

Abstract
para-Aminosalicylic acid (PAS) is a second-line anti-tubercular drug that is used for the treatment of drug-resistant tuberculosis (TB). PAS efficacy in the treatment of TB is limited by its lower potency against Mycobacterium tuberculosis relative to many other drugs in the TB treatment arsenal. It is known that intrinsic metabolites, such as, para-aminobenzoic acid (PABA) and methionine, antagonize PAS and structurally related anti-folate drugs. While the basis for PABA-mediated antagonism of anti-folates is understood, the mechanism for methionine-based antagonism remains undefined. In the present study, we used both targeted and untargeted approaches to identify factors associated with methionine-mediated antagonism of PAS activity. We found that synthesis of folate precursors as well as a putative amino acid transporter, designated MetM, play crucial roles in this process. Disruption of metM by transposon insertion resulted in a ≥30-fold decrease in uptake of methionine in M. bovis BCG, indicating that metM is the major facilitator of methionine transport. We also discovered that intracellular biotin confers intrinsic PAS resistance in a methionine-independent manner. Collectively, our results demonstrate that methionine-mediated antagonism of anti-folate drugs occurs through sustained production of folate precursors.
Keywords: Mycobacterium tuberculosis, anti-folate drug, para-aminosalicylic acid, methionine, para-aminobenzoic acid, biotin, antagonism, methionine transport
Introduction
Mycobacterium tuberculosis is responsible for ~10.4 million new cases of active tuberculosis (TB) and 1.3 million deaths annually (World Health Organization, 2017). While TB chemotherapeutic intervention is highly successful in curing drug-susceptible TB infections, therapy is challenging, in part, because it requires a minimum of 6 months of treatment with drugs associated with adverse reactions. In addition, the emergence of drug-resistant strains of M. tuberculosis has dramatically increased the complexity and cost of TB treatment (Gandhi et al., 2006; Gehre et al., 2016). Therefore, the development of more efficacious TB chemotherapy regimens is imperative to improve treatment outcomes.
para-Aminosalicylic acid (PAS) was the second drug to be developed exclusively for TB chemotherapy (Lehmann, 1946). Although PAS was a cornerstone agent of early multidrug TB therapies, the introduction of more potent anti-tubercular agents into TB treatment regimens greatly diminished its usage (Minato et al., 2015). Emergence of M. tuberculosis strains with resistance to first-line anti-tubercular agents led to the resurgence of PAS as an second-line drug to treat infections that failed to respond to standard short-course therapy (Donald and Diacon, 2015). However, compared to many other anti-tubercular drugs, PAS is less potent and is associated with a high rate of gastrointestinal distress which limits its use to the treatment of multi-drug resistant TB for which there are few other treatment options (Zumla et al., 2013). Thus, it is important to develop novel strategies to enhance PAS potency, limit adverse reactions and improve treatment success rates.
Until recently, little was known regarding the mode of action of PAS. PAS is a selective antimetabolite of the M. tuberculosis folate metabolic pathway acting as a structural analog of the folate precursor para-aminobenzoic acid (PABA) (Chakraborty et al., 2013; Minato et al., 2015). PAS is sequentially converted to 2′-hydroxy-7,8-dihydropteroate and 2′-hydroxy-7,8-dihydrofolate by enzymes in the M. tuberculosis folate metabolic pathway (Figure 1). 2′-hydroxy-7,8-dihydrofolate has been shown to potently inhibit M. tuberculosis dihydrofolate reductase (DHFR), the final step in synthesis of tetrahydrofolate (Zheng et al., 2013; Zhao et al., 2014; Minato et al., 2015; Dawadi et al., 2017). Since PAS and PABA are comparable substrates for the folate biosynthetic pathway, supplementation of M. tuberculosis cultures with PABA antagonizes the inhibitory activity of PAS by outcompeting for ligation to 6-pyrophosphomethyl-7,8-dihydropterin (DHPPP) by dihydropteroate synthase (DHPS) (Youmans et al., 1947). We previously reported that intracellular PABA mediates intrinsic resistance to PAS in M. tuberculosis, and disruption of synthesis of this critical intermediate in folate biosynthesis can potentiate antifolate action, including that of sulfa drugs (Thiede et al., 2016).
Figure 1.

Previously proposed models for PAS activation and methionine-mediated PAS inactivation. As indicated on the left, PAS is an analog of PABA (shown in a dotted box) and is a prodrug that is activated by the M. tuberculosis folate biosynthetic pathway. PAS is incorporated in lieu of PABA by FolP1 and glutamylated by FolC to form the antimetabolite HDHF which inhibits DfrA activity (indicated as red blunted arrows) (Zheng et al., 2013; Zhao et al., 2014; Minato et al., 2015; Dawadi et al., 2017). Previous work has identified N-methyl and N,N-dimethyl PAS species (methyl groups indicated with dotted red boxes) in metabolite extracts from M. tuberculosis treated with PAS (Chakraborty et al., 2013). As N,N-dimethylation of PAS prevents incorporation by FolP1, the resulting metabolite is inactive (shown on the right). This activity is presumed to be dependent upon an as of yet unidentified SAM-dependent methyltransferase(s). Supplementation with methionine may increase SAM pools, which could be utilized by the methyltransferase(s) to inactivate PAS, thereby conferring resistance. PAS, para-aminosalicylic acid; PABA, para-aminobenzoic acid; HDHP, 2′-hydroxy-7,8-dihydropteroate; HDHF, 2′-hydroxy-7,8-dihydrofolate; FolP1, dihydropteroate synthase; FolC, dihydrofolate synthase; DfrA, dihydrofolate reductase; MT, methyltransferase; SAM, S-adenosyl methionine; SAH, S-adenosyl homocysteine.
Methionine is a potent antagonist of PAS in M. tuberculosis (Hedgecock, 1956), yet, the basis for this antagonism remains poorly understood. Because disruption of the folate pathway in M. tuberculosis results in depletion of metabolites within multiple essential folate-dependent pathways (Chakraborty et al., 2013; Nixon et al., 2014), supplementation with methionine alone is not expected to recover loss of folate pathway integrity. A recent study showed that PAS can be converted to N-methyl and N,N-dimethyl PAS species within M. tuberculosis cells (Figure 1; Chakraborty et al., 2013). N-methyl-PAS retains activity against M. tuberculosis, while N,N-dimethyl-PAS shows no anti-tubercular activity since the resulting tertiary amine is incapable of nucleophilically reacting with DHPPP during the first step of PAS bioactivation (Figure 1). Since addition of methionine can potentially enhance the ability of M. tuberculosis to methylate PAS by increasing S-adenosylmethionine (SAM) abundance, it is possible that methionine promotes inactivation of PAS through N,N-dimethylation by an unidentified methyltransferase.
In the present study we screened ~10,000 independent Mycobacterium bovis BCG transposon insertion mutants (BCG::himar1) to identify genetic determinants associated with methionine-mediated PAS antagonism. In parallel to analysis of BCG::himar1 mutants, we characterized factors that affect PAS susceptibility in M. tuberculosis for their involvement in methionine-mediated PAS antagonism. Our findings reveal the importance of folate precursor biosynthesis and methionine transport in methionine-mediated PAS antagonism.
Materials and methods
Chemical reagents
All chemical reagents except for (S)-2-Amino-4-(methylthio-d3) butanoic Acid [L-methionine-(methyl-d3)] (Toronto Research Chemicals) and 2'-hydroxy-pteroate (pterin-PAS) were purchased from Sigma-Aldrich. Pterin-PAS was synthesized by Drs. Richard Lee and Ying Zhao at St Jude Children's Research Hospital by using a similar synthesis method reported elsewhere (Zhao et al., 2016).
Bacterial strains and growth conditions
Bacterial strains utilized in this study are described in Table 1. Unless otherwise indicated, Mycobacterial strains were grown in Middlebrook 7H9 liquid medium supplemented with tyloxapol (0.05% vol/vol) or on Middlebrook 7H10 agar plates at 37°C. For M. bovis BCG and M. tuberculosis H37Ra, oleate-albumin-dextrose-catalase (OADC; Becton Dickinson 10% vol/vol), and glycerol (0.2% vol/vol) were supplemented to Middlebrook 7H9 and Middlebrook 7H10. For Mycobacterium smegmatis mc2155, Middlebrook 7H9, and Middlebrook 7H10 were amended with dextrose (0.2% vol/vol). Escherichia coli DH5α λpir was grown in LB broth or on LB agar plate. When necessary, kanamycin or hygromycin were added to media at 50 and 150 μg/ml respectively for selection of mycobacterial and E. coli strains.
Table 1.
List of bacterial strains used in this study.
| Strain | Relevant features | Reference or source |
|---|---|---|
| M. bovis BCG Pasteur | Pasteur strain of spontaneously attenuated variant of M. bovis | Brosch et al., 2007 |
| M. bovis BCG Pasteur metM::himar1 | Transposon insertion mutant with disruption in BCG_3282c (metM) | This work |
| M. bovis BCG Pasteur metM::himar1/ pUMN002hyg::metM | Complemented BCG_3282c (metM) transposon insertion mutant | This work |
| M. bovis BCG Pasteur bioB::himar1 | Transposon insertion mutant with disruption in bioB | This work |
| M. tuberculosis H37Ra | Spontaneously attenuated variant of M. tuberculosis strain H37Rv | Steenken et al., 1934 |
| M. tuberculosis H37Ra ΔpabB | H37Ra strain with the pabB coding sequence replaced by a hygromycin resistance cassette | Thiede et al., 2016 |
| M. smegmatis mc2155 | Used for propagation of himar1 mycobacteriophage | Snapper et al., 1990 |
| E. coli DH5αλpir | Utilized to replicate self-ligated himar1 plasmids for determination of transposon insertion site | Taylor et al., 1993 |
For sulfur utilization studies, a modified sulfate-free Sautons medium (Allen, 1998) was prepared with all inorganic sulfate salts (MgSO4 and ZnSO4) replaced with inorganic chloride salts (MgCl and ZnCl) keeping the concentrations of Mg2+ and Zn2+ ions the same. For the characterizations of the biotin auxotroph mutant, biotin-free 7H9 medium was prepared. The biotin-auxotrophic strain, M. bovis BCG bioB::himar1, was maintained in the biotin-free 7H9 medium supplemented with 0.5 μg/ml biotin. The PABA-auxotrophic strain H37Ra ΔpabB was maintained in 7H9 medium supplemented with PABA (10 ng/ml). For experiments involving PABA limitation, PABA-free 7H9 medium was prepared in glassware that was baked at 300°C for 1 h to remove residual PABA before use.
M. bovis BCG::himar1 mutant library construction and screening
The phAE180 mycobacteriophage containing a mariner transposable element, himar1, with a kanamycin resistance cassette was used to transduce M. bovis BCG creating a library of BCG::himar1 mutants as described previously (Rubin et al., 1999; Kriakov et al., 2003). Transduced cells were plated onto 7H10 agar containing kanamycin and 10 μg/ml methionine. ~10,000 mutant strains were screened by picking and patching onto 7H10 agar supplemented with methionine (Met plates) and onto 7H10 agar plates additionally amended with 5 μg/ml PAS (Met-PAS plates). Mutant strains that grew on the Met plates, but were inhibited for growth the Met-PAS plates, were selected for secondary screening following the same protocol. PAS susceptibility was assessed for strains that passed the secondary screen. himar1 insertion sites were determined as previously described (Rubin et al., 1999). Briefly, extracted genomic DNA was digested with BssHII and self-ligated to produce circular DNAs. The circularized DNAs that contained ori6K from a part of himar1 transposon were used to transform E. coli DH5αλpir. Plasmids were purified from the transformants. Sequences of genomic DNA adjacent to the 3′ end of the himar1 transposon insertion site were determined by Sanger sequencing (performed by Eurofins) using the KanSeq_Rev (5′-GCATCGCCTTCTATCGCCTTC-3′) primer (Baughn et al., 2010). Insertion site locations were determined by aligning the resulting sequence files with the M. bovis BCG Pasteur genome sequence (GenBank accession number NC_008796).
Construction of metM complemented strain
Complementation of the metM::himar1 strain was accomplished by transforming the mutant with a replicative mycobacterial expression vector constitutively expressing metM under the control of the Psmyc promoter (Ehrt et al., 2005). Since the himar1 transposable element encodes a kanamycin resistance gene, a hygromycin resistant version of the replicative mycobacterial expression vector pUMN002 was constructed (Peterson et al., 2015). The hygromycin resistance cassette was amplified from the plasmid p0004S by PCR with primers p0004s_Hygro+P_F and p0004s_Hygro+P_R (Supplementary Table S1). The entire plasmid except for the kanamycin resistance cassette of pUMN002 was amplified by PCR using primers pTIC6a_F and pTIC6a_R. The linear vector and hygromycin cassette were digested with either Sbf I or AflII and ligated to produce pUMN002hyg. The metM coding region (BCG_3282c) was amplified from M. bovis BCG genomic DNA by PCR with primers BCG3282c_For and BCG3282c_Rev (Supplementary Table S1), digested with NheI and EcoRI, and ligated into pUMN002hyg. The E. coli transformants of the resultant plasmid pUMN002hyg::metM were selected on LB plates containing hygromycin. After verification of the plasmid, the M. bovis BCG metM::himar1 disruption strain was transformed with the pUMN002hyg::metM by electroporation and selected for on supplemented 7H10 agar plates containing hygromycin and kanamycin.
Determination of minimum inhibitory concentrations
The minimum inhibitory concentrations (MIC) of anti-tubercular compounds were determined as previously described (Dillon et al., 2014). Briefly, for determination of the MIC in liquid culture, 2-fold dilution series of drugs in 7H9 medium were prepared. Logarithmically growing Mycobacterium strains were inoculated into the drug-containing 7H9 medium in 30-ml square bottles (Nalgene) to an optical density (OD600) of 0.01. OD600 were measured after shaking (100 rpm) at 37°C for 14 days. The liquid MIC90 was defined as the minimum concentration of drug required to inhibit at least 90% of growth relative to growth in the no-drug control cultures. For determination of the agar plate MIC, logarithmically growing M. bovis BCG strains were serially-diluted and inoculated onto 7H10 agar plates containing drug in 2-fold dilution series. The agar plate MIC was determined by visually inspecting growth relative to growth on the no-drug control plates after grown at 37°C for 21 days. All anti-tubercular compounds employed in this study were dissolved in DMSO. The highest concentration of DMSO in the growth media was 2.5%.
Analysis of growth kinetics
Logarithmically growing Mycobacterium strains were washed twice in an equal volume of fresh medium. Cells were diluted to an OD600 of 0.01 in 30-ml square bottles (Nalgene) and supplements with or without drug were added at the described concentrations. Cultures were shaken (100 rpm) and OD600 were measured at various time points over a 14-day time-course.
Methionine utilization assays
M. bovis BCG strains were grown to mid-log phase in 7H9 broth and washed twice with sulfate-free Sautons medium. Resuspended cells were diluted to an OD600 of 0.01 in sulfate-free Sautons medium. Cultures were then incubated for 5 days to exhaust remaining sulfur. Exhausted cells were aliquoted into 30-ml square bottles (Nalgene) and sulfur-containing metabolites were added at the given concentrations. Cultures were incubated at 37°C and shaken (100 rpm). The fold-change in OD600 (as a ratio of the final OD600/initial OD600) was assessed following 1 week of incubation after the addition of metabolites.
Methionine uptake assays
M. bovis BCG strains were grown until mid-log phase (OD600 0.4–0.6). Cells were then diluted to OD600 0.3 and divided into three 40 ml assay mixtures for each condition tested. Each assay mixture was incubated at 37°C for 2 h with shaking (100 rpm) in the presence or absence of L-methionine-(methyl-d3) and/or carbonylcyanide m-chlorophenylhydrazone (CCCP). L-methionine-(methyl-d3) and CCCP were added at 50 μM and 100 μM, respectively. Following 2 h of incubation, each assay mixture culture was pelleted by centrifugation at 3000 g for 10 min and then washed three times with ice-cold phosphate buffered saline (pH 7.4). Metabolites were extracted by using 1 ml of acetonitrile:methanol:water (40:40:20) extraction buffer as previously described (Chakraborty et al., 2013). Following extraction, 350 μL of each sample was concentrated in vacuo and reconstituted in 90 μL of 10 mM ammonium formate (80:20 MeCN:H2O) containing 1 μM internal standard (N-phenyl glycine). Quantitation was performed using a Shimadzu UFLC-XR system and an AB Sciex QTRAP 5500 mass spectrometer. L-methionine-(methyl-d3) (Toronto Research Chemicals) was used as an authentic standard. N-phenyl glycine (Chem-Impex) was used as an internal standard. LC was performed using a SeQuant ZIC-pHILIC column (4.6 × 150 mm, 5 μm particle size; Millipore Sigma). Mobile phase A was 10 mM ammonium formate in 20:80 MeCN:H2O, and mobile phase B was 10 mM ammonium formate in 80:20 MeCN:H2O. The LC method was as follows: 100% B from 0.0 to 0.10 min, then decreasing from 100% B to 82.5% B from 0.10 to 22.00 min, followed by a decrease from 82.5% B to 0% B from 22.00 to 23.00 min. 0% B was maintained from 23.00 to 24.00 min, then increased to 100% B from 24.00 to 25.00 min. The column was then re-equillibrated at 100% B from 25.00 to 30.00 min, for a total run time of 30 min. The flow rate was 600 μL/min, the injection volume was 10 μL, and the column oven was maintained at 35°C. Analytes were monitored by positive mode MS via multiple reaction monitoring (MRM). MS settings were optimized via infusion of analytes in 10 mM ammonium formate (80:20 MeCN:H2O); L-methionine-(methyl-d3) was infused at 5 μM and 2 μL/min flow rate, and N-phenyl glycine was infused at 1 μM and 2 μL/min. Mass transitions monitored and MS conditions are summarized in Supplementary Table S2. Analyte and internal standard peak areas were quantitated using MultiQuant software (Version 2.0.2); analyte peak areas were normalized to internal standard peak areas. A standard curve was prepared for methionine-(methyl-d3) with 2-fold serial dilutions from 25.00 to 0.39 μM and used to determine concentrations of methionine-(methyl-d3) in samples. Zero concentration standards were also prepared and used to determine the lower limits of detection and quantitation, which were defined as 3:1 and 10:1 signal-to-noise ratios, respectively. Concentrations of methionine-(methyl-d3) were normalized to the protein concentration of each sample as determined by using the BCA protein assay kit (Pierce Biotechnology).
Results
Identification of M. bovis BCG genes involved in methionine-mediated antagonism of PAS
A library of M. bovis BCG transposon insertion mutant strains was constructed using the phAE180 mycobacteriophage containing a mariner-family transposable element. To identify genes associated with methionine-mediated PAS antagonism, ~10,000 BCG::himar1 mutants were screened following the approach outlined in Figure 2. Determination of the PAS MIC on 7H10 agar plates confirmed that 0.25 μg/ml was sufficient to fully inhibit growth of the M. bovis BCG parental strain. Screening was then undertaken on 7H10 agar plates containing 10 μg/ml methionine and 5 μg/ml PAS (Met-PAS plate). Growth of M. bovis BCG on Met-PAS plates was identical to growth seen on control 7H10 plates, which confirmed methionine-mediated PAS antagonism.
Figure 2.
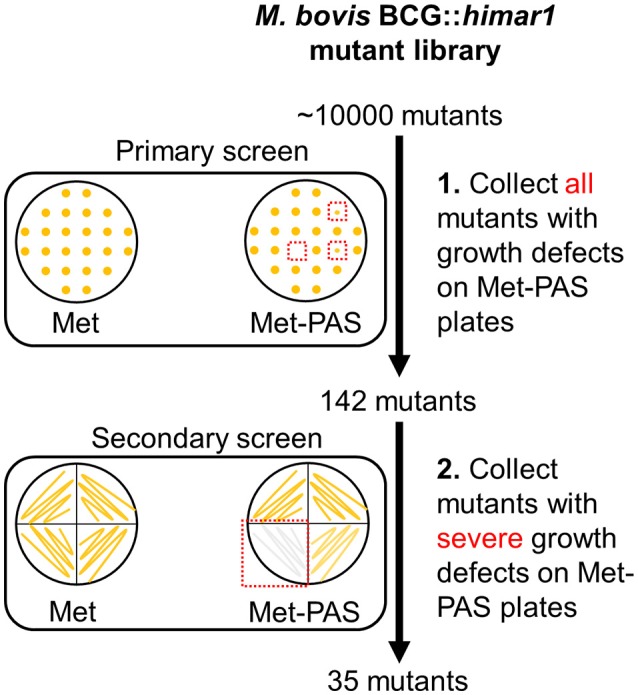
Schematic representation of genome-wide transposon mutagenesis of M. bovis BCG and Met-PAS screening method. M. bovis BCG::himar1 mutants (~10,000 mutants) were patched onto Met and Met-PAS plates. Clones with observable growth defects on Met-PAS plates were subjected to secondary screening. These 35 mutants that passed the secondary screening were collected and insertion site locations were determined.
BCG::himar1 insertion mutants which exhibited observable growth inhibition on the Met-PAS plates in comparison to the growth on 7H10 agar plates containing 10 μg/ml methionine (Met plate) were isolated. We then identified the himar1 insertion sites within the 35 BCG::himar1 mutants that had reproducible growth defects on Met-PAS plates compared to the growth on Met plates (Figure 2 and Table 2). Among these mutants, one strain with a himar1 insertion located within BCG_3282c, encoding a putative amino acid/polyamine/organocation (APC) superfamily transporter of the CAT (cationic amino acid transporter) family (TC 2.A.3.3) (Jack et al., 2000; Elbourne et al., 2017), showed the most severe growth defect on Met-PAS plates suggesting BCG_3282c plays a major role in methionine-mediated antagonism of PAS. We also assessed the susceptibility of each mutant strain to PAS by measuring PAS MICs on 7H10 agar plates (Table 2). We observed the BCG_3282c mutant possessed wild-type PAS susceptibility suggesting this mutation is associated exclusively with methionine-mediated PAS antagonism.
Table 2.
Sequence-validated gene insertions that affect PAS susceptibility in the presence or absence of antagonistic concentrations of methionine.
| Disrupted gene (M. tuberculosis H37Rv homolog) | Predicted function | Increased PAS susceptibilitya | Growth on PAS-Metb |
|---|---|---|---|
| M. bovis BCG | Wild-type | no | ++++ |
| BCG_3282c (Rv3253c) | Amino acid permease | no | – |
| ftsH (ftsH) | Membrane-bound protease (insertion located near upstream folate biosynthesis operon) | yes | + |
| bioB (bioB) | Biotin synthase involved in biotin biosynthesis | yes | + |
| metB (metB) | Cystathionine gamma-synthase involved in methionine biosynthesis | yes | + |
| BCG_1906c (Rv1870c) | Unknown | yes | + |
| cysQ (cysQ) | Sulfate assimilation pathway regulator | no | + |
| accD2 (accD2) | Acetyl-CoA carboxylase involved in mycolic acid biosynthesis | no | + |
| BCG_1988c-1989c (Rv1949c-Rv1950c) | Unknown (conserved hypotheticals) (insertion is located within the intergenic region) | no | + |
| PPE11(PPE11) | Unknown (PPE family protein) | no | + |
| arsC (arsC) | Protein involved in arsenate resistance | no | + |
| mmpL7 (mmpL7) | Phthiocerol dimycocerosate transporter | no | + |
| BCG_2043c (Rv2024c) | Unknown | no | + |
| BCG_3116 (Rv3091) | Unknown | no | + |
| BCG_0914c (Rv0862c) | Unknown | no | + |
| papA2 (papA2) | Protein involved in sulfolipid-1 biosynthesis | no | + |
| BCG_2017 (Rv2000) | Unknown | no | + |
| BCG_1401 (Rv1339) | Unknown | no | + |
| BCG_1635 (Rv1597) | Unknown | no | + |
| BCG_1082 (Rv1026) | Protein involved in polyphosphate regulation | no | + |
| BCG_2497c (Rv2477c) | Macrolide exporter | no | ++ |
| BCG_3185c (Rv3161c) | Dioxygenase | no | ++ |
| BCG_0233 (Rv0196) | Transcriptional regulator | no | ++ |
| BCG_3873 (Rv3811) | Cell surface protein involved in virulence | no | ++ |
| BCG_1897 (Rv1861) | Conserved transmembrane protein | no | ++ |
| BCG_2026 (vapB15) | Antitoxin component of an toxin-antitoxin operon with BCG_2027 (vapC15) | no | ++ |
| kgtP (kgtP) | Ketoglutarate transport protein | no | ++ |
| mbtJ (mbtJ) | Protein involved in mycobactin biosynthesis | no | ++ |
| BCG_1492 (Rv1431) | Conserved membrane protein | no | ++ |
| thiG (thiG) | Protein involved in thiamine biosynthesis | no | ++ |
| BCG_3826c (3767c) | SAM-dependent methyltransferase which may be involved in Polyketide synthesis | no | ++ |
| BCG_0424 (Rv0386) | Transcriptional regulator | no | ++ |
| upp-sapM (upp-sapM) | Proteins involved in pyrimidine the salvage pathway and arresting phagosomal maturation, respectively (insertion is located within the intergenic region of these two genes) | no | ++ |
| fadD2 (fadD2) | Fatty-acid CoA Ligase | no | ++ |
| PPE33a (PPE33a) | Unknown (PPE family protein) | no | ++ |
| esxJ (esxJ) | Unknown | no | ++ |
PAS susceptibility was assessed by determining the minimum concentration (MIC) of drug required to inhibit growth on 7H10 agar plates. The M. bovis BCG PAS MIC was found to be 0.25 μg/ml.
Growth of M. bovis BCG transposants on methionine (10 μg/ml) only plates compared visually to plates containing methionine (10 μg/ml) and PAS (5 μg/ml) to screen for transposon insertion mutants susceptible to PAS-methionine treatment. (++++) represents no growth difference between PAS-methionine and methionine only plates (WT BCG). (++) represents ~50% impairment in growth. (+) represents 25% or less growth. (–) represents no growth observed.
Although most mutant strains that were analyzed showed a similar level of PAS tolerance as the parent M. bovis BCG, four mutants (with transposon insertions in bioB, ftsH, metB, and BCG_1906c) were found to be more susceptible to PAS in the absence of methionine, indicating that the disrupted genes may be involved in intrinsic resistance to PAS (Table 2).
MetM is essential for methionine-mediated antagonism of PAS
Based upon the observation that methionine failed to antagonize PAS activity in the BCG_3282c mutant, we hypothesized that BCG_3282c is the primary methionine transporter. We designated BCG_3282c herein as metM (methionine transporter for Mycobacterium) and further characterized the function of this gene. The himar1 insertion was located near the 5′ end of the coding region for metM resulting in a 401 residue truncation of the 495 residue coding sequence, suggesting functional gene disruption by himar1 insertion. Similar to the majority of transporters within the APC superfamily, MetM is predicted to possess 12 transmembrane spanning α-helices (Elbourne et al., 2017). metM is also highly conserved in the Mycobacterium genus, sharing 100% sequence identity with numerous M. tuberculosis complex organisms including Rv3253c, an ortholog from the virulent reference strain H37Rv. However, no close orthologs of metM have been structurally or functionally characterized thus far.
To confirm whether metM disruption altered methionine antagonism or susceptibility to antimycobacterial drugs, susceptibility testing was conducted in liquid medium (Figure 3A and Supplementary Table S3). Growth of both wild type M. bovis BCG and the metM::himar1 strain was fully inhibited by 5 μg/ml PAS. Addition of methionine restored growth during PAS treatment of wild-type M. bovis BCG in a dose-dependent manner. In contrast, growth of the metM::himar1 strain was still inhibited by PAS even in the presence of 10 μg/ml methionine. When the metM::himar1 strain was transformed with a plasmid expressing metM, addition of methionine growth in the presence of PAS, similar to the wild type strain indicating that MetM plays a role in methionine mediated PAS antagonism (Figure 3A). We were also able to eliminate the possibility that the metM::himar1 strain was auxotrophic for methionine as both strains were able to grow in standard 7H9, a methionine-free medium. These strains were equally inhibited by the first-line antitubercular drugs rifampicin and isoniazid (Supplementary Table S3). PABA, another PAS antagonist, reversed PAS-mediated growth inhibition in both the wild type M. bovis BCG and the metM::himar1 strain, validating that the metM::himar1 strain is specifically impaired for methionine-mediated PAS antagonism.
Figure 3.

MetM is essential for methionine-mediated PAS antagonism and uptake of methionine in M. bovis BCG. (A) Growth kinetics of M. bovis BCG metM::himar1 and M. bovis BCG wild type during PAS exposure when antagonistic metabolites are added. Growth was assessed by OD600 readings every 2–3 days. (B) M. bovis BCG strains were grown to an OD600 of ~0.5, washed three times to remove residual sulfate with sulfate-free Sautons medium, and resuspended in sulfate-free Sautons medium to a starting OD600 of 0.01 and cells were starved for sulfur for 5 days. Following the exhaust period, sulfur-sources were added, and cells were incubated for 7 days to resume growth. The fold-change in growth was assessed as a ratio of the final OD600 over the starting OD600 following the exhaust period (final OD600/starting OD600). (C) Uptake of L-methionine-(methyl-d3) was measured using LC-MS/MS. Samples that fell outside the quantifiable lower limit of the standard curve for L-methionine-(methyl-d3) (25.00–0.39 μM) were labeled #, not quantifiable. (B,C) p-values of pairwise comparisons (denoted by bracketed lines) were calculated using the Student t-test. *p < 0.05, **p < 0.005, ns indicates no significant difference (p > 0.05). Error bars denote standard deviation and are representative of 3 separate experiments.
MetM is a major methionine transporter in M. bovis BCG
M. tuberculosis is known to utilize reverse transsulfuration to assimilate sulfur from methionine which can serve as the sole source of sulfur for this bacterium (Wheeler et al., 2005). Therefore, we tested whether disruption of metM would affect the ability of the bacilli to assimilate sulfur derived from methionine. When M. bovis BCG, the metM::himar1 mutant strain and the metM complemented metM::himar1 mutant strain were grown in sulfate-free Sautons medium, growth of all strains was limited (maximum OD600 = 0.3). Upon addition of sodium sulfate to the medium, strains resumed growth and achieved typical growth yields confirming these strains were previously starved for sulfur (Figure 3B). When methionine was added to sulfur starved M. bovis BCG, growth also resumed in a dose-dependent manner producing similar growth yields as compared to the addition of sulfate alone. In contrast, growth of the metM::himar1 disruption strain could not be restored in the presence of methionine as the sole source of sulfur, but could be fully restored by metM complementation, indicating that MetM is required for methionine utilization.
To confirm our hypothesis that MetM is the primary methionine transporter in M. bovis BCG, we developed a methionine uptake assay using L-methionine-(methyl-d3) as the transport substrate. After M. bovis BCG strains were incubated with L-methionine-(methyl-d3), metabolites were extracted and then analyzed by LC-MS/MS to determine intracellular accumulation of L-methionine-(methyl-d3). We found that wild-type M. bovis BCG showed substantial methionine-(methyl-d3) accumulation compared to the metM::himar1 mutant, (~50–100 fold decrease) whose samples fell below the lower limit of quantitation (Figure 3C). Complementation of the transporter significantly increased accumulation of methionine-(methyl-d3) (2-fold increase) over the same period of as wild-type BCG. Addition of the membrane uncoupler, CCCP, reduced methionine-(methyl-d3) uptake in all strains to a significant degree suggesting that the driving force for L-methionine transport is membrane potential. For all strains tested, control samples without methionine-(methyl-d3) added were found to lack detectable quantities of methionine-(methyl-d3) (data not shown). These findings indicate that MetM is an energy-dependent methionine transporter that plays a major role in methionine transport in M. bovis BCG.
PABA biosynthesis is indispensable for methionine-mediated PAS antagonism in M. tuberculosis
Our large-scale screening failed to identify genes directly involved in methionine-mediated PAS antagonism. Thus, it is possible that genes involved in this process are redundant or are essential for M. bovis BCG under the growth conditions that were employed. Because addition of methionine can increase SAM levels and many M. tuberculosis SAM-dependent methyltransferase genes are essential, we investigated whether the ability to methylate PAS plays a role in methionine-mediated PAS antagonism. To test this, we evaluated whether methionine can antagonize the activated PAS species, 2′-hydroxy-pteroate (pterin-PAS), in M. bovis BCG. It is known that N,N-dimethyl-PAS has no anti-tubercular activity, presumably because N,N-dimethyl-PAS cannot react with DHPPP during the first step of PAS bioactivation (Figure 1). Thus, once PAS is activated to pterin-PAS, N-methylation should not affect its anti-tubercular activity. We confirmed pterin-PAS was active against wild-type M. bovis BCG at a comparable molar concentration to PAS (Table 3). Surprisingly, pterin-PAS was still potently antagonized by methionine suggesting that methionine-mediated PAS antagonism does not occur by N-methylation of PAS.
Table 3.
Antagonism of PAS and pterin-PAS by methionine.
| PAS MIC90a | Pterin-PAS MIC90 | |||
|---|---|---|---|---|
| Strain | –Met | +Metb | –Met | +Met |
| M. bovis BCG | 1 (6.53) | >250 (1630) | 5 (15.1) | >20 (61.5) |
| M. tuberculosis H37Ra | 0.6 (3.92) | >250 (1630) | ND | ND |
| M. tuberculosis H37Ra ΔpabB | 0.15 (0.98) | 0.3 (1.96) | ND | ND |
MIC90 is defined as the minimum concentration of drug required to restrict at least 90% of growth relative to growth seen in the no-drug control cultures. MIC90 are shown in μg/ml (μM).
ND, not determined; Met, methionine; PAS, para-aminosalicylic acid.
Methionine was supplemented at 10 μg/ml.
It is also known that intracellular PABA levels affect PAS susceptibility in M. tuberculosis (Thiede et al., 2016). Since PABA biosynthesis is essential for mycobacterial growth, we hypothesized that PABA biosynthesis could be associated with methionine-mediated antagonism of PAS. PabB, aminodeoxychorismate synthase, is one of the essential enzymes required to convert chorismate to PABA in M. tuberculosis (Figure 4A). Consequently, a M. tuberculosis H37Ra pabB deletion strain is a PABA auxotroph and relies upon exogenous sources of PABA for growth (Figure 4B). The folate precursor dihydropteroate is produced from PABA and DHPPP (Figure 4A). We found that pteroic acid, an oxidized form of dihydropteroate can also support the growth of the M. tuberculosis H37Ra pabB deletion strain (Figure 4B). As expected, unlike PABA and pteroic acid, methionine did not support the growth of the M. tuberculosis H37Ra pabB deletion strain indicating that methionine alone is insufficient to fulfill cellular folate requirements in PABA starved M. tuberculosis cells. Using the M. tuberculosis H37Ra ΔpabB strain, we tested the requirement of PABA biosynthesis on methionine-mediated PAS antagonism. We observed that methionine potently antagonized PAS susceptibility in wild type M. tuberculosis H37Ra. In contrast, PAS susceptibility of the M. tuberculosis H37Ra ΔpabB strain was not antagonized by the addition of methionine (Table 3). Taken together, these data demonstrated that a functional PABA biosynthetic pathway is essential for methionine to antagonize PAS in M. tuberculosis.
Figure 4.

Methionine can affect but not bypass essentiality of upstream folate biosynthetic pathways in M. tuberculosis. (A) New working model of methionine-mediated PAS antagonism. (B) M. tuberculosis ΔpabB was grown to an OD600 of ~0.5, washed three times with PABA-free 7H9 medium to remove residual PABA and resuspended in PABA-free 7H9 medium to a starting OD600 of 0.01. Cultures were then supplemented with the indicated metabolites and incubated for 14 days with OD600 readings taken at the given time points.
Biotin cofactor biosynthesis is essential for intrinsic resistance to PAS and other anti-tubercular drugs
Our screening also identified several mutations that conferred increased susceptibility to PAS even in the absence of methionine. One strain, harboring a himar1 insertion within bioB, encoding biotin synthase, showed increased susceptibility to PAS both in the presence and absence of methionine (Table 2). BioB is a radical SAM-dependent enzyme required for the final step in the synthesis of biotin. We confirmed the bioB::himar1 strain exhibited biotin auxotrophy (Figure 5A). Chemical complementation of biotin revealed that 0.05 μg/ml biotin supplementation was barely sufficient to support near wild-type level growth. We speculated that susceptibility of the bioB::himar1 strain to PAS was dependent upon the intracellular concentration of biotin. Thus, we examined the PAS susceptibility of the bioB::himar1 strain using media containing minimal (0.05 μg/ml) or excess (5 μg/ml) concentrations of biotin (Figure 5B). We observed the bioB::himar1 strain was far more susceptible to PAS (8-fold decrease in MIC90) in minimal biotin medium. In contrast, PAS susceptibility of the bioB::himar1 strain was restored back to near wild-type level in excess biotin medium. Interestingly, the bioB::himar1 strain was also more susceptible to sulfamethoxazole and rifampicin in minimal biotin medium, but maintained wild-type susceptibility to isoniazid, indicating that alterations in susceptibility profiles are drug-specific (Figure 5B). Similar to PAS, sulfamethoxazole and rifampicin susceptibilities of the bioB::himar1 strain were restored to wild-type level in the excess biotin medium. Since PAS, sulfamethoxazole, and rifampicin susceptibilities of the bioB::himar1 were dependent upon the concentration of supplemented biotin, these results indicated that defect in biotin cofactor biosynthesis in the bioB::himar1 strain is responsible for increased susceptibility to PAS, sulfamethoxazole and rifampicin.
Figure 5.
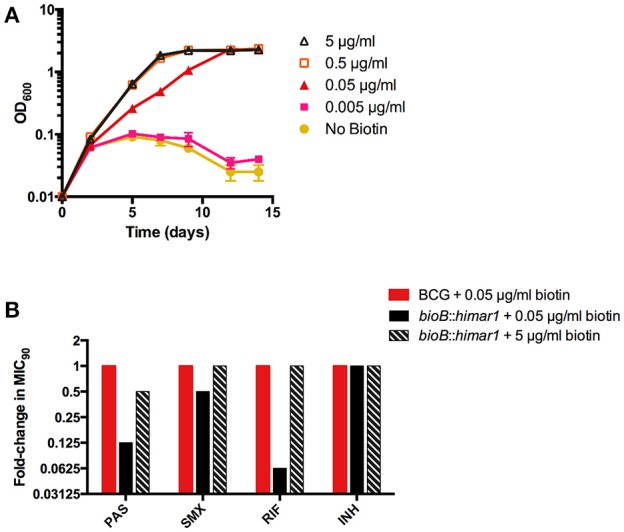
Disruption of bioB is growth inhibitory and potentiates drug action. (A,B) M. bovis BCG bioB::himar1 was grown to an OD600 of ~0.5, washed three times to remove residual biotin with biotin-free 7H9 medium, and resuspended in biotin-free 7H9 medium to a starting OD600 of 0.01. Cultures were then supplemented with biotin and incubated for 14 days with OD600 readings taken at the given time points. Error bars denote standard deviation and are representative of 2 separate experiments. (B) M. bovis BCG and bioB::himar1 were grown to an OD600 of ~0.5, washed three times to remove residual biotin, and resuspended in biotin-free 7H9 medium to a starting OD600 of 0.01. Cultures were then supplemented with biotin (0.05 and 5) and incubated for 14 days with OD600 readings taken at the given time points. MIC90 is defined as the minimum concentration of inhibitor required to restrict at least 90% of growth relative to growth seen in the no-drug control cultures. PAS, para-aminosalicylic acid; SMX, sulfamethoxazole; RIF, rifampin; INH, isoniazid. Results shown are representative of 3 separate experiments.
Discussion
Methionine is the only folate-dependent metabolite known to antagonize certain anti-folate drugs in M. tuberculosis and other bacterial species. Interestingly, anti-folate drugs antagonized by methionine are also antagonized by PABA, a folate precursor. Although the molecular mechanism of PABA-mediated anti-folate antagonism is well-understood, how methionine antagonizes anti-folate drugs has yet to be elucidated. Our findings revealed that methionine-mediated PAS antagonism is linked to synthesis of folate precursors.
One strain isolated in our screen harboring a himar1 disruption within the amino acid permease MetM fully sensitized M. bovis BCG to PAS in the presence of normally antagonistic concentrations of methionine and could be rescued by complementation of the metM disruption. In addition, the himar1 disruption within metM prevented M. bovis BCG from assimilating sulfur derived from methionine and significantly decreased uptake of L-methionine-(methyl-d3). MetM belongs to the APC superfamily of transporters and our data suggested that MetM is principally responsible for uptake of methionine in vitro. Our data also suggest the existence of a secondary transporter for methionine based upon the decreased, but still detectable, accumulation of L-methionine-(methyl-d3) in M. bovis BCG metM::himar1. The most well-studied methionine transport system in bacteria is the MetD ABC transporter system of the methionine uptake transporter family found in numerous organisms including E. coli and even the closely related non-tubercular Mycobacterium, Mycobacterium abscessus (Gál et al., 2002). In E. coli, the MetD ABC transporter is encoded by the metNIQ gene cluster (Merlin et al., 2002). The M. tuberculosis complex has no known orthologs of this system, despite the known bioavailability of methionine in human and mouse serum (Lewis et al., 1980; Rivera et al., 1987). To our knowledge, this study represents the first characterization of a methionine transporter in the M. tuberculosis complex. Orthologs of MetM with high amino acid sequence similarities are found from Gordonia sputi, Bacillus subtilis and Lactococcus lactis and an ortholog from L. lactis has been shown to transport branched-chain amino acids, along with methionine (den Hengst et al., 2006). Existence of a conserved methionine transporter within the mycobacterium complex would be intriguing given that methionine/SAM biosynthesis is indispensable for survival of M. tuberculosis in murine and macrophage models of infection (Berney et al., 2015).
We also found that methionine-mediated PAS antagonism does not appear to occur through N,N-dimethylation by SAM-dependent methyltransferase(s). We addressed this possibility because N,N-dimethyl PAS, an inactive metabolite of PAS, was previously identified in metabolite extracts from PAS treated M. tuberculosis (Chakraborty et al., 2013). In addition, a SAM-dependent methyltransferase (Rv0560c) is induced by salicylate and salicylate analogs, including PAS (Schuessler and Parish, 2012). However, a recent report described that an unmarked in-frame deletion of Rv0560c in M. tuberculosis conferred no alteration in susceptibility to PAS, or other antimicrobials in vitro (Kokoczka et al., 2017). Consistent with this finding, our screen did not identify Rv0560c::himar1 mutants. Together with our observation that pterin-PAS is also antagonized by methionine, we conclud that methionine-mediated PAS antagonism is not likely via PAS inactivation by N,N-dimethylation.
Importantly, methionine was unable to antagonize PAS in a pabB deletion mutant strain indicating that methionine-mediated PAS antagonism is dependent upon a functional PABA biosynthetic pathway. This finding is consistent with past and recent reports that methionine only antagonizes the anti-folate drugs that are also antagonized by PABA (Huang et al., 1997; Zheng et al., 2013; Nixon et al., 2014; Zhao et al., 2016). While the metabolic connections linking methionine to folate precursor biosynthesis remain to be determined, the DHPPP pathway has been shown to modulate susceptibility of E. coli, Salmonella enterica and Burkholderia pseudomallei to sulfamethoxazole (Li et al., 2017; Podnecky et al., 2017; Minato et al., 2018), which is predicted to be metabolically linked with methionine-mediated antagonism (Minato and Baughn, 2017). Further, we recently demonstrated that the biosynthetic pathway to DHPPP is essential for methionine-mediated antagonism of sulfonamide action in E. coli (Minato et al., 2018).
One PAS-sensitive mutant strain with a disruption in biotin synthase (bioB) was found to be auxotrophic for the cofactor biotin. Characterization of this mutant confirmed that disruption of biotin biosynthesis could enhance susceptibility to PAS and rifampicin in biotin-limited conditions. In M. tuberculosis, biotin is a cofactor required for acyl-CoA-carboxylase (ACC) enzymes participating in key metabolic processes in lipid biosynthesis (Takayama et al., 2005; Gago et al., 2011; Salaemae et al., 2011; Woong Park et al., 2011). Biotin biosynthesis and protein biotinylation process have been targeted for novel drug development (Duckworth et al., 2011; Shi et al., 2013; Tiwari et al., 2018). On the basis of our in vitro findings, targeting biotin synthesis may promote accumulation of antimycobacterial drugs by disrupting cell envelope integrity, which could revitalize drug therapies that are unable to overcome the relatively impermeable cell envelope of M. tuberculosis at clinically relevant dosages. Indeed, it was recently reported that disruption of protein biotinylation potentiates rifampicin activity against M. tuberculosis (Tiwari et al., 2018). It was previously reported that biotin has a vital role in methionine-mediated, PAS antagonism, such that supplementation with exogenous biotin was required to observe antagonism by methionine (Hedgecock, 1956). However, our study found that biotin supplementation was non-essential for methionine to antagonize PAS in M. bovis BCG suggesting that the effect of biotin on PAS susceptibility is independent of the precise mechanism of antagonism, and the initial observations in M. tuberculosis by Hedgecock may be a strain specific phenotype.
In summary, the mechanistic basis of methionine-mediated PAS antagonism was examined. Over 30 novel modulators of PAS susceptibility were identified by himar1 transposon mutagenesis. However, with exception of the methionine transporter MetM, none of the functions identified were found to be directly involved in antagonism. Upon closer examination, de novo biosynthesis of PABA was determined as essential for methionine-mediated antagonism, revealing a previously unappreciated relationship between methionine and folate precursor synthesis. Further studies are needed to reveal the precise mechanism of this process. The results presented here also identified tractable drug targets within M. tuberculosis that could be exploited to enhance antimycobacterial drug action.
Author contributions
MH, SK, MC, and AAB performed experiments. ADB, CA, and YM conceived the work. MH, ADB, and YM wrote the manuscript. All authors contributed to analysing data and editing of the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nicholas D. Peterson for assistance in construction of a library of M. bovis BCG transposon insertion mutants. We thank Drs. Richard Lee and Ying Zhao of St Jude Children's Research Hospital for providing pterin-PAS.
Footnotes
Funding. This work was supported by a grants from the University of Minnesota Academic Health Center Faculty Research Development Program to ADB and CA, and the NIH (AI123146) to ADB, and by startup funds from the University of Minnesota to ADB and CA.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00399/full#supplementary-material
References
- Allen B. W. (1998). Mycobacteria. General culture methodology and safety considerations. Methods Mol. Biol. 101, 15–30. 10.1385/0-89603-471-2:15 [DOI] [PubMed] [Google Scholar]
- Baughn A. D., Deng J., Vilchèze C., Riestra A., Welch J. T., Jacobs W. R., et al. (2010). Mutually exclusive genotypes for pyrazinamide and 5-chloropyrazinamide resistance reveal a potential resistance-proofing strategy. Antimicrob. Agents Chemother. 54, 5323–5328. 10.1128/AAC.00529-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney M., Berney-Meyer L., Wong K. W., Chen B., Chen M., Kim J., et al. (2015). Essential roles of methionine and S-adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 112, 10008–10013. 10.1073/pnas.1513033112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch R., Gordon S. V., Garnier T., Eiglmeier K., Frigui W., Valenti P., et al. (2007). Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl. Acad. Sci. U.S.A. 104, 5596–5601. 10.1073/pnas.0700869104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Gruber T., Barry C. E., Boshoff H. I., Rhee K. Y. (2013). para-Aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91. 10.1126/science.1228980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawadi S., Kordus S. L., Baughn A. D., Aldrich C. C. (2017). Synthesis and analysis of bacterial folate metabolism intermediates and antifolates. Org. Lett. 19, 5220–5223. 10.1021/acs.orglett.7b02487 [DOI] [PubMed] [Google Scholar]
- den Hengst C. D., Groeneveld M., Kuipers O. P., Kok J. (2006). Identification and functional characterization of the Lactococcus lactis CodY-regulated branched-chain amino acid permease BcaP (CtrA). J. Bacteriol. 188, 3280–3289. 10.1128/JB.188.9.3280-3289.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N. A., Peterson N. D., Rosen B. C., Baughn A. D. (2014). Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob. Agents Chemother. 58, 7258–7263. 10.1128/AAC.04028-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald P. R., Diacon A. H. (2015). para-Aminosalicylic acid: the return of an old friend. Lancet Infect. Dis. 15, 1091–1099. 10.1016/S1473-3099(15)00263-7 [DOI] [PubMed] [Google Scholar]
- Duckworth B. P., Geders T. W., Tiwari D., Boshoff H. I., Sibbald P. A., Barry C. E., et al. (2011). Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 18, 1432–1441. 10.1016/j.chembiol.2011.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S., Guo X. V., Hickey C. M., Ryou M., Monteleone M., Riley L. W., et al. (2005). Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33:e21. 10.1093/nar/gni013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbourne L. D., Tetu S. G., Hassan K. A., Paulsen I. T. (2017). TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 45, D320–D324. 10.1093/nar/gkw1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gago G., Diacovich L., Arabolaza A., Tsai S. C., Gramajo H. (2011). Fatty acid biosynthesis in actinomycetes. FEMS Microbiol. Rev. 35, 475–497. 10.1111/j.1574-6976.2010.00259.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gál J., Szvetnik A., Schnell R., Kálmán M. (2002). The metD D-methionine transporter locus of Escherichia coli is an ABC transporter gene cluster. J. Bacteriol. 184, 4930–4932. 10.1128/JB.184.17.4930-4932.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N. R., Moll A., Sturm A. W., Pawinski R., Govender T., Lalloo U., et al. (2006). Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 368, 1575–1580. 10.1016/S0140-6736(06)69573-1 [DOI] [PubMed] [Google Scholar]
- Gehre F., Otu J., Kendall L., Forson A., Kwara A., Kudzawu S., et al. (2016). The emerging threat of pre-extensively drug-resistant tuberculosis in West Africa: preparing for large-scale tuberculosis research and drug resistance surveillance. BMC Med. 14:160. 10.1186/s12916-016-0704-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock L. W. (1956). Antagonism of the inhibitory action of aminosalicylic acid on Mycobacterium tuberculosis by methionine, biotin and certain fatty acids, amino acids, and purines. J. Bacteriol. 72, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E. Y., Mohler A. M., Rohlman C. E. (1997). Protein expression in response to folate stress in Escherichia coli. J. Bacteriol. 179, 5648–5653. 10.1128/jb.179.17.5648-5653.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack D. L., Paulsen I. T., Saier M. H. (2000). The amino acid/polyamine/organocation (APC) superfamily of transporters specific for amino acids, polyamines and organocations. Microbiology 146(Pt. 8), 1797–1814. 10.1099/00221287-146-8-1797 [DOI] [PubMed] [Google Scholar]
- Kokoczka R., Schuessler D. L., Early J. V., Parish T. (2017). Mycobacterium tuberculosis Rv0560c is not essential for growth in vitro or in macrophages. Tuberculosis 102, 3–7. 10.1016/j.tube.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriakov J., Lee S., Jacobs W. R. (2003). Identification of a regulated alkaline phosphatase, a cell surface-associated lipoprotein, in Mycobacterium smegmatis. J. Bacteriol. 185, 4983–4991. 10.1128/JB.185.16.4983-4991.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J. (1946). para-Aminosalicylic acid in the treatment of tuberculosis. Lancet 247, 15–16. 10.1016/S0140-6736(46)91185-3 [DOI] [PubMed] [Google Scholar]
- Lewis A. M., Waterhouse C., Jacobs L. S. (1980). Whole-blood and plasma amino acid analysis: gas-liquid and cation-exchange chromatography compared. Clin. Chem. 26, 271–276. [PubMed] [Google Scholar]
- Li K., Li T., Yang S. S., Wang X. D., Gao L. X., Wang R. Q., et al. (2017). Deletion of nudB causes increased susceptibility to antifolates in Escherichia coli and Salmonella enterica. Antimicrob. Agents Chemother. 61:e02378–16. 10.1128/AAC.02378-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C., Gardiner G., Durand S., Masters M. (2002). The Escherichia coli metD locus encodes an ABC transporter which includes Abc (MetN), YaeE (MetI), and YaeC (MetQ). J. Bacteriol. 184, 5513–5517. 10.1128/JB.184.19.5513-5517.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y., Baughn A. D. (2017). Subversion of metabolic wasting as the mechanism for folM-linked Sulfamethoxazole resistance. MBio 8:e01769–17. 10.1128/mBio.01769-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y., Dawadi S., Kordus S. L., Sivanandam A., Aldrich C. C., Baughn A. D. (2018). Mutual potentiation drives synergy between trimethoprim and sulfamethoxazole. Nat. Commun. 9:1003. 10.1038/s41467-018-03447-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y., Thiede J. M., Kordus S. L., McKlveen E. J., Turman B. J., Baughn A. D. (2015). Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob. Agents Chemother. 59, 5097–5106. 10.1128/AAC.00647-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon M. R., Saionz K. W., Koo M. S., Szymonifka M. J., Jung H., Roberts J. P., et al. (2014). Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis. Chem. Biol. 21, 819–830. 10.1016/j.chembiol.2014.04.009 [DOI] [PubMed] [Google Scholar]
- Peterson N. D., Rosen B. C., Dillon N. A., Baughn A. D. (2015). Uncoupling environmental pH and intrabacterial acidification from Pyrazinamide susceptibility in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 7320–7326. 10.1128/AAC.00967-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podnecky N. L., Rhodes K. A., Mima T., Drew H. R., Chirakul S., Wuthiekanun V., et al. (2017). Mechanisms of resistance to folate pathway inhibitors in Burkholderia pseudomallei: deviation from the norm. MBio 8:e01357–17. 10.1128/mBio.01357-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera S., López-Soriano F. J., Azcón-Bieto J., Argilés J. M. (1987). Blood amino acid compartmentation in mice bearing Lewis lung carcinoma. Cancer Res. 47, 5644–5646. [PubMed] [Google Scholar]
- Rubin E. J., Akerley B. J., Novik V. N., Lampe D. J., Husson R. N., Mekalanos J. J. (1999). In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 96, 1645–1650. 10.1073/pnas.96.4.1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaemae W., Azhar A., Booker G. W., Polyak S. W. (2011). Biotin biosynthesis in Mycobacterium tuberculosis: physiology, biochemistry and molecular intervention. Protein Cell 2, 691–695. 10.1007/s13238-011-1100-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler D. L., Parish T. (2012). The promoter of Rv0560c is induced by salicylate and structurally-related compounds in Mycobacterium tuberculosis. PLoS ONE 7:e34471. 10.1371/journal.pone.0034471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Tiwari D., Wilson D. J., Seiler C. L., Schnappinger D., Aldrich C. C. (2013). Bisubstrate inhibitors of biotin protein ligase in Mycobacterium tuberculosis resistant to Cyclonucleoside formation. ACS Med. Chem. Lett. 4, 1213–1217. 10.1021/ml400328a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper S. B., Melton R. E., Mustafa S., Kieser T., Jacobs W. R. (1990). Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4, 1911–1919. 10.1111/j.1365-2958.1990.tb02040.x [DOI] [PubMed] [Google Scholar]
- Steenken W., Oatway W. H., Petroff S. A. (1934). Biological studies of the tubercle bacillus: III. Dissociation and pathogenicity of the R and S variants of the human tubercle bacillus (H(37)). J. Exp. Med. 60, 515–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K., Wang C., Besra G. S. (2005). Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101. 10.1128/CMR.18.1.81-101.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R. G., Walker D. C., McInnes R. R. (1993). E. coli host strains significantly affect the quality of small scale plasmid DNA preparations used for sequencing. Nucleic Acids Res. 21, 1677–1678. 10.1093/nar/21.7.1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiede J. M., Kordus S. L., Turman B. J., Buonomo J. A., Aldrich C. C., Minato Y., et al. (2016). Targeting intracellular p-aminobenzoic acid production potentiates the anti-tubercular action of antifolates. Sci. Rep. 6:38083. 10.1038/srep38083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari D., Park S. W., Essawy M. M., Dawadi S., Mason A., Nandakumar M., et al. (2018). Targeting protein biotinylation enhances tuberculosis chemotherapy. Sci. Transl. Med. 10:eaal1803. 10.1126/scitranslmed.aal1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler P. R., Coldham N. G., Keating L., Gordon S. V., Wooff E. E., Parish T., et al. (2005). Functional demonstration of reverse transsulfuration in the Mycobacterium tuberculosis complex reveals that methionine is the preferred sulfur source for pathogenic Mycobacteria. J. Biol. Chem. 280, 8069–8078. 10.1074/jbc.M412540200 [DOI] [PubMed] [Google Scholar]
- Woong Park S., Klotzsche M., Wilson D. J., Boshoff H. I., Eoh H., Manjunatha U., et al. (2011). Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 7:e1002264. 10.1371/journal.ppat.1002264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2017). Global Tuberculosis Report 2017. Geneva: http://apps.who.int/iris/bitstream/10665/259366/1/9789241565516-eng.pdf?ua/=1 [Google Scholar]
- Youmans G. P., Raleigh G. W., Youmans A. S. (1947). The tuberculostatic action of para-Aminosalicylic acid. J. Bacteriol. 54, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F., Wang X. D., Erber L. N., Luo M., Guo A. Z., Yang S. S., et al. (2014). Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 1479–1487. 10.1128/AAC.01775-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Shadrick W. R., Wallace M. J., Wu Y., Griffith E. C., Qi J., et al. (2016). Pterin-sulfa conjugates as dihydropteroate synthase inhibitors and antibacterial agents. Bioorg. Med. Chem. Lett. 26, 3950–3954. 10.1016/j.bmcl.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Rubin E. J., Bifani P., Mathys V., Lim V., Au M., et al. (2013). para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J. Biol. Chem. 288, 23447–23456. 10.1074/jbc.M113.475798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Nahid P., Cole S. T. (2013). Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12, 388–404. 10.1038/nrd4001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.