

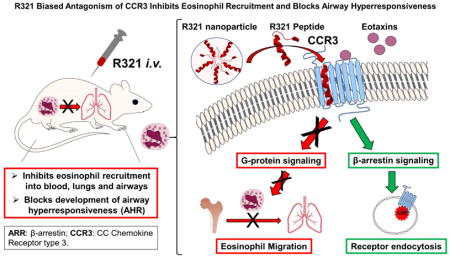
Abstract
Background
Chemokine signaling through CCR3 is a key regulatory pathway for eosinophil recruitment into tissues associated with allergic inflammation and asthma. To date, none of the CCR3 antagonists have shown efficacy in clinical trials. One reason may be their unbiased mode of inhibition that prevents receptor internalization, leading to drug tolerance.
Objective
We sought to develop a novel peptide nanoparticle CCR3 inhibitor (R321) with a biased mode of inhibition that would block G-protein signaling, but enable or promote receptor internalization.
Methods
Self-assembly of R321 peptide into nanoparticles and peptide binding to CCR3 were analyzed by dynamic light scattering and NMR. Inhibitory activity on CCR3 signaling was assessed in vitro using flow cytometry, confocal microscopy, and western blot analysis in a CCR3+ eosinophil cell line and blood eosinophils. In vivo effects of R321 were assessed using a triple allergen mouse asthma model.
Results
R321 self-assembles into nanoparticles and binds directly to CCR3, altering receptor function. IC50 values for eotaxin-induced chemotaxis of blood eosinophils are in the low nanomolar range. R321 inhibits only the early phase of ERK1/2 activation and not the late phase generally associated with β-arrestin recruitment and receptor endocytosis, promoting CCR3 internalization and degradation. In vivo, R321 effectively blocks eosinophil recruitment into the lungs and airways and prevents airway hyperresponsiveness in a mouse eosinophilic asthma model.
Conclusions
R321 is a potent and selective antagonist of the CCR3 signaling cascade. Inhibition through a biased mode of antagonism may hold significant therapeutic promise by eluding the formation of drug tolerance.
Keywords: CCR3, eosinophil, allergic inflammation, asthma, biased antagonist, peptide nanoparticles, airway hyperresponsiveness
Graphical Abstract
INTRODUCTION
In allergic disorders, such as asthma and eosinophilic esophagitis (EoE), eosinophils are recruited into the lung and esophagus, respectively, and activated in excess at these sites of inflammation. In these diseases, eosinophils are both a histologic hallmark and among the major effector cell types contributing to their pathology1, 2. The C-C chemokine receptor 3 (CCR3) signaling pathway is one of the key regulatory pathways involved in eosinophil recruitment and migration into the affected tissues as part of the allergic diathesis.
While CCR3 is most highly expressed by eosinophils, it is also expressed by basophils, subsets of mast cells and Th2 cells, and airway epithelial cells3–5. CCR3 is a promiscuous G-protein-coupled receptor (GPCR), interacting with multiple inflammatory chemokines, including the high affinity agonists eotaxin-1 (CCL11), eotaxin-2 (CCL24), eotaxin-3 (CCL26), and RANTES (CCL5). The receptor is coupled to the pertussis toxin-sensitive G protein Gαi. Upon ligand binding, the receptor is activated and active GTP-bound Gαi and the Gβγ dimer dissociate from CCR3 to trigger downstream signaling cascades including the MAPK (ERK1/2, p38) and the PI3K/AKT pathways6, 7. These intracellular signaling pathways culminate in priming, chemotaxis, activation, and degranulation of eosinophils. Following receptor activation by the ligand, CCR3 is desensitized and internalized8, 9. The mechanism of CCR3 internalization is not yet fully understood, but is thought to occur via β-arrestin recruitment to phosphorylated CCR3 and sequestration of the receptor into endosomes10. In addition, eotaxin-induced CCR3 internalization may be required for actin polymerization and chemotaxis9.
The importance of CCR3 as a potential therapeutic target was established through the observations that CCR3-null mice and eotaxin-1 and eotaxin-2 double knockout mice displayed near complete abolishment (up to ~70%) of allergen-induced airway eosinophil recruitment11. CCR3 transcript and protein levels are increased in the bronchial mucosa of patients with allergic asthma12. In line with this, much effort has been invested in the development of small molecule CCR3 antagonists, yet none have been approved for clinical use to date4.
Most of the currently known CCR3 antagonists are competitive or allosteric inhibitors of CCR3 activation and internalization by chemokines4. The dual inhibitory activity of these molecules classifies them as unbiased antagonists. Reports on the use of unbiased antagonists of other GPCRs, such as CXCR4, suggest that after prolonged exposure, cell surface GPCR accumulates, a phenomenon associated with developing resistance to receptor inhibition13, 14. These findings prompted us to search for antagonists that can “bias” downstream signaling by selectively inhibiting only one of the signaling cascades. There is a growing interest in the development of biased agonists of GPCRs,15 but biased antagonists of GPCRs remain largely unexplored, very few have been identified, and their therapeutic potential remains to be determined.
In the present study, we report the development and validation of R321, a novel peptide inhibitor derived from the second transmembrane helix of CCR3. R321 self-assembles into uniform nanoparticles and inhibits CCR3-mediated chemotaxis of human blood eosinophils with nanomolar potencies. Intravenously administered R321 significantly reduces eosinophil recruitment into the lung and airspaces and diminishes airway hyperresponsiveness (AHR) in a triple allergen (DRA) mouse asthma model of allergic airway inflammation. We propose that the R321 peptide exerts its receptor inhibitory effects on eosinophil function as a biased antagonist by inhibiting G-protein mediated processes and promoting the internalization (endocytosis) and degradation of CCR3.
MATERIALS AND METHODS
Reagents
Small molecule CCR3 antagonists, SB238437 and UCB35625, were purchased from Tocris Bioscience (Bristol, UK).
Peptide synthesis and characterization
Synthesis, purification and evaluation of nanoparticle formation of R321 and R323 peptides were performed as described in the Supplementary Materials.
Cell culture
AML14.3D10-CCR3 cells, an eosinophil-differentiated acute myeloid leukemia cell line stably transfected to express CCR3 (ATCC® CRL-12079), were cultured as previously described.16 Jurkat cells, a T cell leukemia line endogenously expressing CXCR4, but not CCR3, were cultured in RPMI-1640 supplemented with 10% FBS, 1% Penicillin-streptomycin, and 2 mM L-Glutamine.
Eosinophil purification
Eosinophils were purified from blood drawn from mild allergic asthmatic subjects. Peripheral blood was separated over a gradient of Ficoll-Paque Plus (GE Healthcare, Pittsburg, PA). Eosinophils were further purified by negative selection using a commercial Eosinophil Isolation kit (MAC Miltenyi Biotec, Auburn, CA).
Chemotaxis and degranulation assays
Chemotaxis and degranulation assays are described in the Supplementary Materials.
Prolonged exposure to inhibitors
AML14.3D10-CCR3 cells or human peripheral blood eosinophils were incubated for 24, 48, or 72 hours with either vehicle control or 1 μM inhibitors. Cells were resuspended in fresh complete medium with inhibitors every day.
Signal transduction – western blotting and confocal microscopy
Detailed descriptions are provided in the Supplementary Materials.
Receptor expression and internalization
To evaluate CCR3 cell surface expression and ligand-induced internalization, cells were treated for 30 min with vehicle control, R321 (0.01–10 μM) ± CCL11 (12 nM), or R323, SB238437, UCB35625 (all at 1μM) ± CCL11 (12nM). Cells were blocked with 10% heat-inactivated human AB-serum, stained using PE-conjugated anti-human CCR3 antibody (clone 5E8, BioLegend, San Diego, CA) or PE-conjugated isotype-matched control (BioLegend, San Diego, CA) and analyzed on a Quanta SC flow cytometer (Beckman Coulter, Indianapolis, IN). Cell surface staining and gating strategy employed for the enumeration of mouse blood eosinophils and determination of CCR3 surface expression levels is described in the Supplementary Materials.
Mice
Female BALB/cJ mice (10–12 weeks of age) were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal study protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Illinois (Chicago, IL).
Sensitization and airway challenge
Sensitization and intranasal challenges were performed according to the acute asthma protocol previously described by Goplen at al17. In brief, mice were sensitized twice ip with a cocktail of 3 allergens: Dust-mite (D. Farinae) – 5 μg, ragweed (A. artemisifolia) – 50 μg, and Aspergillus fumigatus – 5 μg. All extracts were purchased from Greer Laboratories (Lenoir, NC). One week after the second sensitization, intranasal challenges consisting of 0.15 μg of Aspergillus, 0.15 μg of dust-mite, and 1.5 μg of ragweed extract were given for 3 consecutive days. Control mice were sham-challenged with PBS. For the prophylactic protocol, R321, scrambled R323 peptide control, vehicle, or PBS was delivered by iv injection into the retro-orbital sinus one day before the first challenge and directly prior to each subsequent challenge. For the therapeutic protocol, mice started receiving 12 mg/kg of R321 or R323 on the day after the final allergen challenge and for 3 additional days following the date of the last challenge.
Bronchoalveolar lavage, lung histology and airway responsiveness to methacholine
Bronchoalveolar lavage (BAL) was performed as described in the Supplementary Materials. Whole lungs were fixed in 10% formalin and embedded in paraffin. Lung tissue sections were stained with rat anti-mouse MBP1 antibody (generously provided by the Lee laboratories, Mayo Clinic, Scottsdale, AZ) as previously described.18 Immunostained slides were scanned using Aperio Scanscope CS2 scanner (Aperio, Vista, CA) and analyzed with Aperio's image viewer software. Nuclei were counterstained with Mayer’s Hematoxylin and cell counts were expressed as percent of MBP1 positive cells of the total nucleated cell count. Determination of airway responsiveness to methacholine is described in the Supplementary materials.
NMR
NMR was performed as described in the Supplementary Materials.
Statistical Analysis
Statistical analysis was performed using two-tailed t-tests, one way or two-way ANOVA, followed by Tukey post hoc analysis in GraphPad Prism software (GraphPad, San Diego, CA).
RESULTS
R321 self-assembles into nanoparticles
Peptides containing sequences from each of the seven transmembrane domains and associated extracellular loops of human CCR3 were first screened for inhibition of chemotaxis to CCL11, identifying the second transmembrane domain and first extracellular loop region as the most inhibitory (data not shown). The final design of R321 (Fig. 1A) was based on a previously described self-assembling CXCR4 peptide antagonist19, 20. Twenty-seven units of polyethylene glycol (PEG) were placed on the C-terminus of the peptide to prevent aggregation, and the PEG units were followed by three aspartate residues that ensure homogeneous self-assembly and correct orientation upon membrane fusion (Fig. 1A). The control peptide, R323, was derived by randomly, but separately, scrambling the sequences of the R321 transmembrane and extracellular loop regions (Fig. 1A). DLS analysis (Fig. 1B) showed that R321 and R323 monomers both self-assemble in an aqueous environment into nanospheres with a hydrodynamic radius of 7.1± 0.7 nm and 4.5 ± 0.4 nm, respectively, with R323 smaller and more polydisperse than R321. The size of the R321 particles was maintained over a wide range of monomeric concentrations (Fig. 1C).
Figure 1. The R321 CCR3 peptide and its scrambled control (R323) self-assemble into nanoparticles.

(A) Structures of R321 and the scrambled peptide R323. Alignment with human and mouse CCR3 shows a high degree of identity at the TM2 region. (B) Dynamic Light Scattering (DLS) regularization distribution histograms are shown for 10 μM peptide solutions in PBS. Radii for R321 and R323 are 7.1 ± 0.7 nm and 4.5 ± 0.4 nm, respectively, with R323 somewhat smaller and more polydisperse; the polydispersity index of R321 and R323 were 0.07 and 0.28, respectively. Results represent mean ± SEM from experiments (n=3) performed in duplicate (C). R321 self-assembly into nanoparticles shows no dependence on peptide concentration. TM: transmembrane. ECL: extracellular loop.
R321 specifically inhibits eotaxin-induced eosinophil chemotaxis
Human blood eosinophils and the stable CCR3+ eosinophilic myelocyte cell line, AML14.3D10-CCR3, undergo CCR3-mediated chemotaxis induced by multiple chemokines including CCL11/eotaxin-1, CCL24/eotaxin-2, and CCL26/eotaxin-3 (Fig. 2). R321 was observed to inhibit eotaxin-induced chemotaxis by both primary eosinophils (Fig. 2A) and the AML14.3D10-CCR3 cell line (Fig. 2B) in a dose-dependent manner and with nanomolar potencies. The IC50 and IC90 values are shown in Fig. 2C. When used at a concentration of 1 μM (approximate IC90 value for R321), the scrambled peptide control (R323) failed to significantly inhibit eotaxin-mediated chemotaxis in blood eosinophils (Fig. 2D). R321 failed to inhibit CXCR4-mediated chemotaxis in Jurkat T-cells (Fig. 2E) and platelet-activating factor (PAF)-mediated chemotaxis of blood eosinophils (Supplementary Fig. S1), demonstrating the specificity of R321 inhibition of CCR3. Although CCR3 ligands induce degranulation of cytochalasin-B treated human eosinophils,21 R321 did not promote CCL11-induced degranulation and secretion of eosinophil cationic protein (ECP) in blood eosinophils (Supplementary Fig. S2).
Figure 2. R321 inhibits eotaxin/CCR3-mediated chemotaxis.

R321 (0.001–10 μM) dose-response inhibition of chemotaxis induced by CCL11/Eotaxin-1 (12nM), CCL24/Eotaxin-2 (20nM), and CCL26/Eotaxin-3 (100nM) for 4h of (A) blood eosinophils and (B) AML14.3D10-CCR3 cells. (C) IC50/IC90 inhibitory activity of the R321 peptide on eotaxin-induced chemotaxis of blood eosinophils. (D) Scrambled peptide control – R323 (1 μM) does not significantly inhibit chemotaxis of blood eosinophils. In contrast, R321 inhibits chemotaxis by >90% when tested at the same (1 μM) concentration. (E) R321 does not inhibit CXCL12/CXCR4-mediated chemotaxis of Jurkat-T lymphocytic leukemia cells. Results are normalized to % maximum chemotactic response and are representative of the mean ± SEM from experiments (n=3) performed in triplicate. ns=p>0.05.
Effects of R321 on CCR3 signal transduction pathways
R321 was found to inhibit the activation of Gαi in an immunoprecipitation assay that detects Gαi-GTP (Fig. 3A) but did not inhibit receptor degradation (Fig. 3B). Pretreatment of AML14.3D10-CCR3 cells with 10 μM R321 (a concentration exceeding IC50 and IC90 for chemotaxis) before exposure to ligands was even found to enhance degradation of the CCR3 receptor (Fig. 3B). Both G-protein and β-arrestin mediated signaling pathways can lead to AKT and ERK 1/2 activation, although the time course of activation through the two pathways is different, leading to biphasic phosphorylation.22–25 Following stimulation of AML14.3D10-CCR3 cells with chemokines, biphasic phosphorylation of AKT (Fig. 3C) and ERK 1/2 (Fig. 4A) was observed in western blots. The early (2–5min) phosphorylation is mediated by G-protein signaling. The late sustained phase (30 min) phosphorylation is likely due to β-arrestin signaling as demonstrated for other GPCRs.22, 25, 26 Pretreatment of cells for 30 min with 10 μM R321 prior to stimulation lead to a complete inhibition of the early phase of ERK 1/2 activation (5 min) but had no effect on the prolonged late phase (30 min). The control peptide, R323, had no effect on ERK1/2 phosphorylation patterns. Both SB328437 and UCB35625 fully inhibited the late phase, with SB328437 blocking the early phase as well, and UCB35625 doing so only partially (Fig. 4A). These results indicate that, unlike SB328437 and UCB35625, R321 does not inhibit the β-arrestin signaling that mediates the late phase phosphorylation of ERK1/2.
Figure 3.

(A) R321 inhibits activation of pertussis toxin (PT) sensitive Gαi. AML14.3D10-CCR3 cells were pretreated with PT (200 ng/mL) or R321 (10 μM) before being stimulated with CCL11 (12 nM) for 1 min. Active, GTP-bound Gαi was immunoprecipitated using antibody specific for GαiGTP and detected by western blotting using antibody to total Gαi. The input lysates were blotted for CCR3 as a loading control. (B) R321 does not inhibit β-arrestin signaling by activated CCR3. AML14.3D10-CCR3 cells were treated with CCL11 (12 nM) or RANTES/CCL5 (12 nM) for 3h. Decrease in CCR3 indicates receptor degradation after exposure to ligand. Pretreatment with 10 μM R321 before CCR3 ligands enhances degradation. (C) Eotaxin-mediated activation of CCR3 leads to biphasic activation of AKT. After CCR3 activation by the indicated chemokines (12 nM), biphasic phosphorylation of AKT was observed. Acute (2min) phosphorylation is mediated by G protein signaling. Late phase (30min) phosphorylation is likely due to β-arrestin signaling.
Figure 4. R321 does not inhibit ligand-induced β-arrestin recruitment and signaling by activated CCR3.

(A) Following CCR3 activation with 100 nM CCL11, biphasic ERK1/2 phosphorylation was observed. Acute (2–5min) phosphorylation is mediated by G protein signaling. Late phase (30min) phosphorylation is likely due to β-arrestin signaling. R321 (10μM) inhibits only acute phosphorylation of ERK1/2. Scrambled peptide control – R323 (10μM) does not inhibit acute or late phase phosphorylation. SB328437 (10μM) inhibits both acute and late phase phosphorylation and UCB35625 (10μM) inhibits the late phase to a higher degree than the early phase. (B) Representative confocal images of AML14.3D10-CCR3 cells exposed to vehicle or inhibitors for 30 min and stimulated with CCL11/eotaxin-1 for 30 min. (C) Quantitation by Pearson’s correlation method shows colocalization of CCR3 to β-arrestin2 30 min after stimulation with CCL11/eotaxin-1. R321 and R323 (10 μM) did not inhibit CCL11-induced β-arrestin2 recruitment to CCR3 whereas the CCR3 antagonist SB328437 and UCB35625 strongly inhibited colocalization. Results represent mean (50 cells per treatment group) ± SEM from 3 independent experiments. (*p ≤ 0.05, **p≤ 0.01,****p ≤ 0.0001 as compared to control).
R321 does not antagonize β-arrestin recruitment to CCR3
Co-localization of CCR3 and β-arrestin 2 was observed following stimulation of CCR3 with CCL11 (Fig. 4B), suggesting that β-arrestin plays a role in ligand-induced CCR3 internalization and degradation. Pretreatment of cells with 10 μM R321 or R323 did not significantly (p>0.05) alter the reported co-localization coefficients either before or after CCL11 stimulation (Fig. 4C). In contrast, co-localization of CCR3 and β-arrestin 2 was significantly attenuated after treatment with UCB35625 and SB328437 when compared to untreated cells (p≤0.0001) (Fig. 4C). Representative images of antibody controls can be found in Supplementary Fig. S3.
R321 promotes rather than inhibits CCR3 internalization and degradation
Inhibition of β-arrestin signaling may interfere with effective degradation of CCR3 and lead to receptor accumulation on the eosinophil cell surface. To elucidate the fate of CCR3 upon ligand and inhibitor treatment, surface levels of CCR3 in AML14.3D10-CCR3 cells were determined by flow cytometry. More than 50% of CCR3 present on the cell surface was internalized following 30 min of CCL11 exposure, in keeping with previous reports8, 27. R321 and the scrambled R321 peptide control showed no significant effect on ligand-induced receptor internalization, whereas the small molecule antagonists, UCB35625 and SB328437, partially blocked CCR3 internalization and degradation (Fig. 5A). Of note, R321 at 10 μM was found to promote CCR3 internalization on its own, without the addition of chemokine ligand (Fig. 5B).
Figure 5. R321 does not inhibit CCL11-induced CCR3 internalization and does not induce resistance (tolerance) to inhibition of CCL11-induced chemotaxis.

(A) R321 does not inhibit CCL11-mediated internalization of CCR3. When added concurrently with 12 nM CCL11, R321 (1μM) and R323 (1μM) did not interfere with CCL11-induced receptor internalization. Both SB328437 (1 μM) and UCB35625 (1 μM) significantly inhibited the chemokine’s ability to induce CCR3 internalization. (B) R321 alone decreases CCR3 surface expression. R321 dose-response reduction of surface CCR3 expression on AML14.3D10-CCR3 cells. Significant internalization levels were reached at 1μM R321. (C) R321 maintains prolonged inhibitory activity. AML14.3D10-CCR3 cells were treated for 24h, 48h or 72h with R321 or unbiased antagonists (all at 1 μM) ± CCL11 (12 nM). (D) R321 promotes CCR3 internalization in human blood eosinophils over a prolonged incubation period. Results shown as surface expression of CCR3 as percentage of vehicle expression. Of note, SB328437, when used at equimolar concentrations to R321 and UCB35625 (1μM), was a less effective inhibitor of CCL11/CCR3-mediated chemotaxis and failed to promote CCR3 cell surface accumulation. Results represent mean ± SEM from experiments (n=3) performed in triplicate. Compared to vehicle (B, D) or 24h data point (C): *p ≤ 0.05, **p ≤ 0.01, ***p<0.001, ****p ≤ 0.0001; Error bars = SEM.
R321 maintains its efficacy over 72h in contrast to unbiased antagonists
The effects of prolonged exposure to inhibitors on CCL11-induced chemotaxis and CCR3 surface expression was assessed for up to 72h in AML14.3D10-CCR3 cells and in blood eosinophils (Fig. 5C and 5D). CCL11 (12 nM) alone and CCL11 + R321 reduced CCR3 expression to ~68% and ~55%, respectively, after 24h, and to ~18% and ~12% after 72h (Fig. 5D). Treatment of cells for 72h with R321 alone reduced surface levels of CCR3 to 68%, as compared to untreated cells. In contrast, UCB35625 lead to receptor accumulation on the surface and enhanced CCR3 surface levels up to 133% after 72h. R323 and SB328437 had no significant effect on receptor levels. A similar effect was observed on CCR3 surface expression in AML14.3D10-CCR3 cells (Supplementary Fig. S4). In agreement with the levels of CCR3 expression detected, R321 did not lose any of its inhibitory potency during 72h of treatment (maintaining 90% inhibition of chemotaxis), while UCB35625 and SB328437 inhibition levels dropped by 19.3% and 13.7%, respectively (Fig. 5C). These results indicate that resistance to the small molecule inhibitors, but not to R321, develops over time.
R321 inhibits eosinophil recruitment into the lung and airspaces
In a robust mouse DRA allergic asthma model of eosinophilic airway inflammation (Fig. 6A), prophylactically (Supplementary Fig. S5A) administered iv R321 demonstrated a dose-dependent inhibitory effect on eosinophil recruitment into the airways (Fig. 6B). Significantly, R321 reduced eosinophil counts in the BALF beginning with a dose of 6 mg/kg (44.24 ± 9.33 % of vehicle) and reached 69.33 ± 4.20% inhibition at the maximum dose of 12 mg/kg (Fig. 6C). An IC50 value of 8.16 mg/kg was obtained from linear regression analysis. R323 showed no inhibitory effect at 12 mg/kg. No significant differences were observed in total cell counts of other inflammatory cells including macrophages, neutrophils, or lymphocytes (Supplementary Fig. S6). MBP1-stained lung tissues showed a 36.20 ± 5.28% decrease in eosinophil counts following treatment with 12 mg/kg of R321 (Fig. 6D). In a therapeutic protocol (Supplementary Fig. S5B), R321 (12 mg/kg) successfully reduced airway (BAL) eosinophils by 74.18 ± 6.50%. (Fig. 7A) and lung eosinophils by 83.30 ± 7.29 %, fully reversing both allergen-induced eosinophilia in the blood (Fig. 7B) and in the lung tissue (Fig. 7C and 7E) to levels comparable to PBS-sham challenged mice. As expected, blood eosinophils displayed reduced levels of surface CCR3 upon exposure to allergen when compared to sham-challenged controls (~29% reduction) (Fig. 7D), and R321 treatment further reduced CCR3 levels (~15% reduction compared to vehicle, p=0.01). The protocols for allergen (DRA) sensitization, airway challenge, and peptide treatments are provided in Supplemental Fig. S5.
Figure 6. Prophylactic treatment with R321 significantly reduces eosinophil recruitment into the lung airspaces.

(A) The DRA-allergen challenge induces a robust eosinophilic response in female BALB/cJ mice as demonstrated by increased numbers of eosinophils in the BAL fluid. (B) Total eosinophil cell numbers (x10 ) in the BAL fluid show that R321 significantly inhibits eosinophil recruitment into the lung airspaces starting at an iv dose of 6 mg/kg. (C) The inhibitory effect of R321 is dose-dependent and reaches 69.33 ± 4.20% inhibition at 12 mg/kg. (D) Lungs were stained with anti-mMBP1 antibody to identify eosinophils. R321 (12 mg/kg) treatment reduces lung tissue eosinophil counts by 36.20 ± 5.28%. Results are displayed as % of mMBP1 positive cells as compared to total nucleated cells. The mean ± SEM are shown for 6–7 mice/treatment group from 3 independent experiments. R321 at 12 mg/kg significantly lowers respiratory system (E) and airway (F) responsiveness to methacholine as compared to vehicle or R323 controls. There is no significant difference between R321 treated and sham-challenged mice (n=5, except PBS group where n=4). (****p<0.0001, ***p<0.001,**p<0.01, *p<0.05, ns not significant).
Figure 7. Therapeutic treatment with R321 attenuates established asthmatic lung and airway inflammation in allergen-sensitized/challenged mice.

(A) R321 administered at 12 mg/kg inhibits recruitment of eosinophils to the lung airspaces by 74.18 ± 6.50%. (B) DRA-allergen challenged mice (Vh) develop significant blood eosinophilia as compared to sham-challenged mice (PBS). Treatment with R321 reduces blood eosinophil numbers to levels not significantly different than those observed in allergen sensitized/PBS-sham challenged mice. (C) Following therapeutic treatment with 12 mg/kg of R321, lungs stained for MBP1 positive cells showed tissue eosinophil counts not significantly different from the PBS-sham challenged mice, an 83.30 ± 7.29 % reduction compared to vehicle control. Results are expressed as % MBP1 positive cells compared to total nucleated cells. (D) Surface expression of CCR3 in blood eosinophils is reduced upon allergen challenge. R321 does not inhibit CCR3 internalization, but has a promoting effect (vehicle MFI of 13.7 vs. R321-treated group MFI of 11.7, p=0.01). The mean ± SEM is shown for 5 mice/treatment group. (****p<0.0001, ***p<0.001,**p<0.01, *p <0.05,ns not significant). (E) Representative images of mouse lung airways (top) and blood vessels (bottom) from Fig. 7C immunostained with HRP-conjugated antibodies to MBP1 (positive cells are dark brown). Black bars represent 100 μm.
R321 blocks airway hyperresponsiveness in allergen-challenged mice
DRA-challenged mice showed a ~9 times higher peak system and airway resistance in response to methacholine as compared to sham-challenged (PBS) mice (Fig. 6E and Fig. 6F). R321 treatment had a striking effect on airway responsiveness, reducing both the system and airway pulmonary resistance of challenged mice to levels comparable to those observed in sham-challenged (PBS) mice.
R321 interacts with CCR3 and allows chemokine binding
To study the binding of R321 to CCR3, we used NMR spectroscopy to correlate 13C and 1H frequencies in 13CH3 groups of membrane proteins incorporated by reductive methylation (Supplementary Fig. S7) 28–33. The HSQC spectrum of CCR3 positive membranes contained four discernable signals (Fig. 7A). In contrast, CCR3 null membrane exhibited only two signals (Fig. 7A). These signals overlapped with only two out of four signals of CCR3 positive membranes, suggesting that the remaining two signals belong to CCR3 (Fig. 7A). Immunoblotting only detected CCR3 in the CCR3 positive membranes (Supplementary Fig. S8). Moreover, CCL11 specifically reduced the intensity of CCR3 signals (Fig. 7B, black arrows) but did not affect the spectrum of CCR3 null membranes (Supplementary Fig. S8). Similar to CCL11, R321 shifted only the CCR3 signals (Fig. 7C). This suggests that R321 specifically perturbs CCR3 structure. Next, we investigated the interaction of R321 and CCR3 in the presence of CCL11 (Fig. 7D–F). The chemical shift changes and differences in signal intensities show that neither CCL11 nor R321 interfere with each other’s binding to the receptor. This suggests that R321 and CCL11 interact with CCR3 simultaneously and R321 alters CCL11’s ability to activate signaling (Fig. 7).
Dose-dependent responses of CCR3 at different concentrations of R321 (Supplementary Fig. S7 and Fig. S9) allowed calculation of the dissociation constants (Supplementary Fig. S7C). We found two dissociation constants for R321 binding to the receptor, one in the nanomolar range and the other in the micromolar range. This suggests that R321 might employ two distinct mechanisms for interaction with CCR3, potentially explaining its unique inhibitory profiles in vitro and in vivo.
DISCUSSION
In the present study, we report the development and characterization of a novel peptide inhibitor of CCR3. The described peptide (R321) self-assembles into uniformly sized nanoparticles, essentially functioning as its own carrier and delivery system. Self-assembly protects the peptide from proteolytic degradation, a known issue with peptide-based drugs20. Addition of polyethylene glycol (PEG) to the R321 nanoparticles is recognized to further prevent aggregation, proteolytic degradation, improve pharmacokinetics, and reduce immunogenicity of peptide based drugs34.
In this study, iv administration was used as the most reliable method for delivering R321 peptide nanoparticles to the systemic circulation. However, future studies will involve alternative routes of administration, notably formulating R321 preparations for nebulization or direct airway instillation or inhalation. UCB35625, initially identified as a high affinity unbiased antagonist of CCR3 and CCR1, was subsequently found to be an agonist of CCR2 and CCR5, making it prohibitively complex for in vivo studies35, 36. SB328437 was developed as a specific inhibitor of CCR3 in eosinophils and was shown to successfully suppress OVA-induced accumulation of eosinophils in the lungs of mice adoptively transferred with in vitro-differentiated Th2 cells37, 38. However, a very high subcutaneous dose (100 mg/kg) of SB328437 resuspended with Tween-80 was used in the study, and we have also experienced solubility issues with this compound, making it unsuitable for iv injection.
CCR3 signaling is increasingly implicated in various pathological contexts besides allergic inflammation. These include age-related macular degeneration,39 reproductive malignancies,40–42 eosinophilic myocarditis,43 neurodegenerative diseases,44 renal cell carcinoma,45 Crohn’s disease,46 and glioblastoma.47 Several antagonists that prevent chemokine binding to CCR3 have been developed; however, none of these inhibitors have been FDA approved. Failures of small molecule CCR3 inhibitors in the few clinical trials that have been conducted have called into question the role of CCR3 in airway eosinophilia in asthma, suggesting that CCR3 is not a viable target for drug development. However, a clinical trial of the unbiased CCR3 antagonist, GW766994, showed a trend towards inhibition of sputum eosinophils, with significant inhibition of AHR,48 suggesting treatment duration may not have been sufficient to meet primary study endpoints,48, 49 or eosinophils and other CCR3+ target cells developed resistance (tolerance) to this unbiased antagonist. Furthermore, studies have shown that CCR3 knock-out mice display up to a maximum of 70% reduction in eosinophil recruitment into the airways in an OVA-asthma model.50 In agreement with this finding, our in vivo results from the mouse DRA-asthma model also validate CCR3 as a drug target, since the highest prophylactically administered dose of R321 reached ~70% inhibition of eosinophil recruitment into the airways and strikingly, when delivered therapeutically, completely reversed both blood and lung tissue allergen-induced eosinophilia. The inhibitory effect on blood eosinophil numbers could be explained by either R321 blocking egress of eosinophils from the bone marrow and/or decreasing eosinophil differentiation.51 Further studies are warranted, including in severe chronic murine asthma models. Despite incompletely inhibiting eosinophil recruitment to the airways, R321 delivered at 12 mg/kg fully blocked the development of AHR to methacholine in allergen-challenged mice. Airway hyperresponsiveness is considered a cardinal feature of asthma, and the ability of R321 to completely antagonize the development of AHR in a robust allergic asthma model offers promise of R321 as a therapeutic agent in the treatment of the eosinophilic asthma phenotype. The presence of CCR3 on other non-eosinophil cells relevant to asthma and airway hyperreactivity, such as basophils and subsets of mast cells and Th2-lymphocytes could indicate a wider therapeutic effect of R321 beyond inhibition of eosinophil recruitment and activation.
Drug development thus far has focused on conventional unbiased antagonists, despite growing evidence that chemokine receptors mediate effects both through G protein and non-G protein effectors. An unbiased antagonist of CCR3 acts to inhibit both the activation branch as well as the desensitization and degradation branch of CCR3 signaling following ligand binding. In this scenario, the cell increases its surface receptor density as the basal turnover process continues to produce new receptors.52, 53 Receptor accumulation may potentially explain the limited in vivo success observed with such unbiased antagonists, e.g. in a clinical trial in subjects with eosinophilic asthma,48 as eosinophils may eventually overcome inhibition and become resistant.
Our results demonstrate that the novel R321 peptide effectively inhibits G-protein mediated signal transduction by CCR3, but does not interfere with β-arrestin signaling, receptor internalization and degradation (Supplemental Figure S10). In contrast, small molecule CCR3 antagonists, UCB35625 and SB328437, partially or completely block CCR3 internalization, with this effect becoming more pronounced with longer treatment times. Our observation that R321 by itself appears to promote CCR3 internalization, and without acting as an agonist for chemotaxis, suggests the fate of CCR3 internalized in the presence of R321 is biased towards that of degradation instead of cell activation. As demonstrated by NMR studies, both R321 and CCL11 can bind simultaneously and specifically to the CCR3 receptor, and R321 has two independent binding sites. It is possible that R321 binding at a site different than the eotaxin ligand stabilizes a receptor conformation that induces β-arrestin recruitment, although with a much weaker affinity than observed for the eotaxin ligands. Future structural studies should help clarify the unique inhibitory profile of R321.
Avoiding the pitfall of tolerance development by seeking out novel biased antagonists of the CCR3 signaling cascade may hold significant therapeutic promise for eosinophilic asthma, EoE and other eosinophil-associated diseases. Our results should also prove encouraging in a continuing search for biased antagonists of not only CCR3, but also other chemokine receptors, and point the way toward approaches alternative to classical ligand-displacement compounds.
Supplementary Material
Figure 8. R321 binds CCR3 in plasma membrane in the presence of CCL11.

13C HSQC spectra of 13C-reductively methylated CCR3 positive and CCR3 null membranes were recorded with/without 1 μM CCL11 in the presence/absence of 2 μM R321. Spectral comparisons between (A) CCR3 (CCR3-K-di13CH3)(red) and CCR3 null membranes (blue); (B) CCR3 alone (CCR3-K-di13CH3) (red) and CCR3 + CCL11 (blue); (C) CCR3 alone (CCR3-K-di13CH3) (red) and CCR3 + R321 (blue); (D) CCR3 alone (CCR3-K-di13CH3) (red) and CCR3 + CCL11 and R321 (blue); (E) CCR3 + CCL11 (red) and CCR3 + CCL11 and R321 (blue); and (F) CCR3 + R321 (red) and CCR3 + CCL11 and R321 (blue) show line-broadening and chemical shift changes indicative of binding. Black arrows show significant changes in CCR3-associated signals, but not in the signals that belong to other membrane proteins.
Key Messages.
R321 is a novel biased nanoparticle CCR3 antagonist that inhibits G-protein signaling but not β-arrestin-mediated CCR3 internalization and degradation
R321 blocks eosinophil recruitment into the blood, lungs and airways and prevents airway hyperresponsiveness in a mouse eosinophilic asthma model
Acknowledgments
Funding Sources: This work was supported in part by NIH Grants R21HL118588 (SJA/VG), R01HL126852 (GYP), K01DK106315 (JCM), K24DK100303 (GTF), the LaCache Chair for GI Allergic and Immunologic Diseases (GTF), and a grant from the University of Illinois at Chicago Chancellors Innovation Fund – Proof of Concept program (SJA/VG), and the Intramural Research Program of the National Cancer Institute, NIH (NIT). HA was supported in part by a postdoctoral fellowship from the American Parkinson Disease Association (APDA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. These funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
We would like to thank Dr. Boris Garnier for his help with initial peptide design and characterization, Dr. Jian Du for his help with AML14.3D10-CCR3 cell line maintenance, cryopreservation and experiments, and Dr. S. Tarasov and M. Dyba (Biophysics Resource, SBL, NCI at Frederick) for assistance with mass spectrometry and DLS measurements. We also thank Dr. James Pease, Imperial College, London, for his provision of the 4DE4-CCR3 cell line used in initial studies of CCR3 transmembrane domain peptide inhibition of CCR3/eotaxin-mediated chemotaxis. This work was supported in part by NIH Grants R21HL118588 (SJA/VG), R01HL126852 (GYP), K01DK106315 (JCM), K24DK100303 (GTF), the LaCache Chair for GI Allergic and Immunologic Diseases (GTF), and a grant from the University of Illinois at Chicago Chancellors Innovation Fund – Proof of Concept program (SJA/VG), and the Intramural Research Program of the National Cancer Institute, NIH (NIT). HA was supported in part by a postdoctoral fellowship from the American Parkinson Disease Association (APDA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. These funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Abbreviations
- AHR
Airway Hyperresponsiveness
- BALF
Bronchoalveolar lavage fluid
- CCR3
C-C chemokine receptor 3
- DRA
Dust mite, Ragweed, and Aspergillus
- DLS
dynamic light scattering
- ECP
eosinophil cationic protein
- EoE
Eosinophilic Esophagitis
- GPCR
G-protein-coupled receptor
- NMR
Nuclear Magnetic Resonance
- PAF
platelet-activating factor
Footnotes
Statement of Possible Conflicts of Interest: MG, no conflicts to disclose; KGL, co-inventor of the CCR3 R321 peptide-see below, no other conflicts to disclose; HA, co-inventor of the CCR3 R321 peptide-see below, supported by a postdoctoral fellowship from the American Parkinson Disease Association; BH, co-inventor of the CCR3 R321 peptide-see below; AH, no conflicts to disclose; H-GM, no conflicts to disclose; GYP, supported by NIH grant R01HL126852, no conflicts to disclose; LKR, no conflicts to disclose; JCM, supported by NIH grant K01DK106315, no conflicts to disclose; GTF, supported by NIH grant K24DK100303, LaCache Chair for GI Allergic and Immunologic Diseases, is a co-founder of EnteroTrack, LLC, receives royalties from UpToDate and serves as consultant for Shire; NIT, co-inventor of the CCR3 R321 peptide-see below, supported by the Intramural Research Program of the National Cancer Institute; VG, co-inventor of the CCR3 R321 peptide-see below, supported by NIH Grant R21HL118588 and a grant from the University of Illinois at Chicago Chancellors Innovation Fund–Proof of Concept program; SJA, co-inventor of the CCR3 R321 peptide-see below, supported by NIH Grant R21HL118588 and a grant from the University of Illinois at Chicago Chancellors Innovation Fund – Proof of Concept program, co-founder/co-owner, Chief Scientific Officer and consultant for EnteroTrack, LLC. The IP for the CCR3 R321 peptide nanoparticle biased antagonist was submitted as a US patent entitled “Peptide inhibition of CCR3-mediated diseases and conditions” on Feb. 12, 2016 (PCT/US2016/017714). Co-authors KGL, HA, BH, NIT, VG and SJA are co-inventors; the IP is jointly owned by UIC and NIH/NCI. The PCT application was nationalized (European Patent Office EP16749945.8) on Aug. 12, 2017, and is being filed in Canada.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Busse W, Sedgwick J. Eosinophils in asthma. Annals of Allergy. 1992;68:286–90. [PubMed] [Google Scholar]
- 2.Markowitz JE, Liacouras CA. Eosinophilic esophagitis. Gastroenterology Clinics of North America. 2003;32:949–66. doi: 10.1016/s0889-8553(03)00047-5. [DOI] [PubMed] [Google Scholar]
- 3.Willems LI, Ijzerman AP. Small molecule antagonists for chemokine CCR3 receptors. Medicinal research reviews. 2010;30:778–817. doi: 10.1002/med.20181. [DOI] [PubMed] [Google Scholar]
- 4.Pease JE, Horuk R. Recent progress in the development of antagonists to the chemokine receptors CCR3 and CCR4. Expert opinion on drug discovery. 2014;9:467–83. doi: 10.1517/17460441.2014.897324. [DOI] [PubMed] [Google Scholar]
- 5.Stellato C, Brummet M, Plitt J, Shahabuddin S, Baroody F, Liu M, et al. Expression of the C-C chemokine receptor CCR3 in human airway epithelial cells. Journal of Immunology. 2001;166:1457–61. doi: 10.4049/jimmunol.166.3.1457. [DOI] [PubMed] [Google Scholar]
- 6.Kampen G, Stafford S, Adachi T, Jinquan T, Quan S, Grant J, et al. Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood. 2000;15:1911–7. [PubMed] [Google Scholar]
- 7.Zhu Y, Bertics P. Chemoattractant-induced signaling via the Ras-ERK and PI3K-Akt networks, along with leukotriene C4 release, is dependent on the tyrosine kinase Lyn in IL-5- and IL-3-primed human blood eosinophils. Jornal of Immunology. 2011;186:516–26. doi: 10.4049/jimmunol.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmermann N, Conkright JJ, Rothenberg ME. CC chemokine receptor-3 undergoes prolonged ligand-induced internalization. The Journal of biological chemistry. 1999;274:12611–8. doi: 10.1074/jbc.274.18.12611. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann N, Rothenberg ME. Receptor internalization is required for eotaxin-induced responses in human eosinophils. The Journal of allergy and clinical immunology. 2003;111:97–105. doi: 10.1067/mai.2003.3. [DOI] [PubMed] [Google Scholar]
- 10.Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, et al. The Chemokine Receptor CXCR3 Is Degraded following Internalization and Is Replenished at the Cell Surface by De Novo Synthesis of Receptor. The Journal of Immunology. 2008;180:6713–24. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulkerson PC, Fischetti CA, McBride ML, Hassman LM, Hogan SP, Rothenberg ME. A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16418–23. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ying S, Robinson DS, Meng Q, Rottman J, Kennedy R, Ringler DJ, et al. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. Association with airway hyperresponsiveness and predominant co-localization of eotaxin mRNA to bronchial epithelial and endothelial cells. European journal of immunology. 1997;27:3507–16. doi: 10.1002/eji.1830271252. [DOI] [PubMed] [Google Scholar]
- 13.Uy GL, Retting MP, Motabi IH, McFarland K, Trinkaus kM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–24. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sison EAR, Magoon D, Li L, Annesley CE, Romagnoli B, Douglas GJ, et al. POL5551, a novel and potent CXCR4 antagonist, enhances sensitivity to chemotherapy in pediatric ALL. Oncotarget. 2015;6:30902–18. doi: 10.18632/oncotarget.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, et al. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell. 2017;171:1165–75. doi: 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daugherty BL, Siciliano SJ, DeMartino JA, Malkowitz L, Sirotina A, Springer MS. Cloning, Expression, and Characterization of the Human Eosinophil Eotaxin Receptor. Journal of Experimental Medicine. 1996;183:2349–54. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goplen N, Karim MZ, Liang Q, Gorska MM, Rozario S, Guo L, et al. Combined sensitization of mice to extracts of dust mites, ragweed and aspergillus breaks through tolerance and establishes chronic features of asthma in mice. Journal of Allergy and Clinical Immunology. 2009;123:925–32. doi: 10.1016/j.jaci.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, Justice JP, et al. Extensive Eosinophil Degranulation and Peroxidase-Mediated Oxidation of Airway Proteins Do Not Occur in a Mouse Ovalbumin-Challenge Model of Pulmonary Inflammation. The Journal of Immunology. 2001;167:1672–82. doi: 10.4049/jimmunol.167.3.1672. [DOI] [PubMed] [Google Scholar]
- 19.Tarasov SG, Gaponenko V, Howard OM, Chen Y, Oppenheim JJ, Dyba MA, et al. Structural plasticity of a transmembrane peptide allows self-assembly into biologically active nanoparticles. Proc Natl Acad Sci U S A. 108:9798–803. doi: 10.1073/pnas.1014598108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y, Chen Y, Tarasova NI, Gaponenko V. The structure of monomeric components of self-assembling CXCR4 antagonists determines the architecture of resulting nanostructures. Nanotechnology. 2011;22:505101. doi: 10.1088/0957-4484/22/50/505101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujisawa T, Kato Y, Nagase H, Atsuta J, Terada A, Iguchi K, et al. Chemokines induce eosinophil degranulation through CCR-3. Journal of Allergy and Clinical Immunology. 2000;106:507–13. doi: 10.1067/mai.2000.108311. [DOI] [PubMed] [Google Scholar]
- 22.Kumari P, Srivastava A, Banerjee R, Ghosh E, Gupta P, Ranjan R, et al. Functional competence of a partially engaged GPCR–β-arrestin complex. Nature Communications. 2016;7:13416. doi: 10.1038/ncomms13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. Journal of Biological Chemistry. 2004;279:35518–25. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 24.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. β-Arrestin-dependent, G Protein-independent ERK1/2 Activation by the β2 Adrenergic Receptor. The Journal of biological chemistry. 2006;281:1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 25.Lefkowitz RJ, Shenoy SK. Transduction of Receptor Signals by s-Arrestins. Science. 2005;308:512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. β-arrestin-but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–32. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wise EL, Bonner KT, Williams TJ, Pease JE. A single nucleotide polymorphism in the CCR3 gene ablates receptor export to the plasma membrane. The Journal of allergy and clinical immunology. 2010;126:150–7.e2. doi: 10.1016/j.jaci.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Chavan TS, Abraham S, Gaponenko V. Application of reductive (1)(3)C-methylation of lysines to enhance the sensitivity of conventional NMR methods. Molecules. 2013;18:7103–19. doi: 10.3390/molecules18067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abraham SJ, Hoheisel S, Gaponenko V. Detection of protein-ligand interactions by NMR using reductive methylation of lysine residues. J Biomol NMR. 2008;42:143–8. doi: 10.1007/s10858-008-9274-y. [DOI] [PubMed] [Google Scholar]
- 30.Abraham SJ, Kobayashi T, Solaro RJ, Gaponenko V. Differences in lysine pKa values may be used to improve NMR signal dispersion in reductively methylated proteins. J Biomol NMR. 2009;43:239–46. doi: 10.1007/s10858-009-9306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tripathi A, Vana PG, Chavan TS, Brueggemann LI, Byron KL, Tarasova NI, et al. Heteromerization of chemokine (C-X-C motif) receptor 4 with alpha1A/B-adrenergic receptors controls alpha1-adrenergic receptor function. Proc Natl Acad Sci U S A. 2015;112:E1659–68. doi: 10.1073/pnas.1417564112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, et al. Propagation of conformational changes during mu-opioid receptor activation. Nature. 2015;524:375–8. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–12. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris J, Chess R. Effect of pegylation on pharmaceuticals. Nature reviews Drug discovery. 2003;2:214–21. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 35.Sabroe I, Peck MJ, Keulen BJV, Jorritsma A, Simmons G, Clapham PR, et al. A Small Molecule Antagonist of Chemokine Receptors CCR1 and CCR3. Potent Inhibition of Eosinophil Function and CCR3-mediated HIV-1 Entry. The Journal of Biological Chemistry. 2000;275:25985–92. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- 36.Corbisier J, Huszagh A, Gales C, Parmentier M, Springael J-Y. Partial Agonist and Biased Signaling Properties of the Synthetic Enantiomers J113863/UCB35625 at Chemokine Receptors CCR2 and CCR5. The Journal of Biological Chemistry. 2017;292:575–84. doi: 10.1074/jbc.M116.757559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White John R, Lee Judithann M, Dede Kimberly, Imburgia Christina S, Jurewicz Anthony J, Chan George, et al. Identification of potent, selective non-peptide CCR3 antagonist that inhibits eotaxin-, eotaxin-2 and MCP-4 induced eosinophil migration. The Journal of biological chemistry. 2000;275:36626–31. doi: 10.1074/jbc.M006613200. [DOI] [PubMed] [Google Scholar]
- 38.Mori A, Ogawa K, Someya K, Kunori Y, Nagakubo D, Yoshie O, et al. Selective suppression of Th2-mediated airway eosinophil infiltration by low-molecular weight CCR3 antagonists. International Immunology. 2007;19:913–21. doi: 10.1093/intimm/dxm049. [DOI] [PubMed] [Google Scholar]
- 39.Takeda A, Baffi JZ, Kleinman ME, Cho WG, Nozaki M, Yamada K, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–30. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z, et al. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-κB-mediated MMP-9 upregulation. Stem cells (Dayton, Ohio) 2012;30:2309–19. doi: 10.1002/stem.1194. [DOI] [PubMed] [Google Scholar]
- 41.Agarwal M, He C, Siddiqui J, Wei JT, Macoska JA. CCL11 (eotaxin-1): a new diagnostic serum marker for prostate cancer. The Prostate. 2013;73:573–81. doi: 10.1002/pros.22597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu F, Liu P, Li J, Zhang Y. Eotaxin-1 promotes prostate cancer cell invasion via activation of the CCR3-ERK pathway and upregulation of MMP-3 expression. Oncology reports. 2014;31:2049–54. doi: 10.3892/or.2014.3060. [DOI] [PubMed] [Google Scholar]
- 43.Diny NL, Hou X, Barin JG, Chen G, Talor MV, Schaub J, et al. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur J Immunol. 2016 doi: 10.1002/eji.201646557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber AK, Giles DA, Segal BM, Irani DN. An emerging role for eotaxins in neurodegenerative disease. Clin Immunol. 2016 doi: 10.1016/j.clim.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Johrer K, Zelle-Rieser C, Perathoner A, Moser P, Hager M, Ramoner R, et al. Up-regulation of functional chemokine receptor CCR3 in human renal cell carcinoma. Clinical Cancer Research. 2005;11:2459–65. doi: 10.1158/1078-0432.CCR-04-0405. [DOI] [PubMed] [Google Scholar]
- 46.Masterson JC, McNamee EN, Jedlicka P, Fillon S, Ruybal J, Hosford L, et al. CCR3 Blockade Attenuates Eosinophilic Ileitis and Associated Remodeling. Am J Pathol. 2011;179:2302–14. doi: 10.1016/j.ajpath.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian M, Chen L, Ma L, Wang D, Shao B, Wu J, et al. Expression and prognostic significance of CCL11/CCR3 in glioblastoma. Oncotarget. 2016;7:32617–27. doi: 10.18632/oncotarget.8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neighbour H, Boulet L-P, Lemiere C, Sehmi R, Leigh R, Sousa AR, et al. Safety and efficacy of an oral CCR3 antagonist in patients with asthma and eosinophilic bronchitis: a randomized, placebo-controlled clinical trial. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2014;44:508–16. doi: 10.1111/cea.12244. [DOI] [PubMed] [Google Scholar]
- 49.Thomson NC. Novel therapies targeting eosinophilic inflammation in asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2014;44:462–8. doi: 10.1111/cea.12268. [DOI] [PubMed] [Google Scholar]
- 50.Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-garawi A, et al. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci U S A. 2001;99:1479–84. doi: 10.1073/pnas.261462598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamkhioued B, Abdelilah SG, Hamid Q, Mansour N, Delespesse G, Renzi PM. The CCR3 receptor is involved in eosinophil differentiation and is up-regulated by Th2 cytokines in CD34+ progenitor cells. Journal of Immunology. 2003;171:537–47. doi: 10.4049/jimmunol.170.1.537. [DOI] [PubMed] [Google Scholar]
- 52.Paradisa JS, Lya S, Blondel-Tepaza E, Galana JA, Beautraita A, Scottd MGH, et al. Receptor sequestration in response to β-arrestin-2 phosphorylation by ERK1/2 governs steady-state levels of GPCR cell-surface expression. Proc Natl Acad Sci U S A. 2015;112:E5160–E8. doi: 10.1073/pnas.1508836112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allouche S, Noble F, Marie N. Opioid receptor desensitization: mechanisms and its link to tolerance. Frontiers in Pharmacology. 2014;5:1–6. doi: 10.3389/fphar.2014.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.