

 This work is licensed under a
This work is licensed under a Abstract
This study aims to summarize and analyze the clinical manifestations, genetic characteristics, treatment modalities and long-term prognosis of congenital hyperinsulinemia (CHI) in Chinese children. Sixty children with CHI, who were treated at Beijing Children’s Hospital from January 2014 to August 2017, and their families, were selected as subjects. The CHI-related causative genes in children were sequenced and analyzed using second-generation sequencing technology. Furthermore, the genetic pathogenesis and clinical characteristics of Chinese children with CHI were explored. Among the 60 CHI children, 27 children (27/60, 45%) carried known CHI-related gene mutations: 16 children (26.7%) carried ABCC8 gene mutations, seven children (11.7%) carried GLUD1 gene mutations, one child carried GCK gene mutations, two children carried HNF4α gene mutations and one child carried HADH gene mutations. In these 60 patients, eight patients underwent 18F-L-DOPA PET scan for the pancreas, and five children were found to be focal type. The treatment of diazoxide was ineffective in these five patients, and hypoglycemia could be controlled after receiving partial pancreatectomy. In conclusion, ABCC8 gene mutation is the most common cause of CHI in Chinese children. The early genetic analysis of children’s families has an important guiding significance for treatment planning and prognosis assessment.
Keywords: congenital hyperinsulinemia, KATP channels, gene mutations, diazoxide
Congenital hyperinsulinism (CHI) is a kind of genetically heterogeneous disease, in which intractable, persistent hypoglycemia is induced by excessive insulin secretion and increased serum insulin concentration (1). To date, 14 genes (ABCC8, KCNJ11, GLUD1, GCK, HNF4α, HNF1α, HDAH, UCP2, SLC16A1, HK1, CACNα1D, PGM1, PMM2 and FOXA2) have been found to be associated with the pathogenesis of CHI. Severe and recurrent hypoglycemia may lead to irreversible brain damage and a series of neurological sequelae. Therefore, early diagnosis and effective treatment are essential to improve the prognosis of diseased children. The clinical manifestations, histological types, diagnoses, treatment effects and long-term prognoses of CHI patients with different mutations are diverse (2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 34). Diazoxide, a KATP channel opener, is the first-line treatment for long-term treatment. In case of the diazoxide-unresponsive patient or limitations caused severe side effects, second-line drugs have to be considered, such as the somatostatin analog octreotide, including glucagon, nifedipine and new medicines such as sirolimus, lanreotide (12). KATP-CHI caused by mutations in the ABCC8 or KCNJ11 genes is the most common and most serious type of CHI. Most patients with KATP-CHI are characterized by abnormalities of potassium channels, are ineffective in the treatment of diazoxide, and need to undergo 18F-L-DOPA PET scan for the pancreas to determine the pancreatic histological type, while hypoglycemia can be controlled after the patients receive different degrees of pancreatectomy (13).
There are few studies on CHI in Chinese children. In the present study, 60 CHI children treated in Beijing Children’s Hospital from January 2014 to August 2017 were selected as the research subjects, and the CHI-related causative genes in children were sequenced and analyzed using second-generation sequencing technology. Through the exploration of the genetic pathogenesis and clinical characteristics of Chinese children with CHI, we hope to discover more types of mutations related to CHI in Chinese children, and investigate the diagnosis, treatment and prognosis of CHI.
Materials and methods
Subjects
Sixty children with CHI, who were treated at Beijing Children’s Hospital from January 2014 to August 2017, and their families, were selected as the subjects. All 60 children were diagnosed with CHI through related detection methods after admission to the hospital. The specific diagnostic criteria are as follows: (1) hyperinsulinemia (plasma insulin >2 µIU/mL); (2) hypolipidemia (plasma free fatty acid <1.5 mmol/L); (3) hypoketonemia (serum β-hydroxybutyric acid <2.0 mmol/L); (4) response of glucagon test with 1 mg of intravenous blood: blood glucose changes >30 mg/dL. Patients with temporary CHI were excluded. This study was approved by the Ethics Committee of Beijing Children’s Hospital, Capital Medical University.
After the diagnosis of CHI, 48 of the 60 children were treated with diazoxide, with an initial dose of 5 mg/(kg·day), at 2–3 oral uses per day. The dose was gradually increased according to the patient’s condition, up to a maximum dose of 15 mg (kg·day). Hydrochlorothiazide was added (1–2 mg/(kg·day), orally taken 2–3 times per day) to prevent the side effect of water-sodium retention, and potassium chloride was added (1–2 mL of 10% potassium chloride/(kg·day), orally used three times) to prevent hypokalemia caused by diuretics.
Genetic analysis
Sample acquisition
After these 60 children provided an informed consent, 3 mL of venous blood was collected from them and their parents, and the collected blood was anticoagulated with EDTA. The genomic DNA of the patients and their families was extracted using a BloodGen Midi Kit (CWBIO, China). The operation was carried out strictly in accordance with kit instructions.
Second-generation sequencing technology
According to data obtained from references and the OMIM database, the exon regions of genes associated with hypoglycemia were captured using a capture probe by means of targeted exon capture. The chip used in the total exon capture was the SeqCap EZ MedExome Enrichment Kit (Roche). The sequencing operation flow was standardized on the Illumina hiseq XTen platform.
First-generation sequencing (Sanger) for validation
If the second-generation sequencing technology revealed that children carried mutations in ABCC8, KCNJ11, GLUD1, GCK, HNF4α, HNF1α, HADH, UCP2, SLC16A1, HK1, PGM1, PMM2, CACNα1D and FOXA2, primers were designed according to the sequence of the verified loci in the gene, and the sequence was amplified by PCR: the sequencing was carried out using an ABI 3730XL sequencer, and the primers for sequencing were the same with those used in the PCR. Gene sequences were analyzed and compared using DNASTAR software.
Database analysis
Non-pathogenic variations were excluded based on the SNP Database (dsSNP) and the 1000 Genome Database, and the Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database and NCBI database were searched to determine whether the mutation has been reported or not.
Protein structure prediction software
The effects of the gene mutations on the protein structure were predicted using human genome variation software Provean and SIFT. The criteria for classifying pathogenic variants referred to Richards et al. (14).
Pancreatic tissue type
Among the 60 children with CHI, 8 children underwent an 18F-L-DOPA PET scan of the pancreas in the Hong Kong Sanatorium and Hospital to determine the histological type of the pancreas.
Statistical analysis
To analyze the relationship between phenotype and genotype, chi-squared test and ANOVA were conducted using Statistical Package for the Social Sciences version 19.0 software (IBM Corp.). Descriptive data were expressed as mean ± standard deviation values. Skewed data were shown as median. For all tests, a P value of less than 0.05 was accepted as statistically significant.
Results
Clinical characteristics
Among these 60 patients, 41 patients were male and 19 patients were female. The age of onset ranged between immediately after birth to 7 years and 5 months old. Onset occurred in the neonatal period in 26 children, within 1–6 months after birth in 16 children, and at 6 months after birth in 18 children. The birth weights of these children ranged within 2–5 kg. Among these children, 15 children were macrosomia, 43 children had normal birth weight and 2 were children had low birth weight. All mothers of these children were healthy during pregnancy, and had no a history of impaired glucose tolerance. All patients had no a family history of hypoglycemia (Table 1).
Table 1.
Clinical data of 60 children with CHI.
| Clinical features | Results (n = 60) |
|---|---|
| Male, n (%) | 41 (68.3) |
| Mutation positive, n (%) | 27 (45) |
| Birth weight (kg) | 3.6 ± 0.6 (2–5) |
| Macrosomia, n (%) | 15 (25) |
| Onset age, n (%) | |
| Neonatal period | 26 (43.3) |
| 1–6 month | 16 (26.7) |
| >6 month | 18 (30.0) |
| Blood glucose level (mmol/L) | 1.75 ± 0.54 |
| Insulin level (µIU/mL) | 19.36 ± 21 |
| Diazoxide treatment, n (%) | 48 (80.0) |
| Diazoxide effective, n (%) | 32 (66.7) |
Data statistics using mean value, standard deviation or percentage.
The 18F-L-DOPA PET scan of the pancreas was performed in eight children. These results revealed that three children were found to be diffuse type, while five children were found to be focal type.
Diagnosis and treatment results
The criterion for the ineffective treatment of diazoxide were as follows: when a child was treated with the maximum dose of diazoxide (15 mg/kg·day) for 5 days, blood sugar levels cannot be maintained at more than 70 mg/dL after the child is fasted for 8–10 h, indicating that diazoxide treatment was ineffective.
In the present study, 48 children were treated with diazoxide after the diagnosis of CHI. The other 12 children did not choose diazoxide therapy because of the difficulty in buying medicine or giving up treatment. Among these children, blood glucose in 32 children gradually returned to normal level after diazoxide treatment, suggesting that the diazoxide treatment was effective in these patients, while 16 children were treated with sufficient diazoxide (15 mg/(kg·day)), and their blood sugar presented with no significant improvement after treatment, suggesting that diazoxide treatment was ineffective.
Results of the genetic analysis
Mutations in the ABCC8 gene: Among these 60 children with CHI, 16 children carried 20 mutations in the ABCC8 gene, including 15 missense mutations, three splicing mutations, one nonsense mutation and one frameshift mutation.
Seven children carried GLUD1 gene mutations, one child carried the GCK gene mutation (Fig. 1), two children carried HNF4α gene mutations (Fig. 2) and one child carried a compound heterozygous mutation in the HADH gene (Fig. 3).
Of five cases with focal tissue type, three cases had ABCC8 gene mutation, one case carried HNF4α gene mutation, one case was not found to carry common pathogenic gene mutation, three cases of histological type were diffuse type children: 1 cases with ABCC8 gene mutation, 1 cases with GCK gene mutation, 1 cases not found to carry common pathogenetic gene mutation (Table 2).
Figure 1.
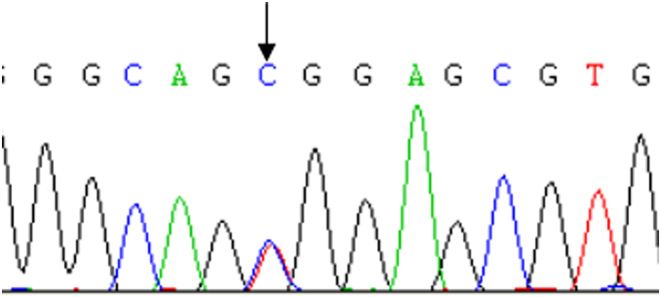
Case 29 carried mutation GCK gene c.295T>C(p.W99R).
Figure 2.
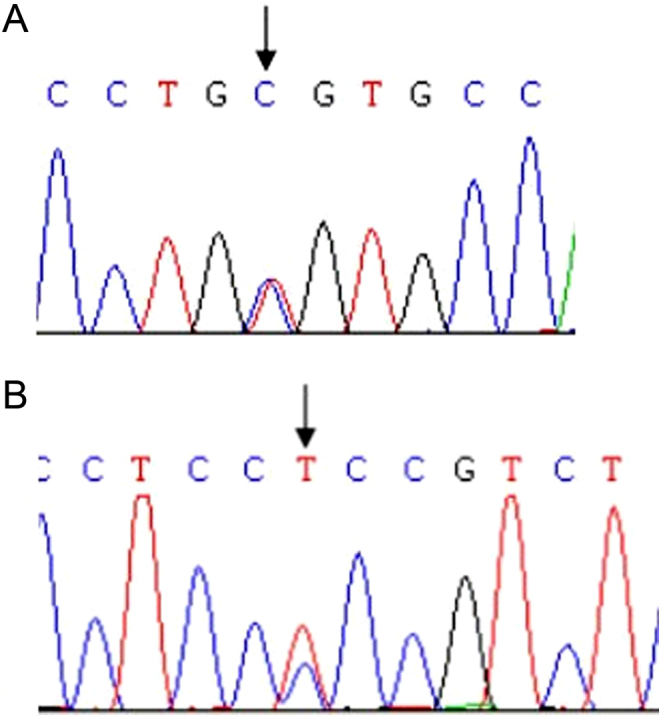
(A) Case 35 carried mutation HNF4α gene c.157T>C(p.C53R); (B) case 50 carried mutation HNF4α gene c.481C>T(p.P161S).
Figure 3.
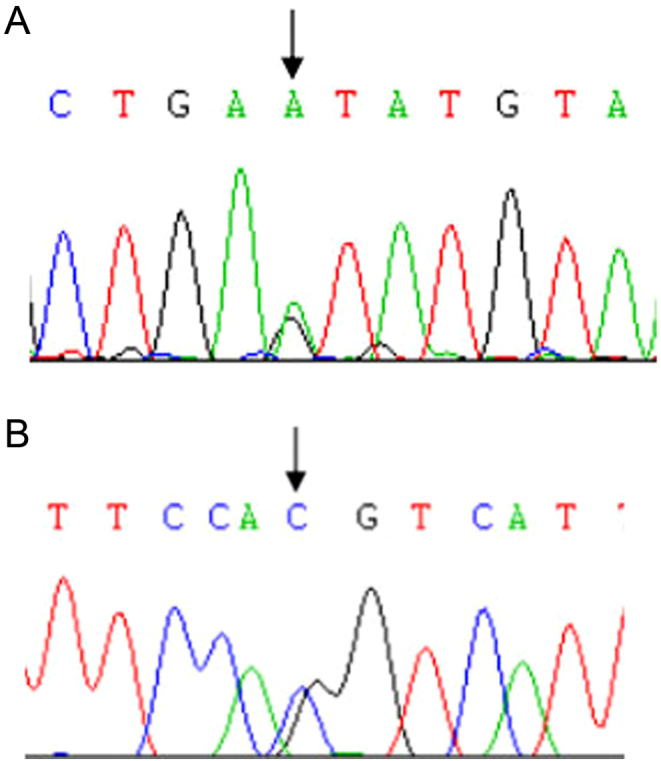
Case 32 carried heterozygous mutations in the HADH gene: two splicing mutations c.419+1G>A (A) and c.547-1G>C (B).
Table 2.
Data of 27 children with known mutations in CHI gene.
| Code | Onset age | Birth weight (kg) | Gene mutation | Protein variation | Efficacy of diazoxide | Source of mutation | 18-F-L-DOPA-PET scan | Has it been reported? | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| ABCC8 gene (NM_000352) | |||||||||
| 2 | 1 day | 4.9 | c.2331G>A | p.W777X | Effective | Unknown | On diazoxied treatment | ||
| 9 | 1 day | 3.95 | c.4316A>G | p.L1439P | Invalid | Father | Focal type | Partial pancreatectomy/still hypoglycemia | |
| 10 | 6 months | 3.75 | c.4411C>T | p.D1471N | Invalid | Father | Already reported | Unable to follow up | |
| 14 | 3 months | 3.25 | c.12Cdel | Frameshift mutation | Invalid | Father | On basal treatment | ||
| 16 | 2 days | 4 | c.382C>T | p.E128K | Invalid | Father | Focal type | Already reported | Partial pancreatectomy/still hypoglycemia |
| 18 | 1 day | 3.8 | c.4198+1G>T | Splice mutation | Unused | Father | No treatment | ||
| c.106C>T | p.H36Y | Mother | |||||||
| 19 | 1 days | 4.7 | c.47G>C | p.R16P | Invalid | Father | On octreotide treatment | ||
| 21 | 2 days | 3.5 | c.331G>A | p.G111R | Invalid | Father | Focal type | Already reported | Partial pancreatectomy |
| 22 | 1 day | 3.84 | c.1775A>C | p.L592R | Effective | Mother | On diazoxied treatment | ||
| 25 | 2 days | 3.7 | c.1671+2T>C | Splice mutation | Invalid | Father | On octreotide treatment | ||
| 26 | 2 years | 4.5 | c.3482C>G | p.P1161R | Invalid | Mother | No treatment | ||
| c.428G>A | p.W143X | Father | Already reported | ||||||
| 30 | 2 days | 3.8 | c.850dupG | p.A284fs | Invalid | Father | On octreotide treatment | ||
| 39 | 1 days | 3.45 | c.536A>G | p.Y179C | Invalid | Mother | Diffuse type | Already reported | On octreotide treatment |
| c.3736T>C | p.W1246R | Father | |||||||
| 40 | 1 days | 4.56 | c.216C>A | p.N72K | Effective | Father | On diazoxied treatment | ||
| c.3733A>G | p.R1245G | Mother | |||||||
| 41 | 3 days | 3.7 | c.149-2A>C | Splice mutation | Invalid | Father | On octreotide treatment | ||
| 49 | 1 day | 4.19 | c.4411C>T | p.D1471N | Invalid | Father | Already reported | On octreotide treatment | |
| c.221G>A | p.R74Q | Mother | Already reported | ||||||
| GLUD1 gene (NM_005271) | |||||||||
| 20 | 8 months | 2.8 | c.1519G>A | p.H507Y | Effective | De novo mutation | Already reported | On diazoxied treatment | |
| 28 | 2 days | 3.65 | c.1388A>T | p.N463I | Effective | De novo mutation | On diazoxied treatment | ||
| 33 | 7 months | 3.3 | c.965C>T | p.R322H | Effective | De novo mutation e | Already reported | On diazoxied treatment | |
| 42 | 6 months | 3.8 | |||||||
| 44 | 1 years 2 months | 4.25 | |||||||
| 45 | 1 day | 2.7 | c.1495C>A | p.G499C | Effective | De novo mutation | Already reported | On diazoxied treatment/epilepsy | |
| 60 | 7 months | 2.9 | c.1388A>G | p.N463S | Effective | De novo mutation | Already reported | On diazoxied treatment | |
| GCK gene (NM_000162.4) | |||||||||
| 29 | 7 years 5 months | 5 | c.295T>C | p.W99R | Effective | De novo mutation | Diffuse type | On diazoxied treatment | |
| HNF4α gene (NM_001258355) | |||||||||
| 35 | 1 days | 4.02 | c.157T>C | p.C53R | Effective | De novo mutation | On diazoxied treatment | ||
| 50 | 1 days | 4.1 | c.481C>T | p.P161S | Invalid | Father | Focal type | Partial pancreatectomy/still hypoglycemia | |
| HADH gene (NM_001184705) | |||||||||
| 32 | 3 days | 3.2 | c.419+1G>A | Splice mutation | Effective | Father | On diazoxied treatment | ||
| c.547-1G>C | Splice mutation | Effective | Mother | ||||||
Associations between genotype and clinical phenotype
No significant differences were found in insulin levels between patient groups with or without mutations. However, children with variant genetics showed significantly higher birth weight, earlier CHI onset and lower diazoxide response rates when compared with the group without identified mutations (P < 0.05) (Table 3).
Table 3.
Clinical and biochemical characteristics of CHI patients.
| KATP-CHI group (n = 16) | Non-mutation group (n = 33) | Statistical test value | P | |
|---|---|---|---|---|
| Onset age (days) | 1 (1–730)* | 150 (1–545)* | F = 4.871 | 0.032 |
| Birth weight (kg) | 3.97 ± 0.47 | 3.36 ± 0.52 | F = 15.804 | 0.000 |
| Blood glucose level (mmol/L) | 1.83 ± 0.47 | 1.68 ± 0.63 | F = 0.713 | 0.403 |
| Insulin level (µIU/mL) | 21.25 (4.76–110.6)* | 8.3 (2.15–69)* | F = 3.424 | 0.071 |
| Positive outcome of diazoxide therapy, n (%) | 3/15 (20.00) | 19/22 (86.36) | χ 2 = 16.295 | 0.000 |
*Median (range).
Discussion
Analysis of ABCC8 gene mutations
In the present study, among the 60 children with CHI, a total of 20 ABCC8 mutations were detected in 16 children, accounting for 26.7% (16/60) of the total number of children. This proportion was lower than that previously reported in China (38%), and more lower than that reported in Norway, Japan, South Korea and Spain. Among these 20 mutations, six mutations have been previously reported (15, 16, 17). Among these 16 children, six children (6/16, 37.5%) had macrosomia, and 13 children (13/16, 81.25%) had onset at the neonatal period. The above two proportions were significantly higher than those in the entire sample (24.2%, 41.9%). Among these 16 children, 15 children were treated with diazoxide, among which 12 children were ineffective, while three children were effective (3/15, 20.0%). The effective rate was significantly lower than that in the whole sample. The above indicates that children with ABCC8 gene mutations have the characteristics of early onset, high birth weight and low effective rate of diazoxide.
In the present study, the mutation source of 16 patients with KATP channel gene mutations was analyzed: five patients carried compound heterozygous mutations, nine patients carried mutations from their fathers, one patient mutation was from the mother, one patient carried mutation had an unknown genetic pattern. Therefore, the probability of compound heterozygous mutations of the KATP channel was 31.3% (5/16), and patients with single-gene mutation accounted for 62.5% (10/16) of patients with mutations. Among these patients, patients with paternal-origin mutations accounted for 90% (9/10), and patients with matrilineal-origin mutations accounted for 10% (1/10). When compared with other countries: South Koreans reported that patients with compound heterozygous mutations accounted for 28% of the total number of patients with mutations, patients with single-gene mutation accounted for 72% of the total number of patients with mutations (in Western countries this proportion was 40–43%, while in Japan, this proportion was 89.5%), and the probability of single mutation from their father was 50% (in Western countries, this was 70%, while in Japan, this was 84.2%) (18, 19).
In the present study, all the five children with compound heterozygous mutations respectively carried one paternal and one maternal mutation in the ABCC8 gene. This is consistent with autosomal recessive inheritance. Among these five children, two children had normal birth weight and three children were macrosomia. Furthermore, four children had onset on the first after birth, and one child had onset at the age of 2 years old. Four children were treated with diazoxide. Among these children, three children were ineffective to the treatment and one child was effective to the treatment. In the present study, the distribution of clinical characteristics of these patients was basically consistent with those reported in the literature. It has been reported in literatures that the histological type of pancreas in children with compound heterozygous mutations was mainly the diffuse type.
Among the nine children with paternal single-gene mutations, seven children had onset in the neonatal period, while two children had onset within 1–6 months after birth. Furthermore, seven children had normal birth weight, while two children had macrosomia. All nine children were treated with diazoxide during the test after the diagnosis of CHI, but they were ineffective to the treatment of diazoxide. Among these children, five children (cases 19, 21, 25 30 and 41) were further treated with octreotide during the test, and all children were effective. Some literatures have revealed that the genetic basis of focal pancreatic histology is as follows: the paternal recessive mutation is accompanied by the deletion of the maternal chromosome 11p15.1 allele in pancreatic lesions; this paternal inherited single parent diploid is adjacent to the mutation site of the Beckwith–Wiedemann syndrome, which includes some imprinted genes (IGF2, H19 and CDKN1C) that promote pancreatic cells to overgrow, and the genetic model is autosomal recessive inheritance. The laboratory of Dr. Arupa Ganguly in Pennsylvania University in USA revealed that the following: patients with a single heterozygous recessive mutation of the ABCC8 or KCNJ11 gene have a 94% possibility that pancreatic histological lesions are focal type (20). Furthermore, if the mutation is inherited from the father, the possibility of a focal type is increased to 98%. In the contrary, if the patient carried the two recessive mutations of the KATP channel, the lesion type was predicted to be diffuse type. In the present study, three children (cases 9, 16 and 21) underwent 18F-L-DOPA PET scan of the pancreas. Results revealed that the histological type of the pancreas were all focal type, and all the gene mutations were inherited from their fathers, which is consistent with that reported in literature. It is a pity that a microsatellite marker analysis of pancreatic lesions was not conducted, and the deletion of the maternal chromosome 11p15.1 allele could not be temporarily confirmed. Three children were ineffective in the treatment of diazoxide and underwent partial pancreatectomy. Among these children, cases 9 and 16 were treated with laparoscopic surgery and continued to have hypoglycemia after operation. However, the degree of hypoglycemia was better than before, and the child required frequent feeding to control blood sugar levels. Case 21 was treated with laparotomy, and blood glucose returned to normal level after the operation.
In the present study, case 22 carried a maternal heterozygous mutation of the ABCC8 gene (c.1775A>C), causing the p.L592R mutation. This mutation has not been reported in any literature. However, bioinformatics analysis software predicted that it may have pathogenicity. The child had a normal birth weight, had onset on day one after birth, and was effective in the treatment of diazoxide. The clinical characteristics were consistent with the clinical manifestations of children with materal autosomal dominant mutations in literature.
SUR1 contains 17 transmembrane spirochaeta, which form the extracellular N-end transmembrane structures TMD0, TMD1 and TMD2, and two nucleic acid-binding structural domains, NBD1 and NBD2, which immediately follow the TMD1 and TMD2 structures in the cell. Nucleotide binding structural domains NBD1 and NBD2 in SUR1 can sense the concentration changes in ATP/ADP and transmit the signal into the cell. NBD1 has a stronger affinity to MgATP and has a weak hydrolysis effect on it. The function of NBD2 was to hydrolyze MgATP into MgADP by binding with MgATP, thereby activating the KATP channel (21). Among the mutations found in the present study, eight mutations were located in TMD0, one mutation was located in NBD1 (amino acids 679–929), three mutations were located in TMD1, four mutations were located in NBD2 (amino acids 1344–1578), two mutations were located in TMD2 and three mutations were located in splice sites (22). The above results suggest that mutations located in TMD0 had the highest number (34.78%, 8/23), while mutations in the NBD2 structural domain accounted for only 17.39% (4/23). This is close to the probability of mutation in this region, as reported in South Korea in 2011 (17).
The reason for the pathogenicity of many mutations in SUR1 is that the mutated KATP channel cannot flow to the cell surface. Therefore, the channel function is impaired. In 2007, Yan FF et al. pointed out in pharmacological and functional studies that sulfonylureas inhibit channel activities. However, these drugs can transport some of the mutated channels to the surface of the cell, channel activities are recovered after drug metabolism, and these mutations were confirmed to be located in the TMD0 region (23). Literatures have revealed that mutations in the NBD1 region, such as G716V and R842G mutations, reduce the affinity of NBD1 to MgATP, affecting the function of KATP channels, and inducing excessive insulin secretion and the according disease (24). Mutations in the NBD2 region do not affect the normal structure or expression ability of KATP channels, but these KATP channels all obviously reduce or even lose the ability to react with MgADP and diazoxide, such as mutation E1506K in the NBD2 region. Due to the ability of the KATP channel to react with MgADP decrease, potassium ion channels are closed, current under the resting state decreases, cells with KATP channels are depolarized and a large amount of Ca ions flow into the cell, increasing insulin release and accordingly causing hyperinsulinemia (21).
Analysis of GLUD1 gene mutations
In the present study, among the 60 children, GLUD1 gene mutations were detected in seven children, accounting for 11.7%, second only to KATP-HI, which is basically consistent with the results reported in a related literature (3). The seven GLUD1 mutations found in the present study were all de novo mutations, and the genetic model was basically consistent with that reported in literatures. The seven mutations were as follows: mutation H507Y (case 20) and mutation N463I (case 28), mutation G499C (case 45) and mutation R322H (cases 33, 42 and 44) and mutation N463S (case 60). Among these mutations, R322H was the most common mutation. All the above mutations have been reported in literatures (3, 25).
Mutation H507Y is located in exon 12 of the GLUD1 gene, encoding the catalytic region of GDH. In 2016, John Odom et al. reported a case of mutation H507Y in the GLUD1 gene. The change in amino acid properties in the region reduced the sensitivity of GDH to the allosteric inhibition of GTP, enhancing the activity of the protease. This increased the amount of α-ketoglutaric acid entering the tricarboxylic acid cycle, increased the ATP/ADP ratio in pancreatic β-cells, caused the closure of ATP-dependent potassium channels on the cell membrane, depolarized the cell membrane, made voltage-dependent Ca channels open, and increased the content of Ca ions in the cytoplasmic matrix, thereby inducing the release of insulin from insulin storage granules. Mutations N463I and N463S are located in exon 10 of the GLUD1 gene. N463 is a mutation hotspot. Mutations in exon 10 of GLUD1 reported in literature also include L466V and N463T (26), and mutations in these two locations can increase the activity of GDH to more than two times the normal level. The difference between mutations L466V and L463T is that the former decreases the sensitivity to the inhibition of GTP, while the latter increases the stimulatory effect of ADP (26). Mutation G499C is located in exon 12 of the GLUD1 gene. The G499 site is also a mutation hotspot. At present, at least five mutation types at this site have been found: G499C, G499S, G499R, G499D and G499V. The pathogenic mechanism is that changes in amino acid properties reduce the sensitivity of GDH to the allosteric inhibition of GTP, increase the activity of the protease and thereby leading to the release of insulin from insulin storage granules. Mutation R322H is located in exon 7 of the GLUD1 gene, which is a common mutation type. In the present study, this mutation was deteccted in three children. The mutation increases the enzyme activity of GDH by reducing the sensitivity of GDH to GTP.
Children with GDH-HI usually have normal birth weights, and rarely have onset in the neonatal period. Children with this type present with repeated episodes of fasting (and postprandial) hypoglycemia, are accompanied with persistent asymptomatic hyperammonemia and are often complicated with epileptic symptoms. The degree of hypoglycemia is mild in children, which is mainly induced by prolonged fasting or protein diet rich in leucine. The ATP-sensitive potassium channels in pancreatic β-cells in this type of patients are normal. Hence, these patients are all effective in the treatment of potassium channel opener diazoxide. In the present study, among the seven children with mutations in GLUD1, three children had onset in the neonatal period, while four children had onset at 6 months after birth. Furthermore, six children had normal birth weights, while one child was macrosomia. In addition, six children had hyperammonemia, while one child had normal blood ammonia level. All seven children were effective in the treatment of diazoxide. It was found through follow-ups that among these seven children, epilepsy was found in three children. Six children continued to orally take diazoxide, and their blood sugar was acceptably controlled. Hypoglycemia was spontaneously relieved at approximately seven years of age in one child. The above onset age and clinical characteristics of these patients were basically consistent with those reported in literature(3).
Analysis of GCK gene mutations
GCK-HI was first discovered by Glaser et al. in 1998. It is induced by function-enhancing mutations in the GCK gene encoding glucokinase. At present, at least 19 types of GCK mutations have been discovered, and the genetic model is autosomal dominant inheritance or novo mutation. Its clinical manifestations such as birth weight, age of onset, severity of hypoglycemia and efficacy of diazoxide in children with GCK-HI are significantly different from each other (4).
In the present study, a mutation c.295C>T (p.W99R) was detected in case 29, and none of the parents of this child carried this mutation. This suggests that this mutation is a de novo mutation. The child had onset at 7 years after birth, the onset was late, and the child was effective in the treatment of diazoxide. The histological type of the pancreas of the child was diffuse type. In 2003, Anna L Gloyn et al. reported this heterozygous mutation type in the GCK gene (27), in which the child developed hypoglycemia after birth. When the child was treated with a dosage of up to 20 mg/kg·day of diazoxide, the symptom of hypoglycemia could be controlled. However, persistent hypoglycemia occurred in the child at the age of 10 months. After additional treatment with 0.45 mg/kg·day of nifedipine, the blood glucose could be maintained at 3.0–3.9 mmol/L. The pancreatic tissue of the child was normal. The father of the child underwent blood sugar monitoring after the child was diagnosed with GCK-HI. Results revealed that blood sugar level was maintained within 2.7–3.1 mmol/L, and no severe hypoglycemia was found.
A scholar analyzed the function of purified glutathione S-transferase to recombine protein after mutation W99R occurred in the GCK gene. Its coding product could increase the enzyme activity index by approximately six times by reducing the threshold of glucose to stimulate insulin secretion. A study revealed that activating mutations in the GCK gene could enhance the affinity between glucokinase and glucose, decrease the threshold of glucose to stimulate insulin secretion (resetting threshold value GSIR), increase metabolic activity, increase the concentration of ATP/ADP in cells and enhance insulin secretion by stimulating KATP channel closure, thereby causing hyperinsulinemia (28). A foreign literature reported that different mutations in the GCK gene could change the curative effects of diazoxide and severity of hypoglycemia by differently decreasing the GSIR value (29).
Analysis of HNF4α gene mutation
The function-deleting mutations in the HNF4α gene can lead to HNF4α-HI. To date, dozens of gene mutations associated with HNF4α-HI have been discovered. The genetic model is usually autosomal dominant inheritance, is occasionally a de novo mutation and the histological type is usually diffuse type. This type of children are mostly macrosomia at birth, the disease usually occurs one week after birth, and the children are effective in the treatment of diazoxide (30).
In the present study, two children carried HNF4α gene mutations. Case 35 carried mutation c.157T>C (p.C53R), but his parents had normal genotypes. This suggests that this mutation is a de novo mutation. Case 50 carried mutation c.481C>T (p.P161S). This mutation was inherited from his father and was an autosomal dominant inheritance. The genetic model of mutations in these two children was consistent with those reported in literature. The above two mutations have not been reported in any literature and were not collected in the SNP database. Hence, the possibility of pathogenicity could not be ruled out (14). Both children were immediately diseased after birth, and were both macrosomia (case 35 was 4.02 kg and case 52 was 4.1 kg). Case 35 was effective in the treatment of diazoxide, and the clinical characteristics were roughly consistent with the clinical manifestations of HNF4α–HI reported in literature. Case 50 was ineffective in the treatment of diazoxide, underwent 18F-L-DOPA PET pancreatic scanning, and it was determined that the histological type of the pancreas was focal type. The child was treated with partial pancreatectomy. Hypoglycemia was not controlled very well after operation, so the patient required frequent feeding to control blood sugar levels. The clinical manifestations and histological type of the pancreas of the child were not completely consistent with those previously reported in literature (30). This suggests that children with HNF4α-HI have complex clinical characteristics.
Existing studies have revealed that HNF4α played an important role in insulin secretion stimulated by glucose in the multiple transcription-factor network of pancreatic β-cells, and function-deleting mutations in HNF4α could lead to abnormalities in glucose-stimulating insulin secretion (31), including hypoglycemia in early childhood and maturity-onset diabetes of the young. However, its specific mechanism remains unknown. A study suggested that this may be related to the interaction among multiple genes. At present, two hypotheses have been relatively recognized: function-deleting mutations in the HNF4α gene can decrease the expression of Kir6.2 subunit in the KATP channel or decrease the expression of PPAR-α. PPAR-α can increase the level of long-chain acyl-CoA in the cytoplasm and act as a signal of abnormal insulin release (32). Another literature revealed that the HNF-4α gene may be related to the amplification of pancreatic β-cells during pregnancy. This suggests that the HNF-4α gene may be related to the proliferation of pancreatic β-cells.
Analysis of HADH gene mutation
Short-chain 3-hydroxamino-CoA dehydrogenase (SCHAD) is a mitochondrial enzyme, which is encoded by the hydroxyacyl-coenzyme A dehydrogenase (HADH) gene. It catalyzes the reciprocal second step reaction of β-oxidation of fatty acid. The function-deleting mutations in the HADH gene can induce SCHAD-HI.
At present, approximately 40 children have been diagnosed with SCHAD-HI. It can manifest as late-onset mild hypoglycemia or severe neonatal hypoglycemia. At present, at least 17 types of mutations have been found, which are all autosomal recessive inheritance, and the histological type is diffuse type. Furthermore, it has certain protein sensitivity, but has no hyperammonemic performance. Children with this type of disease are effective in the treatment of diazoxide. The specific mechanism of mutations in the HADH gene, which leads to abnormal insulin release, remains unknown. It may be involved in the interaction between GDH and SCHAD (33). Under normal conditions, SCHAD can inhibit the activity of GDH. When function-deleting mutations occur in the HADH gene, it can affect the regulation of insulin secretion by GDH, making GDH enhance its sensitivity for its activator leucine or making it free from the regulation of the allosteric inhibition of GTP, accordingly enhancing the activity of GDH and increasing insulin secretion.
In the present study, case 32 carried heterozygous mutations in the HADH gene: two splicing mutations c.419+1G>A (IVS3) and c.547-1G>C (IVS4), which were inherited from the father and mother, respectively, and the genetic model was autosomal recessive inheritance. The child had onset of the disease at 3 days after birth and was effective in the treatment of diazoxide. The clinical characteristics were consistent with those reported in previous literatures. This mutation has not been reported in any literature. Hence, its pathogenicity could not be ruled out based on the prediction of the bioinformatics analysis software.
In summary, the results of the present study revealed that CHI children in China have complex pathogenesis. Mutations in the ABCC8 gene are the most common causative cause of CHI in Chinese children. In more than 50% of CHI patients, no known causative mutations in the CHI gene were detected. This suggests that other unknown genes are involved in the pathogenesis of CHI. In the present study, there were 16 mutations in the ABCC8 gene, two mutations in the HNF4α gene and two mutations in the HADH gene, and these have not been reported, to date. Furthermore, the clinical characteristics of some children were incompletely consistent with those reported in previous literatures. This suggests that CHI has complexity and heterogeneity.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by Beijing Municipal Science & Technology Commission, No. Z141107002514142. Clinical trial registration number: ChiCTR1800016089.
Acknowledgement
The authors thank all the families for their continuous support in this study.
References
- 1.Sakakibara A, Hashimoto Y, Kawakita r, Hosokawa Y, Nagahara K, Hasegawa Y, Hoshino S, Nagasaka H, Yorifuji T. Diagnosis of congenital hyperinsulinism: biochemical profiles during hypoglycemia. Pediatric Diabetes 2018. 19 259–264. ( 10.1111/pedi.12548) [DOI] [PubMed] [Google Scholar]
- 2.Harel S, Cohen AS, Hussain K, Flanagan SE, Schlade-Bartusiak K, Patel M, Courtade J, Li JB, Van Karnebeek C, Kurata H, et al Alternating hypoglycemia and hyperglycemia in a toddler with a homozygous p.R1419H ABCC8 mutation: an unusual clinical picture. Journal of Pediatric Endocrinology and Metabolism 2015. 28 345–351. ( 10.1515/jpem-2014-0265) [DOI] [PubMed] [Google Scholar]
- 3.Odom J, Gieron-Korthals M, Shulman D, Newkirk P, Prijoles E, Sanchez-Valle A. A novel mutation in GLUD1 causing hyperinsulinism-hyperammonemia in a patient with high density of homozygosity on microarray: a case report. Journal of Medical Case Reports 2016. 10 25 ( 10.1186/s13256-016-0811-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, Gloyn AL. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes and hyperinsulinemic hypoglycemia. Human Mutation 2009. 30 1512–1526. ( 10.1002/humu.21110) [DOI] [PubMed] [Google Scholar]
- 5.Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, Siahanidou T, Banerjee I, Akcay T, Rubio-Cabezas O, et al Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. European Journal of Endocrinology 2010. 162 987–992. ( 10.1530/EJE-09-0861) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, Boodhansingh K, Hughes N, Becker S, Givler S, et al Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Hormone Research in Paediatrics 2013. 80 18–27. ( 10.1159/000351943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camtosun E, Flanagan SE, Ellard S, Siklar Z, Hussain K, Kocaay P, Berberoglu M. A deep intronic HADH splicing mutation (c.636+471G>T) in a congenital hyperinsulinemichypoglycemia case: long term clinical course. Journal of Clinical Research in Pediatric Endocrinology 2015. 7 144–147. ( 10.4274/jcrpe.1963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS, Quintos JB, Ganguly A, De Leon DD, Palmieri F, et al Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of uncoupling protein 2. Journal of Clinical Endocrinology and Metabolism. 2017. 102 942–949. ( 10.1210/jc.2016-3164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tosur M, Jeha GS. A novel intragenic SLC16A1 mutation associated with congenital hyperinsulinism. Global Pediatric Health 2017. 17 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, Hussain K, Ellard S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatric Diabetes 2017. 18 320–323. ( 10.1111/pedi.12512) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, Antón-Gamero M, Argente J, Bussell AM, Brandli A, et al Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. Journals of the American Society of Nephrology 2017. 28 2529–2539. ( 10.1681/ASN.2016121312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demirbilek H, Hussain K. Congenital hyperinsulinism: diagnosis and treatment update. Journal of Clinical Research in Pediatric Endocrinology 2017. 30 69–87. ( 10.4274/jcrpe.2017.S007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meintjes M, Endozo R, Dickson J, Erlandsson K, Hussain K, Townsend C, Menezes L, Bomanji J. 18F-DOPA PET and enhanced CT imaging for congenital hyperinsulinism: initial UK experience from a technologist’s perspective. Nuclear Medicine Communications 2013. 34 601–608. ( 10.1097/MNM.0b013e32836069d0) [DOI] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015. 17 405–424. ( 10.1038/gim.2015.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, Ganguly A, Shyng SL, Stanley CA. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the β-cell sulfonylurea receptor. Diabetes 2011. 60 1797–1804. ( 10.2337/db10-1631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong C, Huang S, Su C, Qi Z, Liu F, Wu D, Cao B, Gu Y, Li W, Liang X, et al Congenital hyperinsulinism in Chinese patients: 5-yr treatment outcome of 95clinical cases with genetic analysis of 55 cases. Pediatric Diabetes 2016. 17 227–234. ( 10.1111/pedi.12254) [DOI] [PubMed] [Google Scholar]
- 17.Park SE, Flanagan SE, Hussain K, Ellard S, Shin CH, Yang SW. Characterization of ABCC8 and KCNJ11 gene mutations and phenotypes in Korean patients withcongenital hyperinsulinism. European Journal of Endocrinology 2011. 164 919–926. ( 10.1530/EJE-11-0160) [DOI] [PubMed] [Google Scholar]
- 18.Yorifuji T, Kawakita R, Nagai S, Sugimine A, Doi H, Nomura A, Masue M, Nishibori H, Yoshizawa A, Okamoto S, et al Molecular and clinical analysis of Japanese patients with persistent congenital hyperinsulinism:predominance of paternally inherited monoallelic mutations in the KATP channel genes. Journal of Clinical Endocrinology and Metabolism 2011. 96 141–145. ( 10.1210/jc.2010-1281) [DOI] [PubMed] [Google Scholar]
- 19.Martínez R, Fernández-Ramos C, Vela A, Velayos T, Aguayo A, Urrutia I, Rica I, Castaño L. & Spanish Congenital Hyperinsulinism Group. Clinical and genetic characterization of congenital hyperinsulinism in Spain. European Journal of Endocrinology 2016. 174 717–726. ( 10.1530/EJE-16-0027) [DOI] [PubMed] [Google Scholar]
- 20.De Leon DD, Stanley CA. Congenital hypoglycemia disorders: new aspects of etiology, diagnosis, treatment and outcomes: highlights of the Proceedings of the Congenital Hypoglycemia Disorders Symposium, Philadelphia April 2016. Pediatric Diabetes 2017. 18 3–9. ( 10.1111/pedi.12453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mannikko R, Flanagan SE, Sim X, Segal D, Hussain K, Ellard S, Hattersley AT, Ashcroft FM. Mutations of the same conserved glutamate residue in NBD2 of the sulfonylurea receptor1 subunit of the KATP channel can result in either hyperinsulinism or neonatal diabetes. Diabetes 2011. 60 1813–1822. ( 10.2337/db10-1583) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellanné-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, Valayannopoulos V, Gobrecht S, Sempoux C, Rahier J, et al ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. Journal of Medical Genetics 2010. 47 752–759. ( 10.1136/jmg.2009.075416) [DOI] [PubMed] [Google Scholar]
- 23.Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes 2007. 56 2339–2348. ( 10.2337/db07-0150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez CP, Stagljar M, Muhandiram DR, Kanelis V. Hyperinsulinism-causing mutations cause multiple molecular defects in SUR1 NBD1. Biochemistry 2017. 56 2400–2416. ( 10.1021/acs.biochem.6b00681) [DOI] [PubMed] [Google Scholar]
- 25.Santer R, Kinner M, Passarge M, Superti-Furga A, Mayatepek E, Meissner T, Schneppenheim R, Schaub J. Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism-hyperammonemia syndrome. Human Genetics 2011. 108 66–71. ( 10.1007/s004390000432) [DOI] [PubMed] [Google Scholar]
- 26.Yorifuji T, Kawakita R, Nagai S, Sugimine A, Doi H, Nomura A, Masue M, Nishibori H, Yoshizawa A, Okamoto S, et al Molecular and clinical analysis of Japanese patients with persistent congenital hyperinsulinism:predominance of paternally inherited monoallelic mutations in the KATP channel genes. Journal of Clinical Endocrinology and Metabolism 2011. 96 141–145. ( 10.1210/jc.2010-1281) [DOI] [PubMed] [Google Scholar]
- 27.Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, Midgley P, Shiota C, Buettger C, Magnuson MA, et al Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes 2003. 52 2433–2440. ( 10.2337/diabetes.52.9.2433) [DOI] [PubMed] [Google Scholar]
- 28.Hussain K. Insights in congenital hyperinsulinism. Endocrine Development 2007. 11 106–121. ( 10.1159/000111066) [DOI] [PubMed] [Google Scholar]
- 29.Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM, et al Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes 2009. 58 1419–1427. ( 10.2337/db08-1792) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanescu DE, Hughes N, Kaplan B, Stanley CA, De León DD. Novel presentations of congenital hyperinsulinism due to mutations in MODY genes: HNF1α and HNF4α. Journal of Clinical Endocrinology and Metabolism 2012. 97 E2026–E2030. ( 10.1210/jc.2012-1356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. Hepatocyte nuclear factor 4α gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. Journal of Pediatrics 2011. 158 852–854. ( 10.1016/j.jpeds.2011.01.003) [DOI] [PubMed] [Google Scholar]
- 32.Rieck S, White P, Schug J, Fox AJ, Smirnova O, Gao N, Gupta RK, Wang ZV, Scherer PE, Keller MP, et al The transcriptional response of the islet to pregnancy in mice. Molecular Endocrinology 2009. 23 1702–1712. ( 10.1210/me.2009-0144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, et al Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. Journal of Biological Chemistry 2010. 285 31806–31818. ( 10.1074/jbc.M110.123638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giri D, Vignola ML, Gualtieri A, Scaqliotti V, McNamara P, Peak M, Didi M, Gaston-Massuet C. Novel FOXA2 mutation causes hyperinsulinism, hypopituitarism with craniofacial and endoderm-derived organ abnormalities. Human Molecular Genetics 2017. 26 4315–4326. ( 10.1093/hmg/ddx318) [DOI] [PubMed] [Google Scholar]