

Abstract
Tay-Sachs disease (TSD) is a lethal lysosomal storage disease (LSD) caused by mutations in the HexA gene, which can lead to deficiency of β-hexosaminidase A (HexA) activity and consequent accumulation of its substrate, GM2 ganglioside. Recent reports that progranulin (PGRN) functions as a chaperone of lysosomal enzymes and its deficiency is associated with LSDs, including Gaucher Disease and neuronal ceroid lipofuscinosis, prompted us to screen the effects of recombinant PGRN on lysosomal storage in fibroblasts from 11 patients affected by various LSDs, which led to the isolation of TSD in which PGRN demonstrated the best effects in reducing lysosomal storage. Subsequent in vivo studies revealed significant GM2 accumulation and the existence of typical TSD cells containing zebra-bodies in both aged and ovalbuminchallenged adult PGRN deficient mice. In addition, HexA, but not HexB, was aggregated in PGRN deficient cells. Furthermore, recombinant PGRN significantly reduced GM2 accumulation and lysosomal storage in these animal models. Mechanistic studies indicated that PGRN bound to HexA through granulin G and E domain, and increased the enzymatic activity and lysosomal delivery of HexA. More importantly, Pcgin, an engineered PGRN derivative bearing the granulin E domain, also effectively bound to HexA and reduced the GM2 accumulation. Collectively, these studies not only provide new insights into the pathogenesis of TSD, but may also have implications for developing PGRN-based therapy for this lifethreatening disorder.
Keywords: Tay-Sachs disease, lysosome storage disease, hexosaminidase A, progranulin, Pcgin
Introduction
Tay-Sachs disease (TSD), a subtype of the GM2 gangliosidoses, is an inherited autosomal recessive disease caused by mutations of the gene encoding the α-subunit of β-hexosaminidase A (HexA) that result in the deficiency of the enzyme and the accumulation of its specific substrate, GM2 ganglioside, mainly in the central nervous system[1–3]. The clinical signs and pathologic features of TSD were first described in 1887 according to the combined descriptions of the disease by physicians Warren Tay and Bernard Sachs[3]. Clinical presentation of TSD can be categorized across three subtypes: acute infantile TSD, juvenile (subacute) TSD and chronic or adult-onset TSD[4]. Acute infantile TSD is the most common and aggressive variant with presentation and death occurring in early infancy. Relative to the acute infantile form of the disease, juvenile and adult-onset TSD are rare and have greater variability in neurological presentation. Juvenile (subacute) TSD manifests symptomatically during the first years of childhood and, like acute infantile TSD, is fatal. Adult-onset TSD exists as a more mild form and those affected can survive to adulthood[4].
In the general population of the USA, the incidence of TSD is 1 in 320,000 births; however, incidence of unaffected TSD carriers may reach 1 in 250 births. The Ashkenazi Jewish represent a disproportionately affected subpopulation in which TSD incidence is 1 in 3,600 births in general population and 1 in 30 in unaffected carriers [5,6,4]. Although preventative screening for TSD has progressed, no curative or disease modifying therapies are currently available for TSD and best practices for disease management are limited to supportive care measures[3,7]. Advancements in the understanding of the genetic and pathophysiological basis of TSD have led to the suggestion of several potential treatment strategies including enzyme replacement, substrate reduction therapy, pharmacologic chaperone therapy, cellular infusions and bone marrow transplantation; these strategies are still under investigation [8–10].
Progranulin (PGRN), a 593 amino acid secreted glycoprotein and pleiotropic growth factor-like molecule is widely expressed in distinct tissues and in multiple cell populations, including epithelial, immune, neuronal, chondrocyte, and hematopoietic cells[11–15]. PGRN has wellaccepted roles in cell proliferation, migration and survival, and has demonstrated involvement in pathophysiological processes of inflammation, tumorigenesis, wound healing and tissue repair, bone formation and remodeling, immune disease, atherosclerosis, and neurodegenerative diseases[16–25,15,26,27].
Evidence also suggests that PGRN is crucial in maintaining lysosomal homeostasis. Our recent studies have revealed that PGRN functions as a chaperone molecule to facilitate glucocerebrosidase (GCase) traffic to lysosome, and its deficiency leads to a GD phenotype, including the appearance of lipid-engorged Gaucher cells in mice[28]. Moreover, treatment with either PGRN or its small derivative, Pcgin, is able to ameliorate GD phenotype through protein disaggregation in patient fibroblasts and GD model mice[29]. These findings support a role of PGRN as a disease modifying co-factor in GD[29,28,30], and raise the question of whether PGRN’s chaperone functionality is a general mechanism applicable to multiple lysosomal enzymes[31]. Herein, we screened multiple LSDs and identified TSD as additional LSD in which PGRN showed promising therapeutic effects.
Methods
Mice
All mice were housed in the Skirball Animal Facility of New York University Langone Medical Center. All animal experiments have been performed in accordance with protocols approved by the Institutional Animal Care and use Committee (IACUC) of New York University School of Medicine. All mice were maintained on a C57BL/6 background; genotyping of PGRN knockout (KO) mice and wild type (WT) litter mates was performed as described previously[11,32,22]. Eight-week-old WT and PGRN KO mice and one year old WT and PGRN KO aged mice were used for the experiments.
Reagents and material
Fibroblasts from LSD patients (GD I GM00852, GD II GM08760, GDⅢ GM20272, ML III GM03391, ML IV GM02528, FD GM05752, NPD B GM03393, FABRY GM02769, MPS III GM01094, MPS VI GM02572, TSD GM01110, normal fibroblast GM17152) were purchased from Coriell Cell Repositories (Camden, NJ). Antibodies against PGRN (sc-28928), His-tag (sc-803), GFP (sc-8334), GAPDH (sc-25778), HexA (sc-34577), GFAP (sc-33673), LAMP2 (sc71491) and RIPA Lysis buffer (sc-24948A) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against NeuN was purchased from Abcam (Cat. No. 104224), and CD68 antibody was purchased from AbD Serotec (Cat. No. MCA1957). 4-Methylumbelliferyl 6-Sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside Potassium Salt (MUGS, Cat. No. M335000) was purchased from Toronto Research Chemicals Inc. (Toronto, Ontario, Canada). 2-amino-2-methyl-1-propanol (Cat. No. A65182), bicinchoninic acid solution (BCA, Cat. No. B9643) and Copper (II) sulfate solution (Cat. No. C2284) were purchased from SigmaAldrich (Natick, MA, USA). GM2 (Cat. No. 345759) was purchased from Calbiochem (Merck KGaA, Darmstadt, Germany). Cover glasses (Cat. No. 22293232) and LysoTracker Red DND99 (Cat. No. L7528) were purchased from Thermo Fisher Scientific (Bridgewater, NJ, USA). DAPI (Cat. No. H-1200) was purchased from VECTOR Laboratories (Burlingame, Ca, USA). The plasmids of full-length human HexA in pCMV3 vector (Cat. No. HG12037-UT) were purchased from Sino Biological Inc. (North Wales, PA, USA). Dulbecco’s Modified Eagle Medium (DMEM; Cat. No. 11965–118) and fetal bovine serum (FBS; Cat. No. 16000–044) were purchased from Gibco-BRL (Waltham, MA, USA).
Purification of PGRN and Pcgin
Recombinant PGRN protein was purified from conditioned medium of a HEK293-EBNA stable cell line as reported previously[33,22,3]. Briefly, a HEK293 cells stably expressing His-tagged PGRN were cultured in DMEM supplemented with 10% FBS, 100U/mL penicillin, 10 mg/mL streptomycin and 1 μg/ml geneticin (G418). At 90% confluence, complete medium was replaced with serum-free DMEM with antibiotics for 48 hours, after which the medium was incubated with ProBond™ nickel-chelating resin and purified using ProBond™ Purification System (Cat.K850–01, Life Technologies, Carlsbad, CA, USA) as per the manufacturer’s instructions. Protein was dialyzed against phosphate buffered saline (PBS) pH 7.4 and concentrated using Amicon Ultra-15 Centrifugal Filter Unit (Cat. No. UFC901096, MilliporeSigma, Billerica, MA, USA). Protein concentration was determined via BCA assay and purity of the isolated protein was assessed using 5 μl of purified protein 8% SDS-PAGE and Coomassie Blue staining.
Pcgin, the 98 C-terminal amino acids of PGRN, was prepared on the basis of the procedure described in our previous publication[29]. Briefly, the sequence of Pcgin was inserted into pD444 expression vector with a His-tag (from DNA2.0, Menlo Park, CA) and expanded in the BL21(DE3) E. coli strain after induction by 1mM IPTG. After culture for 3 hours, cells were collected and sonicated to release the fusion protein. His-tagged Pcgin was purified by using ProBond™ Purification System (Cat. No. K850–01, Life Technologies, Carlsbad, CA, USA). The protein was dialyzed against PBS pH 7.4 and concentrated using Amicon Ultra-15 Centrifugal Filter Unit (Cat. No. UFC901096, MilliporeSigma, Billerica, MA, USA). Purity of the protein was analyzed by 15% SDS-PAGE and visualized by Coomassie Blue staining. After endotoxin removal and 0.2µm filter sterilization, recombinant Pcgin protein was used for in vitro and in vivo studies.
Lipid lysate preparation
The preparation of lipid lysate has been described previously[29,28]. Following sacrifice by cervical dislocation, brain tissue was aseptically collected from a C57BL/6 mouse. The brain tissue was weighed and cut into small pieces using a sterile scalpel before homogenization in DMEM. Following homogenization, additional DMEM was added to adjust the concentration to 10mg/mL.
Construction of expression plasmids
The construction of the plasmids containing PGRN, its C-terminal Deletions (CD), its N-terminal Deletions (ND) and the six deletion mutants of Pcgin were described previously [29]. In short, the CD constructs were CD1 (aa 1–521), CD2 (aa 1–444), CD3 (aa 1–376), CD4 (aa 1–284), CD5 (aa 1–209), CD6 (aa 1–127), and CD7 (aa 1–61). The ND constructs were ND1 (aa 45–593), ND2 (aa 113–593), ND3 (aa 179–593), ND4 (aa 261–593), ND5 (aa 336–593), ND6 (aa 416–593), and ND7 (aa 496–593). The six Pcgin deletion mutants were as follows: Δ1 deleted aa496–522, Δ2 deleted aa523–534, Δ3 deleted aa535–539, Δ4 deleted aa540–573, Δ5 deleted aa574–593, and Δ6 deleted last three QLL amino acids.
Treatment effect of PGRN or Pcgin in LSD assessed by fluorescent microscope
Patient fibroblasts were seeded on cover glasses in 24-well plates and treated with PBS (ctrl group), lipid lysis of brain lysate (50 μg/ml), PGRN (0.5 μg/ml) or Pcgin (5 μg/ml) for 24 hours. Cells were stained with LysoTracker Red at the concentration of 300 nM and incubated at 37 °C for one hour. Cells were washed twice with PBS and fixed in 4% paraformaldehyde (PFA) for 10 minutes. The cover glasses were mounted on slides with DAPI. The red fluorescence of lysosomes was visualized by fluorescent microscopy. Ten images were randomly taken for each sample and fluorescent intensities were quantified by ImageJ software.
Fluorescent intensity quantified by plate reader
Patient derived fibroblasts were seeded in 96-well plates. After the treatment with lipid lysates (50 μg/ml), PGRN (0.5 μg/ml) or Pcgin (5 μg/ml) for 24 hours, medium was replaced with fresh medium containing newly made LysoTracker Red (300nM) and plates were incubated at 37 °C for one hour. After aspirating the medium, washing with PBS once and adding 100 µl PBS, plates were read using the SpectraMax i3x system at 559 nm for excitation and 601 nm for emission.
Immunoprecipitation
Plasmids of GFP-tagged PGRN and its C-terminal deletion or N-terminal deletion mutants and His tagged HexA, or constructions of GFP-labelled Pcgin and its amino acid mutants together with His tagged HexA, were co-transfected in 293T cells for 24 hours. Cells were lysed by RIPA lysis and a total of 1 mg protein was used to conduct co-immunoprecipitation (CO-IP) in each sample. Anti-His antibody was used to perform immunoprecipitation and GFP antibody was used to probe the protein complex.
GM2 analysis by mass spectrometry
Mice brain tissues from different groups were collected when mice were sacrificed. The brain tissues were processed at lipid core facility at Cincinnati Children’s Hospital Medical Center to analyze GM2 levels as reported previously[13].
HexA enzymatic activity
Fibroblasts were seeded in 96-well plates. After the treatment, cells were washed twice with PBS, followed by adding 10 mM citrate/phosphate (pH 4.2) buffer 60 μL, containing 0.5% human serum albumin and 0.5% Triton X-100, for 20 minutes on a shaker to lyse the cells. 25 μl of 3.2 mM MUGS was added to the cell lysate and incubated at 37 °C for 1 h. The reaction was stopped by adding 200 μl 0.1 M 2-amino-2-methyl-1-propanol, pH 10.5, and fluorescence was measured at an excitation wavelength of 365 nm and emission wavelength of 450 nm [34,35].
Western blot
Cells were lysed on ice using RIPA lysis buffer and protein concentrations were quantified by BCA assay. Samples were boiled for 5 minutes in SDS sample buffer. Proteins were run on a 12% SDS-polyacrylamide gel and electrotransferred onto a 0.46 μm nitrocellulose membrane. After blocking in 5% nonfat dry milk in Tris buffer-saline-Tween 20 (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; and 0.5% Tween 20; TBS-T) for 30 minutes, blots were incubated with primary antibody for 2 h. Following three washes in TBS-T for 5 minutes each time, the blots were incubated with secondary antibody (horseradish peroxidase conjugated immunoglobulin) for one hour. After washing, visualization of the bound antibody was performed by an enhanced chemiluminescence system (Amersham Life Science, Arlington Heights, IL, USA).
Ovalbumin induced Tay-Sachs Disease mouse model
Eight-week-old C57BL/6 PGRN KO mice were used to establish TSD models through intraperitoneal (IP) injection of ovalbumin (OVA) on Day 1 and Day 15[36]. Intranasal challenge with 1% OVA was administered beginning at Day 29 and continuing for four weeks at a frequency of once every three days. PBS, PGRN (4mg/kg/week), or Pcgin (4mg/kg/week) were administered via IP injection for 4 weeks beginning at Day 29 (n=6 in each group) to test the treatment effects of recombinant PGRN and Pcgin in TSD mouse models. After 4 weeks, mice were sacrificed and brain tissues were fixed and processed by Mass Histology Service (Worcester, MA, USA).
Transmission electron microscope
After OVA treatment, PGRN KO mice were anesthetized and the lung was perfused with fixative containing 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1M sodium cacodylate buffer (pH 7.2) for 2 h. After washing, the samples were post fixed in 1% OsO4 for 1 h, followed by block staining with 1% uranyl acetate for 1 hour. After dehydration, samples were embedded in Embed 812 (Electron Microscopy Sciences, Hatfield, PA, USA). 60 nm sections were cut and stained with uranyl acetate and lead citrate by standard methods. Stained grids were examined under a Philips CM-12 electron microscope (FEI; Eindhoven, Netherlands) and photographed with a Gatan (4 k × 2.7 k) digital camera (Gatan, Inc., Pleasanton, CA, USA).
Immunohistochemistry of GM2
Paraffin-embedded brain slides were deparaffinized in a xylene and ethanol gradient. After antigen retrieval by 0.1% trypsin in DMEM and depletion of endogenous hydrogen peroxidase, the slides were blocked with 3% BSA and 20% goat serum for 30 minutes at room temperature. GM2 antibody in 2% goat serum (1:200) was incubated at 4 °C overnight. After washing with PBS, secondary antibodies were added (1:200) for 1 hour at 37 °C. The staining was visualized with Vector ABC peroxidase kit with DAB as chromagen followed by counter staining and rehydration.
Immunofluorescence staining of GM2
Paraffin-embedded brain sections from PGRN KO mice with OVA challenge were deparaffinized in a xylene and ethanol gradient. After antigen retrieval by 0.1% trypsin in DMEM, slides were fixed with 4% formaldehydrate for 10 minutes at room temperature, permeabilized by 0.1% Triton X-100 in PBS for 5 minutes at room temperature and blocked with donkey serum in PBS (1:50) for one hour at room temperature. GM2 antibody diluted in blocking buffer (1:200) was added at 4 °C overnight, followed by fluorescence-labeled secondary antibodies in blocking buffer (1:200) at 37 °C for one hour. After washing with PBS 3 times, the tissues were mounted with DAPI. The images were taken by immunofluorescence microscope.
Frozen brain sections from PGRN KO mice challenged with OVA were co-stained with GM2 antibody, as well as neuronal marker (NeuN antibody) or microglial marker (CD68) or astrocyte marker (GFAP). After incubation with specific fluorescence labeled secondary antibodies, the slides were imaged by Leica TCS SP5 con-focal system.
Immunofluorescence staining of HexA and LAMP2
Normal and TSD fibroblasts (GM01110) were seeded on cover glasses in 24-well plates and treated with PGRN (0.5 μg/ml) or Pcgin (5 μg/ml) for 3 days. Cells were fixed with 4% formaldehydrate for 10 minutes, permeabilized by 0.1% Triton X-100 in PBS for 5 minutes and blocked with donkey serum in PBS (1:50) for one hour at room temperature. HexA antibody (1:100) and LAMP2 antibody (1:100), a marker of lysosome, diluted in blocking buffer was added at 4 °C overnight, followed by fluorescence-labeled secondary antibodies in blocking buffer (1:200), HexA with Green color and LAMP2 in Red color, at 37 °C for one hour. After washing with PBS 3 times, the tissues were mounted with DAPI. The images were taken by immunofluorescence microscope.
Statistical analysis
Data are reported as the mean with standard error. Comparisons between the treatment groups were performed by unpaired two-tailed t-tests in SPSS software (IBM, Armonk, NY, USA). P value < 0.05 was statistically significant.
Results
PGRN reduces lysosomal storage in patient fibroblasts from multiple LSDs
Previously, we identified PGRN as a novel factor in GD through its function as a co-chaperone required for GCase traffic to lysosome[29,28]. PGRN and its small derivative, Pcgin, are therapeutic against GD in both in vitro and in vivo models[29]. In addition, PGRN’s function as a chaperone may be a generalized mechanism to stabilize and deliver other lysosomal enzymes [37,31,29]. Therefore, we screened PGRN’s potential therapeutic effect in multiple LSDs.
Using a lysotracker approach [38,29], we examined the effects of rPGRN on lysosomal storage in fibroblasts from healthy and patient donors affected by various LSDs, including GD. As expected, PGRN effectively reverted the altered lysosomes in fibroblasts from both Type I and II GD with or without lipid challenge (Fig. 1a, b). We were excited to observe that PGRN also remarkably normalized the altered lysosomes in Tay-Sachs disease, Farber disease, and Mucolipidosis III patient fibroblasts (Fig. 1a, b). In line with these findings, the accumulation of glycosaminoglycan (GAG) and GM2 ganglioside was also observed in the tissues from aged PGRN deficient mice (not shown). In the case of type III GD, Mucopolysacharidosis III and VI, PGRN demonstrated beneficial effects only in the presence of lipid stimulation (Fig. 1c, d). PGRN treatment did not show significant improvements upon Niemann-Pick disease type B, Fabry disease or Mucolipidosis IV (Fig. 1e, f), indicating selective effects of PGRN upon lysosomal storage pathologies. Taken together, these results suggest that PGRN, as a cochaperone of the trafficking pathway, may be also involved in the lysosomal delivery of other lysosome enzymes in addition to GCase.
Fig. 1. PGRN reduces lysosomal storage in multiple LSDs.
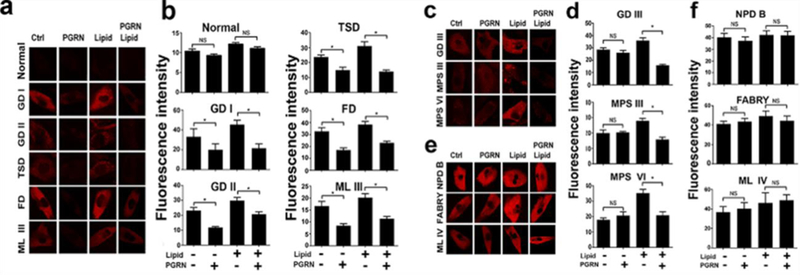
Fibroblasts from healthy controls and 11 different LSD patients were treated with PBS or PGRN (0.5 μg/ml) with or without lipid (50 μg/ml) challenge for 24 hours and cells were stained with LysoTracker Red (300 nM). (a) PGRN effectively mitigated lysosomal storage in Type I and II Gaucher disease, Tay-Sachs disease, Farber disease, and Mucolipidosis III with or without lipid stimulation. (b) The quantification of the red fluorescence of (a). (c) PGRN alleviated lysosomal storage in type III GD, Mucopolysacharidosis III and VI in the presence of lipid stimulation. (d) The quantification of the red fluorescence of (c). (e) PGRN could not improve lysosomal storage in Niemann-Pick disease type B, Fabry disease or Mucolipidosis VI. (f) The quantification of the red fluorescence of (c). Statistical analysis performed by unpaired t test in GraphPad Prism 7. Data are presented as mean ± SEM. *P<0.05.
PGRN deficient mice display the TSD-like phenotypes
The finding that PGRN is therapeutic in the fibroblasts of Tay-Sachs disease, together with our previous studies demonstrating that aged PGRN KO mice develop GD-like phenotypes spontaneously[28] and PGRN is therapeutic against in vivo and in vitro models of GD[29], prompted us to see if PGRN insufficiency also causes a TSD phenotype. In one-year old aged PGRN KO mice, GM2 was aggregated in the brain tissues relative to aged WT mice, as detected by immunohistochemistry (IHC) staining and immunofluorescence staining of GM2 (Fig. 2a, b, c). Under the electronic microscope, we observed tubular-like lysosomes in PGRN KO macrophages, a characteristic of GD phenotype, in addition to round multilayer membranous structures, called zebra bodies (Fig. 2d), which correlate to GM2 ganglioside storage[39]. Since TSD is caused by mutation of the HexA gene, we examined whether HexA distribution is altered in PGRN KO mice after OVA challenge. Interestingly, immunohistochemistry (IHC) staining revealed that HexA was aggregated in PGRN KO mice, a similar observation to those we have reported previously concerning GCase [28]. HexB, however, displayed normal distribution which indicates that the aggregation of the HexA enzyme is specific (Fig. 2e). Since HexA is aggregated in macrophages from peripheral organs in PGRN KO mice after OVA challenge, we also investigated whether OVA-challenged PGRN KO mice develop a TSD-like phenotype in brain tissues. GM2 expression was detected by IHC and immunofluorescence staining and GM2 was accumulated in brain tissues of PGRN KO mice after OVA challenge (Fig. 2f, g, h). In brief, PGRN KO mice display components of the TSD-like phenotype spontaneously, with regard to the appearance of zebra bodies, and under stress-induced conditions, upon which GM2 accumulation is observable in brain tissues.
Fig. 2. PGRN deficiency causes a TSD-like phenotype.

(a) IHC staining of GM2 in paraffinembedded brain tissue slides from aged (one-year old) wild-type and aged PGRN knockout mice. The HexA substrate, GM2, was aggregated in aged PGRN KO mice. (b) Immunofluorescence staining of GM2 in paraffin-embedded brain tissue slides from aged wild-type and PGRN knockout mice. The fluorescence intensity was greatly increased in aged PGRN KO mice relative to aged wild-type mice. (c) Confocal staining of GM2 in frozen brain sections from aged WT and PGRN KO mice. (Red: GM2 Blue: DAPI). (d) Transmission electron microscope analysis of lung tissue from PGRN knockout mice challenged with OVA (upper panel) and fibroblasts from TSD patients (lower panel). The appearance of lysosomes changed from regular round shapes to tubular-like shapes (red arrow) and zebra bodies, multilayer membranous structures (blue arrow), appeared in PGRN KO macrophages. Zebra bodies in TSD fibroblasts served as a positive control (lower panel). (e) IHC staining of HexA and HexB in paraffinembedded lung slides from wild type and PGRN KO mice challenged by OVA. HexA was accumulated while HexB was evenly distributed in macrophages. Aggregation of HexA in macrophages was marked by a red arrow. (f) IHC staining of GM2 in brain tissue from PGRN knockout mice treated with or without OVA. GM2 was dramatically aggregated in PGRN KO mice. (g) Immunofluorescence staining of GM2 in brain tissue from PGRN knockout mice treated with or without OVA. Fluorescence intensity was markedly increased in PGRN KO mice. (h) Confocal staining of GM2 in frozen brain sections from PGRN KO mice, with or without OVA challenge (Red: GM2, Blue: DAPI).
We next determined the cell types in the brains, including neuron, microglia, and astrocyte, in which GM2 accumulated. Briefly, we stained PGRN KO brain tissues with GM2 antibody, as well as antibodies against CD68, GFAP or NeuN, the markers for microglia, astrocytes, and neuron, respectively. Con-focal staining revealed that GM2 was not co-localized with either CD68 or GFAP, but highly co-localized with NeuN, indicated that GM2 is mainly accumulated in the neuron (Fig. 3a–c).
Fig.3. GM2 is mainly accumulated in neurons of PGRN KO brain tissue.

Frozen sections from brain tissues of PGRN KO mice challenged with OVA were co-stained with GM2 antibody (green) and antibodies against different cell type markers (red), including microglia marker CD68 (a), astrocyte marker GFAP (b), and neuron marker NeuN (c). The images were taken under Leica confocal system and the data shown is representative of three individual experiments.
PGRN increases the activities and lysosomal delivery of HexA
To elucidate the potential mechanisms of PGRN’s effect in vitro, we treated TSD fibroblasts with lipid lysate to increase the lysosomal lipid load, and then treated the cells with PGRN protein. Under contrast microscopy, fibroblasts displayed a distinct change in morphology and lipid particles were observed attaching to cells after adding lipid lysate. PGRN treated fibroblasts displayed notably less morphological alteration following lipid stimulation (Data not shown). LysoTracker Red staining further revealed that lysosomal storage was significantly increased following lipid challenge, and it was reduced after PGRN treatment (Fig. 4a, b, c). Immunofluorescence staining confirmed that GM2 was accumulated after lipid challenge and PGRN significantly decreased the lipid-induced fluorescence intensity of GM2 (Fig. 4d, e). In addition, electronic microscope images demonstrated that lipid-induced enhanced zebra bodies and ER engorgement were largely corrected by recombinant PGRN (Fig. 4f). Next, we examined whether PGRN regulated HexA level and enzymatic activity. After the application of PGRN in fibroblasts, the expression level of HexA, its activity, and, in particular, the lysosomal delivery of HexA were significantly elevated (Fig. 4g, h, Fig. 7).
Fig. 4. Therapeutic effect of PGRN acts through increased activity of HexA.
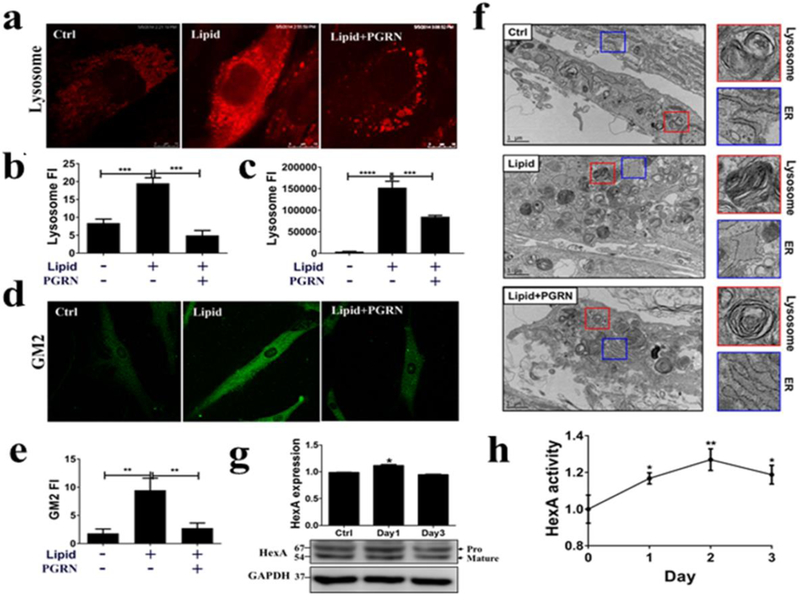
Fibroblasts from TSD patients were treated with PBS, lipid (50 μg/ml) and PGRN (0.5 μg/ml) for 24 hours. (a) The staining of lysosomes by LysoTracker Red (300 nM). PGRN significantly reduced lysosomal storage. (b) The quantification of the red fluorescence of (a). (c) After LysoTracker Red staining, lysosomal signals were detected by the plate reader, SpectraMax ® i3. (d) GM2 staining by immunofluorescence. PGRN greatly reduced GM2 accumulation. (e) The quantification of the GM2 staining shown in (d). (f) Transmission electronic microscope analysis of morphological changes in lysosomes and ER following lipid challenge and PGRN treatment. Fibroblasts were challenged with lipid lysate, or lipid plus recombinant PGRN proteins (0.5 μg/ml) for 3 days. The cells were processed for TEM analysis. The morphological changes of lysosomes (red square) and ER (blue square) were observed following treatment of PGRN. (g) PGRN (0.5 μg/ml) increased HexA expression level in TSD fibroblasts. TSD fibroblasts were treated with PGRN for 3 days. The HexA protein levels were examined by western-blot. The figure is representative of three independent experiments. (h) PGRN enhanced HexA enzymatic activity in TSD fibroblasts. TSD fibroblasts were treated with PGRN (0.5 μg/ml) for 3 days. The enzymatic activities were standardized by control group and measured by processing its specific substrate, MUGS. Data are presented as mean ± SEM. *P<0.05. **P<0.01. ***P<0.001. ****P<0.0001.
Fig.7. PGRN and Pcgin increase the lysosomal delivery of mutant HexA.

Normal and TSD fibroblasts treated with PGRN (0.5 μg/ml) or Pcgin (5 μg/ml) for 3 days were co-stained with HexA antibody (green) and LAMP2 antibody, a marker of lysosome (red). Results showed both PGRN and Pcgin increased the traffic of HexA to lysosome in the patient mutant GM01110 fibroblasts, indicated by white arrows. The images were taken under Leica confocal system and the data shown is representative of three individual experiments.
PGRN binds to HexA through granulin G and E
As PGRN could increase the activity of HexA, we sought to determine whether PGRN bound to HexA. Constructs expressing GFP-tagged PGRN and His-tagged HexA were co-transfected in 293T cells. After 24 hours, cells were lysed with RIPA buffer and Co-IP assay was performed. The cell lysate was immunoprecipitated with His-tag antibody and probed by GFP antibody; results indicated that PGRN bound to HexA under this overexpression condition (Fig. 5a). Next, we examined whether interaction between PGRN and HexA remained in native conditions using a Co-IP assay in RAW264.7 cells without transfection. We found this was the case (Fig. 5b). Immunofluorescence staining, showing the co-localization of PGRN and HexA, further supported our Co-IP results and the conclusion that PGRN and HexA interact with each other (Fig. 5c). To identify the binding site of PGRN to HexA, we first constructed C-and N-terminal deletion mutants of PGRN (Fig. 5d, g). GFP-tagged PGRN and its deletion mutants and Histagged HexA were co-transfected in 293T cells. After 24 hours, cells were lysed and Co-IP was performed. PGRN and all the deletion mutants were successfully expressed after transfection as detected with a GFP antibody (Fig. 5e, h). As shown in Fig 5f, ND1 showed strong binding, and the binding was lost in ND2, which indicates granulin (GRN) G is one binding motif. With further deletion from the N-terminal, binding was restored in ND3 and the binding remained very strong in ND7, which is granulin E domain. Therefore, PGRN binds to HexA at two domains, Grn G and E. The same conclusion is drawn from C-terminal deletions; full-length PGRN binds to HexA, while CD1, which features deletion of Grn E does not display HexA binding activity. Further deletions from the C-terminal show that CD6, which contains Grn-P-G, retains HexA binding activity and that the binding lost with deletion of Grn G (Fig 5i). This binding pattern is very similar to that observed in the PGRN-GCase interaction, in which PGRN binds to GCase through the Grn F and E domains [29].
Fig. 5. PGRN binds to HexA through granulin G and E.
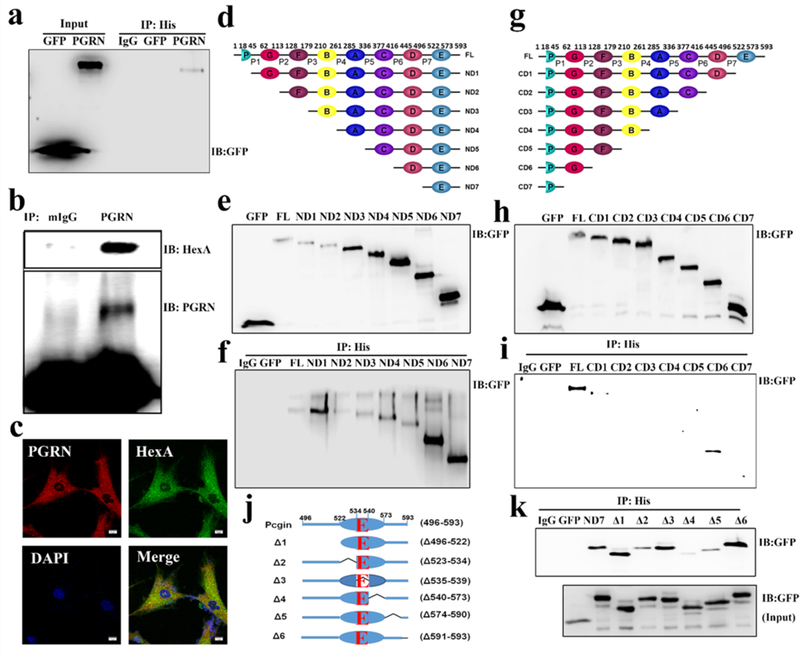
(a) Co-IP assay to test the binding between PGRN and HexA in 293T cells. GFP, GFP tagged PGRN and His-tagged HexA plasmids were co-transfected in 293T cells. Cell lysate was immunoprecipitated with His antibody and probed with GFP antibody. The result showed that PGRN binds to HexA. (b) CoIP assay to test the binding of PGRN to HexA in Raw cells without transfection. Cell lysate was immunoprecipitated with PGRN antibody and probed with HexA and PGRN antibodies. The result shows that PGRN bound to HexA. (c) Fibroblasts from TSD patients were stained with PGRN (Red), HexA (Green) and DAPI (Blue) to test the co-localization of PGRN and HexA. The images were taken under Leica confocal system, which showed the co-localization of PGRN and HexA. (d, g) Schema of N-terminal deletion mutants and C-terminal deletion mutants of PGRN. (e, f, h, i) Co-IP assay to detect the binding site of PGRN to HexA in 293T cells. Plasmids of GFP tagged PGRN and its N-terminal deletion mutants or C-terminal deletion mutants and His-tagged HexA were co-transfected. Cell lysates were immunoprecipitated with His antibody and probed with GFP antibody. (e, h) Expressions of GFP tagged N-terminal deletion or C-terminal deletion mutants of PGRN probed by GFP antibody after the transfection for 24 hours. (f, i) Co-IP assay, immunoprecipitated with His antibody and probed with GFP antibody. The binding sites of PGRN to HexA were granulin E and granulin G. (j, k) Co-IP assay for the binding of Pcgin, the granulin E domain of PGRN, and its mutants to HexA. GFP labelled Pcgin and its mutants together with His tagged HexA were transfected in 293T cells. (j) The construction of Pcgin mutants. (k) Expression of Pcgin and its mutants after transfection (upper panel). Cell lysate was immunoprecipitated with His antibody and probed with GFP antibody. The aa 540–573 was essential for binding between PGRN and HexA (lower panel).
As the Grn E domain contains chaperone activity and has been shown to bind to several lysosomal molecules, such GCase[29], Cathepsin D[37], and Sortilin[3], we further investigated the binding motif important for HexA interaction within Grn E. 293T cells were transfected with serial deletions Δ1-Δ 6 in Grn E and its linker region (Fig. 5j) and Co-IP was performed; results revealed that the Δ4 (deletion of aa540–573) fragment almost abrogated binding, indicating that the aa540–573 sequence is critical for the binding between PGRN and HexA (Fig. 5k).
Pcgin reduces GM2 storage in TSD fibroblasts in vitro
Pcgin is an engineered PGRN derivative bearing the granulin E domain that is capable of binding to HexA. We have shown that Pcgin binds to GCase and is therapeutic against GD[29]. Here, we sought to test whether Pcgin, similar to PGRN, is also therapeutic in TSD. Fibroblasts from TSD patients were cultured in 96-well plates or on cover slips in 24-well plates. Cells were treated with lipid or lipid plus Pcgin or lipid plus PGRN, as a positive control group. After 24 hours, cells were stained with LysoTracker Red to measure lysosomal storage (Fig. 6a, b). Lipid challenge significantly enhanced lysosomal storage, which was reduced following PGRN treatment, and Pcgin also effectively reduced LysoTracker staining (Fig. 6a, b). To confirm the therapeutic effect of Pcgin, we measured GM2 level by immunofluorescence staining, and both Pcgin and PGRN reduced GM2 storage in TSD fibroblasts (Fig. 6c, d). In another experiment, fibroblasts were seeded in 96-well plates, after treatment with lipid, lipid plus PGRN or lipid plus Pcgin, the lysosomal storage was directly read by plate reader; both PGRN and Pcgin shown therapeutic effect in this assay as well (Fig. 6e). Further, electronic microscope images demonstrated that lipid treatment-induced enhancement of zebra bodies and ER swelling were also corrected by recombinant Pcgin (Fig. 6f). Since PGRN functions as a chaperone to stabilize HexA and enhance its enzymatic activity, next we examined whether Pcgin’s therapeutic effect shared a similar mechanism. As shown in Fig. 6g, Pcgin treatment led to the induction of HexA expression at day 1, and particularly the increase of HexA enzymatic activity at all time points tested (Fig. 6h). Therefore, the therapeutic effect of Pcgin appears to be carried out via a mechanism very similar to that of PGRN.
Fig. 6. Pcgin reduces GM2 storage in TSD fibroblasts.

Fibroblasts from TSD patients were treated with PBS, lipid (50 μg/ml), PGRN (0.5 μg/ml) and Pcgin (5 μg/ml) for 24 hours. PGRN was offered as a positive control. (a) The staining of lysosomes by Lysotracker Red (300 nM). Pcgin and PGRN significantly reduced lysosomal storage. (b) The quantification of the lysosomal staining of (a). (c) Immunofluorescence staining of GM2 in TSD fibroblasts following Pcgin treatment, PGRN served as a positive control. (d) The quantification of the GM2 staining of (c). (e) Pcgin reduced lysosomal storage in TSD as evaluated by plate-reader based assay. TSD fibroblasts were seeded in 96-well plates and treated with lipid with or without Pcgin or PGRN. The lysosomes were stained with LysoTracker Red and the fluorescence intensity was read by a plate reader. (f) Lipid induced lysosome storage (red square) and swollen ER (blue square) were ameliorated by Pcgin treatment examined by TEM. TSD fibroblasts were challenged with lipid lysate, or lipid plus recombinant Pcgin proteins (5 μg/ml), processed for TEM. Lysosomes and ER were indicated. (g) The effects of Pcgin on HexA protein level in TSD fibroblasts. TSD fibroblasts were treated with Pcgin (5 μg/ml) for 3 days. The HexA protein levels were examined by Western blotting. The figure is a representative of three independent experiments. (h) Pcgin enhanced HexA enzymatic activity in TSD fibroblasts. TSD fibroblast were treated with Pcgin (5 μg/ml) for 3 days. The enzymatic activities were standardized by control group and measured by processing its specific substrate, MUGS. Data are presented as mean ± SEM. *P<0.05. **P<0.01. ***P<0.001.
Pcgin promotes the lysosomal delivery of HexA
Our previously published data showed PGRN acted as a chaperone in Gaucher diseases to enhance the lysosomal delivery of GCase, together with the findings that both PGRN and Pcgin improve the GM2 storage in vitro and increase HexA activity, promoted us to determine whether Pcgin, similar to PGRN, also enhances HexA translocation to lysosome. To do so, we performed immunofluorescence staining of HexA and LAMP2, a lysosome marker. As demonstrated in Fig. 7, both PGRN and Pcgin increased the lysosomal delivery of HexA in the patient GM01110 mutant fibroblasts.
Pcgin alleviates the GM2 accumulation in mouse model
Pcgin could reduce lysosomal storage and decrease the specific substrate of HexA in fibroblasts from TSD patients. Next, we examined whether Pcgin was also able to ameliorate TSD phenotypes in vivo. HexA KO mice are the current standard of TSD animal models and are not ideal for evaluation of the therapeutic efficacy of PGRN or Pcgin, since PGRN and Pcgin function through increasing mutated HexA’s enzymatic activity, and would be expected to have no effect in HexA KO mice. However, after OVA challenge, our PGRN KO mice displayed a typical TSD-like phenotype, including GM2 accumulation in the central nervous system and typical zebra body inclusions in macrophages as visualized under transmission electron microscopy (TEM). Therefore, we induced a TSD-like phenotype in PGRN KO mice via OVA challenge and treated mice with Pcgin (4mg/kg/week) or sterile vehicle by IP injection. The GM2 level in brain was examined by IHC and immunofluorescence staining (Fig. 8a–f). IHC staining showed many GM2 positive plaques in the cortical region from mice in the OVA challenged group; GM2 staining signal was reduced dramatically following Pcgin treatment (Fig. 8a, b). To further confirm these results, sensitive immunofluorescence staining of GM2 was also conducted and results mirrored those of IHC staining. (Fig.8c, d). These results were confirmed by high-resolution con-focal microscopy, and GM2 was accumulated in PGRN KO after OVA challenge and its level reduced following treatment of Pcgin (Fig. 8e, f). Lipid composition analysis by mass spectrometry demonstrated that GM2 levels were higher in untreated PGRN KO brain than WT brain; however, OVA challenge significantly increased GM2 storage in the brain, and its level reduced following treatment with Pcgin (Fig. 8g). Collectively, these data indicate that Pcgin was effective to reduce GM2 accumulation in both patient fibroblasts and animal models, and might be a promising candidate to treat this devastating disease.
Fig. 8. Pcgin alleviates the GM2 accumulation in mouse model.
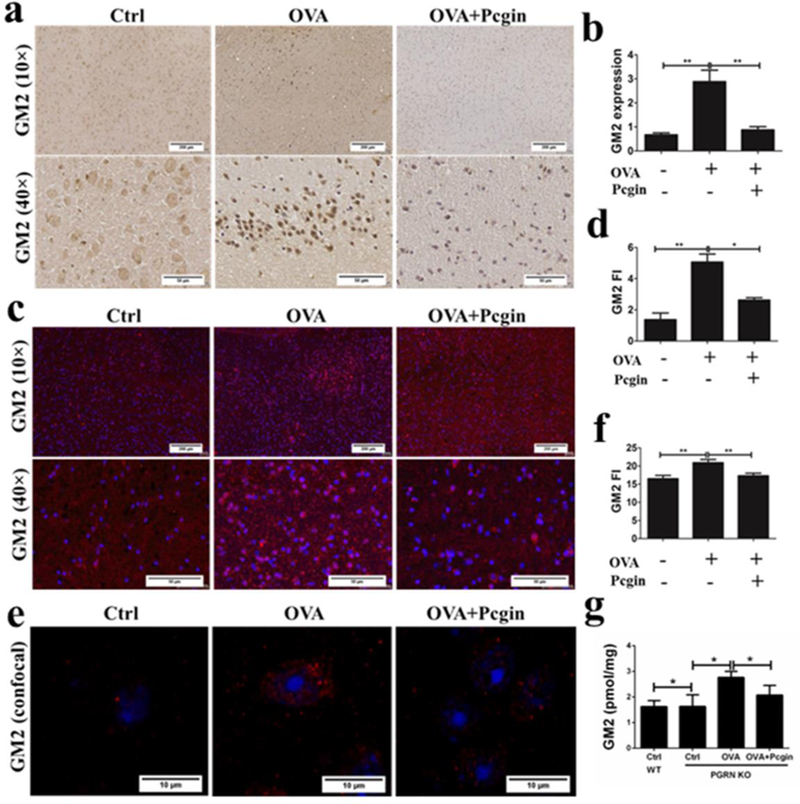
(a) IHC staining of GM2 in paraffin-embedded brain tissue slides from PGRN KO mice challenged with OVA treated with or without Pcgin to detect the GM2 expression. GM2 was aggregated in PGRN KO mice challenged with OVA and the application of Pcgin decreased GM2 accumulation. (b) The quantification of GM2 expression of (a). (c) Immunofluorescence staining of GM2 in paraffinembedded brain tissue from PGRN knockout mice challenged with OVA treated with or without Pcgin. Fluorescent intensity was markedly increased in PGRN KO mice challenged with OVA and Pcgin greatly reduced the fluorescence intensity. (d) The quantification of GM2 fluorescence intensity of (c). (e) Confocal staining of GM2 in frozen brain sections from PGRN KO mice, with or without OVA challenge (Red: GM2, Blue: DAPI). Pcgin alleviates GM2 storage. (f) The quantification of GM2 fluorescence intensity of (e). (g) Lipid analysis of GM2 levels in the brain tissues from WT and PGRN KO mice by mass spectrometry. Pcgin effectively reduced GM2 accumulation induced by OVA challenge. Data are reported as mean ± SEM. *P<0.05. **P<0.01.
Discussion
PGRN consists of seven and a half repeats, P-G-F-B-A-C-D-E in order, of a cysteine-rich granulin motif, where the P domain is half-granulin and the rest are full granulin domains [40,3,23,41]. The granulins F, A and C were identified as the binding domains of PGRN to TNFR[22]. Extracellular PGRN exerts anti-inflammatory function through binding to TNFR1 and inhibiting TNFα activity[42,22,43] and, in particular, through direct binding to and activating TNFR2 anti-inflammatory and protective signaling[32,11,44,45]. Aside from important function in the extracellular space, emerging data have shown that intercellular PGRN is important for maintenance of lysosomal function [29,28,46,47]. PGRN’s importance in lysosome biology is demonstrated by the relationship between PGRN and LSDs; deficiency of PGRN associates with LSDs, including GD[28,29], neuronal ceroid lipofuscinosis[48,49], and TSD (this study). We have reported that PGRN functions as a chaperone molecule that facilitates GCase delivery to the lysosome and this function is mediated through the C-terminal Grn E domain[29]. Further, a recent report has shown that PGRN stabilizes lysosomal enzyme Cathepsin D through action of its C-terminal Grn E domain as a chaperone[37]. Therefore, PGRN is a shared chaperone molecule that facilitates multiple lysosomal enzyme activities[31]. When screening the therapeutic effect of PGRN in multiple LSDs, we were excited to find that PGRN was therapeutic in TSD, Farber’s disease, and ML type 3, in addition to GD (Fig. 1a–b). In aged PGRN KO mice or OVA-challenged adult PGRN KO mice, we found GM2 accumulation in the brain (Fig. 2) and typical zebra bodies in macrophages. Although accumulation of GM2 gangliosides is a common feature associated with a number of lysosomal storage diseases, including Niemann-Pick diseases, mucopolysaccharidoses, prosaposin deficiency, and ceroid lipofuscinoses[50] , we found that HexA, but not HexB, was specifically aggregated in PGRN KO mice (Fig. 2). PGRN KO mice have mixed lipid storage in lysosomes, including lipofuscin, glucosylceramide, and GM2. It is noted that both PGRN or Pcgin significantly increased the HexA activity in the TSD fibroblasts, but is still below the HexA activity in normal fibroblasts (data not shown). These results suggest that increasing the HexA activity might not be the major mechanism underlying PGRN/Pcgin-mediated therapeutic effects, other mechanisms, such as enhancing folding and lysosomal delivery of mutant HexA, as it does for mutant GCase in Gaucher diseases, may be also important for PGRN’s effects in TSD. Indeed, both PGRN and Pcgin increased the lysosomal delivery of HexA (Fig. 7). These findings further support the concept that PGRN is a shared chaperone of lysosomal enzymes and plays an important role in the lysosome. In addition, PGRN KO mice might be useful for modeling multiple LSDs and for testing the therapeutic effects of potential LSD drugs. Although this study specifically focuses on the association of PGRN and its derivative Pcgin with TSD, the potential associations of PGRN/Pcgin with other LSDs, especially Farber’s disease and Mucolipidosis type III in which PGRN also showed potential therapeutic effects (Fig. 1), warrant further investigations.
Interestingly, similar to PGRN binding to GCase, PGRN binds to HexA with two binding sites. Using serial deletions from the either the N-terminal or from C-terminal, we identified that PGRN binds to HexA through Grn G and E domain (Fig. 5 f, i). Grn E domain is a major protein-protein interaction domain for PGRN, as it has been shown to bind to GCase[29], HSP70[29], Cathepsin D[37], and HexA (current report). Grn E binds to HSP70 and has chaperone activity, and we have found that Pcgin (containing Grn E and its c-terminal linker) has beneficial effect through facilitating lysosomal delivery of GCase [29] and enhancing HexA activity (this study). Grn E has also been shown to promote neurite growth in the central nervous system[51]. It is possible that Grn E promotion of neurite growth also relies on its chaperone activity, as HSP70 has been shown to promote neurite growth activity[52]. It is also noted that ND1 differs from FL PGRN and that all the ND-series of constructs possess domain E, some of them (ND2 to ND5) have weak HexA interaction, but the smaller constructs ND6 and ND7 (domains D-E and E) have strong HexA interactions. These paradoxical results may suggest that small N-terminal peptide and other internal domain may inhibit the binding of PGRN to HexA via as acting as the negative regulatory domains, or the structure of the mutants may be different, which also warrants the further investigation.
The results that lysosomal storage phenotypes after ovalbumin stimulation was only observed in KO but not in WT macrophage and mice further indicate that PGRN pays a critical role in lysosome and keeps lysosome function normally in WT mice. Under stressed conditions, such as OVA challenge, and/or HexA mutations, HexA/PGRN complexes aggregate in the cytosol and may undergo protein degradation. Under such conditions, PGRN may be needed to recruit stressinduced HSP70 disaggregation system to HexA aggregates and to unlock the disaggregation of HexA aggregates, as we observed in Gaucher diseases [29]. TSD is a rare autosomal recessive genetic LSD caused by HexA gene mutations. TSD mainly affects the central nervous system and currently there are no effective treatments. Incidence of TSD at birth is low in general populations (carrier frequency 1 in 250) while mutations causing TSD are more common in particular communities. Ashkenazi Jewish is the most affected population with the carrier frequency is 1 in 25[6]. Mutations in HexA gene have been reported to cause HexA protein undergo endoplasmic reticulum-associated degradation[53], which results in reduction of HexA enzymatic activity and leads to GM2 accumulation in lysosomes. Here, we reported that both PGRN and Pcgin increase HexA enzymatic activities, possibly through facilitating the folding of HexA and stabilizing HexA protein. Given that extracellular PGRN acts as a growth factor, we cannot rule out the possibility that extracellular PGRN may affect the transcription, translation, post-translational modifications, synthesis and degradation of HexA, also contributing to the increased levels and activity of HexA.
Pharmacological chaperones, the small molecules acting as competitive suppressors of their target enzyme, are reported to enhance enzyme activity. A good case in point is pyrimethamine, which was selected via drug library screening from 1040 compounds approved by the Food and Drug Administration and it has been used in an open-label phase Ⅰ/ Ⅱclinical trial. Pyrimethamine can improve HexA activity by up to 4-fold at a dose of 50 mg per day or less [54–56]. Recently, Kato et al. reported that several carbohydrate motifs in HexA are activityinhibiting sites. DMDP amide (6) and DNJNAc (1) can bind to these enzyme inhibiting sites and stabilize HexA activity[57]. These reports suggest that chaperone based therapy is a promising approach in the treatment of TSD. PGRN was also reported as a co-chaperone of HSP70 and is therapeutic in GD[29]. Here, we report both PGRN and its derivative, Pcgin, can enhance the HexA activity, which may account for, at least in part, their treatment effect in TSD.
In conclusion, by screening PGRN’s effects on multiple LSDs, we identified TSD as a new LSD in which PGRN could effectively reduce the lysosomal storage and PGRN deficiency led to typical TSD phenotypes, including the existence of zebra bodies. The treatment effect of PGRN in TSD probably results from its binding to HexA, leading to the increased activity and lysosomal delivery of HexA. Pcgin, containing the E domain of PGRN, also showed lysosomal storage reduction ability in TSD. These findings not only provide new insights into the pathogenesis of TSD, but also present evidences demonstrating that PGRN, and in particular the derivative Pcgin, may have potential for treating various kinds of lysosomal storage diseases, particularly TSD, a life-threatening diseases which is currently no cure.
GM2 accumulation and the existence of typical TSD cells containing zebra-bodies are detected in both aged and ovalbumin-challenged adult PGRN deficient mice.
Recombinant PGRN significantly reduces GM2 accumulation and lysosomal storage both in vivo and in vitro, which works through increasing the expression and enzymatic activity of HexA.
Pcgin, an engineered PGRN derivative bearing the granulin E domain, also effectively bound to HexA and reduced the GM2 accumulation.
Acknowledgments
This work was supported partly by NIH research grants 1R01NS103931, R01AR062207, R01AR061484, and a DOD research grant W81XWH-16–1-0482. Yuehong Chen was funded by China Scholarship Council (grant number 201606240021).
Footnotes
Conflict of interest
We herein declare that we have no conflict of interest.
References
- 1.Sandhoff K (2016) Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie 130:146–151. doi:10.1016/j.biochi.2016.05.004 [DOI] [PubMed] [Google Scholar]
- 2.Anheuser S, Breiden B, Schwarzmann G, Sandhoff K (2015) Membrane lipids regulate ganglioside GM2 catabolism and GM2 activator protein activity. Journal of lipid research 56 (9):1747–1761. doi:10.1194/jlr.M061036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandes Filho JA, Shapiro BE (2004) Tay-Sachs disease. Archives of neurology 61 (9):1466–1468. doi:10.1001/archneur.61.9.1466 [DOI] [PubMed] [Google Scholar]
- 4.Kaback MM, Desnick RJ (1993) Hexosaminidase A Deficiency. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews(R) Seattle (WA) [Google Scholar]
- 5.Vallance H, Ford J (2003) Carrier testing for autosomal-recessive disorders. Critical reviews in clinical laboratory sciences 40 (4):473–497. doi:10.1080/10408360390247832 [DOI] [PubMed] [Google Scholar]
- 6.Lew RM, Burnett L, Proos AL, Delatycki MB (2015) Tay-Sachs disease: current perspectives from Australia. Appl Clin Genet 8:19–25. doi:10.2147/TACG.S49628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyakumar M, Butters TD, Dwek RA, Platt FM (2002) Glycosphingolipid lysosomal storage diseases: therapy and pathogenesis. Neuropathology and applied neurobiology 28 (5):343–357 [DOI] [PubMed] [Google Scholar]
- 8.Matsuoka K, Tamura T, Tsuji D, Dohzono Y, Kitakaze K, Ohno K, Saito S, Sakuraba H, Itoh K (2011) Therapeutic potential of intracerebroventricular replacement of modified human betahexosaminidase B for GM2 gangliosidosis. Molecular therapy : the journal of the American Society of Gene Therapy 19 (6):1017–1024. doi:10.1038/mt.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rountree JS, Butters TD, Wormald MR, Boomkamp SD, Dwek RA, Asano N, Ikeda K, Evinson EL, Nash RJ, Fleet GW (2009) Design, synthesis, and biological evaluation of enantiomeric beta-N-acetylhexosaminidase inhibitors LABNAc and DABNAc as potential agents against Tay-Sachs and Sandhoff disease. ChemMedChem 4 (3):378–392. doi:10.1002/cmdc.200800350 [DOI] [PubMed] [Google Scholar]
- 10.Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny EH (2009) Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genetics in medicine : official journal of the American College of Medical Genetics 11 (6):425–433. doi:10.1097/GIM.0b013e3181a1b5c5 [DOI] [PubMed] [Google Scholar]
- 11.Fu W, Hu W, Shi L, Mundra JJ, Xiao G, Dustin ML, Liu CJ (2017) Foxo4-and Stat3dependent IL-10 production by progranulin in regulatory T cells restrains inflammatory arthritis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 31 (4):1354–1367. doi:10.1096/fj.201601134R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jian J, Konopka J, Liu C (2013) Insights into the role of progranulin in immunity, infection, and inflammation. Journal of leukocyte biology 93 (2):199–208. doi:10.1189/jlb.0812429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones EE, Zhang W, Zhao X, Quiason C, Dale S, Shahidi-Latham S, Grabowski GA, Setchell KDR, Drake RR, Sun Y (2017) Tissue Localization of Glycosphingolipid Accumulation in a Gaucher Disease Mouse Brain by LC-ESI-MS/MS and High-Resolution MALDI Imaging Mass Spectrometry. SLAS discovery : advancing life sciences R & D 22 (10):1218–1228. doi:10.1177/2472555217719372 [DOI] [PubMed] [Google Scholar]
- 14.Williams A, Wang EC, Thurner L, Liu CJ (2016) Review: Novel Insights Into Tumor Necrosis Factor Receptor, Death Receptor 3, and Progranulin Pathways in Arthritis and Bone Remodeling. Arthritis & rheumatology (Hoboken, NJ) 68 (12):2845–2856. doi:10.1002/art.39816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YP, Liu B, Tian QY, Wei JL, Richbourgh B, Liu CJ (2015) Progranulin protects against osteoarthritis through interacting with TNF-alpha and beta-Catenin signalling. Annals of the rheumatic diseases 74 (12):2244–2253. doi:10.1136/annrheumdis-2014-205779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang BC, Liu H, Talwar A, Jian J (2015) New discovery rarely runs smooth: an update on progranulin/TNFR interactions. Protein & cell 6 (11):792–803. doi:10.1007/s13238-015-0213-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y, Xu X, Liu L, Mao S, Feng T, Lu Y, Cheng Y, Wang H, Zhao W, Tang W (2016) Progranulin deficiency leads to severe inflammation, lung injury and cell death in a mouse model of endotoxic shock. Journal of cellular and molecular medicine 20 (3):506–517. doi:10.1111/jcmm.12756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu CJ, Bosch X (2012) Progranulin: A growth factor, a novel TNFR ligand and a drug target. Pharmacology & therapeutics 133 (1):124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Z, Ong CH, Halper J, Bateman A (2003) Progranulin is a mediator of the wound response. Nature medicine 9 (2):225–229. doi:10.1038/nm816 [DOI] [PubMed] [Google Scholar]
- 20.Zhao C, Bateman A (2015) Progranulin protects against the tissue damage of acute ischaemic stroke. Brain : a journal of neurology 138 (Pt 7):1770–1773. doi:10.1093/brain/awv123 [DOI] [PubMed] [Google Scholar]
- 21.Johnson J, Yeter K, Rajbhandary R, Neal R, Tian Q, Jian J, Fadle N, Thurner L, Liu C, Stohl W (2017) Serum progranulin levels in Hispanic rheumatoid arthritis patients treated with TNF antagonists: a prospective, observational study. Clinical rheumatology 36 (3):507–516. doi:10.1007/s10067-016-3467-7 [DOI] [PubMed] [Google Scholar]
- 22.Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, Syed NM, Lai Y, Lin EA, Kong L, Su J, Yin F, Ding AH, Zanin-Zhorov A, Dustin ML, Tao J, Craft J, Yin Z, Feng JQ, Abramson SB, Yu XP, Liu CJ (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332 (6028):478–484. doi:10.1126/science.1199214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian Q, Zhao S, Liu C (2014) A Solid-Phase Assay for Studying Direct Binding of Progranulin to TNFR and Progranulin Antagonism of TNF/TNFR Interactions. Methods Mol Biol 1155:163–172. doi:10.1007/978-1-4939-0669-7_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei F, Zhang Y, Zhao W, Yu X, Liu CJ (2014) Progranulin facilitates conversion and function of regulatory T cells under inflammatory conditions. PloS one 9 (11):e112110. doi:10.1371/journal.pone.0112110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei J, Hettinghouse A, Liu C (2016) The role of progranulin in arthritis. Ann N Y Acad Sci doi:10.1111/nyas.13191 [DOI] [PubMed] [Google Scholar]
- 26.Zhao YP, Tian QY, Frenkel S, Liu CJ (2013) The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials 34 (27):6412–6421. doi:10.1016/j.biomaterials.2013.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao YP, Tian QY, Liu CJ (2013) Progranulin deficiency exaggerates, whereas progranulinderived Atsttrin attenuates, severity of dermatitis in mice. FEBS letters 587 (12):1805–1810. doi:10.1016/j.febslet.2013.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jian J, Zhao S, Tian QY, Liu H, Zhao Y, Chen WC, Grunig G, Torres PA, Wang BC, Zeng B, Pastores G, Tang W, Sun Y, Grabowski GA, Kong MX, Wang G, Chen Y, Liang F, Overkleeft HS, Saunders-Pullman R, Chan GL, Liu CJ (2016) Association Between Progranulin and Gaucher Disease. EBioMedicine 11:127–137. doi:10.1016/j.ebiom.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jian J, Tian QY, Hettinghouse A, Zhao S, Liu H, Wei J, Grunig G, Zhang W, Setchell KDR, Sun Y, Overkleeft HS, Chan GL, Liu CJ (2016) Progranulin Recruits HSP70 to betaGlucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine 13:212–224. doi:10.1016/j.ebiom.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choy FYM, Christensen CL (2016) Progranulin as a Novel Factor in Gaucher Disease. EBioMedicine 13:13–14. doi:10.1016/j.ebiom.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jian J, Hettinghouse A, Liu CJ (2017) Progranulin acts as a shared chaperone and regulates multiple lysosomal enzymes. Genes & diseases 4 (3):125–126. doi:10.1016/j.gendis.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei F, Zhang Y, Jian J, Mundra JJ, Tian Q, Lin J, Lafaille JJ, Tang W, Zhao W, Yu X, Liu CJ (2014) PGRN protects against colitis progression in mice in an IL-10 and TNFR2 dependent manner. Scientific reports 4:7023. doi:10.1038/srep07023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng JQ, Guo FJ, Jiang BC, Zhang Y, Frenkel S, Wang DW, Tang W, Xie Y, Liu CJ (2010) Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J 24 (6):1879–1892. doi:10.1096/fj.09-144659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D (2004) Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. The Journal of biological chemistry 279 (14):13478–13487. doi:10.1074/jbc.M308523200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wendeler M, Sandhoff K, Glycoconj J (2009) Hexosaminidase assays. Glycoconjugate Journal 26:945–952 [DOI] [PubMed] [Google Scholar]
- 36.Park SH, Chen WC, Esmaeil N, Lucas B, Marsh LM, Reibman J, Grunig G (2014) Interleukin 13-and interleukin 17A-induced pulmonary hypertension phenotype due to inhalation of antigen and fine particles from air pollution. Pulmonary circulation 4 (4):654–668. doi:10.1086/678511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, Van Damme P (2017) Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Human molecular genetics 26 (15):2850–2863. doi:10.1093/hmg/ddx162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jaattela M (2010) Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463 (7280):549–553. doi:10.1038/nature08710 [DOI] [PubMed] [Google Scholar]
- 39.Stromme P, Mansson JE, Scott H, Skullerud K, Hovig T (1997) Encephaloneuropathy with lysosomal zebra bodies and GM2 ganglioside storage. Pediatric neurology 16 (2):141–144 [DOI] [PubMed] [Google Scholar]
- 40.Hrabal R, Chen Z, James S, Bennett HP, Ni F (1996) The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nature structural biology 3 (9):747–752 [DOI] [PubMed] [Google Scholar]
- 41. .Tian QY, Zhao YP, Liu CJ (2012) Modified yeast-two-hybrid system to identify proteins interacting with the growth factor progranulin. Journal of visualized experiments : JoVE (59). doi:10.3791/3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian Q, Zhao Y, Mundra JJ, Gonzalez-Gugel E, Jian J, Uddin SM, Liu C (2014) Three TNFR-binding domains of PGRN act independently in inhibition of TNF-alpha binding and activity. Frontiers in bioscience (Landmark edition) 19:1176–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mundra JJ, Jian J, Bhagat P, Liu CJ (2016) Progranulin inhibits expression and release of chemokines CXCL9 and CXCL10 in a TNFR1 dependent manner. Scientific reports 6:21115. doi:10.1038/srep21115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jian J, Li G, Hettinghouse A, Liu C (2016) Progranulin: A key player in autoimmune diseases. Cytokine doi:10.1016/j.cyto.2016.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li M, Liu Y, Xia F, Wu Z, Deng L, Jiang R, Guo FJ (2014) Progranulin is required for proper ER stress response and inhibits ER stress-mediated apoptosis through TNFR2. Cellular signalling 26 (7):1539–1548. doi:10.1016/j.cellsig.2014.03.026 [DOI] [PubMed] [Google Scholar]
- 46.Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M (2014) Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulindeficient mice. Acta neuropathologica communications 2:78. doi:10.1186/s40478-014-0078-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evers BM, Rodriguez-Navas C, Tesla RJ, Prange-Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F, Rademakers R, Yu G, White CL 3rd, Herz J (2017) Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell reports 20 (11):2565–2574. doi:10.1016/j.celrep.2017.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lefrancois S, Zeng J, Hassan AJ, Canuel M, Morales CR (2003) The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. The EMBO journal 22 (24):6430–6437. doi:10.1093/emboj/cdg629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. The American journal of pathology 177 (1):311–324. doi:10.2353/ajpath.2010.090915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walkley SU, Vanier MT (2009) Secondary lipid accumulation in lysosomal disease. Biochimica et biophysica acta 1793 (4):726–736. doi:10.1016/j.bbamcr.2008.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjaer A, Petrucelli L (2012) Progranulin regulates neuronal outgrowth independent of sortilin. Molecular neurodegeneration 7:33. doi:10.1186/1750-1326-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frebel K, Wiese S, Funk N, Puhringer D, Sendtner M (2007) Differential modulation of neurite growth by the S-and the L-forms of bag1, a co-chaperone of Hsp70. Neuro-degenerative diseases 4 (2–3):261–269. doi:10.1159/000101850 [DOI] [PubMed] [Google Scholar]
- 53.Dersh D, Iwamoto Y, Argon Y (2016) Tay-Sachs disease mutations in HEXA target the alpha chain of hexosaminidase A to endoplasmic reticulum-associated degradation. Molecular biology of the cell 27 (24):3813–3827. doi:10.1091/mbc.E16-01-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, Tropak MB (2011) An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Molecular genetics and metabolism 102 (1):6–12. doi:10.1016/j.ymgme.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tropak MB, Mahuran D (2007) Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. The FEBS journal 274 (19):4951–4961. doi:10.1111/j.1742-4658.2007.06040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osher E, Fattal-Valevski A, Sagie L, Urshanski N, Sagiv N, Peleg L, Lerman-Sagie T, Zimran A, Elstein D, Navon R, Valevski A, Stern N (2015) Effect of cyclic, low dose pyrimethamine treatment in patients with Late Onset Tay Sachs: an open label, extended pilot study. Orphanet journal of rare diseases 10:45. doi:10.1186/s13023-015-0260-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kato A, Nakagome I, Nakagawa S, Kinami K, Adachi I, Jenkinson SF, Desire J, Bleriot Y, Nash RJ, Fleet GWJ, Hirono S (2017) In silico analyses of essential interactions of iminosugars with the Hex A active site and evaluation of their pharmacological chaperone effects for Tay-Sachs disease. Organic & biomolecular chemistry doi:10.1039/c7ob02281f [DOI] [PubMed] [Google Scholar]