

Abstract
The inflammasome has been linked to metabolic disorders such as obesity and type 2 diabetes. Data now suggest that the crosstalk between the inflammasome and autophagy critically mediates cytoplasmic receptor NLRP3–dependent activation of the inflammasome by the saturated fatty acids contained in a high-fat diet.
Metabolic disorders such as obesity and type 2 diabetes have been linked to chronic inflammatory states1. Nonetheless, the precise mechanisms by which inflammation is triggered and maintained without overt infection in these pathophysiological states are poorly understood. Although macrophages are critically responsible for host defense by detecting and responding to invading pathogens, they can also sense damage-associated molecular patterns that are not pathogens through cytosolic receptors such as the Nod-like receptors (NLRs), which have the potential to lead to autoinflammatory disease. The inflammasome is a multiprotein complex that mediates the cleavage and activation of caspase-1, leading to maturation and release of the inflammatory cytokines interleukin 1β (IL-1β) and IL-18. Cytoplasmic receptors of the NLRP and NLR family are critical components of the inflammasome and interact with the adaptor protein ASC, which recruits the precursor form of caspase-1. Among the reported inflammasomes, the NLRP3 inflammasome has been shown to sense the widest range of targets, not only microbial and pathogen-associated molecular patterns but also damage-associated molecular patterns, as well as other triggers such as ATP, uric acid, necrotic cells and asbestos. Collectively, this suggests that NLRP3 is an important molecule in the regulation of noninfectious sterile inflammatory responses. The mechanism by which the NLRP3 inflammasome is activated remains the focus of great interest, and several key factors such as reactive oxygen species (ROS), ion flux and lysosomal destabilization all have crucial roles in inflammasome activation2. New studies from Wen et al. in this issue of Nature Immunology3 and Vandanmagsar et al. in Nature Medicine4 now demonstrate that NLRP3 is a key molecule in the activation of chronic inflammatory responses after consumption of a high-fat diet and is associated with dampening of insulin signaling and sensitivity. Furthermore, these two papers demonstrate that a macrophage’s ability to activate inflammasomes can induce other cell types such as hepatocytes or T cells to develop insulin resistance.
Because of the rapidly increasing prevalence of type 2 diabetes and its associated high medical costs, it is being rigorously studied to better understand its pathogenesis and the potential for therapeutic strategies. Many studies have demonstrated that proinflammatory cytokines, including IL-1β, tumor necrosis factor (TNF) and IL-6, are linked to the development of insulin resistance and diabetes. A clinical study has shown that inhibition of IL-1β signaling by anakinra, an antagonist of the IL-1β receptor, improves glycemia and beta-cell secretory function as well as diminishing systemic inflammation5. The clinical results obtained with anakinra and the emerging role of the inflammasome as a central mechanism of IL-1β release have prompted investigation of the inflammasome in the pathogenesis of type 2 diabetes. An early clue to its involvement was provided by the improved glucose tolerance and insulin sensitivity seen in NLRP3-deficient mice6. High concentrations of glucose itself also promote IL-1β release mediated by interactions among ROS, thioredoxin-interacting protein and NLRP3 in islets. Subsequent studies have shown that oligomers of islet amyloid polypeptide, a characteristic pancreatic deposition in type 2 diabetes, trigger the NLRP3 inflammasome in both dendritic cells and macrophages7. Furthermore, administration of pralnacasan, a pharmacological inhibitor of caspase-1, improves insulin sensitivity in genetically obese ob/ob mice8. Collectively, therefore, an abundance of data indicates IL-1β and inflammasome activation as underlying factors in insulin resistance. Wen et al. now show that palmitate, a saturated fatty acid whose concentrations are much higher in the plasma after consumption of a high-fat diet, triggers the release of IL-1β and IL-18 by an NLRP3 inflammasome–dependent pathway in macrophages3 (Fig. 1a). In contrast, an unsaturated fatty acid such as oleate (a so-called ‘good’ fat) does not affect inflammasome activation or IL-1β release. Although Vandanmagsar et al. show that ceramide, a lipid molecule composed of sphingosine and a fatty acid, activates the NLRP3 inflammasome, new synthesis of ceramide requires condensation of serine and palmitate to generate 3-keto-dihydrosphingosine4. This suggests that palmitate might also contribute to ceramide-mediated activation of the NLRP3 inflammasome, thereby demonstrating a common mechanistic link between the two studies.
Figure 1.
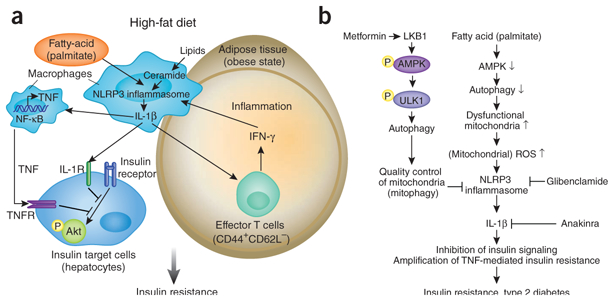
Involvement of the inflammasome in type 2 diabetes pathogenesis. (a) In macrophages, exogenous danger signals such as the saturated fatty acids contained in a high-fat diet and endogenous danger signals such as ceramide activate the NLRP3 inflammasome and trigger release of IL-1β. IL-1β deregulates insulin signaling, which potentially leads to insulin resistance in cells that are a target of insulin by both TNF-dependent and TNF-independent pathways. Enhanced expression of the NLRP3 inflammasome in adipose-tissue macrophages during an obese state is associated with activation of T cells, including production of interferon (IFN-γ) in adipose tissue, which promotes macrophage activation and systemic inflammation. NF-κB, transcription factor; IL-1R, IL-1 receptor; TNFR, TNF receptor; yellow ‘P’, phosphorylation. (b) Regulating the inflammasome via the machinery of autophagy could be a potent therapeutic target in type 2 diabetes. LKB1-mediated activation of AMPK promotes phosphorylation of ULK1, which initiates autophagy. The autophagy machinery controls mitochondrial homeostasis by removing old or damaged mitochondria (mitophagy). Fatty acids (such as palmitate) suppress the activation of AMPK, which leads to inhibition of autophagy and accumulation of dysfunctional mitochondria, along with enhanced generation of ROS. Enhancement of mitochondrial ROS promotes activation of the NLRP3 inflammasome and release of IL-1β. The pharmacological effects of anti-diabetes drugs are potentially linked to regulation of the autophagy and inflammsome pathways: metformin requires LKB1-dependent phosphorylation of AMPK to regulate glucose concentrations, and glibenclamide suppresses activation of the NLRP3 inflammasome in macrophages. Anakinra, an antagonist of the IL-1β receptor, may also be beneficial in type 2 diabetes.
Activation of the NLRP3 inflammasome is also critically linked to autophagy and disruption of mitochondrial homeostasis9. Wen et al. identify a previously unknown upstream mechanism by which fatty acid deregulates autophagy to trigger the inflammasome3. Palmitate activates the NLRP3 inflammasome via mitochondria-derived ROS, which is enhanced by palmitate-induced suppression of autophagy. Activation of AMPK, a key enzyme that controls the oxidation of fatty acids, in macrophages is suppressed by lipopolysac-charide plus palmitate, which results in less autophagic activity. AICAR, an AMPK agonist, can in turn restore the formation of autophagosomes and thereby inhibit both caspase-1 activation and ROS generation. AMPK is therefore an effector molecule upstream of autophagy and a negative regulator of inflammasome (Fig. 1b). Additionally, treatment of macrophages with lipopolysaccharide plus palmitate induces alteration of mitochondrial homeostasis by enhanced generation of mitochondrial ROS. Together these results suggest that activation of AMPK regulates the NLRP3 inflammasome by autophagy-mediated mitochondrial homeostasis (‘mitophagy’; Fig. 1b).
In addition to discovering palmitate and ceramide as additional type 2 diabetes–associated damage-associated molecular patterns, Wen et al. and Vandanmagsar et al. also investigate the potential target cells of inflammasome-activated macrophages3,4. Vandanmagsar et al. link macrophages with T cells specifically in adipose tissue4. Wen et al. focus instead on cells responsible for glucose metabolism that are important targets of insulin; that is, hepatocytes3. Insulin signaling, as measured by phosphorylation of the serine-threonine kinase Akt, is impaired in hepatocytes incubated with conditioned medium from macrophages stimulated with lipopolysaccharide and palmitate but not in those incubated with conditioned medium from macrophages genetically deficient in caspase-1 or the NLRP3 inflammasome. Accordingly, antagonism of the IL-1β receptor or neutralization of TNF partially restores Akt activation. Neutralizing the effects of both of these proinflammatory cytokines together triggers complete recovery of Akt activation, which emphasizes the importance of both TNF and IL-1β in blocking insulin signaling. To further investigate the role of IL-1β in insulin resistance, Wen and colleagues investigate the effect of IL-1β on insulin sensitivity in Tnf−/− and Il1b−/− mice3. Loss of TNF or IL-1β substantially improves insulin sensitivity, and the amelioration of insulin sensitivity in Tnf−/− mice is diminished by the administration of IL-1β. The role of TNF in insulin resistance is well known, but the data from Wen et al. demonstrate IL-1β to be another potentially important participant in this process3.
Although these two studies yield informative insights into how regulation of the inflammasome could be used in the treatment or prevention of insulin resistance, several questions are raised about its mechanism of action in type 2 diabetes. For example, it remains unclear how the adaptive immune system contributes to the development of insulin resistance. How do effector T cells activated via the macrophage inflammasome influence insulin secretion by pancreatic beta cells or the insulin sensitivity of hepatocytes? In addition, although the inflammasome has been investigated extensively in cells of the innate immune system, it will be worthwhile to determine whether the deletion of inflammasome components from cells that are targets of insulin themselves has an effect on insulin sensitivity. It might also be possible that another proinflammatory mediator such as IL-6 contributes to insulin resistance in vivo.
Despite the many studies that have linked inflammation to the pathogenesis of type 2 diabetes, the drugs now used for the treatment of this disease have generally not been based on an anti-inflammation strategy. That being said, anakinra, a drug more normally used for the treatment of rheumatoid arthritis, has shown therapeutic potential for the treatment of type 2 diabetes5. Another drug, glibenclamide, has been widely used as an antidiabetic drug for decades because of its ability to stimulate insulin secretion from beta cells. Glibenclamide operates by inhibiting ATP-sensitive potassium channels but, interestingly, also strongly suppresses caspase-1 activation and IL-1β release by inhibiting signals upstream of NLRP3 (but downstream of the ATP P2X7 receptor)10. These observations suggest that a strategy for the treatment of type 2 diabetes based on anti-inflammation remains not only viable but potentially quite promising, as long as IL-1β signaling is involved as a target. Finally, the data from the study by Wen et al. suggest another potent therapeutic target: autophagy3. This evolutionarily conserved cellular process facilitates the turnover of damaged proteins and organelles such as mitochondria11, and its dysfunction has been associated with various human diseases and hyperinflammation9,11. Metformin, one of the drugs most commonly used for the treatment of type 2 diabetes, controls circulating glucose concentrations through activation of the kinases LKB1 and AMPK12. The present study by Wen et al.3, as well as published studies of tumor cells, have shown that AMPK elicits autophagy. It is possible, therefore, that the beneficial effects of metformin in type 2 diabetes might arise from its ability to regulate the inflammasome by enhancing autophagic activity (Fig. 1b).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Hotamisligil GS Nature 444, 860–867 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Schroder K & Tschopp J Cell 140, 821–832 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Wen H et al. Nat. Immunol 12, 408–415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandanmagsar B et al. Nat. Med 17, 179–188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsen CM et al. N. Engl. J. Med 356, 1517–1526 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Zhou R, Tardivel A, Thorens B, Choi I & Tschopp J Nat. Immunol 11, 136–140 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Masters SL et al. Nat. Immunol 11, 897–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stienstra R et al. Cell Metab 12, 593–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakahira K et al. Nat. Immunol 12, 222–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamkanfi M et al. J. Cell Biol 187, 61–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine B, Mizushima N & Virgin HW Nature 469, 323–335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw RJ et al. Science 310, 1642–1646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]