

 This work is licensed under a
This work is licensed under a Abstract
Pheochromocytoma and paragangliomas (PCC/PGL) are neuroendocrine tumors that arise from chromaffin cells of the adrenal medulla and sympathetic/parasympathetic ganglia, respectively. Of clinical relevance regarding diagnosis is the highly variable presentation of symptoms in PCC/PGL patients. To date, the clear-cut correlations between the genotypes and phenotypes of PCC/PGL have not been entirely established. In this study, we reviewed the medical records of PCC/PGL patients with pertinent clinical, laboratory and genetic information. Next-generation sequencing (NGS) performed on patient samples revealed specific germline mutations in the SDHB (succinate dehydrogenase complex iron-sulfur subunit B) and SDHD (succinate dehydrogenase complex subunit D) genes and these mutations were validated by Sanger sequencing. Of the 119 patients, two were identified with SDHB mutation and one with SDHD mutation. Immunohistochemical (IHC) staining was used to analyze the expression of these mutated genes. The germline mutations identified in the SDH genes were c343C>T and c.541-542A>G in the SDHB gene and c.334-337delACTG in the SDHD gene. IHC staining of tumors from the c.343C>T and c.541-2A>G carriers showed positive expression of SDHB. Tumors from the c.334-337delACTG carrier showed no expression of SDHD and a weak diffused staining pattern for SDHB. We strongly recommend genetic testing for suspected PCC/PGL patients with a positive family history, early onset of age, erratic hypertension, recurrence or multiple tumor sites and loss of SDHB and/or SDHD expression. Tailored personal management should be conducted once a patient is confirmed as an SDHB and/or SDHD mutation carrier or diagnosed with PCC/PGL.
Keywords: PCC/PGL, SDHB, SDHD, genotype–phenotype correlation
Introduction
Pheochromocytomas/paragangliomas (PCC/PGLs) are tumors, arose from neural crest-derived chromaffin cells, produce and secrete catecholamines (1, 2, 3). PCCs are tumors of the adrenal medulla and PGLs originate from sympathetic (e.g. organ of Zuckerkandl) or parasympathetic (e.g. carotid body) paraganglia. The incidence of PCC/PGL is up to 8 per 100,000 with its peak onset around the 4th decade of lives (4, 5, 6). Most PCC/PGLs are benign but with high morbidity and mortality due to hypersecretion of catecholamines and metanephrines, which induce hypertension and cardiovascular diseases. It is estimated that ~30% PCC/PGLs are genetically inherited disease, and this percentage may rise as new PCC/PGL-causing mutations are being identified.
Succinate dehydrogenase (SDH) is a protein complex involving in both citric acid cycle and respiratory electron transfer chain reactions (7). The SDH complex comprises two anchoring subunits SDHC (succinate dehydrogenase subunit C) and SDHD and two catalytic subunits SDHA (succinate dehydrogenase complex flavoprotein subunit A) and SDHB. SDHB, an eight-exon gene localized on chromosome 1p36.13 and part of the mitochondrial electron transport complex II, is the most commonly mutated subunit in hereditary forms of PCC/PGLs. SDHD, the four-exon gene positioned on chromosome 11q23, is another member of the SDH complex (8). If any component of the SDH complex is lost, SDHB IHC becomes negative (9). Loss of SDHB by immunohistochemistry (IHC) in PCC/PGL is strongly correlated with SDH subunit gene mutation. So far, SDH deficiency has been observed in PCC/PGLs, gastrointestinal stromal tumors, pancreatic neuroendocrine tumor, renal carcinoma, pituitary adenoma and pulmonary chondroma (9, 10).
The Cancer Genome Atlas (TCGA) molecular taxonomy divides PCC/PGL into four main clusters: pseudohypoxia, Wnt-signaling, kinase-signaling and cortical mixture (11). The pseudohypoxia group can be divided into at least two subgroups. The tricarboxylic acid (TCA) cycle-related subgroup contains germline mutations in succinate dehydrogenase subunits SDHA, SDHB, SDHC, SDHD as well as SDHAF2 (succinate dehydrogenase complex assembly factor 2), FH (fumarate hydratase), MDH2 (malate dehydrogenase 2) and GOT2 (glutamic-oxaloacetic transaminase 2) (12, 13). The VHL/HIF2A-related subgroup shows both somatic and germline mutations (13). Germline mutations in SDH gene are responsible for 6–9% of sporadic PCC/PGLs, 29% of pediatric cases, 38% of malignant tumors and more than 80% of familial aggregations of PGL and PCC (14). Germline mutations in the SDHB gene are associated with hereditary paraganglioma syndrome type 4 (PGL4), while germline mutations of SDHD are present in hereditary paraganglioma syndrome type 1 (PGL1). The penetrance in SDHB and SDHD mutation-positive non-probands by age 60 years was only 21.8 and 43.2%, respectively (15). Furthermore, maternal transmission and genomic imprinting in SDHD could mask the hereditary nature of paraganglioma in rare cases (16). The difficulty of making a precise diagnosis delays appropriate treatment. Thus, hereditary PCC/PGL poses a significant challenge to clinicians.
Although the genetic basis of PCC/PGL is well characterized, the cancer-driving mutations for all PCC/PGL remain unknown. Here, we report the identification of a nonsense mutation and a splice site mutation in the SDHB gene and an SDHD frameshift mutation by genetic screening and immunohistochemistry.
Materials and methods
Patients and genetic testing
The Institutional Review Board of Daping Hospital of the Third Military Medical University approved this study. Written informed consents were obtained from the patients for use of their medical records and related images. A total of 119 PCC/PGL patients were diagnosed and underwent resection of their tumors in our institute between 2011 and 2018. The diagnoses were confirmed by three licensed pathologists based on H&E-stained tumor specimens (Fig. 1) and tumor-specific expression of CgA (chromogranin A), Syn (synaptophysin), CD56 (neural cell adhesion molecule 1), S-100 (S100 calcium-binding protein B), CK (choline kinase beta), MelanA (protein melan-A), HMB45 (human melanoma black), CD34 (CD34 molecule), SMA (survival of motor neuron 1, telomeric) and Ki-67 (proliferation marker protein Ki-67) (data not shown). For the genetic testing study, inclusion criteria consisted of the early age of onset, extra renal lesions, bilateral adrenal gland lesions, positive family history, recurrent or multifocal disease. To conduct Target Capture-Based Deep Sequencing (BGI Health, Shenzhen, Guangdong, China), total DNA isolated from peripheral blood cells of the patients was used to screen for potential mutations in the following genes: SDHAF2, SDHB, SDHC, SDHD, MAX (MYC associated factor X), NF1 (neurofibromin 1), RET (Ret proto-oncogene), VHL (von Hippel–Lindau) and TMEM127 (transmembrane protein 127). Upon identification of the mutations, Sanger sequencing was conducted on DNA of the probands’ family members to identify the specific mutation. Of these patients, 3 with SDHB or SDHD mutations; 21 in 5 families with VHL mutations; 10 in 4 families with RET mutations and 1 with somatic HIF2A, which has been described in our previous study (17, 18, 19).
Figure 1.

Histopathological features of the tumors. (A, proband 1; B, proband 2; C, proband 3; stained with H&E ×200.)
Immunohistochemistry
Immunohistochemical (IHC) staining was performed as described previously (9, 17, 20, 21). In brief, the tumor specimens were retrieved from the Department of Pathology of Daping hospital and IHC staining was performed on formalin-fixed paraffin-embedded tissues. The sections were deparaffinized and heat antigen retrieved using a citric acid buffer. The antibodies against SDHB (1:200, Proteintech; catalog number: 10620-1-AP) and SDHD (1:200, Bioss, Beijing, China; catalog number: ab08187596; immunogen range: 81–159 amino acid residue) were used. The HRP-labeled secondary goat anti-rabbit antibody was purchased from EnVisio Detection Systems (Dako). A peroxidase-labeled polymer was conjugated to immunoglobulins (DAKO) with 3,3-diaminobenzidine as a chromogen. The GIST (gastrointestinal stromal tumor) tissues were stained and served as an external positive control (9).
Results
Clinical characteristics
Of the 119 cases, 90 (75.6%) developed unilateral neoplasia, 10 (8.4%) developed bilateral tumors, 10 (8.4%) located in bladder, two in carotid body, two in duodenum and one in cerebellum, ear, mediastinum, pleura, rectum, respectively. Of note, four patients (3.4%) presented with malignant PCC/PGL. Among all the patients, three were identified with SDHx mutations.
Proband 1 was a 14-year-old boy. With blurred vision, intermittent headache and high blood pressure (208/156 mmHg), he was diagnosed as hypertensive retinopathic in November 2011. His VMA level was approximately two times of the normal level (72 µmol/24 h urine; normal level <35 µmol/24 h urine) (Table 1). Although craniocerebral MRI revealed no abnormalities, ultrasonography results suggest thyroid nodules and hypertensive heart disease. Enhanced CT scans of the thorax and abdomen revealed a 5.1 × 3.4 cm post-caval mass in the upper part of the abdomen (Fig. 2A and D). He underwent a tumor resection in November 2011 after taking oral alpha-receptor inhibitors for 2 weeks. Results from histopathologic examination of the tumor suggest he had a paraganglioma. His blood pressure became normal 3 days after tumor resection. Enhanced CT scanning of the thorax, abdomen and pelvic cavities showed no recurrence or metastasis. His blood pressure became normal in all the follow-ups and the last one was in August 2017. Briefly, in proband 1’s family, his father died of a stroke at age of 32 years. His mother was tested with Sanger sequencing, but no mutation was identified. His only uncle has hypertension. Therefore, we speculated that the mutation of the proband was inherited from his father. Other family members showed no evidence of PCC/PGL.
Table 1.
Characteristics of patients carrying SDH gene mutations.
| Patient one | Patient two | Patient three | |
|---|---|---|---|
| Gene | SDHB | SDHB | SDHD |
| Gender/age | Male/14 | Male/32 | Female/45 |
| Site | Post-caval | Para-aortic | Bifurcation of abdominal aorta |
| Size (cm) | 5.1 × 3.4 | 3 × 2 × 2 | 2.9 × 2.7 |
| Diagnosis | PGL | PGL | Hereditary PGL |
| Headache | + | – | – |
| Palpitation | + | – | + |
| Diaphoresis | + | – | – |
| Dizziness | – | – | + |
| Nausea | + | – | + |
| Hypertension (mmHg) | 208/156 | 160/100 | 154/75 |
| Nucleotide change | c.343C>T | c.541-2A>G | c.334_337delACTG |
| Mutation | P.Arg115Ter | IVS5-2A>G | p.Asp113Metfs*21 |
| Mutation type | Detrimental mutation | Pathogenic | Pathogenic |
| Heterozygous | Het | Het | Het |
| Aldosterone (erect position/decubitus; ng/mL) | 0.254/0.257 | NA | 0.13/0.14 |
| Cortisol (8/16/24 h; ng/mL) | NA | NA | 215/261/102 |
| Increased E | NA | NA | NA |
| Increased NE | NA | 1.89 ng/mL | NA |
| VMA | 72.0 µmol/24 h | 178.36 µmol/24 h | – |
| MN | 5.1 µmol/24 h | – | – |
| PTH | 41.04 pg/mL | NA | NA |
| 17-OH | 13.5 µmol/24 h | – | – |
| 17-KS | 30.2 µmol/24 h | – | – |
| Renin | NA | 35.71 µIU/mL; 50.38 µIU/mL (2 h after motivated) | 1.08 ng/mL0.75 ng/mL |
| PRA (erect position/decubitus; ng/mL) | NA | NA | 1.08/0.75 |
| Angiotensin II (erect position/decubitus; pg/mL) | NA | NA | 39/35 |
| ARR (erect position/decubitus) | NA | NA | 12/18 |
| ACTH (8/16/24 h; pg/mL) | NA | NA | 10.30/11.15/9.32 |
| Other disease | Thyroid nodule | Atrial flutter | HNPGL; renal cyst |
| Metastasis | – | – | – |
| Follow-up (months) | 76 | 5 | 41 |
| Outcome | NED | NED | NED |
‘+’ represented existence of this phenotype, ‘–’ nonexistence; 17-KS, 17-ketosteroid; 17-OH, 17-OH-corticosteroid; ACTH, adrenocorticotropic hormone; ARR, aldosterone renin ratio; E, epinephrine; MN, noradrenaline; NA, not available; NE, norepinephrine; NED, no evidence of disease; PRA, plasma renin activity; PTH, parathyroid hormone; VMA, vanillylmandelic acid.
Figure 2.

Abdominal CT or MRI scans of the probands. (A and D) Coronal (A) and axial (D) CT images of the 5.1 × 3.4 cm retroperitoneal mass between the aorta and inferior vena cava in proband 1. (B and E) Coronal (B) and axial MRI (E) images of the 3 × 2 × 2 cm retroperitoneal para-aorta mass in proband 2. (C and F) Coronal (C) and axial (F) images of the 2.9 × 2.7 cm mass located at the bifurcation of the abdominal aorta in proband 3.
Proband 2 was a 32-year-old male admitted to our hospital with a history of hypertension for 3 years. His blood pressure was 160/100 mmHg at diagnosis. Physical examination found no abnormalities. Laboratory test showed an elevated urine norepinephrine 1890 µg/L (normal range: 10–70 µg/L). MRI scans showed a 3 × 2 cm para-aortic mass in the middle of his abdomen (Fig. 2B and E). Laparoscopic surgery was attempted initially, but ultimately open surgery was required to remove the mass in December 2017. Pathological examination of the mass revealed a paraganglioma. His blood pressure became normal ten days after the surgery. In proband 2’s family, the father has hypertension for many years, and the mother did not have any abnormality. MRI or CT scan showed no evidence of PCC/PGL. The other family members refused referrals for further medical examination.
Proband 3 was a 45-year-old female with intermittent dizziness, palpitation and nausea for 1 year. History showed that a PGL located in the region of the right jugular foramen was diagnosed 5 years ago and resected at the West China Hospital (Sichuan Province, China). Hyperthyroidism was diagnosed 2 years ago. Enhanced CT scans revealed a 2.7 × 2.9 cm mass located at the bifurcation of the abdominal aorta (Fig. 2C and F). Laboratory tests revealed no abnormalities. She underwent laparoscopic tumor resection on December 12, 2014. Pathological examination revealed a paraganglioma. After surgery, her blood pressure returned to normal without medication. CT scans from the neck to pubic regions on her last follow-up in August of 2016 revealed no lesion. Proband 3’s parents and her two children showed no sign of PGLs and refused to be tested with Sanger sequencing.
Identification of mutations in the SDHB and SDHD genes
We identified two heterozygous germline mutations in the SDHB gene: c.343C>T in proband 1 (Fig. 3A) and c.541-2A>G in proband 2 (Fig. 3B). In addition, a frame-shift variant (c.334_337delACTG, p.Asp113Metfs*21) in exon 4 of the SDHD gene was detected in proband 3 (Fig. 3C). In addition, we identified a somatic point mutation in the SRD5A2 gene (c.578A>G) in proband 2. Of note, all the mutations were further confirmed by Sanger sequencing. There was no mutation in the remaining susceptibility gene panel.
Figure 3.
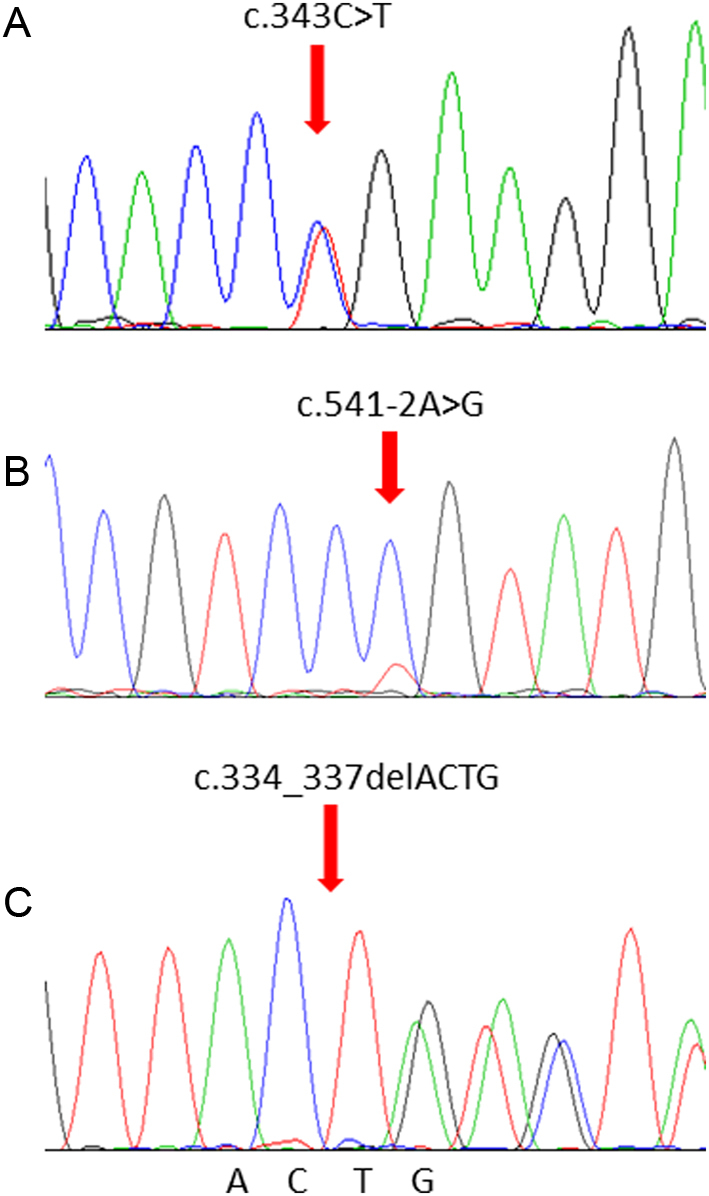
Gene sequencing reveals mutations in the SDHB and SDHD gene. (A) The mutation c.343C>T in proband 1. (B) The mutation c.541-2A>G in proband 2. (C) The mutation c.334_337delACTG in proband 3.
Expression of the mutated SDHB and SDHD
Since multiple lines of evidence indicate that IHC staining of SDHB is a robust and reliable surrogate marker for SDH gene mutations (9, 20, 21, 22, 23, 24), we conducted IHC of SDHB on all the tumor tissues. Positive expression of SDHB was observed using IHC staining in proband 1-derived tumor tissues that harbor the c.343C>T SDHB gene mutation (Fig. 4B). Expression of the c.541_2A>G SDHB mutant allele (proband 2) in PGL cells and surrounding endothelial and inflammatory cells revealed a distinct cytoplasmic granular staining pattern (Fig. 4C). Tissue samples of proband 3 (c.334_337delACTG mutation) were negative for SDHD (Fig. 4H) and showed weak diffused SDHB staining (Fig. 4D).
Figure 4.

IHC staining for SDHB and SDHD in the tumor tissues. (A, B, C and D) IHC staining for SDHB in the GIST positive control tissue (A), in PCC/PGL tissue of proband 1 (B), 2 (C), and 3 (D). (E, F, G and H) IHC staining for SDHD in the tumor tissue of the sporadic PGL patient (used as positive control tissue, E), in PCC/PGL tissue of proband 1 (F), 2 (G), and 3 (H). Magnification ×200.
Discussion
The literature search identified a total of eight reports with 13 c.343C>T mutation carriers in eight families (1, 4, 25, 26, 27, 28, 29, 30). Of which, Ivana Jochmanova reported the c.343C>T as a function affected mutation; van Hulsteijn et al. reported the c.343C>T as a pathologic mutation, which leads to malignant PGL with bone metastasis. In this study, we found this mutation caused an early onset of disease with a broad profile of clinical manifestations. Although the c.343C>T mutation results in the replacement of an arginine by a termination codon (p.Arg115Ter), IHC staining the showed positive SDHB in the tumor from the 14-year-old boy (Fig. 4B). This is consistent with previous studies showing that this nonsense mutation produces a truncated protein of less than half the full-length protein of 280 amino acids (1, 25). A recent nationwide study of 194 SDHB mutation carriers found the prevalence of c.343C>T mutation is about 1.5% (3/194; 1 with PCC and 2 with PGLs) (30), suggesting that this mutation is likely to be underestimated.
Since Timmers et al. first reported the c.541-2A>G mutation in 2007 (26), five additional reports have documented the same mutation. Four probands showed a positive family history of PCC/PGLs, and three had affected relatives while one presented with metastases (1, 26, 31, 32, 33). Noticeably, an infant carrier was diagnosed with leukoencephalopathy without PCC/PGL (33); a 19-year-old female carrier was diagnosed with hereditary oncolytic renal cancer (31) and an 11-year-old boy was diagnosed with polycythemia and abdominal PGL (32). In 2017, our team reported a case with a HIF2A somatic mutation-induced polycythemia and PCC and a case of HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal cell carcinoma (17, 34). It is likely for this reason that the pseudohypoxia-related PCC/PGL is fundamentally a metabolic disease. In our study, the expression of SDHB was similar to the external positive control (Fig. 4C). This is most likely due to the fact that the primary antibody targets only the amino acids present on the truncated protein. Therefore, antibodies specifically against the full-length, the N-terminal or C-terminal portions should be used in future studies. It seems that the c.541-2A>G carriers had a higher penetrance, early onset, more severe and complicated phenotypes, which warrants further investigation.
Though more than 130 unique SDHD gene mutations have been reported in hereditary PGL1 (35), only two studies listed the c.334_337delACTG variant as we report here (4, 36). Amar et al. reported the c.334_337delACTG mutant in a sporadic carrier and a syndromic or familial carrier (36), while Benn et al. reported two carriers of this mutant in a family with PCC, an abdominal PGL and HNPGL (4). Since none of these groups investigated the expression of this mutated gene, we are the first to study the expression of SDHD and SDHB in the c.334-337delATCG carrier. The results showed a weak and diffused SDHB staining pattern and with negative staining for SDHD (Fig. 4D and H). A previous study suggests that a weak-diffused pattern of SDHB may have a stronger correlation with mutations in SDHD rather than SDHB (37). Based on the findings in our study, c.334_337delATCG in the SDHD gene appeared to affect SDHB expression and thus linked to a more grievous phenotype (simultaneous PCC and PGL lesions). In addition, the adjacent mutation (c.337_340delGACT) has the same amino acid change (p.Asp113Metfs*21) with our case, which may indicate it is a hotspot mutation region.
PCC/PGL present as solitary lesions in 90–95% of cases (38). SDHB mutations mainly predispose to extra-adrenal PGLs and to a lesser extent to adrenal PCCs and HNPGLs, while SDHD mutations are typically associated with multifocal HNPGLs and less frequently with adrenal PCCs and extra-adrenal PGLs (39). PGLs are more frequently located in the head and neck region at the carotid bifurcation (carotid body tumor), along with the vagal nerve, in the jugular foramen and the middle ear space. Less common sites are close to the larynx, thyroid, urinary bladder and the upper mediastinum (14). The three probands identified in this study presented with retroperitoneal or pelvic PGLs. Notably, the c.334_337delACTG carrier in this study showed HNPGL in the right jugular foramen five years before entry into our study. In addition, we previously reported on multiple PGL patients with three tumors around the aorta abdominal and the inferior vena cava (17).
Malignant PCC/PGLs are defined by distant metastases commonly found in the liver, lung, bone, and lymph nodes. The term ‘metastatic PCC/PGL’ has been used to replace ‘malignant PCC/PGL’ in the latest WHO endocrine tumors classification (40). Only a minority of PCC/PGL patients harbor malignant tumors. Reported proportions of malignant PGL vary considerably between most genotype-phenotype studies, ranging from 31 to 71.4% in SDHB-mutation carriers to 0 to 22.7% in SDHD-mutation carriers (41). Although death can occur within a year of diagnosis, metastatic disease can be stable for more than 40 years. Detection of metastatic tumors can occur prior to the detection of primary tumors, but metastatic lesions also could be discovered more than 50 years after the primary diagnosis (42). Metastasis is more commonly associated with primary tumors located in the mediastinum (69%) and the infradiaphragmatic para-aortic area, including the organ of Zuckerkandl (66%) (43). In our cohort, 3.4% (4/119) presented with malignant tumors at diagnosis. The two SDHB germline mutation carriers did not present with metastases, but a literature review suggests that patients with such mutations may present with metastases in the neck, lung, mediastinum, abdomen and pelvic region. Rare cases of metastatic HNPGLs have been described within SDHD mutation carriers and their estimated prevalence is 0–10% (39). So far, metastatic lesions have not been recorded in c.334_337delACTG carriers.
SDH-deficient renal carcinoma defined by loss of SDHB expression represents a distinct and rare renal neoplasm subtype (9), showing a strong correlation with germline SDH mutations (44). Though it is likely that not all SDHB IHC-negative tumors will carry SDH mutations, IHC remains a phenotypic test as well as an indirect genotypic test. Though our patients presented with no signs of renal cancer, it is important to note the elevated life-long risk of PGL and renal cancer co-occurrence in such patients. At the same time, it is worthwhile to exclude the possibility of other tumors like GIST, pancreatic neuroendocrine tumor, pituitary adenoma and pulmonary chondroma.
In conclusion, we presented three gene-specific germline mutations in SDH genes and their relevant phenotypes. Findings of our study suggest that the incidence of c.343C>T mutations is likely underestimated in PCC/PGL patients. Patients with the SDHB mutation, c.541-2A>G, had severe and complicated phenotypes. The c.334_337delATCG SDHD mutation appears to influence SDHB expression and associates with a more aggressive phenotype. These specific cases add to our knowledge of PCC/PGLs and may help with the genetic counseling of patients. Genotype-tailored treatment options, follow-up and preventive care are warranted.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Acknowledgment
The author would like to thank all the patients in this study for their collaboration.
References
- 1.Jochmanova I, Wolf KI, King KS, Nambuba J, Wesley R, Martucci V, Raygada M, Adams KT, Prodanov T, Fojo AT, et al SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. Journal of Cancer Research and Clinical Oncology 2017. 143 1421–1435. ( 10.1007/s00432-017-2397-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korpershoek E, van Nederveen FH, Komminoth P, de Krijger RR. Familial endocrine tumours: pheochromocytomas and extra-adrenal paragangliomas – an update. Diagnostic Histopathology 2017. 23 335–345. ( 10.1016/j.mpdhp.2017.06.001) [DOI] [Google Scholar]
- 3.Patócs A, Lendvai NK, Butz H, Liko I, Sapi Z, Szucs N, Toth G, Grolmusz VK, Igaz P, Toth M, et al. Novel SDHB and TMEM127 mutations in patients with pheochromocytoma/paraganglioma syndrome. Pathology and Oncology Research 2016. 22 673–679. ( 10.1007/s12253-016-0050-0) [DOI] [PubMed] [Google Scholar]
- 4.Benn D, Gimenez-Roqueplo A, Reilly J, Bertherat J, Burgess J, Byth K, Croxson M, Dahia P, Elston M, Gimm O, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. Journal of Clinical Endocrinology and Metabolism 2006. 91 827–836. ( 10.1210/jc.2005-1862) [DOI] [PubMed] [Google Scholar]
- 5.Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clinic Proceedings 1983. 58 802–804. [PubMed] [Google Scholar]
- 6.Stenstrom G, Svardsudd K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Medica Scandinavica 1986. 220 225–232. [PubMed] [Google Scholar]
- 7.Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase – assembly, regulation and role in human disease. Mitochondrion 2010. 10 393–401. ( 10.1016/j.mito.2010.03.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000. 287 848–851. ( 10.1126/science.287.5454.848) [DOI] [PubMed] [Google Scholar]
- 9.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018. 72 106–116. ( 10.1111/his.13277) [DOI] [PubMed] [Google Scholar]
- 10.Niemeijer ND, Papathomas TG, Korpershoek E, de Krijger RR, Oudijk L, Morreau H, Bayley JP, Hes FJ, Jansen JC, Dinjens WN, et al Succinate dehydrogenase (SDH)-deficient pancreatic neuroendocrine tumor expands the SDH-related tumor spectrum. Journal of Clinical Endocrinology and Metabolism 2015. 100 E1386–E1393. ( 10.1210/jc.2015-2689) [DOI] [PubMed] [Google Scholar]
- 11.Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017. 31 181–193. ( 10.1016/j.ccell.2017.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Remacha L, Comino-Mendez I, Richter S, Contreras L, Curras-Freixes M, Pita G, Leton R, Galarreta A, Torres-Perez R, Honrado E, et al Targeted exome sequencing of Krebs cycle genes reveals candidate cancer-predisposing mutations in pheochromocytomas and paragangliomas. Clinical Cancer Research 2017. 23 6315–6324. ( 10.1158/1078-0432.CCR-16-2250) [DOI] [PubMed] [Google Scholar]
- 13.Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: towards a molecular classification. Endocrine Reviews 2017. 38 489–515. ( 10.1210/er.2017-00062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasini B, Stratakis C. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. Journal of Internal Medicine 2009. 266 19–42. ( 10.1111/j.1365-2796.2009.02111.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews KA, Vialard L, Ascher DB, Pires DEV, Bradshaw N, Cole T, Cook J, Irving R, Kumar A, Lalloo F, et al Tumour risks and genotype–phenotype–proteotype analysis of patients with germline mutations in the succinate dehydrogenase subunit genes SDHB, SDHC, and SDHD. Lancet 2016. 387 S19 ( 10.1016/S0140-6736(16)00406-2) [DOI] [Google Scholar]
- 16.Taschner P, Jansen J, Baysal B, Bosch A, Rosenberg E, Bröcker-Vriends A, van Der Mey A, van Ommen G, Cornelisse C, Devilee P. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes, Chromosomes and Cancer 2001. 31 274–281. ( 10.1002/gcc.1144) [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Wang Y, Tong D, Liu G, Yuan W, Zhang J, Ye J, Zhang Y, Yuan G, Feng Q, et al A somatic HIF2alpha mutation-induced multiple and recurrent pheochromocytoma/paraganglioma with polycythemia: clinical study with literature review. Endocrine Pathology 2017. 28 75–82. ( 10.1007/s12022-017-9469-4) [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Yuan G, Tong D, Liu G, Yi Y, Zhang J, Zhang Y, Wang LA, Wang L, Zhang D, et al Novel genotype-phenotype correlations in five Chinese families with Von Hippel-Lindau disease. Endocrine Connections 2018. 7 870–878. ( 10.1530/EC-18-0167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Tong D, Yuan W, Liu G, Yuan G, Lan W, Zhang D, Zhang J, Huang Z, Zhang Y, et al. Different RET gene mutation-induced multiple endocrine neoplasia type 2A in 3 Chinese families. Medicine 2017. 96 e5967 ( 10.1097/MD.0000000000005967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, Tissier F, Volante M, Matias-Guiu X, Smid M, et al SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Modern Pathology 2015. 28 807–821. ( 10.1038/modpathol.2015.41) [DOI] [PubMed] [Google Scholar]
- 21.Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, Richardson AL, Sidhu SB, Robinson BG, Clifton-Bligh RJ. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Human Pathology 2010. 41 805–814. ( 10.1016/j.humpath.2009.12.005) [DOI] [PubMed] [Google Scholar]
- 22.van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EMCA, Sleddens HFBM, Derkx P, Rivière J, Dannenberg H, et al An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncology 2009. 10 764–771. ( 10.1016/S1470-2045(09)70164-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mason EF, Sadow PM, Wagner AJ, Remillard SP, Flood TA, Belanger EC, Hornick JL, Barletta JA. Identification of succinate dehydrogenase–deficient bladder paragangliomas. American Journal of Surgical Pathology 2013. 37 1612–1618. ( 10.1097/PAS.0b013e318293d83c) [DOI] [PubMed] [Google Scholar]
- 24.Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, et al SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. Journal of Clinical Endocrinology and Metabolism 2011. 96 E1472–E1476. ( 10.1210/jc.2011-1043) [DOI] [PubMed] [Google Scholar]
- 25.Bayley JP, van Minderhout I, Weiss MM, Jansen JC, Oomen PH, Menko FH, Pasini B, Ferrando B, Wong N, Alpert LC, et al Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma. BMC Medical Genetics 2006. 7 1 ( 10.1186/1471-2350-7-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timmers H, Kozupa A, Eisenhofer G, Raygada M, Adams K, Solis D, Lenders J, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. Journal of Clinical Endocrinology and Metabolism 2007. 92 779–786. ( 10.1210/jc.2006-2315) [DOI] [PubMed] [Google Scholar]
- 27.Hensen EF, van Duinen N, Jansen JC, Corssmit EP, Tops CM, Romijn JA, Vriends AH, van der Mey AG, Cornelisse CJ, Devilee P, et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clinical Genetics 2012. 81 284–288. ( 10.1111/j.1399-0004.2011.01653.x) [DOI] [PubMed] [Google Scholar]
- 28.Lefebvre M, Foulkes WD. Pheochromocytoma and paraganglioma syndromes: genetics and management update. Current Oncology 2014. 21 e8–e17. ( 10.3747/co.21.1579) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Hulsteijn LT, Niemeijer ND, Hes FJ, Bayley JP, Tops CM, Jansen JC, Corssmit EP. Phenotype of SDHB mutation carriers in the Netherlands. Familial Cancer 2014. 13 651–657. ( 10.1007/s10689-014-9738-z) [DOI] [PubMed] [Google Scholar]
- 30.Niemeijer ND, Rijken JA, Eijkelenkamp K, van der Horst-Schrivers ANA, Kerstens MN, Tops CMJ, van Berkel A, Timmers H, Kunst HPM, Leemans CR, et al The phenotype of SDHB germline mutation carriers: a nationwide study. European Journal of Endocrinology 2017. 177 115–125. ( 10.1530/EJE-17-0074) [DOI] [PubMed] [Google Scholar]
- 31.Ricketts C, Shuch B, Vocke C, Metwalli A, Bratslavsky G, Middelton L, Yang Y, Wei M, Pautler S, Peterson J, et al Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. Journal of Urology 2012. 188 2063–2071. ( 10.1016/j.juro.2012.08.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choat H, Derrevere K, Knight L, Brown W, Mack E. SDHB-associated paraganglioma in a pediatric patient and literature review on hereditary pheochromocytoma-paraganglioma syndromes. Case Reports in Endocrinology 2014. 2014 502734 ( 10.1155/2014/502734) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helman G, Caldovic L, Whitehead M, Simons C, Brockmann K, Edvardson S, Bai R, Moroni I, Taylor J, Van Haren K, et al Magnetic resonance imaging spectrum of succinate dehydrogenase-related infantile leukoencephalopathy. Annals of Neurology 2016. 79 379–386. ( 10.1002/ana.24572) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Tong D, Liu G, Yi Y, Zhang D, Zhang J, Zhang Y, Huang Z, Li Y, Chen R, et al HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal-cell carcinoma. Cancer Biology and Therapy 2017. 18 944–947. ( 10.1080/15384047.2017.1394553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benn DE, Robinson BG, Clifton-Bligh RJ. 15 Years Of Paraganglioma: Clinical manifestations of paraganglioma syndromes types 1–5. Endocrine-Related Cancer 2015. 22 T91–T103. ( 10.1530/ERC-15-0268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, et al Genetic testing in pheochromocytoma or functional paraganglioma. Journal of Clinical Oncology 2005. 23 8812–8818. ( 10.1200/JCO.2005.03.1484) [DOI] [PubMed] [Google Scholar]
- 37.Castelblanco E, Santacana M, Valls J, de Cubas A, Cascón A, Robledo M, Matias-Guiu X. Usefulness of negative and weak–diffuse pattern of SDHB immunostaining in assessment of SDH mutations in paragangliomas and pheochromocytomas. Endocrine Pathology 2013. 24 199–205. ( 10.1007/s12022-013-9269-4) [DOI] [PubMed] [Google Scholar]
- 38.Marques RR, Bello CT, Rafael AA, Fernandes LV. Paraganglioma or pheochromocytoma? A peculiar diagnosis. Journal of Surgical Case Reports 2018. 2018 rjy060 ( 10.1093/jscr/rjy060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bardella C, Pollard P, Tomlinson I. SDH mutations in cancer. Biochimica et Biophysica Acta 2011. 1807 1432–1443. ( 10.1016/j.bbabio.2011.07.003) [DOI] [PubMed] [Google Scholar]
- 40.Lam A. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocrine Pathology 2017. 28 213–227. ( 10.1007/s12022-017-9484-5) [DOI] [PubMed] [Google Scholar]
- 41.van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EP. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. Journal of Medical Genetics 2012. 49 768–776. ( 10.1136/jmedgenet-2012-101192) [DOI] [PubMed] [Google Scholar]
- 42.Hamidi O, Young WF, Jr, Iniguez-Ariza NM, Kittah NE, Gruber L, Bancos C, Tamhane S, Bancos I. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. Journal of Clinical Endocrinology and Metabolism 2017. 102 3296–3305. ( 10.1210/jc.2017-00992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, Busaidy N, Cote GJ, Perrier N, Phan A, et al Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. Journal of Clinical Endocrinology and Metabolism 2011. 96 717–725. ( 10.1210/jc.2010-1946) [DOI] [PubMed] [Google Scholar]
- 44.Gill AJ, Hes O, Papathomas T, Šedivcová M, Tan PH, Agaimy A, Andresen PA, Kedziora A, Clarkson A, Toon CW, et al Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity a clinicopathologi series of 36 tumors from 27 patients. American Journal of Surgical Pathology 2014. 38 1588–1602. ( 10.1097/PAS.0000000000000292) [DOI] [PMC free article] [PubMed] [Google Scholar]