

Abstract
The abundantly expressed calcium/calmodulin‐dependent protein kinase II (CAMK2), alpha (CAMK2A), and beta (CAMK2B) isoforms are essential for learning and memory formation. Recently, a de novo candidate mutation (p.Arg292Pro) in the gamma isoform of CAMK2 (CAMK2G) was identified in a patient with severe intellectual disability (ID), but the mechanism(s) by which this mutation causes ID is unknown. Here, we identified a second, unrelated individual, with a de novo CAMK2G p.Arg292Pro mutation, and used in vivo and in vitro assays to assess the impact of this mutation on CAMK2G and neuronal function. We found that knockdown of CAMK2G results in inappropriate precocious neuronal maturation. We further found that the CAMK2G p.Arg292Pro mutation acts as a highly pathogenic gain‐of‐function mutation, leading to increased phosphotransferase activity and impaired neuronal maturation as well as impaired targeting of the nuclear CAMK2G isoform. Silencing the catalytic site of the CAMK2G p.Arg292Pro protein reversed the pathogenic effect of the p.Arg292Pro mutation on neuronal maturation, without rescuing its nuclear targeting. Taken together, our results reveal an indispensable function of CAMK2G in neurodevelopment and indicate that the CAMK2G p.Arg292Pro protein acts as a pathogenic gain‐of‐function mutation, through constitutive activity toward cytosolic targets, rather than impaired targeting to the nucleus.
Keywords: calcium/calmodulin‐dependent protein kinase type 2, functional genomics, in utero electroporation, intellectual disability, neurodevelopmental disorders, neuronal arborization
1. INTRODUCTION
Intellectual disability (ID), characterized by an IQ score of <70 and moderate to severe limitations in adaptive functioning, often has a genetic cause. Next generation sequencing (NGS) offers a powerful diagnostic modality for undiagnosed ID patients. Currently, despite the recent identification of more than 700 genes associated with ID, in approximately half of ID cases, the genetic pathogenicity cannot be established with certainty, since the mutation has not been identified before (Vissers, Gilissen, & Veltman, 2016).
Calcium/calmodulin‐dependent protein kinase type II (CAMK2) is a Ser/Thr protein kinase family consisting of four isozymes (CAMK2A, CAMK2B, CAMK2G, and CAMK2D). CAMK2A and CAMK2B have a highly abundant and neuron‐selective expression. Targeted deletion of CAMK2A or CAMK2B in mice has revealed their indispensable requirement for normal cognition (Borgesius et al., 2011; Elgersma et al., 2002; Giese, Fedorov, Filipkowski, & Silva, 1998; Hell, 2014; Lisman, Schulman, & Cline, 2002; Lisman, Yasuda, & Raghavachari, 2012; Silva, Paylor, Wehner, & Tonegawa, 1992a; Silva, Stevens, Tonegawa, & Wang, 1992b; van Woerden et al., 2009). Moreover, human CAMK2A and CAMK2B mutations have recently been established as a cause of ID (Küry et al., 2017). In contrast to CAMK2A and CAMK2B, much less is known about the other CAMK2 isoforms, CAMK2G and CAMK2D. However, recently a de novo mutation in the CAMK2G gene (c.875G > C p.Arg292Pro, henceforth denoted as CAMK2GArg292Pro) was identified in a patient with severe ID (de Ligt et al., 2012). In addition, here, we describe a second, unrelated individual with ID carrying the c.875G > C p.Arg292Pro mutation, strongly suggesting a causative role for CAMK2G in ID.
Even though the expression of CAMK2G in the adult brain is relatively low compared to CAMK2A and CAMK2B (Tobimatsu & Fujisawa, 1989), CAMK2G is highly abundant in the developing nervous system in rats (Bayer, Löhler, Schulman, & Harbers, 1999) and in humans, where it is the dominant isoform in the first trimester in the hippocampus (Figure 1A). Other than a few studies, reporting associations of common variation in CAMK2G with reduced memory performance (de Quervain & Papassotiropoulos, 2006), depression (Li et al., 2013) and chronic inflammatory pain (Pan et al., 2014), and a recent study proposing that CAMK2G functions as a calcium/calmodulin (Ca2+/CaM) shuttle (Ma et al., 2014), the mechanistic function of CAMK2G during neurodevelopment has remained largely unknown.
Figure 1.
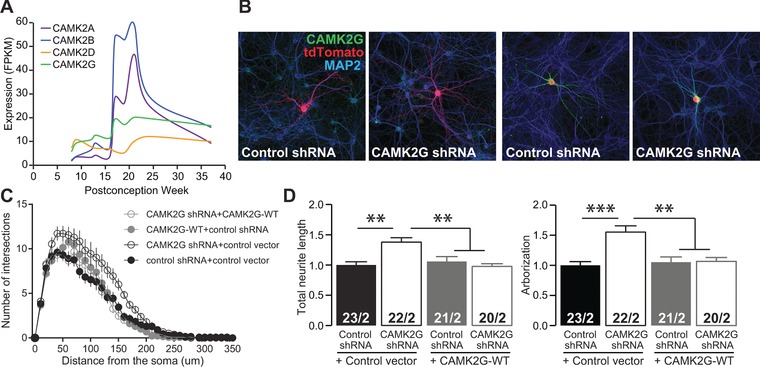
CAMK2G knockdown in primary hippocampal neurons results in increased arborization. (A) Expression levels of human CAMK2A, CAMK2B, CAMK2D, and CAMK2G in the hippocampus during gestation, showing that CAMK2G is the dominant isoform in the first trimester, and contributes substantially in the second and the third trimester. Data was extracted from the BrainSpan database (https://www.brainspan.org/). (B) Representative confocal images of hippocampal neurons co‐transfected on DIV7 with combinations of control shRNA, with control vector or CAMK2GWT and an RFP plasmid or shRNA against Camk2g with control vector or CAMK2GWT and an RFP plasmid. Transfected neurons are identified by the RFP plasmid (red). See also Supplement 1 related to Figure 1. (C) Sholl analysis of neurite complexity. (D) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control shRNA (total neurite length: one‐way ANOVA, F[3,82] = 8.42, P = 6.05E‐05; control vector+control shRNA vs. control vector+Camk2g‐shRNA, P = 0.0004; CAMK2GWT+control shRNA, P = 0.99; CAMK2GWT+Camk2g‐shRNA, P = 0.99; control vector+Camk2g‐shRNA vs. CAMK2GWT+control shRNA, P = 0.005; CAMK2GWT+Camk2g‐shRNA, P = 0.0002; CAMK2GWT+control shRNA vs. CAMK2GWT+Camk2g‐shRNA, P = 0.99; arborization: one‐way ANOVA, F[3,82] = 10.08, P = 1.00E‐05; control vector+control shRNA vs. control vector+Camk2g‐shRNA, P = 0.0001; CAMK2GWT+control shRNA, P = 0.99; CAMK2GWT+Camk2g‐shRNA, P = 0.99; control vector+Camk2g‐shRNA vs. CAMK2GWT+control shRNA, P = 0.0003; CAMK2GWT+Camk2g‐shRNA, P = 0.0005; CAMK2GWT+control shRNA vs. CAMK2GWT+Camk2g‐shRNA, P = 0.99)
Here, we describe the clinical features of both individuals carrying the CAMK2G p.Arg292Pro mutation, the contribution of CAMK2G to neuronal migration in vivo and neuronal maturation in vitro and how the CAMK2GArg292Pro mutation affects CAMK2G function. We found that reduced expression of CAMK2G leads to inappropriate precocious neuronal maturation. Moreover, we demonstrate that the p.Arg292Pro mutation is a pathogenic gain‐of‐function mutation, which renders the CAMK2G protein constitutively active, and causes a severe impairment of neuronal migration and maturation. Although the mutation also interferes with nuclear import, we found that the pathogenic effect we describe is not related to Ca2+/CaM shuttling. Taken together, our findings establish CAMK2G as a critical regulator of neurodevelopment and propose a novel mechanism by which the ID mutation, CAMK2GArg292Pro exerts its pathogenic effect.
2. MATERIAL AND METHODS
2.1. Identification of the CAMK2G p.Arg292Pro patients
Patient 1 was part of a whole‐exome sequencing study performed on patients with severe intellectual disability in Nijmegen, the Netherlands (de Ligt et al., 2012). Written consent for use of the patients’ photos were obtained from the parents. Patient 2 was identified by clinical whole‐exome sequencing done as part of a diagnostic evaluation through the Greenwood Genetic Center in South Carolina.
2.2. Constructs
The cDNA sequence from human CAMK2GWT (NM_172171.2, variant without NLS) was obtained from a human brain cDNA library by PCR (Phusion high fidelity, Thermo Fisher) using the following primers: Fw 5′ ACATCTGGCGCGCCACCATGGCCACCACCGCCACCTG 3′ and Rev 5′ AAGTCCTTAATTAATTATCACTGCAGCGGTGCGGCAG 3′ and cloned into our dual promoter expression vector (Suppl. Fig. S1). The cDNA sequence from rat CAMK2AWT and CAMK2BWT were kindly provided by Tobias Meyer and was cloned into our expression vector. The NLS version of CAMK2GWT was generated using the following primers: Fw 5′ CGAGTTCCAGCGTGCACCTAATGCCACAGAGCAACAACAAAAACAGTCTCG 3′ and Rev 5′ CATTAGGTGCACGCTGGAACTCGACTTCCTTTTCTTGACACCGCCATCCGAC 3′. The p.Thr287Ala point mutation cDNA was synthesized by and ordered from GeneCust. The different point mutations were introduced with site‐directed mutagenesis (Invitrogen) using the following primers: CAMK2G–c.875G > C (p.Arg292Pro), Fw 5′ CTGTGGAGTGTTTGCCCAAGTTCAATGCCCG 3′ and Rev 5′ CGGGCATTGAACTTGGGCAAACACTCCACAG 3′; CAMK2G–c.128A > G (p.Lys43Arg), Fw 5′ ACGCAGGAGTACGCAGCAAGAATCATCAATACCAAGAAG 3′ and Rev 5′ GTATTGATGATTCTTGCTGCGTACTCCTGCGT 3′; CAMK2G–c.859‐860AC > GA (p.Thr287Asp), Fw 5′ GCATCGTCAGGAGGATGTGGAGTGTTTG 3′ and Rev 5′ CAAACACTCCACATCCTCCTGACGATGC 3′; CAMK2G–c.916‐919ACGA > GTGG (p.Thr306Val/Thr307Ala), Fw 5′ CTGAAGGGTGCCATCCTCGTGGCCATGCTTGTCTCCAGGA 3′ and Rev 5′ TCCTGGAGACAAGCATGGCCACGAGGATGGCACCCTTCAG 3′; CAMK2G–c.907‐909GCC > AGA (p.Ala303Arg), Fw 5′ GAAAACTGAAGGGTAGAATCCTCACGACCAT 3′ and Rev 5′ ATGGTCGTGAGGATTCTACCCTTCAGTTTTC 3′. CAMK2G–c.1000T > G (p.Ser334Ala), Fw 5′ GGTGTCAAGAAAAGGAAGGCGAGTTCCAGCGTGCACCT 3′ and Rev 5′ AGGTGCACGCTGGAACTCGCCTTCCTTTTCTTGACACC 3′. CAMK2A–c.871‐873 AAG > CCT (p.Lys291Pro), Fw 5′ GGAGACCGTGGACTGCCTGCCTAAGTTCAATGCCAGGAGGA 3′ and Rev 5′ TCCTCCTGGCATTGAACTTAGGCAGGCAGTCCACGGTCTCC 3′; CAMK2B–c.874‐876AAG > CCT (p.Lys292Pro), Fw 5′ GGAGACTGTGGAATGTCTGCCTAAGTTCAATGCAAGGAGGA 3′ and Rev 5′ TCCTCCTTGCATTGAACTTAGGCAGACATTCCACAGTCTCC 3′. The dual promoter expression vector was generated from the pCMV–tdTomato vector (Clonetech), where the CMV promoter was replaced with a CAGG promoter followed by a multiple cloning site (MCS) and transcription terminator sequence. To assure expression of the tdTOMATO independent from the gene of interest, a PGK promoter was inserted in front of the tdTomato sequence. For all the in vivo and in vitro experiments, the vector without a gene inserted in the MCS was taken along as control (control vector). shRNA constructs were obtained from the MISSION shRNA library for mouse genomes of Sigma Life Sciences and The RNAi Consortium (TRC). For knockdown of CAMK2G, we used three different shRNA plasmids, each targeting a different sequence: (1) GCCCGAGATCATCAGAAACTA, (2) CCTGAGGTCTTGAGGAAAGAT, and (3) CTACGCAGGAATATGCTGCAA. The control shRNA plasmid is the MISSION non‐target shRNA control vector: CAACAAGATGAAGAGCACCAA.
2.3. Mice
For the neuronal cultures, FvB/NHsD females were crossed with FvB/NHsD males (both ordered at 8–10 weeks old from Envigo). For the in utero electroporation, female FvB/NHsD (Envigo) were crossed with male C57Bl6/J (ordered at 8–10 weeks old from Charles River). All mice were kept group‐housed in IVC cages (Sealsafe 1145T, Tecniplast) with bedding material (Lignocel BK 8/15 from Rettenmayer) on a 12/12 h light/dark cycle in 21°C (±1°C), humidity at 40–70% and with food pellets (801727CRM(P) from Special Dietary Service) and water available ad libitum. All animal experiments were approved by the Local Animal Experimentation Ethical Committee, in accordance with Institutional Animal Care and Use Committee guidelines.
2.4. HEK‐293T cell transfections
To test the expression vector with the CAMK2 constructs, to measure the phosphorylation levels of CAMK2, and for immunoprecipitation (IP) experiments, we chose HEK‐293T cells, a cell line that is easy to transfect and culture. These cells were mycoplasma‐free but not authenticated. HEK‐293T cells were cultured in DMEM/10% Fetal Calf Serum (FCS)/1% penicillin/streptomycin in 6‐well plates (or 10 cm dishes for IP experiments) and transfected when 50% confluent with the following DNA constructs: control vector, CAMK2GWT, CAMK2G‐NLSWT, CAMK2GArg292Pro, CAMK2G‐NLSArg292Pro, CAMK2GAla303Arg, CAMK2AWT, CAMK2ALys291Pro, CAMK2BWT, and CAMK2BLys292Pro (3 ug per 6‐well and 18 ug per 10 cm dish). Transfection of the plasmids was done using polyethylenimine (PEI) according to the manufacturer instructions (Sigma). Six to twelve hours after transfection, the medium was changed to reduce toxicity. Transfected cells were then used for western blot and Immunoprecipitation experiments.
2.5. Western blot
Two to three days after transfection, HEK cells were harvested and homogenized in lysis buffer (10 mM Tris‐HCl 6.8, 2.5% SDS, 2 mM EDTA), containing protease inhibitor cocktail (#P8340, Sigma), phosphatase inhibitor cocktail 2 (#P5726, Sigma) and phosphatase inhibitor cocktail 3 (#P0044, Sigma). Protein concentration in the samples was determined using the BCA protein assay kit (Pierce) and then lysate concentrations were adjusted to 1 mg/ml. Western blots were probed with primary antibodies against CAMK2G (C‐18; raised against the 478–495 C‐terminal peptide, 1:1000, sc‐1541, Santa Cruz; validated in this study by overexpression experiments), CAMK2A (6G9, 1:40.000, Abcam; validated in Elgersma et al. (2002)), CAMK2B (CB‐β1, 1:10.000, Invitrogen; validated in van Woerden et al. (2009)), Actin (MAB1501R, 1:20.000, Chemicon; validated in Antibodypedia (https://Antibodypedia.com), Ph‐Thr286/Thr287 (auto‐phosphorylated CAMK2 antibody; #06‐881; 1:1000; Upstate Cell Signaling Solutions; validated in Elgersma et al. (2002)) and RFP (#600401379, 1:2000, Rockland, validated in this study by overexpression experiments), and secondary antibodies (goat anti‐mouse (#926‐32210), goat anti‐rabbit (#926‐68021), and donkey anti‐goat (#926‐68074), all 1:15.000, LI‐COR). Blots were quantified using LI‐COR Odyssey Scanner and Odyssey 3.0 software.
2.6. Immunoprecipitation
Two to three days after HEK transfection in 10 cm dishes, cells were harvested in PBS and cell pellets were taken up in 500 ul of IP lysis buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1% NP40) supplemented with protease inhibitor cocktail (#P8340, Sigma), phosphatase inhibitor cocktail 2 (#P5726, Sigma), and phosphatase inhibitor cocktail 3 (#P0044, Sigma). Protein concentration in the samples was determined using the BCA protein assay kit (Pierce) and 600 ug of total protein adjusted to a volume of 800 ul was used for immunoprecipitation. 20 ul of the total volume was saved as “input.” Cell lysates were incubated with 50 ul of prewashed 50 % slurry Protein A Sepharose CL‐4B beads (#17‐0780‐01, GE Healthcare) at 4°C for >1 h. Beads were then isolated and discarded by spinning down samples at 1000 rpm for 1′. Lysates were placed in a new tube with 3 ul of mouse calmodulin antibody (C3545, Sigma) and allowed to mix overnight at 4°C. The next morning, lysates were incubated with 50 ul of prewashed 50% slurry Protein G Sepharose beads (#17‐0618‐01, GE Healthcare) at 4°C for > 1 h. Beads were then washed 3× with cold lysis buffer and after final wash proteins were eluted adding 20 ul of 2× Laemmli sample buffer and boiling for 5′ at 95°C before processing by western blot.
2.7. Primary hippocampal cultures
Primary hippocampal and cortical neuronal cultures were prepared from FvB/NHsD wild‐type mice according to the procedure described in Goslin and Banker (1991). Briefly, hippocampi were isolated from brains of E16.5 embryos and collected altogether in 10 ml of neurobasal medium (NB, Gibco) on ice. After two washings with NB, the samples were incubated in pre‐warmed trypsin/EDTA solution (Invitrogen) at 37°C for 20 min. After 2× washing in pre‐warmed NB, the cells were resuspended in 1.5 ml NB medium supplemented with 2% B27, 1% penicillin/streptomycin, and 1% glutamax (Invitrogen), and dissociated using a 5 ml pipette. Following dissociation, neurons were plated in a small drop on poly‐d‐lysine (25 mg/ml, Sigma) coated 15 mm glass coverslips at a density of 1×106 cells per coverslip in 12 well plates containing 1 ml of supplemented NB for each coverslip. The plates were stored at 37°/5% CO2 until the day of the transfection.
2.8. Neuronal transfection and immunocytochemistry
Neurons were transfected after 7 days in vitro (DIV) with the following DNA constructs: control vector (1.8 ug per coverslip), CAMK2GWT, CAMK2GArg292Pro, CAMK2GLys43Arg, CAMK2GLys43Arg/Arg292Pro, CAMK2GThr287Ala, CAMK2GThr287D/Thr306Val/Thr307Ala, CAMK2GAla303Arg, CAMK2G‐NLSWT, CAMK2G‐NLSArg292Pro, CAMK2G‐NLSSer334Ala, CAMK2G‐NLSSer334Ala/Arg292Pro, CAMK2G‐NLSLys43Arg/Arg292Pro, CAMK2AWT, CAMK2ALys291Pro, CAMK2BWT, and CAMK2BLys292Pro (all 2.5 ug per coverslip) or for knockdown experiments with a pool of the CAMK2G shRNAs with an RFP plasmid (Addgene) or the control shRNA with an RFP plasmid (all in total 4 ug per coverslip). Lipofectamine 2000 was used to transfect neurons, according to the manufacturer instructions (Invitrogen). For the neuronal morphology analysis, neurons were fixed 5 days post‐transfection with methanol and 4% paraformaldehyde (PFA)/4% sucrose, when stained for CAMK2G and 4% paraformaldehyde/4% sucrose, when stained for CAMK2A or CAMK2B and incubated overnight at 4°C with primary antibodies in GDB buffer (0.2% BSA, 0.8 M NaCl, 0.5% Triton X‐100, 30 mM phosphate buffer (PB), pH 7.4). To measure level of knockdown of CAMK2G and protein localization, neurons were fixed 5 days post‐transfection with methanol and 4% paraformaldehyde/4% sucrose and stained for CAMK2G, but for morphological analysis neurons were fixed with 4% paraformaldehyde/4% sucrose and processed without staining for CAMK2G. The following primary antibodies were used: MAP2 (1:500, #188004, Synaptic System, validation can be found on the manufacturers website: https://www.sysy.com/products/map2/ref.php), CAMK2G (C‐18, 1:100, Santa Cruz), CAMK2A (6G9, 1:200, Abcam), CAMK2B (CB‐β1, 1:100, Invitrogen). For secondary antibodies, anti‐mouse‐Alexa488 (#715‐545‐150), anti‐goat‐Alexa488 (#705‐545‐003), and anti‐guinea‐pig‐Alexa647 (#706‐605‐148) conjugated antibodies (all 1:200, Jackson ImmunoResearch) were used. For the localization experiments, neurons were also stained with 4′,6‐diamidino‐2‐phenylindole solution (DAPI, 1:10000, Invitrogen) for 15′ before being mounted on glass. Slides were mounted using Mowiol‐DABCO (Sigma) mounting medium. Confocal images were acquired using a LSM700 confocal microscope (Zeiss).
For the analysis of the neuronal transfections, at least 10 confocal images (20× objective, 0.5 zoom, 1024 × 1024 pixels) of different transfected neurons (identified by the red staining) were taken from each coverslip for each experiment with at least two independent replications. For the analysis of the neuronal morphology, the NeuronJ plugin for ImageJ software was used to trace the dendrites with their branches. Total neurite length and arborization were measured and analyzed. All values were normalized against the mean value for each parameter of the control (control vector). Sholl analysis for the in vitro knockdown experiment was performed by counting the number of neurite intersections between the traced neurons and a series of concentric circumferences at an interval of 10 um from each other with the center on the soma. For the analysis of the protein levels upon shRNA transfection, the “Measure RGB” plugin from ImageJ software was used to measure the intensity of the fluorescent signal of the transfected cell, which was normalized against non‐transfected cells on the same coverslip and then normalized against the mean value of control shRNA transfected cells. For the analysis of the protein localization experiments, the DAPI staining was used to identify the nucleus and define a region of interest (ROI). To measure the intensity of the fluorescent signal relative to the protein of interest of the ROI nucleus and the ROI cytoplasm of each transfected cell, we used the “Measure” plugin of Fiji software. The intensity signal (Integrated density) was corrected for the area of the ROI and the background signal by using the following formula: IntDen–(area of the ROI × mean fluorescence of background reading). The ratio of the nucleus/cytoplasm corrected intensity value for each construct was averaged and normalized against the averaged ratio of the WT of the same neuronal batch. Analysis was done by an experimenter blinded for the transfection conditions.
2.9. In utero electroporation
The procedure was performed in pregnant FvB/NHsD mice at E14.5 of gestation to target mainly the progenitor cells giving rise to pyramidal cells of the layer 2/3 (Saito, 2006; Taniguchi, Young‐Pearse, Sawa, & Kamiya, 2012). The DNA construct (1.5–3 ug/ul) was diluted in fast green (0.05%) and injected in the lateral ventricle of the embryos while still in uterus, using a glass pipette controlled by a Picospritzer III device. To ensure the proper electroporation of the injected DNA constructs (1–2 ul) into the progenitor cells, five electrical square pulses of 45 V with a duration of 50 ms per pulse and 150 ms inter‐pulse interval were delivered using tweezer‐type electrodes connected to a pulse generator (ECM 830, BTX Harvard Apparatus). The electrodes were placed in such a way that the positive pole was targeting the developing somatosensory cortex. The following plasmids were injected: control vector, CAMK2GWT, CAMK2GArg292Pro, CAMK2GLys43Arg, CAMK2GLys43Arg/Arg292Pro, CAMK2GThr287Ala, CAMK2GThr287Asp/Thr306Val/Thr307Ala, CAMK2GAla303Arg, CAMK2G‐NLSWT, CAMK2G‐NLSArg292Pro, CAMK2G‐NLSLys43Arg/Arg292Pro, CAMK2AWT, CAMK2ALys291Pro, CAMK2BWT, and CAMK2BLys292Pro or for knockdown experiments: a pool of the CAMK2G shRNAs with an RFP plasmid (Addgene) or the control shRNA with an RFP plasmid. After birth, pups (M/F) were sacrificed at P18‐P22 for histochemical processing.
2.10. Immunohistochemistry
Mice were deeply anesthetized with an overdose of Nembutal and transcardially perfused with 4% paraformaldehyde. Brains were extracted and post‐fixed in 4% PFA. Brains were then embedded in gelatin and cryoprotected in 30% sucrose in 0.1 M phosphate buffer, frozen on dry ice, and sectioned using a freezing microtome (40–50 μm thick). Free‐floating coronal sections were washed in 0.1 M PB and a few selected sections were counterstained with 4′,6‐diamidino‐2‐phenylindole solution (DAPI, 1:10,000, Invitrogen) before being mounted with Mowiol on glass. Overview images of the coronal sections were acquired by tile scan imaging using a LSM700 confocal microscope (Zeiss) with a 10× objective. Zoom in images of the targeted area were taken using a 20× objective.
For the migration analysis, confocal images (10× objective, 0.5 zoom, 1024 × 1024 pixels) were taken from 2 to 3 non‐consecutive sections from at least three successfully targeted animals per plasmid. Images were rotated to correctly position the cortical layers, and the number of cells in different layers were counted using ImageJ (Analyze particles option), and the results were exported to a spreadsheet for further analysis. Cortical areas from the pia to the ventricle were divided in 10 equal‐sized bins and the percentage of tdTOMATO‐positive cells per bin was calculated.
2.11. Statistical analysis
All data was assumed to be normally distributed. Statistical difference between the conditions for the in vitro overexpression experiments was determined using one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc test for multiple comparisons and for the knockdown experiments using the two‐tailed unpaired t‐test (dual comparison). For the western blot analysis, one‐tailed and two‐tailed unpaired t‐test was used (dual comparison). For the in vivo experiments on neuronal migration, the analysis was performed on the number of targeted cells, measured as sum of the first four bins, considered to correspond to the layers 2/3 of the somatosensory cortex. Statistical analysis was performed using one‐way ANOVA, followed by Bonferroni's multiple comparison tests. Based on previous experiments performed in our lab, we considered that for the neuronal experiments in vitro at least two replicates, with 10 neurons each were necessary. For the western blot analysis, we considered at least three replicates, and for the IUE experiments, previous experiments in our laboratory showed that at least three targeted pups were necessary to draw any conclusion about the migration. P‐values are indicated in the figures with “*”: * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.0001.
3. RESULTS
3.1. CAMK2G p.Arg292Pro mutation causes severe intellectual disability
Patient 1 carrying the CAMK2G p.Arg292Pro mutation has already briefly been described in a whole‐exome sequencing study describing several individuals with severe ID (de Ligt et al., 2012). Here, we identified a second, unrelated carrier of the heterozygous de novo CAMK2G p.Arg292Pro mutation (patient 2). He presented in infancy with generalized hypotonia and developmental delay and has remained hypotonic with markedly delayed gross motor and speech/language skills. He is currently diagnosed with severe ID and autism at 5 years of age. His physical exams have been notable for relative macrocephaly, prominent capillary vascular malformations on the forehead and glabella, minor dysmorphic facial features, and severe hypotonia. A detailed description of both patients is provided in Suppl. Fig. S2. Both patients suffer from severe ID and severe hypotonia. They both have some facial dysmorphisms (Suppl. Fig. S2), though not strikingly similar. By comparison, although individuals with mutations in CAMK2A or CAMK2B all suffer from ID and in most cases hypotonia, facial dysmorphisms are not commonly observed among individuals carrying mutations in CAMK2A or CAMK2B (Küry et al., 2017).
3.1.1. Knockdown of CAMK2G causes increased neuronal arborization
Identifying two unrelated patients with a severe neurodevelopmental disorder carrying the same mutation in CAMK2G, strongly suggests that CAMK2G plays a role in neurodevelopment and neuronal functioning. However, whereas the indispensable roles of CAMK2A and CAMK2B for neuronal plasticity and cognitive function are well established, the neuronal function of CAMK2G remains poorly characterized. Considering the highly abundant expression of CAMK2G in the first trimester during which the nervous system starts to develop and the stable expression during the rest of the prenatal period (Figure 1A), we set out to investigate the role of CAMK2G during neuronal maturation in vitro, using shRNAs to effect knockdown of the CAMK2G protein. We transfected mouse primary hippocampal neurons with shRNAs directed against Camk2g or a control plasmid, together with a red florescent protein (RFP) reporter plasmid to visualize transfected neurons. Despite the CAMK2G antibody cross‐reactivity with CAMK2A and CAMK2B (Suppl. Fig. S3), we nevertheless observed a 35% reduction of immunofluorescent signal 5 days after transfection, without any change in CAMK2A or CAMK2B levels (Suppl. Fig. S3) consistent with a robust decrease specifically of CAMK2G. Transfection of Camk2g shRNA resulted in a substantial alteration of neuronal morphology, with a clear increase in the complexity of the neurite tree compared to the control condition, as measured by Sholl analysis (Figure 1C). Additional quantification of the neurite length and arborization revealed that neurons with reduced levels of CAMK2G exhibited a 38% increase in total neurite length and 55% increase of arborization compared to the control condition (Figure 1B and D, see figure legend for statistics). Mice express three CAMK2G isoforms, one of which contains a nuclear localization signal (NLS) (Suppl. Fig. S4A). This isoform is present in both cytosol and nucleus and was recently proposed to shuttle Ca2+/CaM into the nucleus (Ma et al., 2014). In order to assess to what extent the observed effect on neurite length and arborization was specific for the down‐regulation of CAMK2G and whether there is differential contribution of the CAMK2G isoforms, we co‐transfected either the human CAMK2G isoform lacking the NLS (CAMK2G), the CAMK2G isoform containing the NLS (CAMK2G‐NLS), CAMK2A or CAMK2B together with the shRNA against mouse Camk2g. Whereas co‐transfection of CAMK2A or CAMK2B with Camk2g shRNA had no effect on total neurite length (Suppl. Fig. S4B and C), co‐transfection of CAMK2G or CAMK2G‐NLS with Camk2g shRNA both resulted in a complete rescue of the neuronal morphology phenotype caused by the down‐regulation of CAMK2G (Figure 1B–D and Suppl. Fig. S4B and C). These results indicate that CAMK2G is indispensable for regulating neuronal morphology, but that this function does not require its nuclear import.
3.1.2. The CAMK2GArg292Pro mutation is pathogenic both in vitro and in vivo
To study the effect of the ID‐associated CAMK2G missense variant on neuronal function, we cloned the human CAMK2G gene with and without the c.875G > C mutation in a dual promoter expression vector (see Section “Material and Methods”). Transfection of HEK‐293T cells with CAMK2GArg292Pro achieved a 10‐fold lower CAMK2G protein signal compared to that achieved by transfection with CAMK2GWT, despite equal tdTomato levels, indicating that the p.Arg292Pro mutation affects protein stability (Figure 2A and B). Next, we investigated the effect of expression of CAMK2GWT and CAMK2GArg292Pro in primary hippocampal neurons. Despite the reduced stability of CAMK2GArg292Pro, levels of CAMK2G were increased in neurons transfected (tdTomato‐positive) with CAMK2GArg292Pro (Figure 2C). Whereas expression of CAMK2GWT yielded no discernible morphological changes with respect to total neurite length or arborization, expression of CAMK2GArg292Pro strongly reduced total neurite length as well as arborization, which was notably in the opposite direction as observed following Camk2g knockdown (Figure 2D). These results suggest that the p.Arg292Pro mutation functions as a pathogenic gain‐of‐function.
Figure 2.
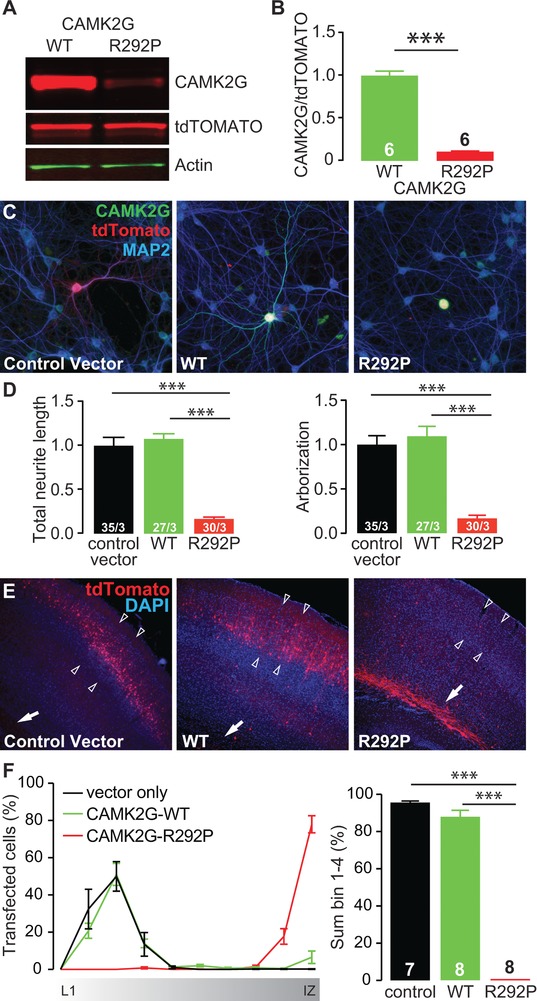
CAMK2GArg292Pro overexpression is severely disruptive for neurons in vitro and in vivo. (A) Representative western blot of CAMK2GWT or CAMK2GArg292Pro (R292P) transfected HEK‐293T cells. (B) Quantification of the levels of CAMK2G expression, normalized against tdTOMATO in the different conditions (CAMK2GWT vs. CAMK2GArg292Pro: t[10] = 16.62, P = 1.30E‐08, two‐tailed unpaired t‐test). (C) Representative confocal images of hippocampal neurons transfected on DIV7 with control vector (lacking CAMK2G), CAMK2GWT or CAMK2GArg292Pro. See also Supplement 1 related to Figure 2. Transfected neurons are identified by the tdTOMATO (red). (D) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control vector (total neurite length: one‐way ANOVA F[2,89] = 49.35, P = 3.79E‐15; control vector vs. CAMK2GWT, P = 0.9; CAMK2GArg292Pro, P = 0.0001; CAMK2GArg292Pro vs. CAMK2GWT, P = 0.0001; arborization: one‐way ANOVA F[2,89] = 31.8, P = 3.80E‐11; control vector vs. CAMK2GWT, P = 0.9; CAMK2GArg292Pro, P = 0.0001; CAMK2GArg292Pro vs. CAMK2GWT, P = 0.0001). (E) Representative images of P20‐P22 pups in utero electroporated at E14.5 with control vector, CAMK2GWT or CAMK2GArg292Pro tdTOMATO positive cells indicate the successfully targeted neurons. DAPI (blue) counterstaining is used to identify general cortical structure. (F) Left: quantification of the neuronal migration pattern from the Layer 1 (L1) to the intermediate zone (IZ); Right: analysis of the percentage of targeted cells that reach the outer layers of the cortex measured as sum of bin 1–4 (one‐way ANOVA, F[2,20] = 58.88, P = 4.16E‐09; control vector vs. CAMK2GWT, P = 0.07; CAMK2GArg292Pro, P = 0.0001; CAMK2GWT vs. CAMK2GArg292Pro, P = 0.0001). Arrowheads indicate layer 2/3 of the somatosensory cortex, whereas the arrow indicates the subventricular zone (SVZ). Data in (B), (D), and (F) are presented as mean ± SEM. Numbers (X/Y) depicted in the bar graphs represent the number of samples (B), the total number of cells (X) and number of independent cultures (Y) (D) or the number of pictures (F) analyzed
To evaluate the effect of CAMK2GArg292Pro expression in vivo, we used in utero electroporation at E14.5. This assay measures the ability of targeted neural cells in the subventricular zone to successfully migrate to layer 2/3 of somatosensory cortex. This assay is very sensitive for changes that perturb normal neural cell function (Saito & Nakatsuji, 2001; Tabata & Nakajima, 2001; Taniguchi et al., 2012). Strikingly, cells expressing CAMK2GArg292Pro exhibited a complete block of migration from the subventricular zone, despite more than 80% of the cells expressing CAMK2GWT having migrated normally to layer 2/3 of the somatosensory cortex (Figure 2E and F). These results further confirm the severe dominant effect of the p.Arg292Pro mutation on CAMK2G function.
Taken together these data suggest that although CAMK2GArg292Pro has reduced protein stability, this mutation does not lead to a loss of function of CAMK2G, but instead appears to result in a paradoxical gain‐of‐function.
3.1.3. CAMK2GArg292Pro has increased phosphotransferase activity
Considering that a gain‐of‐function mediates the effects of CAMK2GArg292Pro and that the p.Arg292Pro mutation is located within the α‐helical auto‐regulatory domain of CAMK2G that maintains the kinase in an inactive state in the absence of Ca2+/CaM (Hudmon & Schulman, 2002; Rellos et al., 2010), we hypothesized that the mechanism for the gain‐of‐function elicited by CAMK2GArg292Pro might involve enhanced phosphotransferase or even constitutive activity. Therefore, we first assessed the level of Thr287 phosphorylation in HEK‐293T cells as readout for the phosphotransferase activity by expressing either CAMK2GWT or CAMK2GArg292Pro. To allow for a direct comparison of their relative phosphorylation, we obtained equal protein levels by titrating the amount of transfected DNA. We found a significant increase of Thr287 phosphorylation in cells expressing CAMK2GArg292Pro compared to CAMK2GWT (Figure 3A and B), suggesting that the p.Arg292Pro mutation enhances phosphotransferase activity and acts as a gain‐of‐function mutation.
Figure 3.
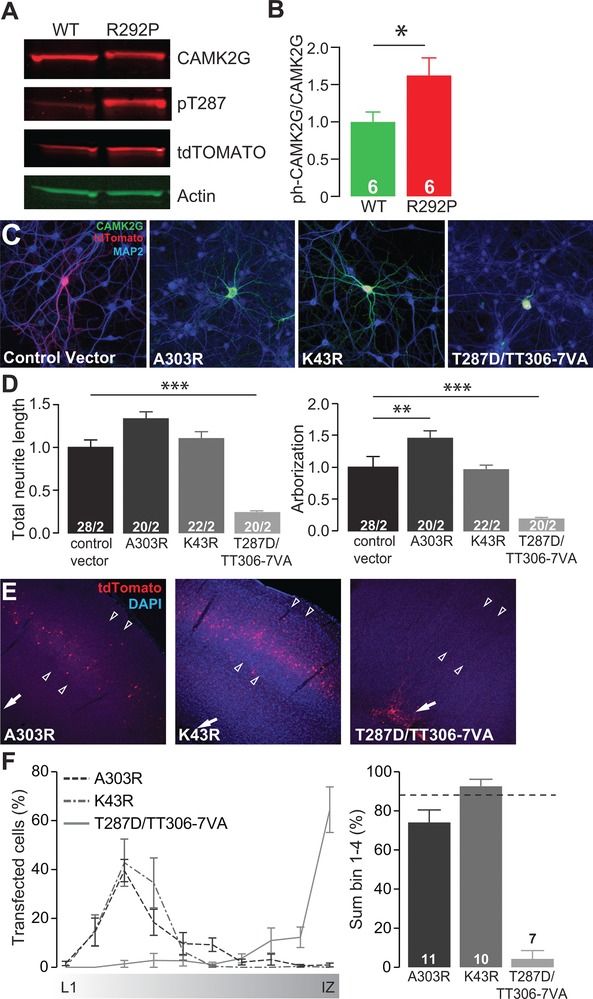
Overexpression of a dominant active form of CAMK2G is severely disruptive for neurons in vitro and in vivo. (A) Representative western blot of CAMK2GWT or CAMK2GArg292Pro (R292P) transfected HEK‐293T cells. (B) Quantification of the levels of CAMK2G–Thr287 phosphorylation, normalized against total CAMK2G in the different conditions (t[10] = 2.29, P = 0.05 two‐tailed unpaired t‐test). (C) Representative confocal images of hippocampal neurons transfected on DIV7 with control vector, CAMK2GLys43Arg (K43R), CAMK2GAla303Arg (A303R) or CAMK2GThr287Asp/Thr306Val/Thr307Ala (T287D/TT306‐7VA). Transfected neurons are identified by the tdTOMATO (red). (D) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control vector (total neurite length: one‐way ANOVA, F[3,86] = 24.75, P = 1.23E‐11; control vector vs. CAMK2GAla303Arg, P = 0.03; CAMK2GLys43Arg, P = 0.99; CAMK2GThr287Asp/Thr306Val/Thr307Ala, P = 0.0001; arborization: one‐way ANOVA, F[3,86] = 28.29, P = 8.03E‐13; control vector vs. CAMK2GAla303Arg, P = 0.0003; CAMK2GLys43Arg, P = 0.99; CAMK2GThr287Asp/Thr306Val/Thr307Ala, P = 0.0001). (E) Representative images of P20‐P22 pups in utero electroporated at E14.5 with control vector, CAMK2GLys43Arg, CAMK2GAla303Arg or CAMK2GThr287Asp/Thr306Val‐Thr307Ala. tdTOMATO positive cells represent neurons successfully targeted. DAPI (blue) counterstaining is used to identify general cortical structure. (F) Left: quantification of the neuronal migration pattern from the Layer 1 (L1) to the intermediate zone (IZ); Right: analysis of the percentage of targeted cells that reach the outer layers of the cortex measured as sum of bin 1–4 (one‐way ANOVA, F[3,32] = 56.92, P = 6.31E‐13; CAMK2GWT vs. CAMK2GAla303Arg, P = 0.15; CAMK2GLys43Arg, P = 0.99, CAMK2GThr287Asp/Thr306Val/Thr307Ala, P = 0.0001). The dotted line represents the level reached by the CAMK2G‐WT (see Figure 2F for comparison). Arrowheads indicate layer 2/3 of the somatosensory cortex, whereas the arrow indicates the subventricular zone (SVZ). Data in (B), (D), and (F) are presented as mean ± SEM. Numbers (X/Y) depicted in the bar graphs represent the number of samples (B), the total number of cells (X) and number of independent cultures (Y) (D) or number of pictures (F) analyzed
To further test whether the p.Arg292Pro mutation affects neuronal function by acting as a gain of function mutation, we first analyzed the effect of two mechanistically well‐established loss‐of‐function CAMK2 mutations on neuronal migration and morphology. The CAMK2GAla303Arg mutation prevents binding of Ca2+/CaM, and therefore Ca2+/CaM‐dependent activation (Fink et al., 2003; Payne et al., 1988; Shen & Meyer, 1999). In contrast, the CAMK2GLys43Arg mutation impairs phosphotransferase activity (Fink et al., 2003; Shen & Meyer, 1999). Notably, neither CAMK2GAla303Arg nor CAMK2GLys43Arg altered neurite length when expressed in cultured primary hippocampal neurons. Moreover, although a small but statistically significant increase of arborization was observed in CAMK2GAla303Arg expressing neurons, no effect of arborization was found in neurons expressing CAMK2GLys43Arg (Figure 3C and D). Finally, in vivo neural cell migration was also unaffected by expression of CAMK2GAla303Arg or CAMK2GLys43Arg (Figure 3E and F). Taken together these results show that expression of these phosphotransferase loss‐of‐function variants does not phenocopy the severe effects of the ID‐associated CAMK2GArg292Pro, indicating that the CAMK2GArg292Pro mutation is not a loss‐of‐function mutation.
We next tested the effect of the CAMK2GThr287Asp/Thr306Val/Thr307Ala mutant, which has previously been shown to significantly enhance autonomous activity due to the combination of a Threonine 287 substitution by the phospho‐mimetic aspartic amino acid (Mayford, Wang, Kandel, & O'Dell, 1995; Waldmann, Hanson, & Schulman, 1990) together with prevention of inhibitory phosphorylation at the Thr306 and 307 sites by substituting conserved but non‐phosphorylatable amino acids (Elgersma et al., 2002; Pi et al., 2010a; Pi, Otmakhov, Lemelin, De Koninck, & Lisman, 2010b). Interestingly, expression of this CAMK2GThr287Asp/Thr306Val/Thr307Ala mutant in HEK‐293T cells also achieved severe reduction of CAMK2G protein signal compared to cells transfected with CAMK2GWT (data not shown). Consistent with the hypothesis that increased phosphotransferase activity is a critical mediator of the gain‐of‐function mechanism underlying the p.Arg292Pro mutation, expression of the CAMK2GThr287Asp/Thr306Val/Thr307Ala mutant phenocopied CAMK2GArg292Pro with similarly severe reductions of total neurite length (Figure 3C), arborization (Figure 3D), and neuronal migration (Figure 3E and F). These findings lend additional support to a model whereby the CAMK2GArg292Pro mutation acts as a gain‐of‐function mutation by rendering CAMK2G constitutively active.
3.1.4. CAMK2G‐NLSArg292Pro has impaired nuclear translocation while permitting calmodulin binding
Recently, it was shown that CAMK2G functions as a shuttle for Ca2+/CaM into the nucleus (Ma et al., 2014). Hence, we investigated if the p.Arg292Pro mutation impacts this shuttling function. Using the nuclear localization signal‐containing isoform of CAMK2G, we first confirmed the functional impact of CAMK2G‐NLSArg292Pro on neuronal maturation and migration, which yielded the similarly severe neurodevelopmental impairments as CAMK2GArg292Pro (Suppl. Fig. S5). We then investigated whether CAMK2G‐NLSArg292Pro affects CaM binding by performing a CaM immunoprecipitation. Despite reduced expression levels, we found that more CAMK2G‐NLSArg292Pro is pulled down using the CaM antibody compared to CAMK2G‐NLSWT (Figure 4), suggesting that CaM binding is not disrupted, and even possibly that the affinity of CAMK2G‐NLSArg292Pro for CaM is increased.
Figure 4.
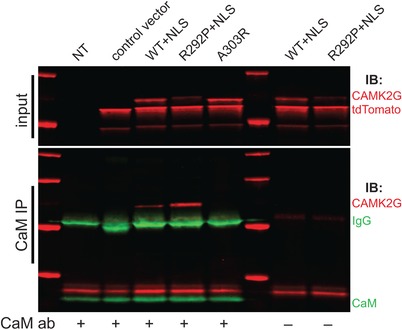
The CAMK2G‐NLSArg292Pro does not disrupt calmodulin binding. Representative western blot of a calmodulin immunoprecipitation experiment performed in HEK‐293T cells transfected with either control vector, CAMK2G‐NLSWT, CAMK2G‐NLSArg292Pro (R292P), CAMK2GAla303Arg (A303R), or non‐transfected (NT), probed with an antibody against CAMK2G, tdTOMATO, and calmodulin (CaM). Note the clear increase in CAMK2G pull‐down (in the presence of CaM antibody, left blot) in the mutant CAMK2G‐NLSArg292Pro lane compared to the WT, despite the reduced stability of the mutant (as observed in the input). As expected, no detectable CAMK2G is observed in the CAMK2GAla303Arg mutant lane. The “+” or “—” sign indicates the presence or absence, respectively of the calmodulin antibody
Next, we assessed whether CAMK2G‐NLSArg292Pro has impaired nuclear translocation. Neurons were transfected with CAMK2G‐NLSArg292Pro and CAMK2G‐NLSWT to determine their respective subcellular localization. As expected, we found that CAMK2G‐NLSWT was located in the nucleus as well as the cytoplasm. In contrast, CAMK2G‐NLSArg292Pro was almost exclusively localized to the cytoplasm (Figure 5A and B), suggesting that the p.Arg292Pro mutation disrupts the nuclear localization of CAMK2G‐NLS.
Figure 5.
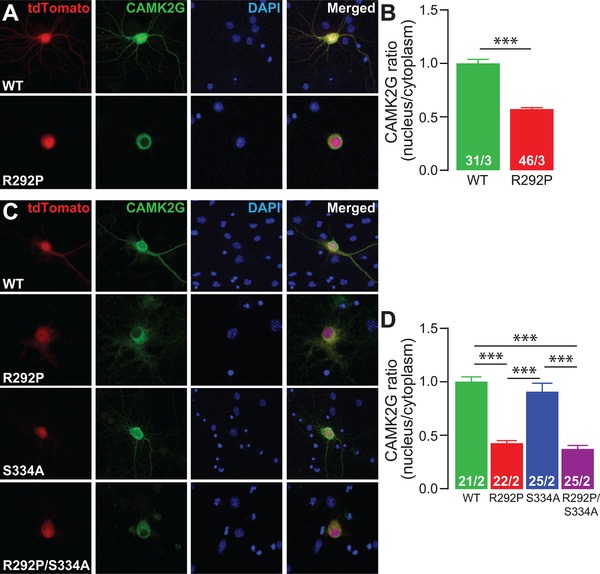
The CAMK2G‐NLSArg292Pro mutation interferes with nuclear translocation of CAMK2G‐NLS. (A and C) Representative confocal images of hippocampal (A) or cortical (C) neurons transfected on DIV7 with CAMK2G‐NLSWT, CAMK2G‐NLSArg292Pro (R292P), CAMK2G‐NLSSer334Ala (S224A), or CAMK2G‐NLSArg292Pro/Ser334Ala (R292P/S334A). Transfected neurons are identified by the tdTOMATO (red). (B and D) Summary bar graphs of CAMK2G expression level measured as a ratio nucleus vs. cytoplasm and normalized to the WT level. (B: CAMK2GWT vs. CAMK2GArg292Pro, P = 6.81E‐19, t(75) = 11.87, two‐tailed unpaired t‐test; D: one‐way ANOVA, F[3,86] = 43.58, P = 3.19E‐17; CAMK2G‐NLSWT vs. CAMK2G‐NLSArg292Pro, P = 0.0001; CAMK2G‐NLSSer334Ala, P = 0.99; CAMK2G‐NLSArg292Pro/Ser334Ala, P = 0.0001; CAMK2GArg292Pro vs. CAMK2G‐NLSSer334Ala, P = 0.0001; CAMK2G‐NLSArg292Pro/Ser334Ala, P = 0.99; CAMK2G‐NLSSer334Ala vs. CAMK2G‐NLSArg292Pro/Ser334Ala, P = 0.0001). Data are presented as mean ± SEM. Numbers (X/Y) depicted in the bar graphs represent the total number of cells (X) and number of independent cultures (Y) analyzed
Nuclear translocation of NLS‐containing CAMK2 isoforms requires a conformational change previously been shown to be dependent upon dephosphorylation of a Serine immediately adjacent to the NLS sequence (Ser334 in CAMK2G), resulting in exposure of the NLS domain (Heist, Srinivasan, & Schulman, 1998; Ma et al., 2014; Shioda, Sawai, Ishizuka, Shirao, & Fukunaga, 2015). Given that the p.Arg292Pro mutation enhances the Serine/Threonine kinase activity of CAMK2G, we sought to investigate whether increased phosphorylation of the Ser334 residue is a critical mediator of the abnormal localization of CAMK2G‐NLSArg292Pro. Therefore, we generated the CAMK2G‐NLSSer334Ala mutant and the CAMK2G‐NLSArg292Pro/Ser334Ala double mutant constructs to assess their respective subcellular localization. Whereas CAMK2G‐NLSSer334Ala was localized in both the nucleus and the cytoplasm, CAMK2G‐NLSArg292Pro/Ser334Ala remained exclusively cytoplasmic, indicating that the mislocalization resulting from the p.Arg292Pro mutation is not the result of excessive Ser334 phosphorylation (Figure 5C and D). Taken together, these data show that even though the p.Arg292Pro mutation does not reduce CaM affinity, it strongly impairs the nuclear localization of CAMK2G.
3.1.5. Pathogenicity of CAMK2GArg292Pro is not related to nuclear shuttling of Ca2+/CaM
Our findings indicate that the p.Arg292Pro mutation has two major effects on CAMK2G: it renders the protein constitutively active and interferes with its nuclear targeting. Therefore, we next aimed to investigate whether the pathogenicity is caused by the constitutive activity or by the impairment of nuclear targeting.
To examine whether the constitutive activity results from the observed increase of Thr287 auto‐phosphorylation (Figure 3A and B), we mutated Threonine 287 to Alanine, a mutation that strongly reduces autonomous activity (Fong, Taylor, Means, & Soderling, 1989; Giese et al., 1998; Hanson, Kapiloff, Lou, Rosenfeld, & Schulman, 1989; Ohsako, Nakazawa, Sekihara, Ikai, & Yamauchi, 1991; Waxham, Aronowski, Westgate, & Kelly, 1990). Expression of CAMK2GThr287Ala resulted in no discernible effects on morphology or neural cell migration (Figure 6). In contrast, expression of CAMK2GThr287Ala/Arg292Pro was comparably disruptive as CAMK2GArg292Pro on neuronal morphology and migration (Figure 6). These results indicate that functional consequences of the CAMK2GArg292Pro mutation are independent of Thr287 phosphorylation.
Figure 6.
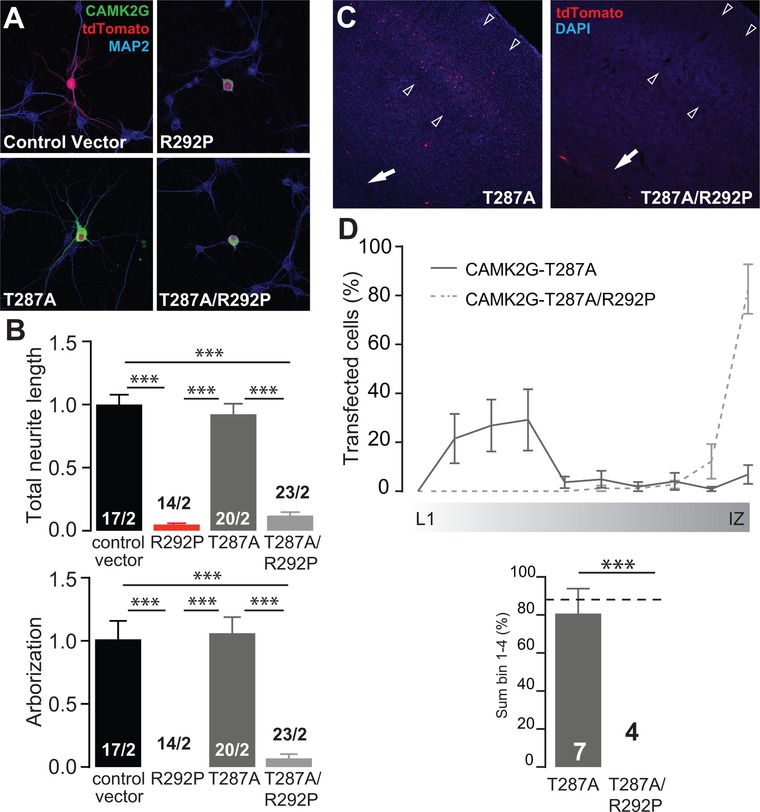
Auto‐phosphorylation at the Thr287 site is dispensable for the phenotype caused by the p.Arg292Pro mutation. (A) Representative confocal images of hippocampal neurons transfected on DIV7 with control vector, CAMK2GArg292Pro (R292P), CAMK2GThr287Ala (T287A), CAMK2GThr287Ala/Arg292Pro (T287A/R292P). Transfected neurons are identified by the tdTOMATO (red). (B) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control vector (total neurite length: one‐way ANOVA, F[3,70] = 69.33, P = 6.39E‐21; control vector vs. CAMK2GArg292Pro, P = 0.0001; CAMK2GThr287Ala, P = 0.9; CAMK2GThr287Ala/Arg292Pro, P = 0.0001; CAMK2GArg292Pro vs. CAMK2GThr287Ala, P = 0.0001; CAMK2GThr287Ala/Arg292Pro, P = 0.99; CAMK2GThr287Ala vs. CAMK2GThr287Ala/Arg292Pro, P = 0.0001; arborization: one‐way ANOVA, F[3,71] = 34.88, P = 5.74E‐14; control vector vs. CAMK2GArg292Pro, P = 0.0001; CAMK2GThr287Ala, P = 0.9; CAMK2GThr287Ala/Arg292Pro, P = 0.0001; CAMK2GArg292Pro vs. CAMK2GThr287Ala, P = 0.0001; CAMK2GT287A/R292P, P = 0.99; CAMK2GThr287Ala vs. CAMK2GThr287Ala/Arg292Pro, P = 0.0001). (C) Representative image of a P20 pup in utero electroporated at E14.5 with CAMK2GThr287Ala or CAMK2GThr287Ala/Arg292Pro. tdTOMATO positive cells represent neurons successfully targeted. DAPI (blue) counterstaining is used to identify general cortical structure. (D) Upper graph: quantification of the neuronal migration pattern from the Layer 1 (L1) to the intermediate zone (IZ); Lower graph: analysis of the percentage of targeted cells that reach the outer layers of the cortex measured as sum of bin 1–4 (one‐way ANOVA, F[2,16] = 24.34, P = 1.40E‐05; CAMK2GWT vs. CAMK2GThr287Ala, P = 0.99; CAMK2GThr287Ala/Arg292Pro, P = 0.0001; CAMK2GThr287Ala vs. CAMK2GThr287Ala/Arg292Pro, P = 0.0001). The dotted line represents the level reached by the CAMK2G‐WT (see Figure 2F for comparison). Arrowheads indicate layer 2/3 of the somatosensory cortex, whereas the arrow indicates the subventricular zone (SVZ). Data in (B) and (D) are presented as mean ± SEM. Numbers (X/Y) depicted in the bar graphs represent the total number of cells (X) and number of independent cultures (Y) (B) or number of pictures (D) analyzed
We next examined whether the p.Arg292Pro mutation gain‐of‐function requires the catalytic function of the kinase by introducing a p.Lys43Arg mutation, known to severely disable CAMK2G catalytic function. Compared to the severe neurodevelopmental impairments resulting from expression of CAMK2GArg292Pro, CAMK2GArg292Pro/Lys43Arg had a significantly milder impact on total neurite length and fully rescued arborization (Figure 7A). Moreover, CAMK2GArg292Pro/Lys43Arg expressing cells targeted at E14.5 by in utero electroporation revealed no discernible migration deficits compared to CAMK2GWT (Figure 2F, Figure 7C and D), indicating that the pathogenicity of CAMK2GArg292Pro requires its kinase activity.
Figure 7.
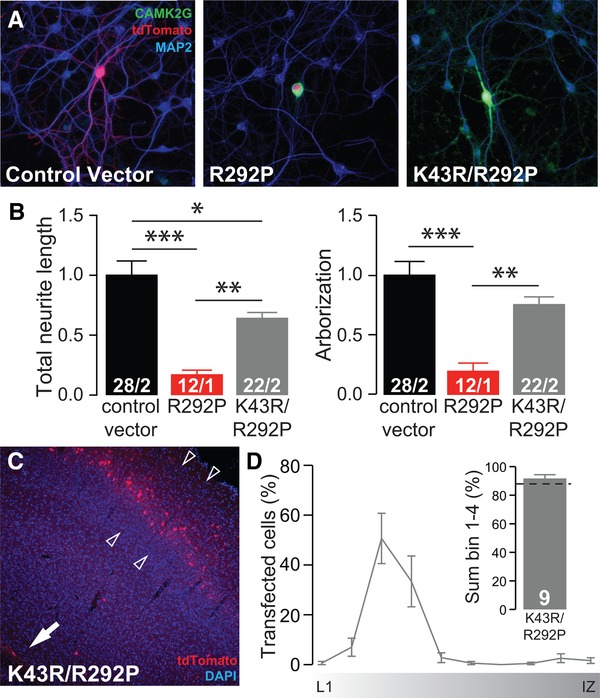
Silencing the catalytic activity of CAMK2GArg292Pro normalizes neuronal maturation and migration. (A) Representative confocal images of hippocampal neurons transfected on DIV7 with control vector or CAMK2GLys43Arg/Arg292Pro (K43R/R292P). Transfected neurons are identified by the tdTOMATO (red). (B) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control vector (total neurite length: one‐way ANOVA, F[2,61] = 18.50, P = 5.25E‐07; CAMK2GLys43Arg/Arg292Pro vs. control vector: P = 0.05; CAMK2GArg292Pro: P = 0.01; arborization: one‐way ANOVA, F[2,61] = 15.02, P = 4.97E‐06; CAMK2GLys43Arg/Arg292Pro vs. control vector, P = 0.16; CAMK2GArg292Pro, P = 0.01). (C) Representative image of a P20 pup in utero electroporated at E14.5 with CAMK2GLys43Arg/Arg292Pro. tdTOMATO positive cells represent neurons successfully targeted. DAPI (blue) counterstaining is used to identify general cortical structure. (D) Left: quantification of the neuronal migration pattern from the Layer 1 (L1) to the intermediate zone (IZ); Right: analysis of the percentage of targeted cells that reach the outer layers of the cortex measured as the sum of bin 1–4 (CAMK2GWT vs. CAMK2GLys43Arg/Arg292Pro, t[15] = 0.83, P = 0.42, two‐tailed unpaired t‐test).The dotted line indicates the CAMK2GWT level, see Figure 2F for comparison. Arrowheads indicate layer 2/3 of the somatosensory cortex, whereas the arrow indicates the subventricular zone (SVZ). Data in (B) and (D) are presented as mean±SEM. Numbers (X/Y) depicted in the bar graphs represent the total number of cells (X) and number of independent cultures (Y) (B) or number of pictures (D) analyzed
Since the introduction of the p.Lys43Arg mutation into the exclusively cytosolic CAMK2GArg292Pro isoform strongly attenuates the pathogenicity of the Arg292Pro mutation, we hypothesized that the pathogenicity of the p.Arg292Pro mutation is independent of its function as a nuclear Ca/CaM shuttle. To investigate this, we introduced the p.Lys43Arg mutation into the CAMK2G that carries an NLS, resulting in CAMK2G‐NLSArg292Pro. Analysis of the CAMK2G‐NLSLys43Arg/Arg292Pro double mutation showed that the Lys43Arg mutation did not alter the nuclear targeting deficit (Suppl. Fig. S6A and B). Nevertheless, both neuronal maturation and migration were again fully rescued by introducing the p.Lys43Arg mutation (Suppl. Fig. S6C–E).
Taken together these experiments suggest that the p.Arg292Pro mutation renders CAMK2G constitutively active and impairs neuronal maturation and migration independent of its role as a Ca2+/CaM shuttle.
3.1.6. The CAMK2GArg292Pro analogous mutations in CAMK2A and CAMK2B exhibit similar neurodevelopmental pathogenicity as CAMK2GArg292Pro
Given that CAMK2GArg292Pro exerts the pathogenic phenotype we describe via a non‐nuclear function, we hypothesized that the analogous mutations in CAMK2A or CAMK2B lacking an NLS might be similarly disruptive for neurodevelopment, given that they are almost exclusively localized to the cytoplasm. Therefore, we generated the analogous mutations of CAMK2GArg292Pro in CAMK2A and CAMK2B (Figure 8A). We first investigated the basal Thr286 and Thr287 phosphorylation levels of CAMK2A and CAMK2B, respectively. Whereas basal auto‐phosphorylation in HEK‐293T cells of CAMK2GWT was clearly evident, that of CAMK2AWT or CAMK2BWT was hardly detectible (Figure 8B). In contrast, both CAMK2ALys291Pro and CAMK2BLys292Pro exhibited robust basal Thr286/287 phosphorylation (Figure 8C), indicating that this Arg/Lys residue functions critically across multiple CAMK2 isoforms for constraining phosphotransferase activity.
Figure 8.
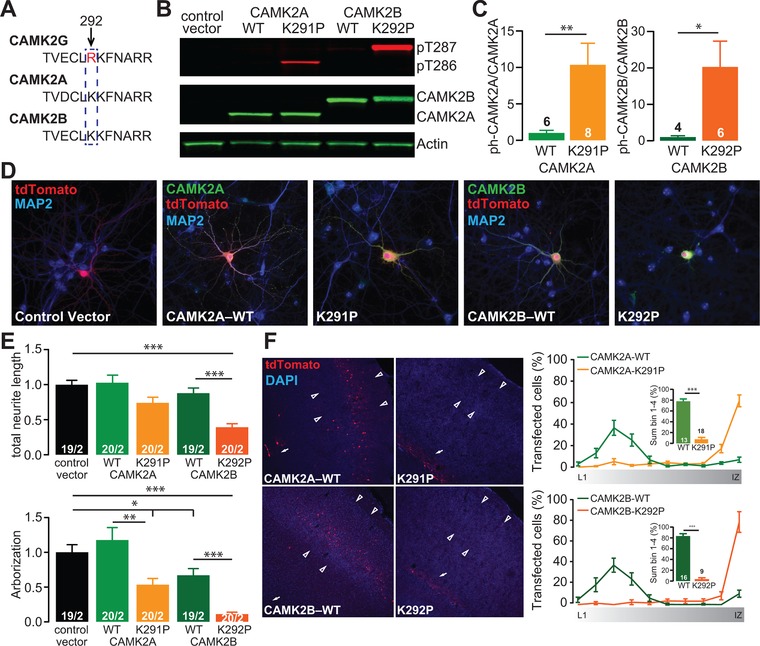
CAMK2ALys291Pro and CAMK2BLys292Pro exhibit similar neurodevelopmental pathogenicity as CAMK2GArg292Pro. (A) Alignment of the protein sequence of CAMK2A, CAMK2B, and CAMK2G showing that the Arginine (R) at 292 in CAMK2G is a Lysine (K) in CAMK2A and CAMK2B. (B) Representative western blot of HEK‐293T cells transfected with control vector, CAMK2AWT, CAMK2ALys291Pro (K291P), CAMK2BWT, or CAMK2BLys292Pro (K292P). (C) Quantification of the levels of CAMK2–Thr286/Thr287 phosphorylation, normalized against total CAMK2 in the different conditions (CAMK2AWT vs. CAMK2ALys291Pro, t[12] = 2.7, P = 0.01; CAMK2BWT vs. CAMK2BLys292Pro, t(8) = 2.18, P = 0.03, one‐tailed unpaired t‐test). (D) Representative confocal images of hippocampal neurons transfected on DIV7 with control vector, CAMK2AWT, CAMK2ALys291Pro, CAMK2BWT, or CAMK2BLys292Pro. Transfected neurons are identified by the tdTOMATO (red). (E) Summary bar graphs of total neurite length and arborization measured for each condition and normalized to the control vector (CAMK2A total neurite length: one‐way ANOVA, F[2,56] = 3.38, P = 0.04; control vector vs. CAMK2AWT, P = 0.9; CAMK2ALys291Pro, P = 0.1, CAMK2AWT vs. CAMK2ALys291Pro, P = 0.06; CAMK2A arborization: one‐way ANOVA F[2,56] = 6.37, P = 0.003; control vector vs. CAMK2AWT, P = 0.6; CAMK2ALys291Pro, P = 0.04, CAMK2AWT vs. CAMK2ALys291Pro, P = 0.003; CAMK2B total neurite length: one‐way ANOVA, F[2,55] = 27.26, P = 5,9E‐09; control vector vs. CAMK2BWT, P = 0.5; CAMK2BLys292Pro, P = 0.0001, CAMK2BWT vs. CAMK2BLys292Pro, P = 0.0001; CAMK2B arborization: one‐way ANOVA F[2,55] = 29.07, P = 2.4E‐09; control vector vs. CAMK2BWT, P = 0.02; CAMK2BLys292Pro, P = 0.0001, CAMK2BWT vs. CAMK2BLys292Pro, P = 0.0001). (F) Representative images of P20–P22 pups in utero electroporated at E14.5 with control vector, CAMK2AWT, CAMK2ALys291Pro, CAMK2BWT, or CAMK2BLys292Pro. tdTOMATO positive cells represent neurons successfully targeted. Arrowheads indicate layer 2/3 of the somatosensory cortex, whereas the arrowhead indicates the subventricular zone (SVZ). Right: quantification of the neuronal migration pattern from the Layer 1 (L1) to the intermediate zone (IZ); insets represent the percentage of targeted cells that reach the outer layers of the cortex measured as the sum of bin 1–4 (CAMK2AWT vs. CAMK2ALys291Pro, t(29) = 12.97, P = 1.34E‐13, unpaired two‐tailed t‐test; CAMK2BWT vs. CAMK2BLys292Pro, t(23) = 13.33, P = 2.64E‐12, unpaired two‐tailed t‐test). Data in (C) and (E) are presented as mean ± SEM. Numbers (X/Y) depicted in the bar graphs represent the number of samples (C) or the total number of cells (X) and number of independent cultures (Y) (E) analyzed
We next tested whether the expression of CAMK2ALys291Pro and CAMK2BLys292Pro impacts the morphological development of cultured primary hippocampal neurons (Figure 8D). No changes in neuronal morphology were observed with expression of CAMK2AWT (Figure 8E). Expression of CAMK2BWT did result in a small but significant decrease in arborization, but without changes in total neurite length (Figure 8E). In contrast, expression of either CAMK2ALys291Pro or CAMK2BLys292Pro each resulted in substantial alterations of neuronal morphology (Figure 8D and E).
Lastly, we evaluated the impact of CAMK2ALys291Pro and CAMK2BLys292Pro on in vivo neural cell migration. The majority of cells expressing either CAMK2AWT or CAMK2BWT migrated normally to layer 2/3 of the somatosensory cortex (Figure 8F). However, expression of either CAMK2ALys291Pro or CAMK2BLys292Pro caused a severe disruption of migration, analogous to the effect of CAMK2GArg292Pro (Figure 8F).
Therefore, our findings suggest a strong functional conservation across the major brain‐expressed CAMK2 isoforms of the ID‐associated CAMK2G p.Arg292Pro mutation for which neurodevelopmental pathogenicity is mediated by a gain‐of‐function through enhanced cytoplasmic phosphotransferase activity.
4. DISCUSSION
In the present study, we describe two unrelated patients with severe ID, carrying a de novo CAMK2G p.Arg292Pro mutation and made use of an in vitro morphology and in vivo migration assay to investigate the pathogenicity of this mutation.
Mutations in CAMK2A and CAMK2B have recently been shown to cause ID, severely delayed speech and behavioral issues (Küry et al., 2017). Apart from these common features, there is much variability between the different CAMK2 patients. The two patients described here carrying a mutation in CAMK2G share similar features with CAMK2A/CAMK2B‐mutated patients (e.g., ID and hypotonia), but also show clear differences such as facial dysmorphisms. The continuous identification of individuals with ID carrying mutations in the CAMK2 genes, may enable further characterization of specific characteristics for the CAMK2‐dependent syndrome.
We have established several lines of mechanistic evidence indicating that the p.Arg292Pro mutation acts as a gain‐of‐function, with regard to catalytic activity of the kinase, but as a loss‐of‐function with regard to its nuclear localization. First, we showed that expression of CAMK2G mutants that reduce kinase activity by interfering with Ca2+/CaM binding or rendering the protein kinase‐dead, do not recapitulate the neurodevelopmental phenotypes exhibited by expression of CAMK2GArg292Pro. Second, we showed that the CAMK2G p.Arg292Pro mutation causes increased phosphotransferase activity as well as increased CaM affinity, resulting in increased Thr287 phosphorylation. Third, we showed that expression of the constitutively active mutant CAMK2GThr287Asp/Thr306Val/Thr307Ala phenocopies the neurodevelopmental impairments seen with expression of CAMK2GArg292Pro. Finally, we showed that CAMK2G‐NLSArg292Pro is deficient in nuclear localization. Taken together, we propose a model whereby the p.Arg292Pro mutation renders CAMK2G constitutively active albeit with a reduced protein half‐life, either due to intrinsic instability or targeted degradation. Notably, expression of the constitutively active CAMK2GThr287Asp/Thr306Val/Thr307Ala mutant in HEK‐293T cells also resulted in lower CAMK2G protein expression but the higher activity of the expressed protein led to the gain‐of‐function phenotype. Additionally, mice expressing the CAMK2AThr305Val/Thr306Ala protein, which lacks auto‐inhibitory function, exhibits a two‐fold reduction in CAMK2A protein but similarly produces a net gain‐of‐function phenotype (Elgersma et al., 2002). Since CAMK2 is highly dependent on a Ca2+ stimulus for activity, even low expression of a constitutively active form such as CAMK2GArg292Pro may elicit gain‐of‐function effects. Future studies using a targeted mouse mutant and genome‐edited human pluripotent stem cells would provide further insights regarding the endogenous regulation of CAMK2GArg292Pro protein levels.
Although the effect of the CAMK2GArg292Pro mutation has not previously been studied, our finding that its pathogenicity derives from a gain‐of‐function is consistent with the rich knowledge of CAMK2 biochemistry (for reviews see, Hudmon and Schulman (2002); Lisman et al. (2002)). In the basal state, the auto‐regulatory domain functions to suppress catalytic activity by maintaining the kinase in an inactive conformation and blocking access of external substrates to the binding pocket. Binding of Ca2+/CaM to the auto‐regulatory domain displaces it to allow the active conformation and opening the substrate binding pocket. In the process, it exposes Thr286 (CAMK2A)/Thr287 (CAMK2B and CAMK2G) for auto‐phosphorylation, which disables the inhibitory function of the auto‐regulatory domain and results in Ca2+/CaM independent (autonomous) activity. The auto‐regulatory domain consists of several residues that participate in auto‐inhibition, one of which is located at amino acid position 291 in CAMK2A (Smith, Colbran, Brickey, & Soderling, 1992). Indeed, for CAMK2A it has previously been shown that mutation of Lysine 291 (equivalent to Arginine 292 in CAMK2G) to Alanine increases the Ki for inhibitory peptides and reduced the inhibitory potency of the auto‐regulatory domain (Smith et al., 1992). Notably, mutation of CAMK2A just three residues downstream, from Asparagine 294 to aspartic acid, generates an even greater enhancement of constitutive activity than the phosphomimetic p.Thr286Asp mutation. Therefore, it is possible that the disruptive nature of the Proline substitution in p.Arg292Pro reduces the contribution of the critical Asparagine for auto‐inhibition resulting in enhanced constitutive activity (Yang & Schulman, 1999). Additionally, mutation of CAMK2A Lysine 291 to Glutamic acid reduced the relative KCaM by 50%, thereby increasing CaM binding (Yang & Schulman, 1999). Hence, these structural and biochemical findings are entirely consistent with our finding that the p.Arg292Pro mutation would generates a constitutively active kinase that facilitates CaM binding and promotes Thr287 phosphorylation that delinks kinase activity from complete dependence on Ca2+ stimulus for activity.
Our finding that knockdown of CAMK2G affects neuronal arborization, which could be rescued by CAMK2G expression with or without an NLS, but not by heterologous CAMK2A or CAMK2B overexpression indicates that cytosolic CAMK2G plays a unique role in neuronal development, and that loss‐of‐function mutations or haploinsufficiency could potentially be disruptive. Indeed, according to ExAC, the probability of CAMK2G being LoF intolerant is high (pLI = 0.99). However, to our knowledge no patient with a neurodevelopmental disorder has yet been found carrying an indel or a nonsense mutation in the CAMK2G gene. Interestingly, another CAMK2G candidate variant (CAMK2G c.1075G > A p.Val359Met) was found in a patient with a developmental disorder in the Deciphering Developmental Disorders Study (Deciphering Developmental Disorders Study, 2017). In contrast to the p.Arg292Pro mutation, this variant is located in the association domain of the protein, which makes it difficult to predict a priori if and how it affects CAMK2G protein function.
Very few studies have looked at the function of CAMK2G in neurons. Only recently, a first hint for CAMK2G having a unique function in spinal cord ganglion cells was published (Ma et al., 2014). This study indicated that CAMK2G functions as a shuttle to transport Ca2+/CaM from the cell surface and cytoplasm to the nucleus, a function that does not require its catalytic activity but depends on phosphorylation of its Thr287 by a second CAMK2 holoenzyme, and on the integrity of its NLS to initiate changes in gene expression in response to specific Ca2+ signals. Our study further expands the mechanistic evidence underlying the function of CAMK2G in neurons by demonstrating that reduced levels of CAMK2G resulted in a significant elaboration of neuronal morphology. Notably, this effect could be reversed by expression of CAMK2G with or without an NLS, but not by expression of CAMK2A or CAMK2B. Therefore, we conclude that cytosolic CAMK2G functions to constrain dendritic arborization. In contrast, whereas CAMK2A knockdown does not affect dendritic arborization, CAMK2B knockdown has been shown to reduce dendritic arborization of primary hippocampal neurons (Fink et al., 2003), together suggesting distinct neurodevelopmental functions of the various CAMK2 isoforms.
Even though a subset of CAMK2G isoforms contains an NLS (Takeuchi, Fukunaga, & Miyamoto, 2002), we did not observe differential effects on neuronal morphology or neural cell migration that were dependent on the presence of the NLS domain. However, we did find that CAMK2G‐NLSArg292Pro is localized predominantly to the cytosol, whereas CAMK2G‐NLSWT is located in both the nucleus and cytosol, indicating that the p.Arg292Pro mutation interferes with its nuclear localization. However, given that abrogation of the phosphotransferase activity (via the Lys43Arg mutation) fully rescued the pathogenic effect of the p.Arg292Pro mutation, independently of the presence or absence of the NLS domain, we conclude that the pathogenicity of the p.Arg292Pro mutation is most likely unrelated to its nuclear function.
Our results also demonstrate that the combination of in vitro and in vivo neurodevelopmental assays provide a robust platform for investigating the functional pathogenicity of candidate CAMK2 mutations for ID. Furthermore, by exploiting the wealth of previous data that has been gathered for the CAMK2 protein (Colbran, 1992; Colbran & Brown, 2004; Hanson, Meyer, Stryer, & Schulman, 1994; Hudmon & Schulman, 2002; Rellos et al., 2010; Yang & Schulman, 1999), we were subsequently able to carefully dissect the biochemical mechanism underlying the pathogenicity of the mutation.
In conclusion, we show that CAMK2G is indispensable for normally developing neurons. Moreover, we have demonstrated that the pathogenicity of the de novo CAMK2G p.Arg292Pro mutation is mediated by a gain‐of‐function through enhanced constitutive enzymatic activity.
CONFLICT OF INTEREST STATEMENT
The Greenwood Genetic Center receives fee income from clinical laboratory testing.
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Erika Goedknegt, Minetta Elgersma, Mehrnoush Aghadavoud Jolfaei, Christian G. Bouwkamp, Charlotte de Konink, and Randall de Joode for technical support.
Proietti Onori M, Koopal B, Everman DB, et al. The intellectual disability‐associated CAMK2G p.Arg292Pro mutation acts as a pathogenic gain‐of‐function. Human Mutation. 2018;39:2008–2024. 10.1002/humu.23647
Communicated by María‐Jesús Sobrido
Contributor Information
Ype Elgersma, Email: y.elgersma@erasmusmc.nl.
Geeske M. van Woerden, Email: g.vanwoerden@erasmusmc.nl.
REFERENCES
- Bayer, K. U. , Löhler, J. , Schulman, H. , & Harbers, K. (1999). Developmental expression of the CaM kinase II isoforms: Ubiquitous gamma‐ and delta‐CaM kinase II are the early isoforms and most abundant in the developing nervous system. Brain Research. Molecular Brain Research, 70(1), 147–154. [DOI] [PubMed] [Google Scholar]
- Borgesius, N. Z. , van Woerden, G. M. , Buitendijk, G. H. S. , Keijzer, N. , Jaarsma, D. , Hoogenraad, C. C. , & Elgersma, Y. (2011). βCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31(28), 10141–10148. 10.1523/JNEUROSCI.5105-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran, R. J. (1992). Regulation and role of brain calcium/calmodulin‐dependent protein kinase II. Neurochemistry International, 21(4), 469–497. [DOI] [PubMed] [Google Scholar]
- Colbran, R. J. , & Brown, A. M. (2004). Calcium/calmodulin‐dependent protein kinase II and synaptic plasticity. Current Opinion in Neurobiology, 14(3), 318–327. 10.1016/j.conb.2004.05.008 [DOI] [PubMed] [Google Scholar]
- de Ligt, J. , Willemsen, M. H. , van Bon, B. W. M. , Kleefstra, T. , Yntema, H. G. , … Kroes, T. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. The New England Journal of Medicine, 367(20), 1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- de Quervain, D. J.‐F. , & Papassotiropoulos, A. (2006). Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proceedings of the National Academy of Sciences of the United States of America, 103(11), 4270–4274. 10.1073/pnas.0510212103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study . (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma, Y. , Fedorov, N. B. , Ikonen, S. , Choi, E. S. , Elgersma, M. , Carvalho, O. M. , … Silva, A. J. (2002). Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron, 36(3), 493–505. [DOI] [PubMed] [Google Scholar]
- Fink, C. C. , Bayer, K.‐U. , Myers, J. W. , Ferrell, J. E. , Schulman, H. , & Meyer, T. (2003). Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron, 39(2), 283–297. [DOI] [PubMed] [Google Scholar]
- Fong, Y. L. , Taylor, W. L. , Means, A. R. , & Soderling, T. R. (1989). Studies of the regulatory mechanism of Ca2+/calmodulin‐dependent protein kinase II. Mutation of threonine 286 to alanine and aspartate. The Journal of Biological Chemistry, 264(28), 16759–16763. [PubMed] [Google Scholar]
- Giese, K. P. , Fedorov, N. B. , Filipkowski, R. K. , & Silva, A. J. (1998). Autophosphorylation at Thr286 of the alpha calcium‐calmodulin kinase II in LTP and learning. Science, 279(5352), 870–873. [DOI] [PubMed] [Google Scholar]
- Goslin, K. , & Banker, G. (1991). Culturing nerve cells. MIT Press: Cambridge, pp. 251–281. [Google Scholar]
- Hanson, P. I. , Kapiloff, M. S. , Lou, L. L. , Rosenfeld, M. G. , & Schulman, H. (1989). Expression of a multifunctional Ca2+/calmodulin‐dependent protein kinase and mutational analysis of its autoregulation. Neuron, 3(1), 59–70. [DOI] [PubMed] [Google Scholar]
- Hanson, P. I. , Meyer, T. , Stryer, L. , & Schulman, H. (1994). Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron, 12(5), 943–956. [DOI] [PubMed] [Google Scholar]
- Heist, E. K. , Srinivasan, M. , & Schulman, H. (1998). Phosphorylation at the nuclear localization signal of Ca2+/calmodulin‐dependent protein kinase II blocks its nuclear targeting. The Journal of Biological Chemistry, 273(31), 19763–19771. [DOI] [PubMed] [Google Scholar]
- Hell, J. W. (2014). CaMKII: Claiming center stage in postsynaptic function and organization. Neuron, 81(2), 249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon, A. , & Schulman, H. (2002). Neuronal Ca2+/calmodulin‐dependent protein kinase II: The role of structure and autoregulation in cellular function. Annual Review of Biochemistry, 71, 473–510. 10.1146/annurev.biochem.71.110601.135410 [DOI] [PubMed] [Google Scholar]
- Küry, S. , van Woerden, G. M. , Besnard, T. , Proietti Onori, M. , Latypova, X. , … Towne, M. C. (2017). De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. American Journal of Human Genetics, 101(5), 768–788. 10.1016/j.ajhg.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Zhou, T. , Liao, L. , Yang, Z. , Wong, C. , Henn, F. , … Hu, H. (2013). βCaMKII in lateral habenula mediates core symptoms of depression. Science, 341(6149), 1016–1020. 10.1126/science.1240729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman, J. , Schulman, H. , & Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nature Reviews. Neuroscience, 3(3), 175–190. 10.1038/nrn753 [DOI] [PubMed] [Google Scholar]
- Lisman, J. , Yasuda, R. , & Raghavachari, S. (2012). Mechanisms of CaMKII action in long‐term potentiation. Nature Reviews. Neuroscience., 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. , Groth, R. D. , Cohen, S. M. , Emery, J. F. , Li, B. , Hoedt, E. , … Tsien, R.W. (2014). γCaMKII shuttles Ca²⁺/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell, 159(2), 281–294. 10.1016/j.cell.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford, M. , Wang, J. , Kandel, E. R. , & O'Dell, T. J. (1995). CaMKII regulates the frequency‐response function of hippocampal synapses for the production of both LTD and LTP. Cell, 81(6), 891–904. [DOI] [PubMed] [Google Scholar]
- Ohsako, S. , Nakazawa, H. , Sekihara, S. , Ikai, A. , & Yamauchi, T. (1991). Role of threonine‐286 as autophosphorylation site for appearance of Ca2(+)‐independent activity of calmodulin‐dependent protein kinase II alpha subunit. Journal of Biochemistry, 109(1), 137–143. [DOI] [PubMed] [Google Scholar]
- Pan, Z. , Zhu, L.‐J. , Li, Y.‐Q. , Hao, L.‐Y. , Yin, C. , Yang, J.‐X. , … Cao J. L. (2014). Epigenetic modification of spinal miR‐219 expression regulates chronic inflammation pain by targeting CaMKIIγ. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 34(29), 9476–9483. 10.1523/JNEUROSCI.5346-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, M. E. , Fong, Y. L. , Ono, T. , Colbran, R. J. , Kemp, B. E. , Soderling, T. R. , & Means, A. R. (1988). Calcium/calmodulin‐dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. The Journal of Biological Chemistry, 263(15), 7190–7195. [PubMed] [Google Scholar]
- Pi, H. J. , Otmakhov, N. , Gaamouch, El, F. , Lemelin, D. , De Koninck, P. , & Lisman, J. (2010a). CaMKII control of spine size and synaptic strength: Role of phosphorylation states and nonenzymatic action. Proceedings of the National Academy of Sciences of the United States of America, 107(32), 14437–14442. 10.1073/pnas.1009268107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi, H. J. , Otmakhov, N. , Lemelin, D. , De Koninck, P. , & Lisman, J. (2010b). Autonomous CaMKII can promote either long‐term potentiation or long‐term depression, Depending on the state of T305/T306 phosphorylation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30(26), 8704–8709. 10.1523/JNEUROSCI.0133-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rellos, P. , Pike, A. C. W. , Niesen, F. H. , Salah, E. , Lee, W. H. , Delft, von, F. , & Knapp, S. (2010). Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biology, 8(7), e1000426 10.1371/journal.pbio.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, T. (2006). In vivo electroporation in the embryonic mouse central nervous system. Nature Protocols, 1(3), 1552–1558. 10.1038/nprot.2006.276 [DOI] [PubMed] [Google Scholar]
- Saito, T. , & Nakatsuji, N. (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Developmental Biology, 240(1), 237–246. 10.1006/dbio.2001.0439 [DOI] [PubMed] [Google Scholar]
- Shen, K. , & Meyer, T. (1999). Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science, 284(5411), 162–166. 10.1126/science.284.5411.162 [DOI] [PubMed] [Google Scholar]
- Shioda, N. , Sawai, M. , Ishizuka, Y. , Shirao, T. , & Fukunaga, K. (2015). Nuclear translocation of calcium/calmodulin‐dependent protein kinase IIδ3 promoted by protein phosphatase‐1 enhances brain‐derived neurotrophic factor expression in dopaminergic neurons. Journal of Biological Chemistry, 290(35), 21663–21675. 10.1074/jbc.M115.664920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, A. J. , Paylor, R. , Wehner, J. M. , & Tonegawa, S. (1992a). Impaired spatial learning in alpha‐calcium‐calmodulin kinase II mutant mice. Science, 257(5067), 206–211. [DOI] [PubMed] [Google Scholar]
- Silva, A. J. , Stevens, C. F. , Tonegawa, S. , & Wang, Y. (1992b). Deficient hippocampal long‐term potentiation in alpha‐calcium‐calmodulin kinase II mutant mice. Science, 257(5067), 201–206. [DOI] [PubMed] [Google Scholar]
- Smith, M. K. , Colbran, R. J. , Brickey, D. A. , & Soderling, T. R. (1992). Functional determinants in the autoinhibitory domain of calcium/calmodulin‐dependent protein kinase II. Role of His282 and multiple basic residues. The Journal of Biological Chemistry, 267(3), 1761–1768. [PubMed] [Google Scholar]
- Tabata, H. , & Nakajima, K. (2001). Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience, 103(4), 865–872. [DOI] [PubMed] [Google Scholar]
- Takeuchi, Y. , Fukunaga, K. , & Miyamoto, E. (2002). Activation of nuclear Ca(2+)/calmodulin‐dependent protein kinase II and brain‐derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108‐15 cells. Journal of Neurochemistry, 82(2), 316–328. [DOI] [PubMed] [Google Scholar]
- Taniguchi, Y. , Young‐Pearse, T. , Sawa, A. , & Kamiya, A. (2012). In utero electroporation as a tool for genetic manipulation in vivo to study psychiatric disorders: From genes to circuits and behaviors. The Neuroscientist, 18(2), 169–179. 10.1177/1073858411399925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobimatsu, T. , & Fujisawa, H. (1989). Tissue‐specific expression of four types of rat calmodulin‐dependent protein kinase II mRNAs. The Journal of Biological Chemistry, 264(30), 17907–17912. [PubMed] [Google Scholar]
- van Woerden, G. M. , Hoebeek, F. E. , Gao, Z. , Nagaraja, R. Y. , Hoogenraad, C. C. , Kushner, S. A. , … Elgersma, Y. (2009). betaCaMKII controls the direction of plasticity at parallel fiber‐Purkinje cell synapses. Nature Neuroscience, 12(7), 823–825. 10.1038/nn.2329 [DOI] [PubMed] [Google Scholar]
- Vissers, L. E. L. M. , Gilissen, C. , & Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nature Reviews. Genetics, 17(1), 9–18. 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- Waldmann, R. , Hanson, P. I. , & Schulman, H. (1990). Multifunctional Ca2+/calmodulin‐dependent protein kinase made Ca2+ independent for functional studies. Biochemistry, 29(7), 1679–1684. [DOI] [PubMed] [Google Scholar]
- Waxham, M. N. , Aronowski, J. , Westgate, S. A. , & Kelly, P. T. (1990). Mutagenesis of Thr‐286 in monomeric Ca2+/calmodulin‐dependent protein kinase II eliminates Ca2+/calmodulin‐independent activity. Proceedings of the National Academy of Sciences of the United States of America, 87(4), 1273–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, E. , & Schulman, H. (1999). Structural examination of autoregulation of multifunctional calcium/calmodulin‐dependent protein kinase II. The Journal of Biological Chemistry, 274(37), 26199–26208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information