

Abstract
Gaucher disease is an autosomal recessive lysosomal storage disorder resulting from mutations in the gene GBA1 that lead to a deficiency in the enzyme glucocerebrosidase. Accumulation of the enzyme’s substrates, glucosylceramide and glucosylsphingosine, result in symptoms ranging from skeletal and visceral involvement to neurological manifestations. Nonetheless, there is significant variability in clinical presentations amongst patients, with limited correlation between genotype and phenotype. Contributing to this clinical variation are genetic modifiers that influence the phenotypic outcome of the disorder. In this review, we explore the role of genetic modifiers in Mendelian disorders, and describe methods to facilitate their discovery. In addition, we provide examples of candidate modifiers of Gaucher disease, explore their relevance in the development of potential therapeutics, and discuss the impact of GBA1 and modifying mutations on other more common diseases like Parkinson disease. Identifying these important modulators of Gaucher phenotype may ultimately unravel the complex relationship between genotype and phenotype and lead to improved counseling and treatments.
Keywords: genetic modifiers, Mendelian disorders, Gaucher disease, genotype-phenotype correlation, glucocerebrosidase, Parkinson disease
Introduction
While thousands of diseases are classified as single-gene Mendelian disorders, it has become increasingly clear that this definition is often an oversimplification. Were this distinction completely true, single gene disorders should manifest similarly in patients with the same genotype. Instead, many monogenic diseases, including Gaucher disease, are characterized by a wide variety of clinical presentations, even within families and among those carrying the same mutations in the primary gene of interest (Weatherall, 2001; Jan de Beur et al., 2003; Lachmann et al., 2004; Cammarata et al., 2015). This inherent variability in presentation of monogenic disorders is likely due to a multitude of factors, including genetic background, environment, and epigenetic status, which are specific to each patient (Sidransky, 2004).
Genetic background refers to the genetic make-up of an individual, and includes the genotype across all loci. Within genetic background, there may be many single nucleotide variants or even more common single nucleotide polymorphisms (SNPs) that affect the presentation of traits associated with disease, and hence the resulting phenotype. These disease-modifying genetic variants have been a focus of study since the concept was first introduced (Haldane, 1941), and are often used to understand unexpected phenotypes, as well as to identify new therapeutic targets. While the definition of a modifier gene has varied throughout time, it can be summarized as a genetic variant that interacts with a primary variant through a variety of different mechanisms impacting the RNA or protein. These range from changing gene expression to altering protein function, resulting in varied phenotypic expression (Génin et al., 2008).
Mechanisms of Action of Genetic Modifiers
In general, modifier genes can alter clinical phenotype through four basic mechanisms: changes in penetrance, expressivity, dominance, and/or pleiotropy (Nadeau, 2001). Penetrance is the ratio of all individuals carrying a disease allele to those individuals affected by the disease. An example of reduced genetic penetrance due to modifiers occurs in autosomal-dominant retinitis pigmentosa (MIM# 600138). This disease is caused by a mutation in the gene PRPF31 (pre-mRNA processing factor 31), which typically results in severe visual impairment. However, some patients who carry the disease-causing allele remain asymptomatic. This is often due to a mutation in the gene CNOT3 (CCR4-NOT transcription complex subunit 3), which leads to an increase in wild-type PRPF31 protein expression and rescues the disease phenotype (Venturini et al., 2012).
Similarly, genes that alter expressivity can affect the phenotypic severity of a disease. Expressivity refers to the degree to which expression of a trait differs among individuals. An example of a gene modifying expressivity is seen in the disorder familial hypertrophic cardiomyopathy (MIM# 115195, 192600), caused by autosomal dominant mutations in MYBPC3 (myosin binding protein C), resulting in thickened heart walls, chest pain, aortic arrhythmia, and sudden cardiac arrest in some patients. Since myosin binding protein C, the protein encoded by MYBPC3, functions with myosin binding protein H (encoded by MYBPH) to regulate cardiomyocyte contraction (Mouton et al., 2015), the effect of MYBPH on disease severity was subsequently investigated. A MYBPH isoform was found to increase the thickness of heart walls in individuals with specific MYBPC3 mutations, and thus accentuate the disease manifestations (Mouton et al., 2016).
The presence of expected dominance relationships in diseases thought to be autosomal recessive or dominant could also be modified by genetic background, i.e. mutations in a locus separate from the disease-causing locus can change the effects of alleles at the disease-causing locus. For example, homozygous mutations in HFE (homeostatic iron regulator), usually lead to hereditary haemochromatosis, or iron overload (MIM# 235200). However, even heterozygotes for a HFEC282Y allele can develop increased iron absorption when inherited alongside a heterozygous loss of function mutation in HAMP (hepcidin antimicrobial peptide). This gene encodes hepcidin, a protein critical in the maintenance of iron homeostasis through its roles in regulation of iron storage in macrophages and in intestinal iron absorption (Merryweather-Clarke et al., 2003).
Pleiotropy occurs when a single gene has two or more different effects. Examples of pleiotropic effects occur in Bardet-Biedl syndrome (MIM# 209900, 600151, 605231, 615981 615996, 617406, 617119) and Meckel-Gruber syndrome (MIM# 249000, 267010, 603194, 607361, 611561, 611134, 612284, 613885, 614175, 614209, 615397, 616258). Both disorders result from single mutations in multiple genes and manifest with ciliary dysfunction. However, a mutation in MKS1 (Meckel syndrome type 1 protein), a gene typically associated with Meckel-Gruber syndrome, was found to promote seizures in patients with Bardet-Biedl syndrome, although seizures are not a typical manifestation of either disease (Leitch et al., 2008).
Gaucher Disease: A Monogenic Disorder with Clinical Heterogeneity
Modifier genes and the prospective mechanisms underlying their ability to alter clinical outcome are well established in some Mendelian diseases. While the genetic modifiers found have not always resulted in new therapeutics, they have advanced their respective fields forward, and allowed researchers to better understand the heterogeneity in diseases termed monogenic, sometimes explaining disease phenotypes, disease severity, or drug efficacy. Modifiers elucidated in these different monogenic disorders, as well as the strategies employed in their identification, may inform studies in other disorders such as Gaucher disease. The role of modifier genetics in select well-characterized diseases has been previously reviewed (Gusella et al., 2014; Cutting, 2015; Chang et al., 2018).
The clinical manifestations of Gaucher disease (GD; MIM# 230800, 230900, 231000, 231005, 606463, 608013) are known to be affected by modifier genes. GD is an autosomal recessive lysosomal storage disorder caused by mutations in the GBA1 (glucosylceramidase beta 1) gene, resulting in a deficiency of the enzyme glucocerebrosidase (GCase). As a result, the glycolipids glucosylceramide and glucosylsphingosine accumulate within the lysosomes of macrophages. Macrophages are particularly vulnerable to this buildup due to their role in phagocytizing the glycolipid-rich cell membranes of erythrocytes and leukocytes. Some aspects of GD pathogenesis have been directly attributed to glycolipid accumulation within these macrophages, as it results in increased expression of inflammatory cytokines and the storage of toxic species resulting from alternative breakdown pathways. However, many pathogenic mechanisms are yet unrealized.
GD presents as a multi-system disorder with non-neuronopathic (type 1) and acute and subacute neuronopathic (types 2 and 3, respectively) forms. The clinical outcomes range from asymptomatic adults to critically ill infants, with sequelae including hepatosplenomegaly, bone crises, horizontal gaze palsy, myoclonic epilepsy, and difficulty swallowing among others (Rosenbloom and Weinreb, 2013; Stirnemann et al., 2017). Current FDA-approved treatments for GD include enzyme replacement and substrate reduction therapies, both of which ameliorate the visceral symptom, but fail to cross the blood barrier, and thus do not alleviate neurological manifestations. To fill this need, treatments that can reach the brain are in development, and are becoming a major research focus, particularly since GD and GBA1 dysfunction have come into the spotlight as a risk factors for Parkinson disease (PD; MIM# 168600) (Siebert et al., 2014).
GD is characterized by significant genotypic and phenotypic heterogeneity; the division of Gaucher disease into several types illustrates this spectrum of pathology. However, these classifications are not rigid, as a range of manifestations exist between and within types. Multiple genotypes and over 500 different GBA1 mutations have been identified in patient samples, and while specific genotypes have been associated with the different forms of the disease, patients with the same genotype can display a variety of symptoms (Beutler, 2001; Lachmann et al., 2004; Fairley et al., 2008). For example, it was shown that patients who all shared the genotype p.L483P/p.L483P have different levels of residual GCase enzymatic activity, and moreover, the associated phenotypic spectrum includes children with autism or severe intellectual impairment, patients with myoclonic epilepsy, and highly successful college graduates (Goker-Alpan et al., 2005). This vast genotypic and phenotypic variation is a feature shared by many lysosomal storage disorders, and monogenic diseases in general. Thus, lessons learned from other disorders may provide insights into factors contributing to phenotypic variation, as well as strategies for the identification of these factors in GD. In turn, mechanisms contributing to phenotype in GD may inform studies of unrelated monogenic disorders. Elucidating these unknown modifiers and their functions may provide a pathway leading to the missing heritability of Gaucher phenotypes.
Approaches to Identify Genetic Modifiers
Investigators have utilized a variety of methods to search for potential modifiers of different diseases and biological processes. As highlighted in Figure 1, the allele frequency and effect size of a genetic variant can complicate our ability to uncover specific genes which modify clinical phenotypes (Fig. 1). Variants with low frequency, but with an intermediate or low effect size, have been hypothesized to drive missing heritability and collectively cause large shifts in penetrance, but their identification through conventional approaches has proved the most challenging (McCarthy et al., 2008; Stratton and Rahman, 2008). Although the type of variants/modifiers found depends heavily on the technique used for their identification, the method employed is usually driven by data availability (linkage or association studies) and/or preferred approach (systematic or biology driven) (Génin et al., 2008).
Figure 1.
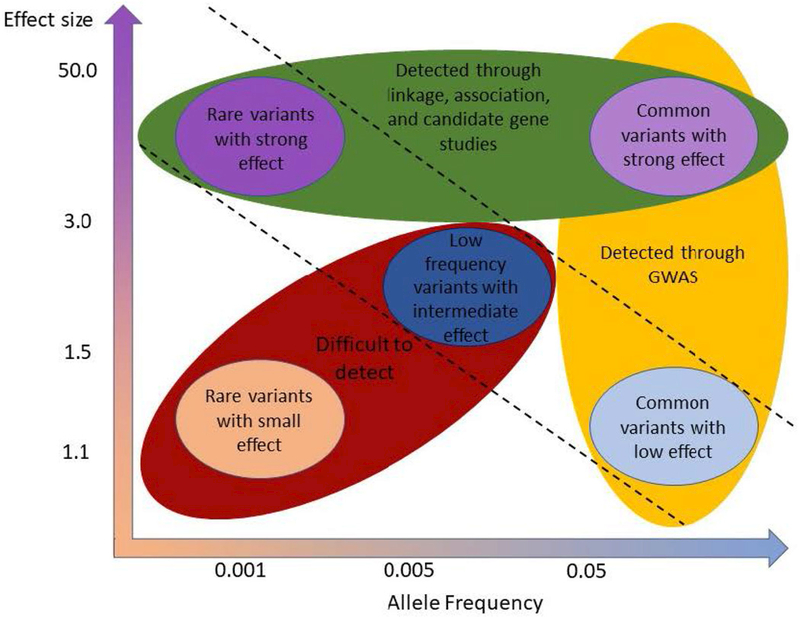
Methods for and challenges of discovering modifiers, incorporating the influence of effect size and allele frequency. Rare or common variants with a large effect size (green) can often be appreciated through linkage and association studies. GWAS can often successfully identify common variants (yellow), even those with a small effect. However rare or low frequency variants with a small or intermediate effect size (red) can often prove difficult to identify, and techniques for their detection are in development/refinement. Adapted from McCarthy et al., 2008 and Manolio et al., 2009.
Linkage analysis is based on the concept that genes in close proximity are more likely to be inherited together, and as such, SNPs can be used to track certain gene regions that may segregate with phenotype. This systematic technique is commonly performed when family genetic data is available and is particularly valuable when allelic heterogeneity is present, as linkage analysis identifies causal loci instead of alleles. For example, Wright et al. performed a linkage study on 486 sibling pairs with Cystic Fibrosis (CF, MIM# 2097000) and found a locus at 20q13.2 that may contribute to 50% of the variance in lung function between siblings (Wright et al., 2011). However, these studies have often failed to detect non-Mendelian disease-causing variants.
In addition to linkage studies, investigators have used genome-wide association studies (GWAS) to identify modifiers. This technique has advantages over linkage studies in terms of the data required and specificity of outputs; association studies do not require family data, and can identify variants which are causal of phenotype. In a GWAS, DNA from both controls and patients is analyzed on microarrays containing extremely large numbers of SNPs, and the SNPs which are found more often among patients are thought to be associated with phenotype. However, large sample sizes are needed in order to determine the contribution of rare variants to phenotype (Lindquist et al., 2013). As such, considerable effort is being used to improve GWAS techniques, including microarrays with a greater number of SNPs, and the rise of imputation, both of which allow for the analysis of increasingly more rare variants (Hoffman and Witte, 2015). In imputation, genotypes of rare variants which were not determined through microarray can be assumed, based on the matching of haplotypes to those of reference genomes where the sequence is known. Using this method, many more rare variants than those assayed directly can be screened at once, and sample sizes covering these variants can be increased. For example, Hill-Burns et al. used an imputed GWAS to screen the association of 7.2 million SNPs with PD age of onset in those with first or second-degree relatives with PD. The study yielded one directly genotyped variant that mapped to LHFPL2 (LHFPL tetraspan subfamily member 2) on 5q14.1 and one imputed variant that mapped to TPM1 (tropomyosin 1) on 15q22.2 at the most stringent significance level (Hill-Burns et al., 2016). However, imputation is still a method under development, and problems such as controlling for genetic background, de novo mutations, and non-genic regions are currently being addressed (Hoffman and Witte, 2015). Yet, the use of next-generation sequencing to sequence target loci related to GWAS-defined associations and candidate genes associated with severe phenotypes may be robust (Landis et al., 2017), and whole-exome sequencing may further reveal genetic modifiers. The advantages of such systematic screens over smaller-scale approaches is that more data will be derived, and unanticipated genes may be found. However, this is a double-edged sword, as working at such a large population level may introduce confounding variables that limit the ability to draw clear conclusions from variant-phenotype correlations and most of the variants identified may lack clear functional connections to the phenotype of interest (Choudhry et al., 2006; Manolio et al., 2009; Martin and Eskin, 2017).
Aside from GWAS, other global screens for genetic modifiers have also been explored. Global RNAi screens are designed to knock-down random genes using siRNAs in order to explore how they affect phenotype, and are most often performed using cell models in a high-throughput manner (Mohr and Perrimon, 2012). Genome-wide screens in in vivo animal models, ranging from Caenorhabditis elegans to mice, have similarly been used to identify genetic modifiers by altering expression in a variety of genes through techniques ranging from N-ethyl-N-nitrosourea mutagenesis to RNAi and CRISPR, and determining how these alterations might affect phenotype. One pitfall of such screens is the possibility for both false negative and positives, leading to a need for validation. However, a variety of diseases have been probed for modifiers through such expression altering screens, including Parkinson disease, Huntington disease (MIM# 143100), amyotrophic lateral sclerosis (MIM# 105400), and metastatic cancer. (Zhang et al., 2010; Fernandes and Rao, 2011; Van Hoecke et al., 2012; Chen et al., 2015). Proteomic analyses also provide powerful tools to explore genetic modification of disease, with techniques including comparisons of protein expression in model organisms with different genetic backgrounds through two-dimensional gel electrophoresis, as well as determining protein interactions through mass spectrometry (De Castro et al., 2012; Shirasaki et al., 2012).
An alternative for such wide-scale screens for modifiers is to use a more targeted candidate gene approach by searching for modifiers based on biological relevance, starting with related pathways or indirectly connected biological processes (Génin et al., 2008). For example, when exploring modifiers in a given “simple” disorder, both the processes related to the expression of the mutated gene’s protein product, and the genes involved in downstream effects of resultant pathology would be relevant. When searching for modifiers of autosomal dominant polycystic kidney disease (ADPKD, MIM# 173900; caused by mutations in PKD1 (polycystin 1), Nikonova et al. hypothesized that NEDD9 (neural precursor cell expressed developmentally downregulated protein 9) may influence disease outcome based on its roles in activating Src pathways and regulating cellular attachment and migration, processes disrupted in patients with ADPKD. They tested this hypothesis in mice, creating knockouts for nedd9, pkd1, and a combined knockout of both genes and searched for phenotypic differences. The compound knockouts displayed far more kidney expansion and cystogenesis, implicating nedd9 as a modulator of pathology (Nikonova et al., 2014). Such studies illustrate the vital role of secondary pathways on disease pathogenesis and their capacity to modify disease outcome.
Modifiers of Gaucher Disease: Results of Non-Targeted Studies
Several “agnostic” or non-targeted techniques have been employed to identify genetic modifiers of GD, including in-vitro, animal, and human studies. Zhang et al. performed a GWAS in Ashkenazi Jewish patients with type 1 GD (N370S homozygotes). They genotyped >500,000 SNPs scattered throughout the genome, pinpointing SNPs within CLN8 (transmembrane ER and ERGIC protein), which they implicated as a potential modifier. While its full function is still unknown, CLN8 is involved in GD-related pathways such as lipid trafficking, membrane trafficking, and autophagy/mitophagy. Mutations in CLN8 cause a neuronal ceroid lipofuscinosis known as Northern epilepsy (MIM# 610003) (Passantino et al., 2013). The authors proposed that this gene may play a protective role in GD, noting higher expression levels in fibroblasts of patients with milder symptoms (Zhang et al., 2012). Despite this functional assessment, further confirmation is necessary, especially since this potential modifier of GD expressivity did not meet significance after Bonferroni corrections.
Klein et al. performed a GWAS analysis on 15 different strains of mice that were treated with conduritol-β-epoxide, an inhibitor of GCase, yielding SNPs in 17 genes. The researchers found great variability in disease severity among the different murine strains despite the same injection doses and regimens, including differences in lifespan and levels of glucosylceramide. Many of the genes identified were involved in glutamate-related pathways, including grin2B, which encodes a subunit of the NMDA receptor. To validate the hypothesis that glutamate pathology was affecting disease expressivity, they treated groups of short lived mice with an antagonist of the NMDA receptor, which mediated an increase in lifespan. Further, GRIN2B (glutamate ionotropic receptor NMDA type subunit 2B) expression was found to be significantly elevated in human brain tissue derived from a patient with type 2 GD, further substantiating the role of the glutamate pathway in neuronopathic GD. (Klein et al., 2016). Glutamate-related pathways have been previously functionally implicated in neuronopathic GD (Korkotian et al., 1999; Pelled et al., 2000, 2005), but the therapeutic potential of modulation of the glutamate pathway remains unknown.
Epigenetic tools have also been used to evaluate modifiers in GD. Siebert et al. performed a screen of 875 miRNA mimics in Gaucher fibroblasts to identify miRNAs that modulate GCase activity. They found that miR-127-5p decreased GCase activity and protein expression via downregulation of LIMP-2, a known GCase transporter (Siebert et al., 2014). Furthermore, multiple miRNAs were found to increase GCase expression and activity, meriting further investigation on both a basic and translational level, as their mechanism of action and their potential as eventual therapeutics are unknown. Non-protein coding regions such as enhancer/silencer regions and those that encode non-coding RNAs have been increasingly implicated in the pathogenesis of diseases as GWASs continue to find hits in these non-coding regions, and great effort is being placed in creating new techniques which allow study of their function (Cech and Steitz, 2014; Elkon and Agami, 2017). The contribution of epigenetics to GD and lysosomal storage disorders as a whole, has been recently reviewed (Hassan et al., 2017).
Modifiers of Gaucher Disease: Results of Candidate Gene Studies
In addition to genome-wide approaches, some investigators have pursued specific candidate genes (summarized in Table 1) hypothesized to affect the Gaucher phenotype. These include genes encoding proteins involved in the activation or transport of glucocerebrosidase, glycolipid synthesis or degradation, or regulatory functions, as well as downstream pathways related to GD pathology. However, the most well-studied and highly-relevant modifiers of GD are PSAP (prosaposin, a precursor protein) and SCARB-2 (Scavenger receptor class B member 2, coding for lysosomal integral membrane protein 2, LIMP-2.)
Table 1.
Summary of articles covering proposed modifiers for Gaucher disease and related diseases.
Prosaposin (PSAP)
Among the proteins known to impact the processing of gluocosylceramide that serve as candidate modifiers is Saposin C (Sap C), a GCase activator. A product of the cleavage of prosaposin,in the lysosome, Sap C is encoded by the gene PSAP. Sap C was initially discovered as an activator of GCase activity by Ho and O’Brien in 1971, and has been implicated in GD pathology since, through clinical and functional studies (Ho and O’Brien, 1971; Christomanou et al., 1986; Tamargo et al., 2012). Deficiencies in either Sap C or GCase can affect the interaction of these proteins, disrupting GCase function and causing Gaucher manifestations. The exact interaction between the two proteins is not fully understood, although some hypothesize a liftase model in which Sap C attaches to the lysosomal membrane and presents lipid substrates to GCase for subsequent degradation (Rossmann et al., 2008; Tamargo et al., 2012). Certain GBA1 mutations, particularly p.N409S, may cause pathology by disrupting the interaction with Sap C, which could be exacerbated by a mutation in PSAP. Salvioli et al. observed that when anionic phospholipids were reduced in the lysosomal membrane, wildtype Sap C activated wildtype GCase, but not p.N409S mutant GCase (Salvioli et al., 2005).
The GCase/Sap C interaction has been shown to play a role in modulating expressivity in mouse models. Sun et al. created a mouse model with a knock-in point mutation in exon 11 of the PSAP gene, creating Sap C deficient mice. These mice exhibited central nervous system (CNS) abnormalities including ataxia, axonal impairment, reduced neuromotor functioning, and a moderate increase in glucosylceramide storage (Sun et al., 2010b). They then crossed these Sap C deficient mice with p.V433L/ p.V433L GD mice that did not have a neuronopathic phenotype. The offspring, deficient for both gba and psap, exhibited decreased GCase activity, increased brain levels of glucosylceramide and glucosylsphingosine, and earlier CNS manifestations, compared to mice with mutations in gba or psap alone (Sun et al., 2010a). Thus, deficient Sap C may increase GD-related symptoms, particularly CNS abnormalities and glycosphingolipid storage.
Mutations in PSAP have also been noted in a subset of patients with GD symptoms. Multiple patients with PSAP mutations in the Sap C domain have been reported: some exhibited features of type 1 GD, while others presented with specific neurological symptoms characteristic of patients with type 3 GD (Tamargo et al., 2012). Additionally, Kang et al. published a case report highlighting a patient with GD resulting from biallelic PSAP mutations who had Gaucher cells and increased chitotriosidase activity, a biomarker elevated in GD, despite normal GCase activity. The authors proposed that the PSAP mutations were responsible for the patient’s GD-like manifestations (Kang et al., 2018). Thus, the contribution of Sap C levels to patient manifestations merits further evaluation.
Sap C may play a crucial role in ameliorating GCase deficiency, as synthetic Sap C increases the activity of GCase in-vitro (Yoneshige et al., 2015). Thus, it has been proposed that in GD, co-administration of Sap C and enzyme replacement therapy may provide a synergistic effect and improve treatment. Even though the role of Sap C has been well studied in GD, further evaluation is necessary to better define its interaction with GCase and whether it can be exploited as a treatment option.
SCARB2
Clinical and functional studies suggest that LIMP-2, a GCase transporter encoded by the gene SCARB2, may modify GD expressivity. LIMP-2 transports GCase to the lysosome either independently or as part of a complex with mannose-6-phosphate, and, as a result, is critical to GCase function. Until recently it was thought that LIMP-2 functioned only independently of mannose-6-phosphate for lysosomal transport; this has recently been challenged as it was found to covalently bind to a P-Man9GlcNAc2 moiety that attaches to a mannose phosphate receptor, forming a ternary complex between GCase, LIMP-2, and the mannose phosphate receptor (Zhao et al., 2014). Nevertheless, the interaction between LIMP-2 and GCase is key for proper GCase processing, since mice deficient in LIMP-2 have reduced GCase activity and protein expression (Reczek et al., 2007).
Alterations in LIMP-2 may have a variety of consequences. Gonzalez et al. reviewed 14 mutations in SCARB2, either associated with action myoclonus, renal failure, or progressive myoclonic epilepsy, demonstrating the diverse function of LIMP-2. Neurological consequences have been studied in relation to GD, as deficient mice exhibit neurodegeneration, partial paralysis, and alpha-synuclein accumulation. Meanwhile, overexpression of LIMP-2 in neuronal cell lines resulted in increased GCase activity and alpha-synuclein clearance (Rothaug et al., 2014). LIMP-2 was also explored as a potential genetic modifier in a pair of siblings who presented with GD who were discordant for myoclonic epilepsy. Molecular analyses showed a novel heterozygous SCARB2 mutation in the sibling with myoclonic epilepsy, who also had decreased LIMP-2 and GCase levels compared to his sibling and controls (Velayati et al., 2011). The fact that the brother with the same GBA1 genotype, but without the SCARB2 mutation, had mild type 1 GD, suggests that the SCARB2 mutation resulted in the type 3 phenotype. These studies show that LIMP-2 is critical for proper functioning of GCase, and may contribute to certain GD manifestations, such as myoclonic epilepsy.
Neighboring genes (contiguous genes)
Other genes that could modify GD phenotype are those located in close proximity to GBA1. The human GBA1 gene, located on chromosome 1q21, has a pseudogene that shares 96% exonic sequence homology, located 16 kb downstream of the functional gene (Horowitz et al., 1989). The entire 1q21 region is gene rich, with seven genes and two pseudogenes very closely clustered (Fig. 2) (Winfield et al., 1997). MTX1 (metaxin 1) is located only 430 nucleotides downstream of the GBA1 pseudogene and is convergently transcribed, encoding a protein of 317 amino acids that appears to be part of a preprotein import complex in the outer membrane of mitochondria (Armstrong et al., 1997). MTX1 also has a pseudogene located 670 nucleotides downstream at the 3’-UTR (enhancer region) of the functional GBA1 gene (Long et al., 1996). The presence of contiguous genes and their pseudogenes increases the chance of unequal chromosomal pairing and recombination between each gene and its pseudogene. Therefore, recombination between the MTX1 gene and its pseudogene has the potential to interrupt the 3’-UTR of GBA1 (which is in the promotor region of the MTX1 pseudogene) (Winfield et al., 1997; Tayebi et al., 2003). These recombination events can contribute to GBA1 expression and the phenotypic heterogeneity in GD, signaling an avenue of study for how adjacent genes may modify disease progression (Tayebi et al., 2000, Tayebi et al., 2003). As a consequence, the interruption of the enhancer, caused by these potential crossovers between gene and pseudogen may influence the regulation of gene expression and impact epigentic influences. Additionally, Blech et al. shows that the region encompassing the 3’ UTR of GBA1 through intron 3 of the MTX1 pseudogene is highly conserved, and includes three possible transcription factor binding sites. This study also explored the potential activity of the 3’ UTR of the GBA1/MTX1 pseudogene, which may be disrupted with any recombination between MTX1 and the pseudogene, therefore resulting in changes inGBA1 expression (Blech-Hermoni et al., 2010).
Figure 2.
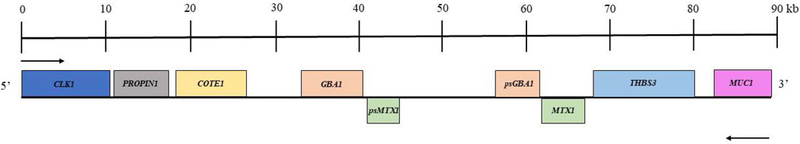
Scaled map of a 90kb gene-rich region surrounding GBA1. Genes represented above the line are transcribed right to left, while the genes below are transcribed left to right. Adapted from Winfield et al., 1997.
Furthermore, alteration in MTX1 itself can have phenotypic consequences. Bornstein et al. demonstrated that murine metaxin (mtx) may be critical for mouse embryonic development. After introducing a mild gba mutation and a phosphoglycerate kinase-neomycin gene cassette in the coding region of the terminal exon of mtx, they observed that the mice died in early embryonic development. The authors hypothesized that the mtx mutation was responsible for the embryonic death, although they did not present a homozygous mtx mutation-only control (Bornstein et al., 1995). As such, it is possible that this resulted in a pleiotropic effect, where neither of the mutations alone cause embryonic lethality. LaMarca et al. identified a p.F202L mutation in the MTX1 gene in some patients with GD, most commonly found in association with the p.N409S mutation. The authors extrapolated that this MTX1 mutation could dampen the burden of disease in patients with the p.N409S mutation (LaMarca et al., 2004). Moreover, Gan-Or et al. observed that the MTX1 c.184T>A mutation was more frequently found in patients with PD carrying GBA1 mutations, compared to non-carriers, suggesting that a homozygous MTX1 c.184A/A genotype was associated with an earlier onset of motor symptoms (Gan-Or et al., 2011). Additional functional and clinical studies are necessary to determine the role of MTX1 and other adjacent genes in modifying GD, as most of the genes in this area have not been well studied. Only very recently was it shown that the GBA1 pseudogene may express competing-endogenous RNA, thereby indirectly upregulating GBA1 expression by sponging miRNAs which may have bound and inhibited the translation of GBA1 mRNA (Straniero et al., 2017).
Non-Lysosomal Glucosylceramides
Several proteins other than GCase are known to degrade glucosylceramides, rendering them biological candidates for the modification of GD. GBA2 (glucosylceramidase beta 2) encodes a protein localized to the endoplasmic reticulum and known initially for its role as a microsomal bile acid β-glucosidase. However, its primary function as an ER membrane-bound glucocerebrosidase was appreciated when a GBA2-deficient mouse model developed storage of glucosylceramide without any changes in lipid and bile acid metabolism (Yildiz et al., 2006). It was then found that expression of GBA2 is increased when GCase from GBA1 is deficient (Aureli et al., 2012; Burke et al., 2013). These results were recapitulated in a subsequent murine study showing that mice with both a gba1 and gba2 knockout stored more glucosylceramide than either single knockout. However, single and multi-marker genetic analyses showed that GBA2 did not affect disease severity. (Yildiz et al., 2013).
This work led to the hypothesis by Mistry et al. that GBA2 contributes to pathology by processing glucosylsphingosine and glucosylceramide into sphingosine, since this species has potential to be toxic. To test this hypothesis, they developed a Mx1–Cre+: GD1:gba2−/− mouse, and observed that deletion of gba2 partially rescued aspects of the pathology observed in the Mx1–Cre+:GD1 mice, although storage of substrate in the spleen persisted. Thus, it was proposed that targeting GBA2 and sphingosine production are potential therapeutic targets (Mistry et al., 2014). However, some of these results have not been validated in subsequent studies, as Schonauer et al. found GBA2 to be downregulated or at control levels in the absence of GCase activity, and that sphingosine itself binds to GBA2 and inhibits the further formation of sphingosine (Schonauer et al., 2017). These conflicting results indicate that while GBA2 may play a role in GD pathogenesis, its exact role is unclear.
Similarly, the role of GBA3 (glucosylceramidase beta 3), encoding a cytosolic glucocerebrosidase, has been debated. Initially, GBA3 was discounted as a modifier when the presence of mutations was not found to not correlate to GD1 disease severity (Beutler et al., 2004). However, it was then shown that GBA3 could hydrolyze glucosylceramide, albeit at low levels, which particularly spills into the cytoplasmic compartment in cells from patients with GD types 2 and 3 (Korkotian et al., 1999; Hayashi et al., 2007), suggesting that it could play a role in neuronopathic GD. This was further investigated by Dekker et al., who determined that this gene is unlikely to be a modifier of GD1 by verifying GBA3’s low activity towards native glucosylceramide, and again finding a lack of correlation between mutations in GBA3 and GD1 disease severity (Dekker et al., 2011). Thus, while GBA3 has been shown to have no effect on type 1 GD pathogenesis, it is possible that it might still impact neuronopathic GD. Further, there may be additional GBAs that play a role in disease pathogenesis, as fibroblasts deficient for GBA1 and GBA2 were still found to have 50% of control enzyme activity and some, but not all, residual activity was attributed to GBA3 (Harzer and Yildiz, 2015).
Other Candidate Modifiers Explored
Many other genes have been proposed as modifiers of GD, and while not well validated, could contribute to missing heritability and discordant genotype-phenotype relationships. One example is the glucosylceramide synthase gene UGCG (UDP-glucose ceramide glucosyltransferase), whose product catalyzes the chemical reaction that creates glucosylceramide. Alfonso et al. examined seven polymorphisms in UGCG, identifying the rs200460111 and rs3780520 variants as modulators of GD severity, implicated in increasing and decreasing severity respectively (Alfonso et al., 2013). However, Beutler et al. did not find a significant association between polymorphisms in UGCG and disease severity in patients with GD (Beutler and West, 2002). Thus, it is difficult to determine whether glucosylceramide synthase modifies GD phenotype, although its leverage in substrate reduction therapy suggests that such an association may exist.
Furthermore, a study by Maor et al. proposed that ITCH, an E3 ubiquitin ligase, encoded by ITCH (itchy E3 ubiquitin protein ligase), modified GD pathology (Maor et al., 2013). Typically, misfolded GCase is eliminated by cells through endoplasmic reticulum associated degradation (ERAD). However, it has been purported that with higher levels of ERAD, less correctly folded GCase reaches the lysosome in disease conditions, resulting in increased disease severity (Ron and Horowitz, 2005). They demonstrated that ITCH interacts with mutated GCase variants, inducing their polyubiquitination and proteasomal degradation. Interestingly, studies have shown that other E3 ubiquitin ligases, parkin and c-CbI, also interact with mutant GCase (Lu et al., 2010; Ron et al., 2010). Since the amount of GCase degradation could be a marker of disease severity, these E3 ubiquitin ligases might further be investigated as modifiers of GD (Maor et al., 2013).
The possible modifying role of tumor necrosis factor (TNF), encoding TNF-α, a cytokine implicated in inflammation has been explored. Michelakakis et al. described higher levels of TNF-α in patients with GD2 and GD3 relative to GD1 and control cases. Up-regulation of TNF-α was also reported in brains of fetal GD mice (Michelakakis et al., 1996; Hong et al., 2006). Altarescu et al. studied polymorphisms in TNF in patients with GD, finding that homozygosity for the common rs1800629 polymorphism in the promoter region of TNF seemed to be associated with increased TNF-α expression and non-neuronopathic forms of GD (Altarescu et al., 2005). These studies suggest that TNF-α may mediate aspects of GD expressivity, specifically with respect to inflammation, overall disease severity and potentially neuropathology.
Studies also show that transcription factor EB (TFEB), encoding TFEB, may be important for proper GCase function, due to its role as a master regulator of lysosomal genes. Song et al. showed that TFEB activation improved mutant lysosomal GCase activity by enhancing its folding and trafficking, partially by increasing the expression of LIMP-2. This approach was also shown to increase activity of Hexosaminidase A, a lysosomal enzyme dysregulated in Tay-Sach disease (Song et al., 2013). Further, Awad et al. investigated TFEB in iPSC-derived neuronal cells from patients with GD types 2 and 3, finding the cells to have decreased numbers of lysosomes, an accumulation of autophagosomes, and decreased levels of TFEB as compared to GD type 1 and control cells. While recombinant GCase administration restored the number of lysosomes, this effect was enhanced when TFEB was overexpressed (Awad et al., 2015). The purported pharmacological chaperone ambroxol was shown to increase the expression of TFEB, as well as several lysosomal genes including GCase (McNeill et al., 2014; Narita et al., 2016).
Progranulin, encoded by GRN (granulin precursor), linked to a variety of cellular functions, ranging from neurodegeneration to inflammation and including lysosomal function, has been considered as a modifier of GD. In a study of 115 patients with GD and 99 healthy controls, Jian et al. initially determined that serum progranulin levels were significantly lower in patients with GD. They found that this decrease corresponded to mutations in GRN in patients with GD. They further found that a grn knockout mouse model showed hepatosplenomegaly, and was unable to localize GCase to the lysosome. Treatment with recombinant progranulin reduced the GD-related pathology. The established p.D448V/– GD mouse also showed a similar amelioration of symptoms when administered progranulin (Jian et al., 2016b). The authors suggested that progranulin deficiency was a risk factor for GD, and that progranulin supplementation could become a therapeutic strategy. In further studies, the authors determined that progranulin is required to mobilize GCase to the lysosome in stress conditions, and that it binds directly to GCase and recruits HSP70 to form a complex involving LIMP-2 to allow for final trafficking to the lysosome. A modified progranulin was found to reduce storage and increase lysosomal localization of GCase in patient fibroblasts (Jian et al., 2016a). Progranulin has been proposed as a chaperone of other lysosomal enzymes, supporting the possible role of progranulin as a modifier of GD and lysosomal function in general (Jian et al., 2017).
Modifiers May Mediate Other Gaucher-Associated Disorders
Genetic modifiers may also work to mediate the association between GD and other disorders, including PD, cancer, and bone disease (summarized in Table 1). In PD and cancers such as multiple myeloma, having Gaucher disease is a risk factor, but it is not clear why only a minority of patients with GD go on to develop the second diagnosis.
Parkinson Disease
Multiple studies have established a link between mutations in GBA1 and PD (Sidransky et al., 2009; Lesage et al., 2011; Winder-Rhodes et al., 2013; Gegg and Schapira, 2018). Although underlying mechanisms and pathways have been pursued, the basis for this association is not fully understood. Several studies have highlighted genes that may moderate this link. Studying the gene BIN1 (bridging integrator 1), which encodes a protein important in endocytosis and apoptosis, the minor allele of the polymorphism rs13403026 was associated with an older age-at-onset of PD in 153 patients with GBA1-associated PD (Gan-Or et al., 2015). This implies that the BIN1 locus may serve a protective function for patients with both GD and PD. Another potential modifier gene, TMEM175 (transmembrane protein 175), a lysosomal K+ channel, was shown to destabilize lysosomal pH, decrease GCase activity, and increase susceptibility to exogenous α-synuclein fibrils. These observations suggest a direct role for TMEM175 in lysosomal dysfunction and increased susceptibility for PD (Jinn et al., 2017). Though such studies are intriguing, more research is required to further validate these genes and to elucidate additional modifier genes.
Cancer
Patients with GD have a modestly increased risk of developing hematologic malignancies, particularly multiple myeloma. The GD-cancer association may occur through a variety of mechanisms, including immune dysregulation, lipid accumulation, and alternate activation of macrophages (Mistry et al., 2013). The prevailing current hypothesis is that this relationship is mediated by sphingosine-related pathways, as sphingosine-1-phosphate has been shown to have tumor promoting properties, and some patients with GD and monoclonal gammopathy of unknown significance produce anti-glucosylsphingosine antibodies. It has been proposed that those that do may be at a higher risk of developing multiple myeloma (Nair et al., 2016). Several genes have been noted to mediate the relationship between GD and cancer. In a study of 29 individuals with GD, Lieblich et al. observed a significant association between GD patients with cancer and the ApaI (rs7975232) polymorphism of VDR (the vitamin D receptor), when compared to GD patients without cancer (Lieblich et al., 2011). Polymorphisms of this receptor have also been associated with a rare cardiac variant of GD and osteoporosis in type 1 GD (Greenwood et al., 2010a, 2010b). Additionally, Lo et al. performed whole-exome capture and massively parallel sequencing on DNA samples of two siblings with type 1 GD and T-cell acute lymphoblastic lymphoma. They found a novel homozygous mutation in MSH6 (mutS homolog 6), which could disrupt the patients’ mismatch repair system and contribute to their cancer (Lo et al., 2012). As Mistry et al. highlight, the association between GD and cancer is complex, but can be further unraveled through such studies of modifier gene(s) that may mediate the incidence of some cancers in patients with GD (Mistry et al., 2013).
Bone Disease
There have also been several modifiers proposed that may impact the degree of bone disease seen in GD. Patients with GD, even with the same genotype, exhibit varying degrees of bone involvement, (van Dussen et al., 2014) and therefore, identifying modifier genes may lead to a better understanding of the bone pathology in GD. As previously mentioned, polymorphisms of the vitamin D receptor gene have been associated with osteoporosis in type 1 GD, and Greenwood et al. suggest that VDR genotype may be predictive of bone mineral density and bone involvement in GD (Greenwood et al., 2010b). Another gene postulated to impact bone in GD is IL6 (interleukin 6). The SNP rs1800795 of IL6 was evaluated, but no significant association with bone mineral density or GD disease severity was evident. Yet, it was suggested that homozygosity for the C/C genotype at this SNP correlated to less severe Gaucher phenotype (Altarescu et al., 2003). Moreover, Gervas-Arruga et al., studied polymorphic sites in three genes, specifically rs2234693 in ESRI (estrogen receptor 1), rs2073618 in TNFRSF11B (TNF receptor superfamily member 11b) and rs11868820 in VDR, finding that they were associated with protection from loss of bone mineral density in splenectomized patients with GD. These results indicate that genetic variability contributes to susceptibility to bone disease in GD (Gervas-Arruga et al., 2015).
Modifiers as Targets for Therapeutic Strategies
The discovery of modifiers is vital to improving the understanding of disease pathophysiology, which may then yield new or improved therapies relevant to the primary disease. In a recent commentary, McCabe discusses the importance of protective modifiers in improving disease phenotypes, citing the examples of glycerol kinase deficiency, spinal muscular atrophy, and Duchenne muscular dystrophy (McCabe, 2017). Modifiers of GD affect a variety of interconnected pathways, including glycolipid degradation, lysosomal function, and downstream effects such as inflammation, as well as yet unidentified pathways; each gene and each pathway as a whole provide opportunities for intervention (Fig. 3). Candidate modifiers have helped guide research into potential new therapeutic directions. For example, it was suggested that progranulin might serve as a target for treatment, based on preliminary studies in patient fibroblasts and PGRN-deficient mice (Jian et al., 2016a). Considering its critical role in GCase activation, LIMP-2 also can be a potential target for therapy in not only GD, but also other neurological conditions. As previously highlighted, overexpression of LIMP-2 in neurons led to improved lysosomal activity and increased clearance of alpha-synuclein (Rothaug et al., 2014). Further, synthetic Sap C increased the activity of recombinant GCase 14 to 22- fold, and prevented GCase degradation by cathepsins. This work indicates that Sap C treatment may synergize with enzyme replacement therapy (Yoneshige et al., 2015). Thus, modifiers may have utility not only in developing new therapies, but also in modifying existing ones. Some new Gaucher treatments under development are already leveraging identified modifier genes, such as substrate reduction therapy, by decreasing the activity of glucosylceramide synthase, and ambroxol, by increasing the activity of TFEB.
Figure 3.
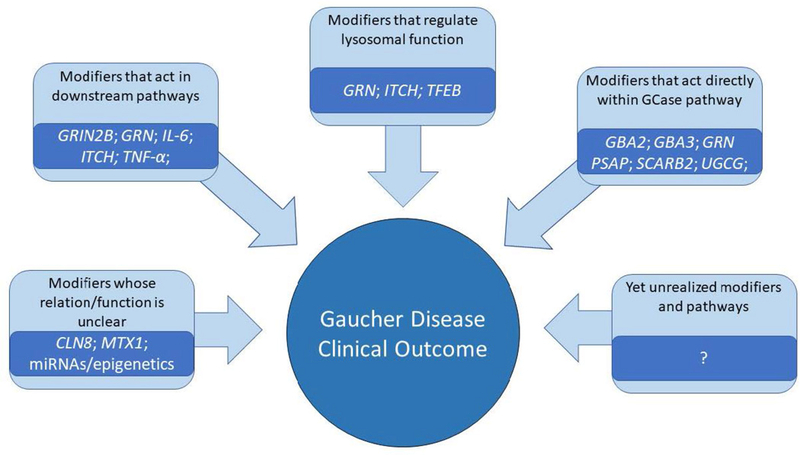
Potential genetic modifiers and the proposed mechanisms by which they may regulate clinical outcome of Gaucher disease.
Although several of these new avenues are promising, more research is needed to assess the therapeutic value of these modifiers fin GD. The current body of modifier genetics research in GD does not yet explain the wide phenotypic spectrum in patients, nor has it resulted in new treatments for patients with neurological manifestation. However, building a network of genes that relate to pathology may eventually elucidate unknown disease mechanisms, and lead to new treatment options.
Conclusion
When the GBA1 gene was first identified and sequenced, there was great hope that the phenotypic heterogeneity in GD would be explained by the different gene mutations. Today, although more than 500 GBA1 mutations have been identified, it is clear that genetic modifiers must play a role. Identifying these modifiers is the next frontier in Mendelian diseases, but it is particularly challenging especially in rare disorders. In this review, we have highlighted several strategies for identifying modifiers using GD as a model, and provided examples of different proposed modifiers and their potential impact on disease pathology. Additionally, we have reviewed modifiers that may explain the link between GD and other associated disorders, such as PD and multiple myeloma. However, even after combining all the genes discussed, the proposed modifiers account for only a fraction of the observed phenotypic heterogeneity and further exploration of both these and additional unknown modifiers is essential. Because GD is a rare disorder, assembling the number of cases needed to obtain adequate power for genome-wide screens will require large international collaborations and the use of novel and developing techniques to address this challenge. While therapies are currently being developed that leverage GD modifier genes, the hunt for additional genes to explain the heterogeneity seen in the disease population will likely yield exciting new therapeutic avenues to pursue.
Acknowledgements
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institutes of Health.
References
- Alattia JR, Shaw JE, Yip CM, Prive GG. 2007. Molecular imaging of membrane interfaces reveals mode of beta-glucosidase activation by saposin C. Proc Natl Acad Sci U S A 104:17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso P, Navascués J, Navarro S, Medina P, Bolado-Carrancio A, Andreu V, Irún P, Rodríguez-Rey JC, Pocoví M, España F, Giraldo P. 2013. Characterization of Variants in the Glucosylceramide Synthase Gene and their Association with Type 1 Gaucher Disease Severity. Hum Mutat 34:1396–1403. [DOI] [PubMed] [Google Scholar]
- Altarescu G, Phillips M, Foldes AJ, Elstein D, Zimran A, Mates M. 2003. The interleukin-6 promoter polymorphism in Gaucher disease: a new modifier gene? QJM An Int J Med 96:575–578. [DOI] [PubMed] [Google Scholar]
- Altarescu G, Zimran A, Michelakakis H, Elstein D. 2005. TNF-α levels and TNF-α gene polymorphism in type I Gaucher disease. Cytokine 31:149–152. [DOI] [PubMed] [Google Scholar]
- Armstrong LC, Komiya T, Bergman BE, Mihara K, Bornstein P. 1997. Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J Biol Chem 272:6510–6518. [DOI] [PubMed] [Google Scholar]
- Aureli M, Bassi R, Loberto N, Regis S, Prinetti A, Chigorno V, Aerts JM, Boot RG, Filocamo M, Sonnino S. 2012. Cell surface associated glycohydrolases in normal and Gaucher disease fibroblasts. J Inherit Metab Dis 35:1081–1091. [DOI] [PubMed] [Google Scholar]
- Awad O, Sarkar C, Panicker LM, Miller D, Zeng X, Sgambato JA, Lipinski MM, Feldman RA. 2015. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum Mol Genet 24:5775–5788. [DOI] [PubMed] [Google Scholar]
- Beutler E, Beutler L, West C. 2004. Mutations in the gene encoding cytosolic β-glucosidase in Gaucher disease. J Lab Clin Med 144:65–68. [DOI] [PubMed] [Google Scholar]
- Beutler E, West C. 2002. Polymorphisms in glucosylceramide (glucocerebroside) synthase and the Gaucher disease phenotype. Isr Med Assoc J 4:986–988. [PubMed] [Google Scholar]
- Blech-Hermoni YN, Ziegler SG, Hruska KS, Stubblefield BK, Lamarca ME, Portnoy ME, NISC Comparative Sequencing Program NCS, Green ED, Sidransky E 2010. In silico and functional studies of the regulation of the glucocerebrosidase gene. Mol Genet Metab 99:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P, McKinney CE, LaMarca ME, Winfield S, Shingu T, Devarayalu S, Vos HL, Ginns EI. 1995. Metaxin, a gene contiguous to both thrombospondin 3 and glucocerebrosidase, is required for embryonic development in the mouse: implications for Gaucher disease. Proc Natl Acad Sci U S A 92:4547–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DG, Rahim AA, Waddington SN, Karlsson S, Enquist I, Bhatia K, Mehta A, Vellodi A, Heales S. 2013. Increased glucocerebrosidase (GBA) 2 activity in GBA1 deficient mice brains and in Gaucher leucocytes. J Inherit Metab Dis 36:869–872. [DOI] [PubMed] [Google Scholar]
- Cammarata G, Fatuzzo P, Rodolico MS, Colomba P, Sicurella L, Iemolo F, Zizzo C, Alessandro R, Bartolotta C, Duro G, Monte I. 2015. High variability of Fabry disease manifestations in an extended Italian family. Biomed Res Int 2015:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. 2014. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 157:77–94. [DOI] [PubMed] [Google Scholar]
- Chang AK, Ginter Summarell CC, Birdie PT, Sheehan VA. 2018. Genetic modifiers of severity in sickle cell disease. Clin Hemorheol Microcirc 68:147–164. [DOI] [PubMed] [Google Scholar]
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, Lee H, Zhang F, et al. 2015. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160:1246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry S, Coyle NE, Tang H, Salari K, Lind D, Clark SL, Tsai H-J, Naqvi M, Phong A, Ung N, Matallana H, Avila PC, et al. 2006. Population stratification confounds genetic association studies among Latinos. Hum Genet 118:652–664. [DOI] [PubMed] [Google Scholar]
- Choy FYM, Christensen CL. 2016. Progranulin as a novel factor in Gaucher Disease. EBioMedicine 13:13–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christomanou H, Aignesberger A, Linke RP. 1986. Immunochemical characterization of two activator proteins stimulating enzymic degradation in vitro. Absence of one of them in a human Gaucher disease variant. Biol Chem Hoppe Seyler 367:879–90. [DOI] [PubMed] [Google Scholar]
- De Castro SCP, Malhas A, Leung KY, Gustavsson P, Vaux DJ, Copp AJ, Greene NDE. 2012. Lamin B1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet 8:e1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker N, Voorn-Brouwer T, Verhoek M, Wennekes T, Narayan RS, Speijer D, Hollak CEM, Overkleeft HS, Boot RG, Aerts JMFG. 2011. The cytosolic β-glucosidase GBA3 does not influence type 1 Gaucher disease manifestation. Blood Cells, Mol Dis 46:19–26. [DOI] [PubMed] [Google Scholar]
- Elkon R, Agami R. 2017. Characterization of noncoding regulatory DNA in the human genome. Nat Biotechnol 35:732–746. [DOI] [PubMed] [Google Scholar]
- Fairley C, Zimran A, Phillips M, Cizmarik M, Yee J, Weinreb N, Packman S. 2008. Phenotypic heterogeneity of N370S homozygotes with type I Gaucher disease: An analysis of 798 patients from the ICGG Gaucher Registry. J Inherit Metab Dis 31:738–744. [DOI] [PubMed] [Google Scholar]
- Fernandes C, Rao Y. 2011. Genome-wide screen for modifiers of Parkinson’s disease genes in Drosophila. Mol Brain 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamp AC, Tanaka Y, Lüllmann-Rauch R, Wittke D, D’Hooge R, De Deyn PP, Moser T, Maier H, Hartmann D, Reiss K, Illert AL, Von Figura K, et al. 2003. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet 12:631–646. [PubMed] [Google Scholar]
- Gan-Or Z, Amshalom I, Bar-Shira A, Gana-Weisz M, Mirelman A, Marder K, Bressman S, Giladi N, Orr-Urtreger A. 2015. The Alzheimer disease BIN1 locus as a modifier of GBA-associated Parkinson disease. J Neurol 262:2443–2447. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Bar-Shira A, Gurevich T, Giladi N, Orr-Urtreger A. 2011. Homozygosity for the MTX1 c.184T>A (p.S63T) alteration modifies the age of onset in GBA-associated Parkinson’s disease. Neurogenetics 12:325–332. [DOI] [PubMed] [Google Scholar]
- Gegg ME, Schapira AHV. 2018. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J: 10.1111/febs.14393. [DOI] [PubMed]
- Génin E, Feingold J, Clerget-Darpoux F. 2008. Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet 124:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervas-Arruga J, Cebolla JJ, Blas I de, Roca M, Pocovi M, Giraldo P 2015. The influence of genetic variability and proinflammatory status on the development of bone disease in patients with Gaucher disease. PLoS One 10:e0126153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker-Alpan O, Hruska KS, Orvisky E, Kishnani PS, Stubblefield BK, Schiffmann R, Sidransky E. 2005. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J Med Genet 42:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Valeiras M, Sidransky E, Tayebi N. 2014. Lysosomal integral membrane protein-2: a new player in lysosome-related pathology. Mol Genet Metab 111:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood A, Altarescu G, Zimran A, Elstein D. 2010a. Vitamin D Receptor (VDR) polymorphisms in the cardiac variant of Gaucher disease. Pediatr Cardiol 31:30–32. [DOI] [PubMed] [Google Scholar]
- Greenwood A, Elstein D, Zimran A, Altarescu G. 2010b. Effect of vitamin D receptor (VDR) genotypes on the risk for osteoporosis in type 1 Gaucher disease. Clin Rheumatol 29:1037–1041. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME, Lee JM. 2014. Genetic modifiers of Huntington’s disease. Mov Disord 29: 1359–65. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. 1941. The relative importance of principal and modifying genes in determining some human diseases. J Genet 41:149–157. [Google Scholar]
- Harzer K, Yildiz Y. 2015. High β-glucosidase (GBA) activity not attributable to GBA1 and GBA2 in live normal and enzyme-deficient fibroblasts may emphasise the role of additional GBAs. Biol Chem 396:1241–1246. [DOI] [PubMed] [Google Scholar]
- Hassan S, Sidransky E, Tayebi N. 2017. The role of epigenetics in lysosomal storage disorders: Uncharted territory. Mol Genet Metab 122:10–18. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Okino N, Kakuta Y, Shikanai T, Tani M, Narimatsu H, Ito M. 2007. Klotho-related protein is a novel cytosolic neutral beta-glycosylceramidase. J Biol Chem 282:30889–30900. [DOI] [PubMed] [Google Scholar]
- Hill-Burns EM, Ross OA, Wissemann WT, Soto-Ortolaza AI, Zareparsi S, Siuda J, Lynch T, Wszolek ZK, Silburn PA, Mellick Beate Ritz GD, Scherzer CR, Zabetian CP et al. 2016. Identification of genetic modifiers of age-at-onset for familial parkinson’s disease. Hum Mol Genet 25: 3849–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MW, O’Brien JS. 1971. Gaucher’s disease: deficiency of “acid” β-glucosidase and reconstitution of enzyme activity in vitro. Proc Natl Acad Sci USA 68: 2810–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman TJ, Witte JS. 2015. Strategies for imputing and analyzing rare variants in association studies. Trends Genet 31:556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y Bin, Kim EY, Jung S-C 2006. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J Korean Med Sci 21:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. 1989. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4:87–96. [DOI] [PubMed] [Google Scholar]
- Jan de Beur S, Ding C, Germain-Lee E, Cho J, Maret A, Levine MA. 2003. Discordance between Genetic and Epigenetic Defects in Pseudohypoparathyroidism Type 1b Revealed by Inconsistent Loss of Maternal Imprinting at GNAS1. Am J Hum Genet 73:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Hettinghouse A, Liu C. 2017. Progranulin acts as a shared chaperone and regulates multiple lysosomal enzymes. Genes Dis 4:125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Tian Q-Y, Hettinghouse A, Zhao S, Liu H, Wei J, Grunig G, Zhang W, Setchell KDR, Sun Y, Overkleeft HS, Chan GL, et al. 2016a. Progranulin Recruits HSP70 to β-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine 13:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Zhao S, Tian Q-Y, Liu H, Zhao Y, Chen W-C, Grunig G, Torres PA, Wang BC, Zeng B, Pastores G, Tang W, et al. 2016b. Association Between Progranulin and Gaucher Disease. EBioMedicine 11:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Drolet RE, Cramer PE, Wong AH-K, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin-Strapps M, Stone DJ. 2017. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 114:2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Zhan X, Ye J, Han L, Qiu W, Gu X, Zhang H. 2018. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells, Mol Dis 68:60–65. [DOI] [PubMed] [Google Scholar]
- Klein AD, Ferreira N-S, Ben-Dor S, Duan J, Hardy J, Cox TM, Merrill AH, Futerman AH. 2016. Identification of modifier genes in a mouse model of Gaucher disease. Cell Rep 16:2546–2553. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Schwarz A, Pelled D, Schwarzmann G, Segal M, Futerman AH. 1999. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J Biol Chem 274:21673–21678. [DOI] [PubMed] [Google Scholar]
- Lachmann RH, Grant IR, Halsall D, Cox TM. 2004. Twin pairs showing discordance of phenotype in adult Gaucher’s disease. QJM 97:199–204. [DOI] [PubMed] [Google Scholar]
- LaMarca ME, Goldstein M, Tayebi N, Arcos-Burgos M, Martin BM, Sidransky E. 2004. A novel alteration in metaxin 1, F202L, is associated with N370S in Gaucher disease. J Hum Genet 49:220–222. [DOI] [PubMed] [Google Scholar]
- Landis BJ, Schubert JA, Lai D, Jegga AG, Shikany AR, Foroud T, Ware SM, Hinton RB. 2017. Exome sequencing identifies candidate genetic modifiers of syndromic and familial thoracic aortic aneurysm severity. J Cardiovasc Transl Res 10:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieblich M, Altarescu G, Zimran A, Elstein D. 2011. Vitamin D Receptor (VDR) polymorphic variants in patients with cancer and Gaucher disease. Blood Cells, Mol Dis 46:92–94. [DOI] [PubMed] [Google Scholar]
- Lindquist KJ, Jorgenson E, Hoffman TJ, Witte JS. 2013. The impact of improved microarray coverage and larger sample sizes on future genome-wide association studies. Genet Epidemiol 4:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SM, Choi M, Liu J, Jain D, Boot RG, Kallemeijn WW, Aerts JMFG, Pashankar F, Kupfer GM, Mane S, Lifton RP, Mistry PK. 2012. Phenotype diversity in type 1 Gaucher disease: discovering the genetic basis of Gaucher disease/hematologic malignancy phenotype by individual genome analysis. Blood 119:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SM, Liu J, Chen F, Pastores GM, Knowles J, Boxer M, Aleck K, Mistry PK. 2011. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy. J Inherit Metab Dis 34:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GL, Winfield S, Adolph KW, Ginns EI, Bornstein P. 1996. Structure and Organization of the Human Metaxin Gene (MTX) and Pseudogene. Genomics 33:177–184. [DOI] [PubMed] [Google Scholar]
- Lu J, Chiang J, Iyer RR, Thompson E, Kaneski CR, Xu DS, Yang C, Chen M, Hodes RJ, Lonser RR, Brady RO, Zhuang Z. 2010. Decreased glucocerebrosidase activity in Gaucher disease parallels quantitative enzyme loss due to abnormal interaction with TCP1 and c-Cbl. Proc Natl Acad Sci U S A 107:21665–21670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, et al. 2009. Finding the missing heritability of complex diseases. Nature 461:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor G, Filocamo M, Horowitz M. 2013. ITCH regulates degradation of mutant glucocerebrosidase: implications to Gaucher disease. Hum Mol Genet 22:1316–1327. [DOI] [PubMed] [Google Scholar]
- Martin LS, Eskin E. 2017. Review: Population Structure in Genetic Studies: Confounding Factors and Mixed Models bioRxiv 092106. [DOI] [PMC free article] [PubMed]
- McCabe ERB. 2017. Modifier genes: Moving from pathogenesis to therapy . Mol Genet Metab 122:1–3. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN. 2008. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9:356–369. [DOI] [PubMed] [Google Scholar]
- McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AH. 2014. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137:1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VLC, Livesey KJ, Tanphaichitr V, et al. 2003. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet 12:2241–2247. [DOI] [PubMed] [Google Scholar]
- Michelakakis H, Spanou C, Kondyli A, Dimitriou E, Van Weely S, Hollak CE, Van Oers MH, Aerts JM. 1996. Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease. Biochim Biophys Acta 1317:219–222. [DOI] [PubMed] [Google Scholar]
- Michelakakis H, Xiromerisiou G, Dardiotis E, Bozi M, Vassilatis D, Kountra P-M, Patramani G, Moraitou M, Papadimitriou D, Stamboulis E, Stefanis L, Zintzaras E, et al. 2012. Evidence of an association between the scavenger receptor class B member 2 gene and Parkinson’s disease. Mov Disord 27:400–405. [DOI] [PubMed] [Google Scholar]
- Mistry PK, Liu J, Sun L, Chuang W-L, Yuen T, Yang R, Lu P, Zhang K, Li J, Keutzer J, Stachnik A, Mennone A, et al. 2014. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc Natl Acad Sci U S A 111:4934–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry PK, Taddei T, Vom Dahl S, Rosenbloom BE. 2013. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog 18:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Perrimon N. 2012. RNAi screening: new approaches, understandings, and organisms. Wiley Interdiscip Rev RNA 3: 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta M, Camerini S, Tatti M, Casella M, Torreri P, Crescenzi M, Tartaglia M, Salvioli R. 2014. Gaucher disease due to saposin C deficiency is an inherited lysosomal disease caused by rapidly degraded mutant proteins. Hum Mol Genet 23:5814–5826. [DOI] [PubMed] [Google Scholar]
- Mouton J, Loos B, Moolman-Smook JC, Kinnear CJ. 2015. Ascribing novel functions to the sarcomeric protein, myosin binding protein H (MyBPH) in cardiac sarcomere contraction. Exp Cell Res 331:338–351. [DOI] [PubMed] [Google Scholar]
- Mouton JM, Van der Merwe L, Goosen A, Revera M, Brink PA, Moolman-Smook JC, Kinnear C. 2016. MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients. Hum Genet 135:477–483. [DOI] [PubMed] [Google Scholar]
- Nadeau JH. 2001. Modifier genes in mice and humans. Nat Rev Genet 2:165–174. [DOI] [PubMed] [Google Scholar]
- Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. 2016. Clonal immunoglobulin against lysolipids in the origin of myeloma. N Engl J Med 374:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita A, Shirai K, Itamura S, Matsuda A, Ishihara A, Matsushita K, Fukuda C, Kubota N, Takayama R, Shigematsu H, Hayashi A, Kumada T, et al. 2016. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann Clin Transl Neurol 3:200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova AS, Plotnikova OV, Serzhanova V, Efimov A, Bogush I, Cai KQ, Hensley HH, Egleston BL, Klein-Szanto A, Seeger-Nukpezah T, Golemis EA. 2014. Nedd9 restrains renal cystogenesis in Pkd1−/− mice. Proc Natl Acad Sci USA 111: 12859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passantino R, Cascio C, Deidda I, Galizzi G, Russo D, Spedale G, Guarneri P. 2013. Identifying protein partners of CLN8, an ER-resident protein involved in neuronal ceroid lipofuscinosis. Biochim Biophys Acta - Mol Cell Res 1833:529–540. [DOI] [PubMed] [Google Scholar]
- Pelled D, Shogomori H, Futerman AH. 2000. The increased sensitivity of neurons with elevated glucocerebroside to neurotoxic agents can be reversed by imiglucerase. J Inherit Metab Dis 23:175–184. [DOI] [PubMed] [Google Scholar]
- Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky E, Schiffmann R, Futerman AH. 2005. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol Dis 18:83–88. [DOI] [PubMed] [Google Scholar]
- Reczek D, Schwake M, Schröder J, Hughes H, Blanz J, Jin X, Brondyk W, Van Patten S, Edmunds T, Saftig P. 2007. LIMP-2 Is a receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 131:770–783. [DOI] [PubMed] [Google Scholar]
- Ron I, Horowitz M. 2005. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14:2387–2398. [DOI] [PubMed] [Google Scholar]
- Ron I, Rapaport D, Horowitz M. 2010. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet 19:3771–3781. [DOI] [PubMed] [Google Scholar]
- Rosenbloom BE, Weinreb NJ. 2013. Gaucher Disease: a comprehensive review. Crit Rev Oncog 18:163–175. [DOI] [PubMed] [Google Scholar]
- Rossmann M, Schultz-Heienbrok R, Behlke J, Remmel N, Alings C, Sandhoff K, Saenger W, Maier T. 2008. Crystal structures of human Saposins C and D: Implications for lipid recognition and membrane interactions. Structure 16:809–817. [DOI] [PubMed] [Google Scholar]
- Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lüllmann-Rauch R, Kallemeijn WW, Gaspar P, Aerts JM, Glatzel M, Saftig P, Krainc D, et al. 2014. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A 111:15573–15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvioli R, Tatti M, Scarpa S, Moavero SM, Ciaffoni F, Felicetti F, Kaneski CR, Brady RO, Vaccaro AM. 2005. The N370S (Asn370 →Ser) mutation affects the capacity of glucosylceramidase to interact with anionic phospholipid-containing membranes and saposin C. Biochem J 390:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonauer S, Körschen HG, Penno A, Rennhack A, Breiden B, Sandhoff K, Gutbrod K, Dörmann P, Raju DN, Haberkant P, Gerl MJ, Brügger B, et al. 2017. Identification of a feedback loop involving β-glucosidase 2 and its product sphingosine sheds light on the molecular mechanisms in Gaucher disease. J Biol Chem 292:6177–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, Yang XW. 2012. Network organization of the Huntingtin proteomic interactome in the mammalian brain. Neuron 75: 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E 2004. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 83:6–15. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, et al. 2009. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s Disease. N Engl J Med 361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert M, Westbroek W, Chen Y-C, Moaven N, Li Y, Velayati A, Saraiva-Pereira ML, Martin SE, Sidransky E. 2014. Identification of miRNAs that modulate glucocerebrosidase activity in Gaucher disease cells. RNA Biol 11:1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Wang F, Savini M, Ake A, Di Ronza A, Sardiello M, Segatori L. 2013. TFEB regulates lysosomal proteostasis. Hum Mol Genet 22:1994–2009. [DOI] [PubMed] [Google Scholar]
- Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, Rose C, Billette de Villemeur T, et al. 2017. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straniero L, Rimoldi V, Samarani M, Goldwurm S, Di Fonzo A, Krüger R, Deleidi M, Aureli M, Soldà G, Duga S, Asselta R. 2017. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22–3p. Sci Rep 7:12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR, Rahman N. 2008. The emerging landscape of breast cancer susceptibility. Nat Genet 40:17–22. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liou B, Ran H, Skelton MR, Williams MT, Vorhees C V, Kitatani K, Hannun YA, Witte DP, Xu YH, Grabowski GA. 2010a. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum Mol Genet 19:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ran H, Zamzow M, Kitatani K, Skelton MR, Williams MT, Vorhees C V., Witte DP, Hannun YA, Grabowski GA. 2010b. Specific saposin C deficiency: CNS impairment and acid β-glucosidase effects in the mouse. Hum Mol Genet 19:634–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamargo RJ, Velayati A, Goldin E, Sidransky E. 2012. The role of saposin C in Gaucher disease. Mol Genet Metab 106:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayebi N, Park J, Madike V SE. 2000. Gene rearrangement on 1q21 introducing a duplication of the glucocerebrosidase pseudogene and a metaxin fusion gene. Hum Genet 107:400–403. [DOI] [PubMed] [Google Scholar]
- Tayebi N, Stubblefield BK, Park JK, Orvisky E, Walker JM, Lamarca ME, Sidransky E. 2003. Reciprocal and Nonreciprocal Recombination at the Glucocerebrosidase Gene Region: Implications for Complexity in Gaucher Disease. Am J Hum Genet 72:519–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dussen L, Lips P, Van Essen HW, Hollak CEM, Bravenboer N. 2014. Heterogeneous pattern of bone disease in adult type 1 Gaucher disease: Clinical and pathological correlates. Blood Cells, Mol Dis 53:118–123.Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, Peeters E, Philips T, Goris An, Dubois B, Andersen PM, Al-Chalabi A et al. 2012. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 18: 1418–1422. [DOI] [PubMed] [Google Scholar]
- Velayati A, DePaolo J, Gupta N, Choi JH, Moaven N, Westbroek W, Goker-Alpan O, Goldin E, Stubblefield BK, Kolodny E, Tayebi N, Sidransky E. 2011. A mutation in SCARB2 is a modifier in Gaucher disease. Hum Mutat 32:1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. 2012. CNOT3 is a modifier of PRPF31 mutations in Retinitis Pigmentosa with Incomplete Penetrance. PLoS Genet 8:e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ. 2001. Phenotype—genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet 2:245–255. [DOI] [PubMed] [Google Scholar]
- Weiler S, Tomich JM, Kishimoto Y, O’Brien JS, Barranger JA. 2008. Identification of the binding and activating sites of the sphingolipid activator protein, saposin C, with glucocerebrosidase. Protein Sci 4:756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, Duran R, Mencacci NE, Sawcer SJ, Barker RA. 2013. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136:392–399. [DOI] [PubMed] [Google Scholar]
- Winfield SL, Tayebi N, Martin BM, Ginns EI, Sidransky E. 1997. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: implications for Gaucher disease. Genome Res 7:1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, Berthiaume Y, Cutler D, Cojocaru A, Collaco JM, Corey M, Dorfman R, et al. 2011. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 43:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz Y, Hoffmann P, vom Dahl S, Breiden B, Sandhoff R, Niederau C, Horwitz M, Karlsson S, Filocamo M, Elstein D, Beck M, Sandhoff K, et al. 2013. Functional and genetic characterization of the non-lysosomal glucosylceramidase 2 as a modifier for Gaucher disease. Orphanet J Rare Dis 8:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz Y, Matern H, Thompson B, Allegood JC, Warren RL, Ramirez DMO, Hammer RE, Hamra FK, Matern S, Russell DW. 2006. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J Clin Invest 116:2985–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshige A, Muto M, Watanabe T, Hojo H, Matsuda J. 2015. The effects of chemically synthesized saposin C on glucosylceramide-β-glucosidase. Clin Biochem 48:1177–1180. [DOI] [PubMed] [Google Scholar]
- Zhang CK, Stein PB, Liu J, Wang Z, Yang R, Cho JH, Gregersen PK, Aerts JMFG, Zhao H, Pastores GM, Mistry PK. 2012. Genome-wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation. Am J Hematol 87:377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ren J, Padilla-Parra S, Fry EE, Stuart DI. 2014. Lysosome sorting of β-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat Commun 5:4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunke F, Andresen L, Wesseler S, Groth J, Arnold P, Rothaug M, Mazzulli JR, Krainc D, Blanz J, Saftig P, Schwake M. 2016. Characterization of the complex formed by β-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc Natl Acad Sci U S A 113:3791–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]