitized and challenged; Pla, placebo; Mel100, melittin 100 μg; Mel200, melittin 200 μg; Mel500, melittin 500 μg.
Figure 3.
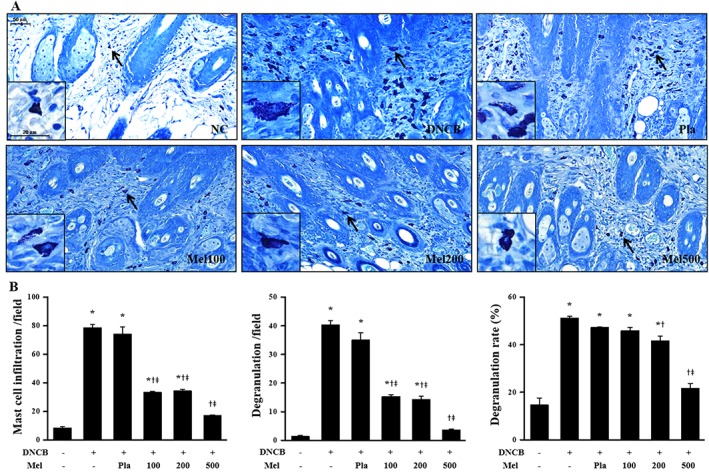
Effects of melittin on DNCB‐induced changes in mast cell infiltration and degranulation. The representative mast cells were indicated by arrows (A). (B) The number of mast cells and degranulated mast cells were counted from all fields per section (sections were taken from 5 mice). Scale bar = 50 μm. The enlarged frames scale bar = 20 μm. *P < 0.05 versus NC; + P < 0.05 versus DNCB; ǂ P < 0.05 versus Pla; NC, normal control; DNCB, DNCB‐sensitized and challenged; Pla, placebo; Mel100, melittin 100 μg; Mel200, melittin 200 μg; Mel500, melittin 500 μg.
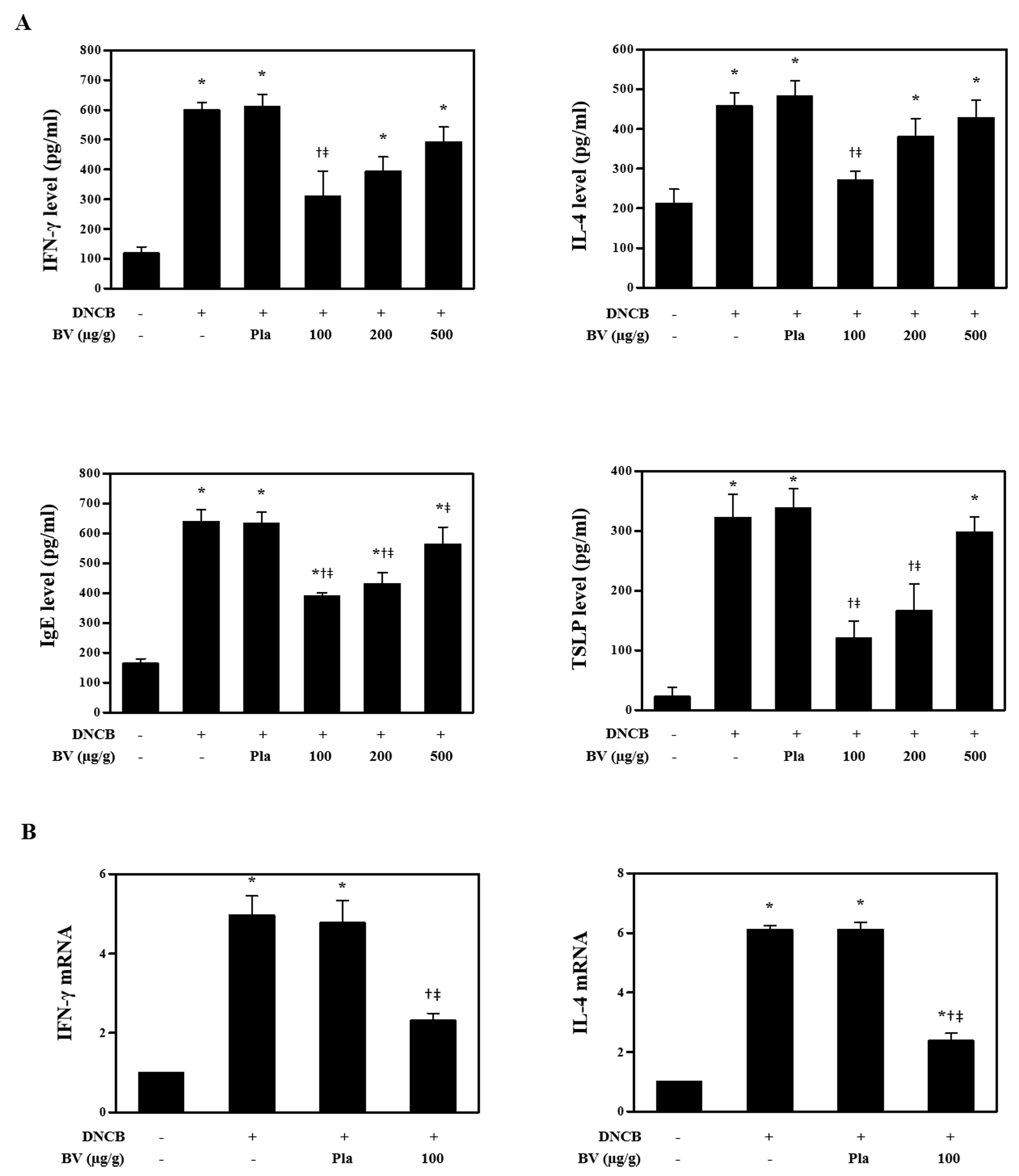
Effects of bee venom and melittin on serum levels of IFN‐γ, IL‐4, IgE and TSLP in mice
AD is characterized by two distinct phases, the chronic and acute phases, resulting in either Th1‐ or Th2‐dominant inflammation (Kim et al., 2015). Th1‐specific signature cytokines IFN‐γ and TNF‐α activate mononuclear phagocytes including macrophages, which are implicated in organ‐specific autoimmunity by the infection of intracellular pathogens, such as bacteria and protozoa (Guo et al., 2009). In contrast, IL‐4 is associated with the Th2 response. Therefore, this study investigated whether bee venom and melittin affected the Th1 or Th2 responses in an AD‐like model, in Balb/c mice. The levels of IFN‐γ, IL‐4, IgE and TSLP in the serum increased in response to DNCB treatment, relative to normal control mice. However, treatment with bee venom or melittin markedly decreased the serum IFN‐γ, IL‐4, IgE and TSLP levels in DCNB‐sensitized Balb/c mice (Figure 4A, Supporting Information Figure S3A). The effect of melittin on DNCB‐induced IgE change in splenocytes was further investigated. The concentration of IgE in the spleen tissue of the DNCB‐treated group mice was increased, but this increase was inhibited by melittin in both the 200 and 500 μg dose groups (Figure 4B). Next, we examined whether bee venom or melittin could regulate immune responses. Real‐time PCR was used to measure the gene expression of IFN‐γ and IL‐4. The mRNA expression levels of IFN‐γ and IL‐4 in the skin of the DNCB‐treated group mice were increased, but this increase was inhibited by bee venom or melittin in the 100 and 500 μg doses, respectively (Figure 4C, Supporting Information Figure S3B).
Figure 4.
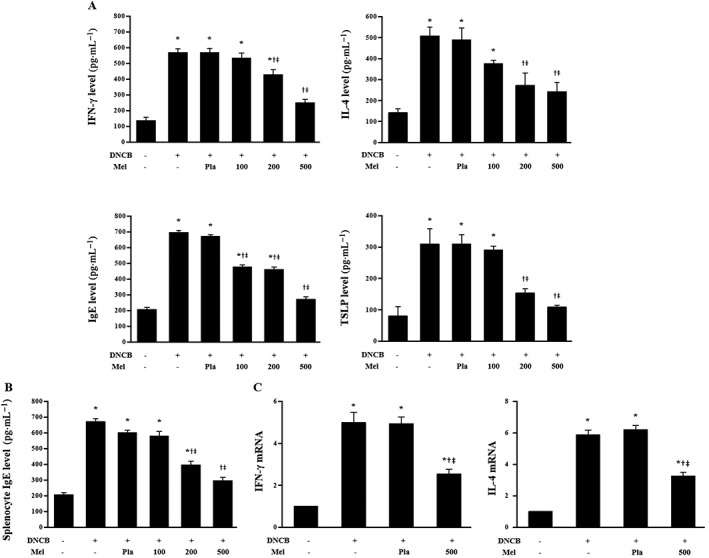
Effects of melittin on DNCB‐induced changes in the serum levels of IFN‐γ, IL‐4, IgE and TSLP, which were determined by ELISA kits (n = 5) (A). (B) Effects of melittin on DNCB‐induced IgE in splenocytes of Balb/c. The IgE level in cultured splenocytes was measured by ELISA (n = 5). (C) Real‐time PCR analyses show that melittin treatment inhibited the mRNA levels of IFN‐γ and IL‐4 (n = 5). *P < 0.05 versus NC; + P < 0.05 versus DNCB; ǂ P < 0.05 versus Pla; NC, normal control; DNCB, DNCB‐sensitized and challenged; Pla, placebo; Mel100, melittin 100 μg; Mel200, melittin 200 μg; Mel500, melittin 500 μg.
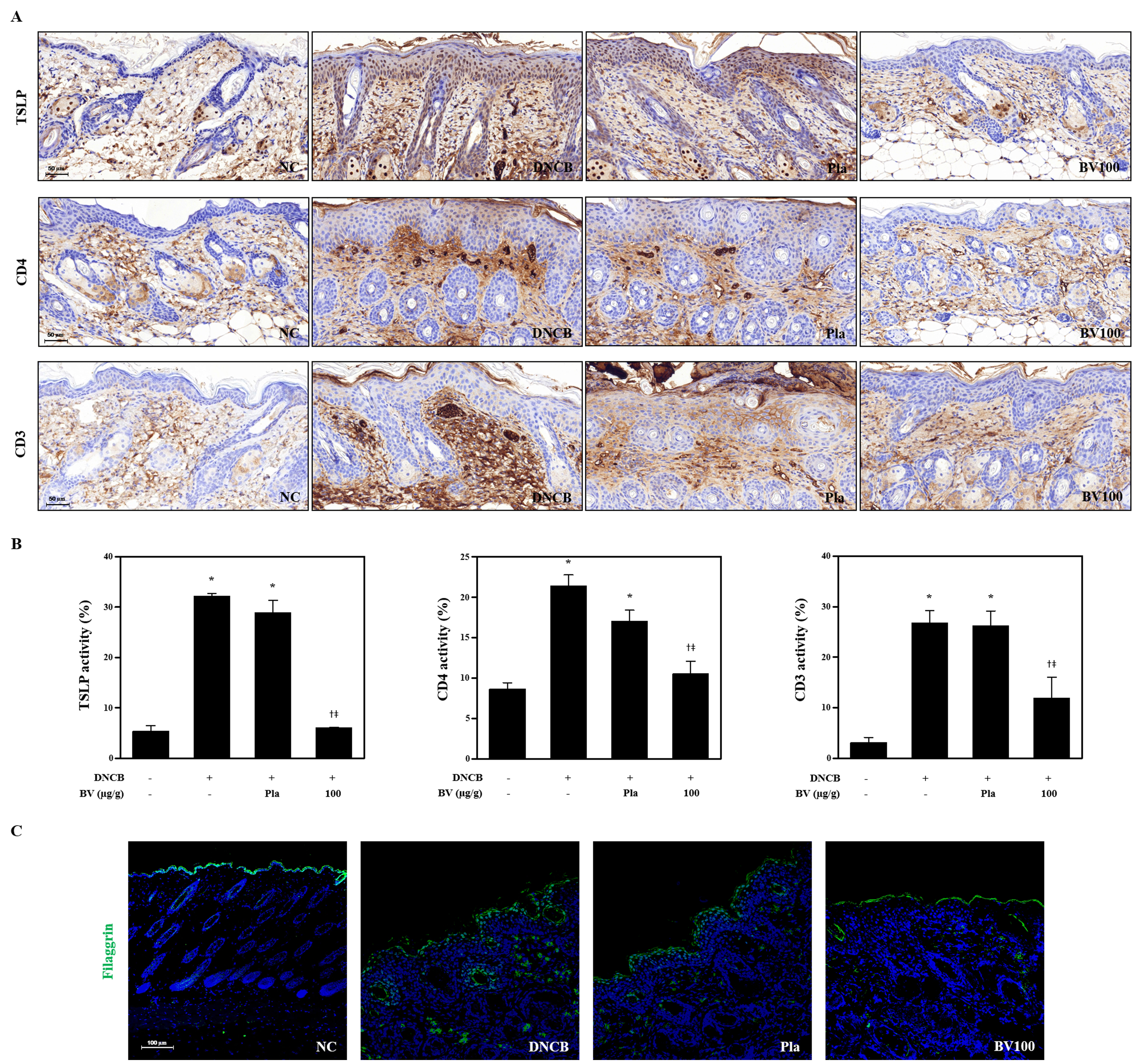
Effects of bee venom and melittin on TSLP expression, CD4+ and CD3+ T cells in DNCB‐sensitized mice
TSLP expression plays an important role in regulating Th2 cell‐mediated immunity and AD. In addition, environmental stimuli trigger keratinocytes for the production of TSLP with the subsequent activation of dendritic cells, epithelial cells and mast cells, which results in Th2‐type allergic immune responses (Lee et al., 2011). Treatment with bee venom or melittin significantly inhibited the DNCB‐induced expression of TSLP, suggesting that these agents have the potential to regulate Th2 cell‐mediated immunity in the epidermis. As TSLP expression appeared to be distinctly localized in the extracellular and cytoplasmic domains of the epidermal layer, they seem to be triggered by keratinocytes. The infiltration of CD4+ T cells and the up‐regulation of Th2 cytokines are typical features of AD lesions (Choi et al., 2013). We performed immunohistochemical and histological analysis to examine whether bee venom or melittin reduced the levels of CD4+ and CD3+ T cells, respectively, in the dorsal skin. The levels of CD4+ and CD3+ T cells in DNCB‐induced AD lesions in mice were higher than those in normal mice. Bee venom or melittin, however, decreased the levels of CD4+ and CD3+ (Figure 5A,B, Supporting Information Figure S4A,B) T cells in the dorsal skin.
Figure 5.
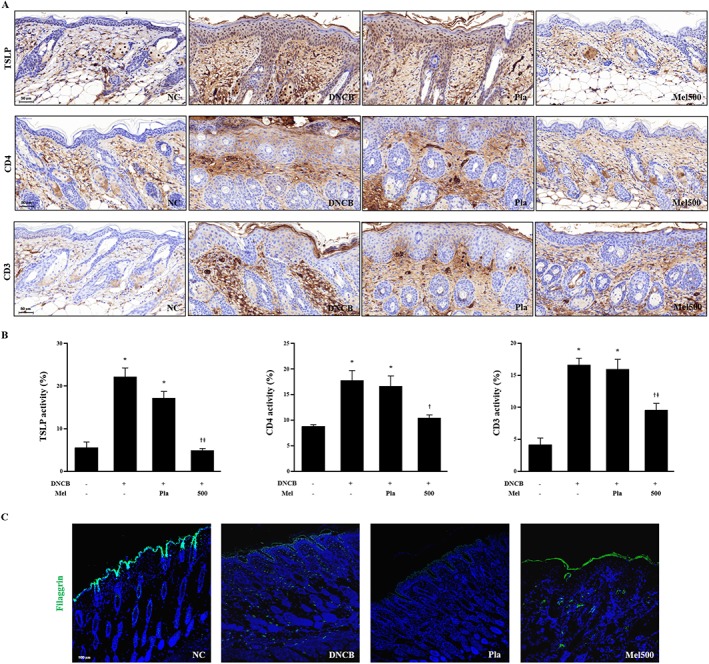
Effects of melittin on TSLP expression, CD4+ and CD3+ T cells in DNCB‐sensitized mice. (A) Histological images and (B) graphs indicating the relative percentage of TSLP expression and CD4+ and CD3+ immunopositive cells of the dorsal skin sections (n = 5). (C) Effects of melittin on abnormal epidermal differentiation. Immunofluorescence staining of sections of skin with antibody specific for filaggrin shows the expression of epidermal differentiation markers as labelled with FITC, green. Cells were counterstained with Hoechst 33342 (blue). Scale bar = 100 μm. *P < 0.05 versus NC; + P < 0.05 versus DNCB; ǂ P < 0.05 versus Pla; NC, normal control; DNCB, DNCB‐sensitized and challenged; Pla, placebo; Mel500, melittin 500 μg.
Bee venom and melittin improve abnormal epidermal differentiation
Filaggrin‐related barrier disruption can be a primary cause of AD (Elias and Schmuth, 2009). Th2 cytokines, such as IL‐4, IL‐13 and IL‐22, have been implicated in the down‐regulation of filaggrin expression in AD‐induced lesions (Jung et al., 2014). We investigated the therapeutic effects of bee venom or melittin on the abnormal epidermal differentiation. The apparent decrease in the filaggrin level shown as the expression of the epidermal differentiation marker using immunofluorescence staining after DNCB stimulation was reversed with bee venom or melittin treatment (Figure 5C, Supporting Information Figure S4C).
Effects of bee venom and melittin on pro‐inflammatory chemokines and cytokines in TNF‐α/IFN‐γ‐stimulated HaCaT cells
To investigate the cytotoxicity of bee venom and melittin, CCK‐8 assay was performed to analyse the cell viability of the HaCaT cell line. Cells were treated with bee venom or melittin at different concentrations and assessed at different time points. Keratinocytes were relatively stable with no significant change in viability up to 9 h. Cells treated with 100 ng·mL−1 bee venom or 1 μg·mL−1 melittin showed a significant decrease in viability by approximately 10–15% at 24 h compared to no treatment (Figure 6A, Supporting Information Figure S5A). These data suggest that both bee venom and melittin exhibited no cytotoxicity to HaCaT cells up to 9 h. Accordingly, 9 h cell culture was used in subsequent in vitro experiments. AD lesions are capable of producing CCL17 and CCL22 through the activated keratinocytes (Brandt and Sivaprasad, 2011). As such, a decrease in the production of these two chemokines in keratinocytes may be an effective target for AD treatment (Qi et al., 2009). Therefore, we investigated the effects of bee venom or melittin on the expression of TNF‐α/IFN‐γ‐induced pro‐inflammatory chemokines in HaCaT cells. TNF‐α/IFN‐γ treatment significantly induced the mRNA expression of CCL17 and CCL22, which was then inhibited by bee venom or melittin treatment (Figure 6C, Supporting Information Figure S5C). Consistent with these findings, TNF‐α/IFN‐γ treatment significantly provoked the release of IL‐1β, IL‐6 and IFN‐γ from HaCaT cells. However, bee venom or melittin treatment significantly inhibited the secretion of pro‐inflammatory cytokines, induced by TNF‐α/IFN‐γ (Figure 6B, Supporting Information Figure S5B). These results suggest that bee venom or melittin can modulate the expression of chemokines via the suppression of pro‐inflammatory cytokines in TNF‐α/IFN‐γ‐stimulated HaCaT cells.
Figure 6.
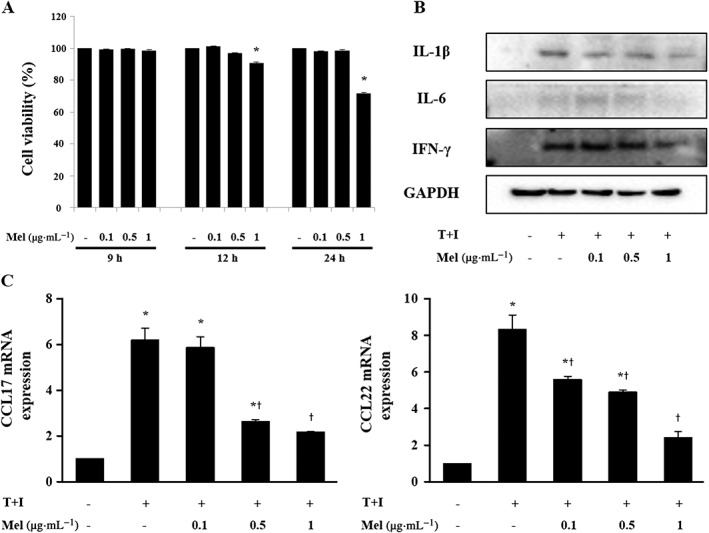
Cytotoxic effects of (A) melittin on HaCaT cells. Cell viability was determined by CCK‐8 assay (n = 5). Effects of melittin on the chemokine expressions of cytokines (B) and chemokines (n = 5) (C) in HaCaT cells that were stimulated by TNF‐α/IFN‐γ. T+I, TNF‐α/IFN‐γ‐stimulated; *P < 0.05 versus no treatment. + P < 0.05 versus TNF‐α/IFN‐γ alone.
Effects of bee venom and melittin on JAK/STAT signal pathways in TNF‐α/IFN‐γ‐activated HaCaT cells
The JAK/STAT pathway plays a critical role in the disruption of the epidermis barrier function and can be a therapeutic target in AD (Amano et al., 2015). In addition, cytokine‐mediated activation of STAT proteins plays an essential role in CD4+ T cell differentiation and the regulation of immune responses (O'Shea et al., 2011). We addressed the effects of bee venom and melittin on JAK/STAT signalling. HaCaT cells were incubated in the presence or absence of bee venom or melittin for 2 h and then stimulated with TNF‐α/IFN‐γ for 1 h. Cytosolic and nuclear fractions were evaluated for the expression of JAK2, STAT1 and STAT3 by Western blotting. The phosphorylation of JAK2 and nuclear translocation of p‐STAT1 and p‐STAT3 were markedly increased by TNF‐α/IFN‐γ treatment. However, treatment with bee venom or melittin decreased the levels of p‐JAK2 as well as the nuclear translocation of p‐STAT1 and p‐STAT3 (Figure 7A, Supporting Information Figure S6A). The suppression of the phosphorylation of JAK2, STAT1 and STAT3 following bee venom or melittin treatment was further confirmed by immunofluorescent staining. When cells were treated with bee venom (100 ng·mL−1) or melittin (1 μg·mL−1), STAT1 and STAT3 were predominantly localized in the cytoplasm, rather than in the nucleus, suggesting a role of STAT1 and STAT3 as transcriptional activators (Figure 7B, Supporting Information Figure S6B). The inhibition of these signalling pathways by bee venom and melittin could be responsible for the decrease in the production of pro‐inflammatory chemokines and cytokines.
Figure 7.
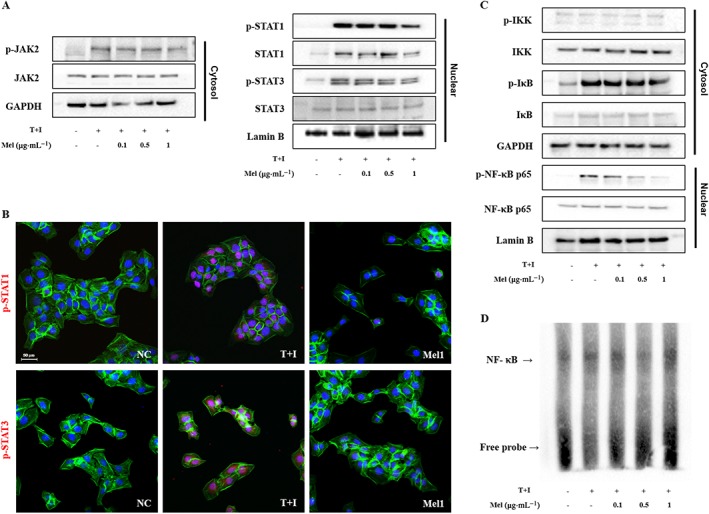
Effects of melittin on activation of the (A) JAK2, STAT1 and STAT3 signalling pathways in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (B) Immunofluorescence staining for p‐STAT1 and p‐STAT3 (labelled with Alexa Fluor 555, red), and F‐actin (labelled with Alexa Fluor 488, green). Cells were counterstained with Hoechst 33342 (blue). Effects of melittin on activation of the (C) NF‐κB signalling pathway in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (D) NF‐κB DNA binding activity in the nuclear extract was measured by EMSA. Representative images from each group. Scale bar = 50 μm. NC, normal control; T+I, TNF‐α/IFN‐γ‐stimulated; Mel1, TNF‐α/IFN‐γ‐stimulated +1 μg·mL−1 Mel.
Effects of bee venom and melittin on NF‐κB pathways in TNF‐α/IFN‐γ‐stimulated HaCaT cells
TNF‐α/IFN‐γ induces the production of chemokines and cytokines in keratinocytes by activating NF‐κB (Ju et al., 2009). The NF‐κB signalling pathway contributes to the production of CCL17 and CCL22 in TNF‐α/IFN‐γ‐stimulated HaCaT cells (Qi et al., 2012). Accordingly, we focused our analysis on IKK, IκB and NF‐κB in TNF‐α/IFN‐γ‐stimulated HaCaT cells, as shown in Figure 7C, Supporting Information Figure S6C, TNF‐α/IFN‐γ treatment significantly increased the phosphorylation of cytosolic IκB, and NF‐κB and nuclear p‐NF‐κB. However, bee venom or melittin treatment decreased the phosphorylation levels of IκB, and NF‐κB in a dose‐dependent manner. To further evaluate the effects of bee venom and melittin on NF‐κB DNA‐binding activity in HaCaT cells, nuclear extracts were analysed by EMSA (Figure 7D, Supporting Information Figure S6D). TNF‐α/IFN‐γ increased the NF‐κB DNA‐binding activity in HaCaT cells. In contrast, this increased binding activity was markedly reduced after treatment with bee venom and melittin. These results suggest that the inhibition, by bee venom and melittin, of the production of chemokines CCL17 and CCL22 induced by TNF‐α/IFN‐γ, was mediated by the suppression of both NF‐κB and STAT1 and STAT3 activation.
Discussion
AD is the most common allergic inflammatory skin disease (Sugiura et al., 2003). Interactions of genetic, environmental and immunological factors result in the initiation and progress of AD (Novak and Bieber, 2005). Although many possible mechanisms of AD have been researched and proposed, its aetiology remains unclear (Lee et al., 2010). In addition, despite the many studies on biological and pharmacological activities of bee venom and melittin, the therapeutic effects of these two substances against AD‐related mechanisms remain unknown. We found that bee venom and melittin possessed potent anti‐atopic activities, as shown by improvements in the DNCB‐induced AD‐like skin lesions in a mouse model. In vitro studies using TNF‐α/IFN‐γ‐stimulated human keratinocytes revealed that bee venom and melittin inhibited the increased expression of two chemokines, CCL17 and CCL22, and pro‐inflammatory cytokines, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974, IL‐6 and IFN‐γ, through the blockade of the NF‐κB and STAT signalling pathways. As a result, this study demonstrated that bee venom and melittin have immunomodulatory activity, and such activity was associated with the regulation of Th cell differentiation, thereby ameliorating the inflammatory skin lesions caused by AD.
The Th cells are important components of the regulation of the immune response to intracellular and extracellular pathogens, through activating macrophages and cytotoxic T cells, releasing T‐cell cytokines, and B‐cell antibody class switching (Hahn and Erb, 1999). Mature Th cells are also known as CD4+ T cells because they express the CD4 glycoprotein on their surface (Finlayson, 2012). These CD4+ T cells play a key role in eliciting and maintaining AD inflammation. The differentiation of CD4+ T cells leads to the generation of Th1 cells that secrete IFN‐γ, Th2 cells that secrete IL‐4, IL‐5 and IL‐13, or Th17 cells that secrete http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4982 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4988. Therefore, modulating T cell‐elicited immune responses is a promising therapeutic approach for AD (Biedermann et al., 2015). In this study, we observed that DNCB induced the expression of IFN‐γ and IL‐4 via the activation of CD4+ T cells. However, bee venom and melittin significantly suppressed both IFN‐γ and IL‐4 production by CD4+ T cells.
Numerous studies have suggested that the cytokine TSLP acts as a master switch that triggers both the initiation and maintenance of AD (Ziegler et al., 2013). Apart from this, TSLP can act directly on naive CD4+ T cells to promote cell proliferation and Th2 differentiation (Yao et al., 2013). TSLP is expressed in the epidermis after mechanical stimulation and plays a principal role in the activation of Th2 cytokine‐secreting invariant natural killer cells and serum IgE, which are increased in AD skin (Wu et al., 2010). IgE production is associated with a predominant Th2 cellular response. Our study showed that TSLP and IgE were increased in DNCB‐induced AD‐like mice, and bee venom and melittin decreased TSLP and IgE production. The inhibitory effects of bee venom and melittin on the expression of TSLP were confirmed by immunohistochemical staining. In addition, several types of immune cells are activated by TSLP, including NK cells, dendritic cells, T cells, B cells, basophils, eosinophils and mast cells, which promote allergic inflammation (Ziegler et al., 2013). In allergic inflammation, IgE is synthesized by B cells and induces the activation of mast cells, leading to the secretion of Th2 cytokines and histamine (Kim et al., 2012). In this study, we could observe the infiltration and degranulation of mast cells in DNCB‐induced AD‐like skin lesions. The topical application of bee venom and melittin to mice diminished DNCB‐induced mast cell infiltration and degranulation.
As Th1 and Th2 cell differentiation and their functions as well as keratinocyte activation are critical to AD development, an ideal therapeutic approach to AD may be treatment with inhibitors that modulate T cell and keratinocyte activation and thus ameliorate AD symptoms. Keratinocytes play a critical role in atopic skin inflammation (Giustizieri et al., 2001). Many studies have shown that the chemokines CCL17 and CCL22 play critical roles in the migration of lymphocytes to the skin. Therefore, a decrease in the release of these chemokines in keratinocytes may be an effective target for the treatment of inflammatory skin diseases (Hatano et al., 2013). In this study, bee venom and melittin suppressed TNF‐α/IFN‐γ‐induced expression of CCL17, CCL22, IL‐1β and IL‐6 in HaCaT cells. The modulation of AD‐associated chemokines and cytokines may provide therapeutic efficacy.
In addition, NF‐κB is a crucial factor in the immune‐inflammatory responses involved in various skin diseases, including AD (Andrés et al., 2013). The activation of the NF‐κB pathway leads to the transcription of numerous genes, including cytokines, chemokines and growth factors that are involved in the initiation of the inflammatory response (Perera et al., 2012). The molecular mechanisms of bee venom and melittin on AD‐related NF‐κB and STAT pathways have not yet been fully delineated. Our findings show that bee venom and melittin inhibited the TNF‐α/IFN‐γ‐stimulated phosphorylation of NF‐κB. In fact, the down‐regulation of this transcriptional factor may be the cause of the decrease of the cytokines measured in skin samples, as their transcription is dependent on NF‐κB activation. The role of JAK/STAT signalling in AD was further evaluated in the present study.
JAK/STAT signalling pathways are key transcriptional mechanisms that are essential for a variety of cell functions, and they are activated by several cytokines (Amano et al., 2015). Cytokine‐mediated activation of STAT proteins plays an essential role in CD4+ T cell differentiation and the regulation of immune responses (O'Shea et al., 2011). For example, STAT1 is induced by IFN‐γ, IL‐6 and EGF, whereas STAT2 is induced by IFN‐α, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2994&familyId=990&familyType=OTHER by EGF and IL‐6, and STAT4 by IL‐12 (Shi et al., 2011). STAT3 is a major player in cutaneous inflammatory diseases, as well as in normal keratinocyte function. The increased phosphorylation of STAT3 has been observed in skin lesions where pro‐inflammatory cytokines are involved (Andrés et al., 2013). Both bee venom and melittin prevented TNF‐α/IFN‐γ‐stimulated phosphorylation of JAK2 and the nuclear translocation of p‐STAT1 and p‐STAT3. NF‐κB and JAK/STAT signalling pathways are involved in the regulation of CCL17 and CCL22 release in HaCaT cells. Thus, our results suggest that suppressed activation of NF‐κB and JAK/STAT signalling pathways by bee venom and melittin might decrease the production of chemokines and cytokines in keratinocytes and thus mitigate AD.
In summary, this study demonstrated the ameliorating effects of bee venom and melittin on the DNCB‐induced AD‐like skin lesions of Balb/c mice and TNF‐α/IFN‐γ‐stimulated human keratinocyte HaCaT cells. Both bee venom and melittin reduced inflammatory symptoms in AD‐like skin lesions by suppressing the production of Th1/Th2‐associated and pro‐inflammatory cytokines with a subsequent decrease of CD4+ T cells, mast cell infiltration and the release of serum IgE. In addition, bee venom and melittin improved the abnormal epidermal differentiation by restoring filaggrin expression. Bee venom and melittin prevented the production of chemokines, most likely through the inhibition of NF‐κB, STAT1 and STAT3 transcriptional activity in TNF‐α/IFN‐γ‐stimulated keratinocytes. In conclusion, these results suggest that bee venom and melittin would be suitable for epicutaneous application, as topical administration is often applicable for the treatment of AD.
Author contributions
H.‐J.A. and K.‐K.P. designed the study and prepared the manuscript. H.‐J.A., J.‐Y.K., W.‐H.K., M.‐G.G., H.M.G., M.J.J., S.‐M.H. and I.S.P. performed the overall experiments and analysed the data. S.C.P. and C.‐K.L. discussed the study. All authors have read and approved the final version of this manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of BV on DNCB‐induced changes in (A) epidermal hyperplasia and a heavy dermal cell infiltration and (B) epidermal thickness and dermal thickness (n = 5). Scale bar = 100 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S2 Effects of BV on DNCB‐induced changes in mast cell infiltration and degranulation. The representative mast cells were indicated by arrows (A). (B) The number of mast cells and degranulated mast cells were counted from all fields per section (n = 5). Scale bar = 50 μm. The enlarged frames scale bar = 20 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S3 Effects of BV on DNCB‐induced changes in the serum levels of IFN‐γ, IL‐4, IgE, and TSLP, which were determined by ELISA kits (n = 5) (A). (B) Real‐time PCR analyses show that BV treatment inhibited the mRNA levels of IFN‐γ and IL‐4 (n = 5). *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S4 Effects of BV on TSLP expression, CD4+ and CD3+ T cells in DNCB‐sensitized mice. (A) Histological images and (B) graphs indicating the relative percentage of TSLP expression and CD4+ and CD 3+ immunopositive cells of the dorsal skin sections (n = 5). (C) Effects of BV on abnormal epidermal differentiation. Immunofluorescence staining of sections of skin with antibody specific for filaggrin shows the expression of epidermal differentiation markers as labelled with FITC, green. Cells were counterstained with Hoechst 33342 (blue). Scale bar = 100 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg.
Figure S5 Cytotoxic effects of (A) BV on HaCaT cells. Cell viability was determined by CCK‐8 assay (n = 5). Effects of BV on the chemokine expressions of cytokines (B) and chemokines (n = 5) (C) in HaCaT cells that were stimulated by TNF‐α/IFN‐γ. T + I: TNF‐α/IFN‐γ‐stimulated; *P < 0.05 vs. no treatment. + P < 0.05 vs. TNF‐α/IFN‐γ alone.
Figure S6 Effects of BV on activation of the (A) JAK2, STAT1, and STAT3 signalling pathway in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (B) Immunofluorescence staining for p‐STAT1 and p‐STAT3 (labelled with Alexa Fluor 555, red), and F‐actin (labelled with Alexa Fluor 488, green). Cells were counterstained with Hoechst 33342 (blue). Effects of BV on activation of the (C) NF‐κB signalling pathway in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (D) NF‐κB DNA binding activity in the nuclear extract was measured by EMSA. Representative images from each group. Scale bar = 50 μm. NC: normal control; T + I: TNF‐α/IFN‐γ‐stimulated; Mel1: TNF‐α/IFN‐γ‐stimulated +100 ng/mL BV.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgement
This work was carried out with the support of ‘Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01316601)’ Rural Development Administration, Republic of Korea.
An, H.‐J. , Kim, J.‐Y. , Kim, W.‐H. , Gwon, M.‐G. , Gu, H. M. , Jeon, M. J. , Han, S.‐M. , Pak, S. C. , Lee, C.‐K. , Park, I. S. , and Park, K.‐K. (2018) Therapeutic effects of bee venom and its major component, melittin, on atopic dermatitis in vivo and in vitro . British Journal of Pharmacology, 175: 4310–4324. 10.1111/bph.14487.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G et al (2015). The Janus kinase inhibitor JTE‐052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol 136: 667–677.e7. [DOI] [PubMed] [Google Scholar]
- An HJ, Kim JY, Kim WH, Han SM, Park KK (2016). The protective effect of melittin on renal fibrosis in an animal model of unilateral ureteral obstruction. Molecules 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés RM, Montesinos MC, Navalón P, Payá M, Terencio MC (2013). NF‐κB and STAT3 inhibition as a therapeutic strategy in psoriasis: in vitro and in vivo effects of BTH. J Invest Dermatol 133: 2362–2371. [DOI] [PubMed] [Google Scholar]
- Berke R, Singh A, Guralnick M (2012). Atopic dermatitis: an overview. Am Fam Physician 86: 35–42. [PubMed] [Google Scholar]
- Biedermann T, Skabytska Y, Kaesler S, Volz T (2015). Regulation of T cell immunity in atopic dermatitis by microbes: the Yin and Yang of cutaneous inflammation. Front Immunol 6: 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely K, Gooderham M, Papp K (2016). Dupilumab, a monoclonal antibody for atopic dermatitis: a review of current literature. Skin Therapy Lett 21: 1–5. [PubMed] [Google Scholar]
- Boguniewicz M, Leung DY (2011). Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev 242: 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt EB, Sivaprasad U (2011). Th2 cytokines and atopic dermatitis. J Clin Cell Immunol 2: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Liou CJ, Xu PY, Shen JJ, Kuo ML, Len WB et al (2013). Effect of dehydroepiandrosterone on atopic dermatitis‐like skin lesions induced by 1‐chloro‐2,4‐dinitrobenzene in mouse. J Dermatol Sci 72: 149–157. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim SH (2013). Rutin suppresses atopic dermatitis and allergic contact dermatitis. Exp Biol Med (Maywood) 238: 410–417. [DOI] [PubMed] [Google Scholar]
- Choi JK, Oh HM, Lee S, Park JW, Khang D, Lee SW et al (2013). Oleanolic acid acetate inhibits atopic dermatitis and allergic contact dermatitis in a murine model. Toxicol Appl Pharmacol 269: 72–80. [DOI] [PubMed] [Google Scholar]
- Coondoo A (2012). The role of cytokines in the pathomechanism of cutaneous disorders. Indian J Dermatol 57: 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnowicki T, Krueger JG, Guttman‐Yassky E (2017). Novel concepts of prevention and treatment of atopic dermatitis through barrier and immune manipulations with implications for the atopic march. J Allergy Clin Immunol 139: 1723–1734. [DOI] [PubMed] [Google Scholar]
- Del Rosso J, Friedlander SF (2005). Corticosteroids: options in the era of steroid‐sparing therapy. J Am Acad Dermatol 53: S50–S58. [DOI] [PubMed] [Google Scholar]
- Elias PM, Schmuth M (2009). Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol 9: 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson M (2012). Multiple Sclerosis Rehabilitation: From Impairment To Participation. Crc Press. [Google Scholar]
- FröHlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J et al (2009). IL‐21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324: 1576–1580. [DOI] [PubMed] [Google Scholar]
- Galli SJ, Tsai M (2012). IgE and mast cells in allergic disease. Nat Med 18: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustizieri ML, Mascia F, Frezzolini A, De Pit O, Chinni LM, Giannetti A et al (2001). Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell–derived cytokines. J Allergy Clin Immunol 107: 871–877. [DOI] [PubMed] [Google Scholar]
- Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K et al (2009). IL‐1 family members and stat activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A 106: 13463–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, Erb P (1999). The immunomodulatory role of CD4‐positive cytotoxic T‐lymphocytes in health and disease. Int Rev Immunol 18: 449–464. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano Y, Adachi Y, Elias PM, Crumrine D, Sakai T, Kurahashi R et al (2013). The Th2 cytokine, interleukin‐4, abrogates the cohesion of normal stratum corneum in mice: implications for pathogenesis of atopic dermatitis. Exp Dermatol 22: 30–35. [DOI] [PubMed] [Google Scholar]
- Irvine AD, Mclean WH, Leung DY (2011). Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 365: 1315–1327. [DOI] [PubMed] [Google Scholar]
- Ju SM, Song HY, Lee SJ, Seo WY, Sin DH, Goh AR et al (2009). Suppression of thymus‐ and activation‐regulated chemokine (TARC/CCL17) production by 1,2,3,4,6‐penta‐O‐galloyl‐β‐d‐glucose via blockade of NF‐κB and STAT1 activation in the HaCat cells. Biochem Biophys Res Commun 387: 115–120. [DOI] [PubMed] [Google Scholar]
- Jung KH, Baek H, Kang M, Kim N, Lee SY, Bae H (2017). Bee venom phospholipase A2 ameliorates house dust mite extract induced atopic dermatitis like skin lesions in mice. Toxins (Basel) 9: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Choi J, Lee SA, Kim H, Hwang J, Choi EH (2014). Pyrrolidone carboxylic acid levels or caspase‐14 expression in the corneocytes of lesional skin correlates with clinical severity, skin barrier function and lesional inflammation in atopic dermatitis. J Dermatol Sci 76: 231–239. [DOI] [PubMed] [Google Scholar]
- Jung MR, Lee TH, Bang MH, Kim H, Son Y, Chung DK et al (2012). Suppression of thymus‐ and activation‐regulated chemokine (TARC/CCL17) production by 3‐O‐β‐d‐glucopyanosylspinasterol via blocking NF‐κB and STAT1 signaling pathways in TNF‐α and IFN‐γ‐induced HaCat keratinocytes. Biochem Biophys Res Commun 427: 236–241. [DOI] [PubMed] [Google Scholar]
- Kalinin AE, Kajava AV, Steinert PM (2002). Epithelial barrier function: assembly and structural features of the cornified cell envelope. Bioessays 24: 789–800. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H et al (2012). Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin‐null mice. J Allergy Clin Immunol 129: 1538–46 E6. [DOI] [PubMed] [Google Scholar]
- Kidd P (2003). Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev 8: 223–246. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GD, Park YS, Ahn HJ, Cho JJ, Park CS (2015). Aspartame attenuates 2, 4‐dinitrofluorobenzene‐induced atopic dermatitis‐like clinical symptoms in NC/Nga mice. J Invest Dermatol 135: 2705–2713. [DOI] [PubMed] [Google Scholar]
- Kim TH, Kim GD, Jin YH, Park YS, Park CS (2012). Omega‐3 fatty acid‐derived mediator, resolvin E1, ameliorates 2,4‐dinitrofluorobenzene‐induced atopic dermatitis in NC/Nga mice. Int Immunopharmacol 14: 384–391. [DOI] [PubMed] [Google Scholar]
- Lariviere WR, Melzack R (1996). The bee venom test: a new tonic‐pain test. Pain 66: 271–277. [DOI] [PubMed] [Google Scholar]
- Lee G, Bae H (2016). Bee venom phospholipase A2: yesterday's enemy becomes today's friend. Toxins (Basel) 8: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Lee SH (2014). Epidermal permeability barrier defects and barrier repair therapy in atopic dermatitis. Allergy Asthma Immunol Res 6: 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K‐S, Jeong E‐S, Heo S‐H, Seo J‐H, Jeong D‐G, Choi Y‐K (2010). A novel model for human atopic dermatitis: application of repeated DNCB patch in Balb/C mice, in comparison with NC/Nga mice. Lab Anim Res 26: 95–102. [Google Scholar]
- Lee KH, Cho KA, Kim JY, Baek JH, Woo SY, Kim JW (2011). Filaggrin knockdown and Toll‐like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp Dermatol 20: 149–151. [DOI] [PubMed] [Google Scholar]
- Lee WR, Kim KH, An HJ, Kim JY, Chang YC, Chung H et al (2014). The protective effects of melittin on propionibacterium acnes‐induced inflammatory responses in vitro and in vivo. J Invest Dermatol 134: 1922–1930. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misery L (2011). Therapeutic perspectives in atopic dermatitis. Clin Rev Allergy Immunol 41: 267–271. [DOI] [PubMed] [Google Scholar]
- Mizutani N, Sae‐Wong C, Kangsanant S, Nabe T, Yoshino S (2015). Thymic stromal lymphopoietin‐induced interleukin‐17A is involved in the development of IgE‐mediated atopic dermatitis‐like skin lesions in mice. Immunology 146: 568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak N, Bieber T (2005). The role of dendritic cell subtypes in the pathophysiology of atopic dermatitis. J Am Acad Dermatol 53: S171–S176. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y (2011). Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol 11: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyoshi MK, He R, Kumar L, Yoon J, Geha RS (2009). Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol 102: 135–226. [DOI] [PubMed] [Google Scholar]
- Park KD, Pak SC, Park KK (2016). The pathogenetic effect of natural and bacterial toxins on atopic dermatitis. Toxins (Basel) 9: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera GK, Di Meglio P, Nestle FO (2012). Psoriasis. Annu Rev Pathol 7: 385–422. [DOI] [PubMed] [Google Scholar]
- Qi XF, Kim DH, Yoon YS, Li JH, Song SB, Jin D et al (2009). The adenylyl cyclase‐cAMP system suppresses TARC/CCL17 and MDC/CCL22 production through p38 MAPK and NF‐κB in HaCat keratinocytes. Mol Immunol 46: 1925–1934. [DOI] [PubMed] [Google Scholar]
- Qi XF, Kim DH, Yoon YS, Song SB, Teng YC, Cai DQ et al (2012). Bambusae caulis in liquamen suppresses the expression of thymus and activation‐regulated chemokine and macrophage‐derived chemokine in human keratinocytes due to antioxidant effect. Evid Based Complement Alternat Med 2012: 617494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman H, Chattopadhyay A (2007). Melittin: a membrane‐active peptide with diverse functions. Biosci Rep 27: 189–223. [DOI] [PubMed] [Google Scholar]
- Shah PP, Desai PR, Singh M (2012). Effect of oleic acid modified polymeric bilayered nanoparticles on percutaneous delivery of spantide II and ketoprofen. J Control Release 158: 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Jin L, Dang E, Chang T, Feng Z, Liu Y et al (2011). IL‐17A upregulates keratin 17 expression in keratinocytes through STAT1‐ and STAT3‐dependent mechanisms. J Invest Dermatol 131: 2401–2408. [DOI] [PubMed] [Google Scholar]
- Sohn EH, Jang SA, Lee CH, Jang KH, Kang SC, Park HJ et al (2011). Effects Of Korean Red Ginseng extract for the treatment of atopic dermatitis‐like skin lesions in mice. J Ginseng Res 35: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son DJ, Lee JW, Lee YH, Song HS, Lee CK, Hong JT (2007). Therapeutic application of anti‐arthritis, pain‐releasing, and anti‐cancer effects of bee venom and its constituent compounds. Pharmacol Ther 115: 246–270. [DOI] [PubMed] [Google Scholar]
- Sugiura K, Shamoto M, Sakamoto N, Shinzato M, Osada A, Sugiura M et al (2003). It is true that, when langerhans cells migrate from the skin to the lymph node, they are transported via lymph vessels. Dermatology 206: 222–224. [DOI] [PubMed] [Google Scholar]
- Tan RA, Corren J (2011). The relationship of rhinitis and asthma, sinusitis, food allergy, and eczema. Immunol Allergy Clin North Am 31: 481–491. [DOI] [PubMed] [Google Scholar]
- Wu WH, Park CO, Oh SH, Kim HJ, Kwon YS, Bae BG et al (2010). Thymic stromal lymphopoietin‐activated invariant natural killer T cells trigger an innate allergic immune response in atopic dermatitis. J Allergy Clin Immunol 126: 290–299, 299.e1–4. [DOI] [PubMed] [Google Scholar]
- Yao W, Zhang Y, Jabeen R, Nguyen ET, Wilkes DS, Tepper RS et al (2013). Interleukin‐9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity 38: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H (2013). The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol 66: 129–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of BV on DNCB‐induced changes in (A) epidermal hyperplasia and a heavy dermal cell infiltration and (B) epidermal thickness and dermal thickness (n = 5). Scale bar = 100 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S2 Effects of BV on DNCB‐induced changes in mast cell infiltration and degranulation. The representative mast cells were indicated by arrows (A). (B) The number of mast cells and degranulated mast cells were counted from all fields per section (n = 5). Scale bar = 50 μm. The enlarged frames scale bar = 20 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S3 Effects of BV on DNCB‐induced changes in the serum levels of IFN‐γ, IL‐4, IgE, and TSLP, which were determined by ELISA kits (n = 5) (A). (B) Real‐time PCR analyses show that BV treatment inhibited the mRNA levels of IFN‐γ and IL‐4 (n = 5). *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg; BV200: BV 200 μg; BV500: BV 500 μg.
Figure S4 Effects of BV on TSLP expression, CD4+ and CD3+ T cells in DNCB‐sensitized mice. (A) Histological images and (B) graphs indicating the relative percentage of TSLP expression and CD4+ and CD 3+ immunopositive cells of the dorsal skin sections (n = 5). (C) Effects of BV on abnormal epidermal differentiation. Immunofluorescence staining of sections of skin with antibody specific for filaggrin shows the expression of epidermal differentiation markers as labelled with FITC, green. Cells were counterstained with Hoechst 33342 (blue). Scale bar = 100 μm. *P < 0.05 vs. NC; + P < 0.05 vs. DNCB; ǂ P < 0.05 vs. Pla; NC: normal control; DNCB: DNCB‐sensitized and challenged; Pla: placebo; BV100: BV 100 μg.
Figure S5 Cytotoxic effects of (A) BV on HaCaT cells. Cell viability was determined by CCK‐8 assay (n = 5). Effects of BV on the chemokine expressions of cytokines (B) and chemokines (n = 5) (C) in HaCaT cells that were stimulated by TNF‐α/IFN‐γ. T + I: TNF‐α/IFN‐γ‐stimulated; *P < 0.05 vs. no treatment. + P < 0.05 vs. TNF‐α/IFN‐γ alone.
Figure S6 Effects of BV on activation of the (A) JAK2, STAT1, and STAT3 signalling pathway in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (B) Immunofluorescence staining for p‐STAT1 and p‐STAT3 (labelled with Alexa Fluor 555, red), and F‐actin (labelled with Alexa Fluor 488, green). Cells were counterstained with Hoechst 33342 (blue). Effects of BV on activation of the (C) NF‐κB signalling pathway in TNF‐α/IFN‐γ‐stimulated HaCaT cells. (D) NF‐κB DNA binding activity in the nuclear extract was measured by EMSA. Representative images from each group. Scale bar = 50 μm. NC: normal control; T + I: TNF‐α/IFN‐γ‐stimulated; Mel1: TNF‐α/IFN‐γ‐stimulated +100 ng/mL BV.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item