

Abstract
Duchenne muscular dystrophy (DMD) is the most severe form of muscular dystrophy affecting 1 in 3500 live male births. Although there is no cure for DMD, therapeutic strategies aimed at enhancing calcineurin signalling and promoting the slow fibre phenotype have shown promise in mdx mice, which is the classical mouse model for DMD. Sarcolipin (SLN) is a small protein that regulates the sarco(endo)plasmic reticulum Ca2+-ATPase pump and its expression is highly upregulated in dystrophic skeletal muscle. We have recently shown that SLN in skeletal muscle amplifies calcineurin signalling thereby increasing myofibre size and the slow fibre phenotype. Therefore, in the present study we sought to determine the physiological impact of genetic Sln deletion in mdx mice, particularly on calcineurin signalling, fibre-type distribution and size and dystrophic pathology. We generated an mdx/Sln-null (mdx/SlnKO) mouse colony and hypothesized that the soleus and diaphragm muscles from these mice would display blunted calcineurin signalling, smaller myofibre sizes, an increased proportion of fast fibres and worsened dystrophic pathology compared with mdx mice. Our results show that calcineurin signalling was impaired in mdx/SlnKO mice as indicated by reductions in utrophin, stabilin-2 and calcineurin expression. In addition, mdx/SlnKO muscles contained smaller myofibres, exhibited a slow-to-fast fibre-type switch that corresponded with reduced expression of mitochondrial proteins and displayed a worsened dystrophic pathology compared with mdx muscles. Altogether, our findings demonstrate a critical role for SLN upregulation in dystrophic muscles and suggest that SLN can be viewed as a potential therapeutic target.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscle wasting disorder affecting 1 in 3500 live male births (1). Currently, there is no cure for DMD and most therapeutic strategies aim to improve quality of life while slowing disease trajectory. Calcineurin, a serine/threonine Ca2+-dependent phosphatase, has emerged as a therapeutic target as studies using mdx mice, the classical DMD mouse model (2), demonstrate that enhancing calcineurin signalling alleviates the dystrophic phenotype (3–10). Specifically, calcineurin positively impacts muscular dystrophy in a multifactorial manner including an increase in the slow twitch type I fibres that are less prone to the dystrophic phenotype (11,12), an increase in utrophin expression that enhances sarcolemmal membrane stability (3,4,9,13–16) and an increase in the muscles’ regenerative capacity (8,17–21). Furthermore, calcineurin may also alleviate the dystrophic phenotype by increasing myofibre size (22), however, there are some discrepant results regarding calcineurin’s role in regulating muscle mass (23–28).
We have recently shown that sarcolipin (SLN), a regulator of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pump (29–32), amplifies calcineurin signalling in muscle thereby increasing the number of type I fibres, augmenting average myofibre cross-sectional area (CSA) and ultimately improving muscle function and structure in atrophic/myopathic conditions (33,34). In addition, we have recently shown that SLN’s activation of calcineurin is critical for the adaptive responses that occur within a functionally overloaded plantaris (34). In the absence of SLN, functionally overloaded plantaris muscles fail to hypertrophy and transition towards the slow-oxidative phenotype as a result of impaired calcineurin signalling (34). Thus, our recent work has uncovered an important role for SLN in regulating adaptive changes in fibre-type composition and size through its effects on calcineurin signalling, which could prove beneficial for muscular dystrophy.
Interestingly, we and others have found SLN protein to be highly upregulated in skeletal muscles from mdx mice (Supplementary Material, Fig. S1 and 35); however, the physiological importance of this increase in SLN expression in the mdx model remains unknown. In the present study, we sought to determine the physiological role of SLN in mdx skeletal muscles by generating an mdx/Sln-null (mdx/SlnKO) mouse line. Based on our previous findings demonstrating the importance of SLN in amplifying calcineurin signalling and muscle remodelling (33,34), we hypothesized that genetic deletion of Sln in mdx mice would result in blunted calcineurin signalling, reduced type I fibre counts, smaller myofibres and worsened dystrophic pathology.
Results
SLN deletion enhances SERCA’s affinity for Ca2+ in soleus and diaphragm muscles
Western blotting for SLN protein in mdx soleus and diaphragm muscles shows that Sln was successfully deleted from mdx mice (Fig. 1A). Assessments of SERCA activity in soleus and diaphragm homogenates show that while Sln deletion did not improve maximal SERCA activity in either muscle (Fig. 1B), it did improve SERCA’s apparent affinity for Ca2+ as indicated by a leftward shift in the activity curves and a significant increase in the Ca2+ concentration required to elicit 1/2 Vmax (presented as the negative logarithm of Ca2+, pCa50) pCa50 in both muscles (Fig. 1C–E). Finally, western blot analyses did not reveal any significant differences in SERCA1a or SERCA2a protein expression between the mdx and mdx/SlnKO mice (Fig. 1F and G).
Figure 1.
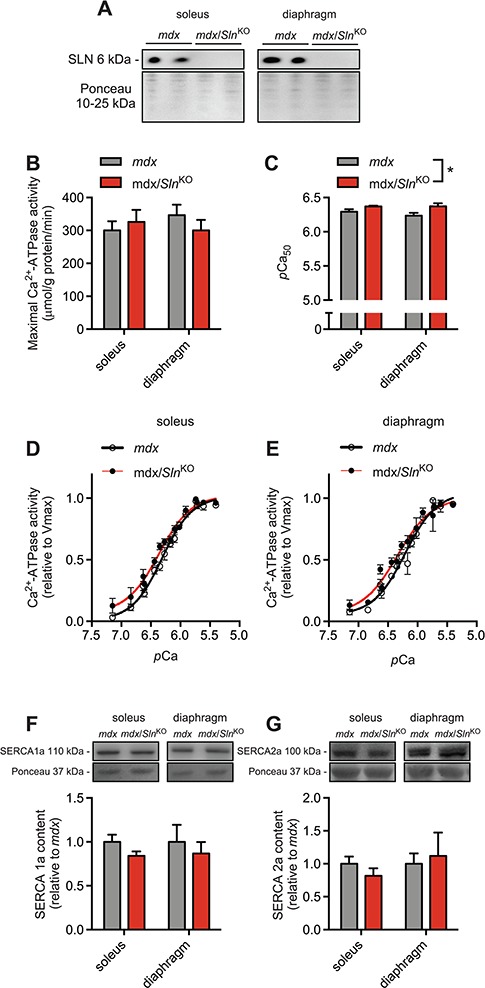
Sln deletion as depicted by western blotting (A) does not alter maximal SERCA activity (B) but does enhance SERCA’s apparent affinity for Ca2+ in soleus and diaphragm muscles (C–E) from mdx mice. Western blot analyses of SERCA1a (F) and SERCA2a (G) did not reveal any significant differences. For (C), *denotes a significant main effect when using a two-way ANOVA, P ≤ 0.05, n = 5−6 per group.
Calcineurin signalling is blunted in muscles from mdx/SlnKO mice
Utrophin and stabilin-2 protein expressions are largely controlled by calcineurin signalling in muscle (13,21), and therefore, we used them as indicators of calcineurin activation. Our western blot analyses show that both utrophin and stabilin-2 expression were significantly lowered in soleus and diaphragm muscles from mdx/SlnKO mice compared with mdx (Fig. 2A and B). Furthermore, we also found a significant reduction in calcineurin expression in muscles from mdx/SlnKO mice compared with mdx (Fig. 2C).
Figure 2.
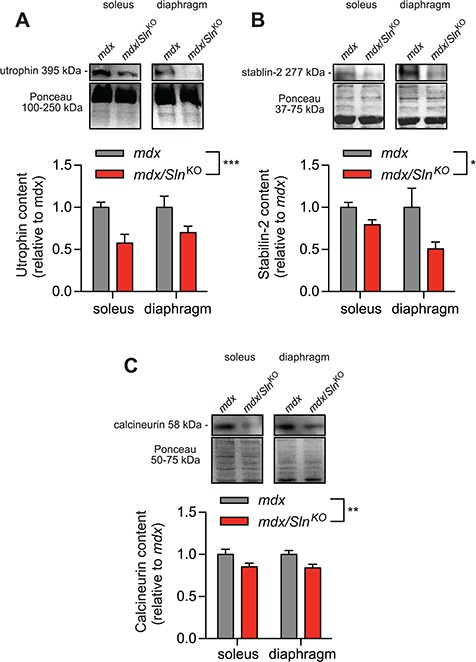
Calcineurin signalling in mdx/SlnKO muscles is impaired as revealed with lowered utrophin, (A) stabilin-2 (B) and calcineurin (C) protein expression (n = 4−6 per group). *Denotes a significant main effect when using a two-way ANOVA, P ≤ 0.05.
SLN deletion alters fibre-type composition and size in mdx mice
We next examined the effects of Sln deletion on fibre-type composition and CSA within the mdx mouse model using immunofluorescent microscopy (Fig. 3A and B). With respect to fibre size, we found that myofibre CSA was significantly reduced in the mdx/SlnKO soleus and diaphragm across all fibre types (Fig. 3C and D). With respect to fibre-type composition, both the soleus and diaphragm muscles underwent a slow-to-fast fibre-type shift in the mdx/SlnKO mice compared with mdx. Specifically, we saw a significant reduction in type I fibre counts in the mdx/SlnKO soleus (Fig. 3E), whereas, in the diaphragm, we observed a significant reduction in type IIA fibres and a significant increase in type IIX in the mdx/SlnKO mice compared with mdx (Fig. 3F). Corresponding with the slow-to-fast fibre-type shift, we also found a significant reduction in the mitochondrial protein cytochrome c oxidase subunit IV (COX IV) (Fig. 3G) and a trending reduction in cytochrome c (Fig. 3H) in mdx/SlnKO mice compared with mdx.
Figure 3.
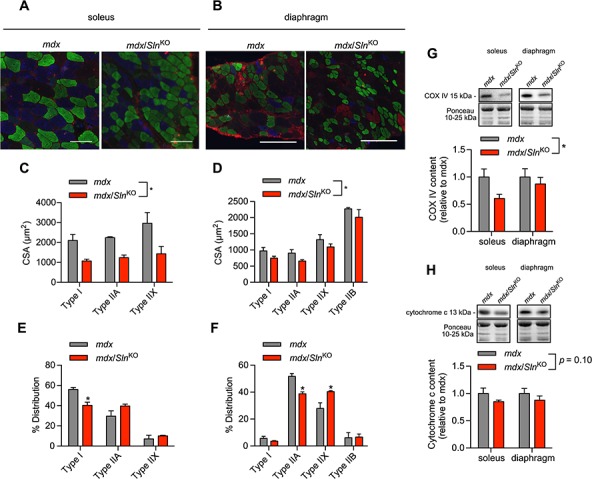
Immunofluorescent fibre typing (A and B) revealed a significant reduction in myofibre CSA (C and D) and a slow-to-fast fibre-type shift (E and F) in response to Sln deletion in the mdx mice (n = 3−4 per group). Western blotting demonstrated a significant reduction in COX IV (G) and a trending reduction in cytochrome c (H) in muscles from mdx/SlnKO mice compared with mdx. For (C), (D), (G) and (H), *denotes a significant main effect when using a two-way ANOVA, P ≤ 0.05.for (E) and (F), *denotes significance using a Student’s t-test comparing mdx versus mdx/SlnKO muscles, P ≤ 0.05.
Worsened dystrophic pathology in muscles from mdx/SlnKO mice
Finally, we tested whether dystrophic pathology would be worse in mdx/SlnKO muscles compared with mdx. In this respect, serum creatine kinase (CK) was significantly elevated in mdx/SlnKO mice compared with mdx (Fig. 4A). In addition, haematoxylin and eosin (H&E) staining revealed greater variability in fibre size with a significant increase in the coefficient of variation of the minimal Feret’s diameter, which is a strong indicator of dystrophic pathology (36) (Fig. 4B and C). We also found a significant reduction in central nuclei counts (Fig. 4D) and a trending increase in muscle degeneration across the soleus and diaphragm from mdx/SlnKO mice compared with mdx (Fig. 4E). After examining cage activity patterns with comprehensive laboratory animal monitoring system (CLAMS; Oxymax series; Columbus Instruments, Columbus, OH, USA), we found that mdx/SlnKO mice were significantly less active compared with mdx mice (Fig. 4F).
Figure 4.
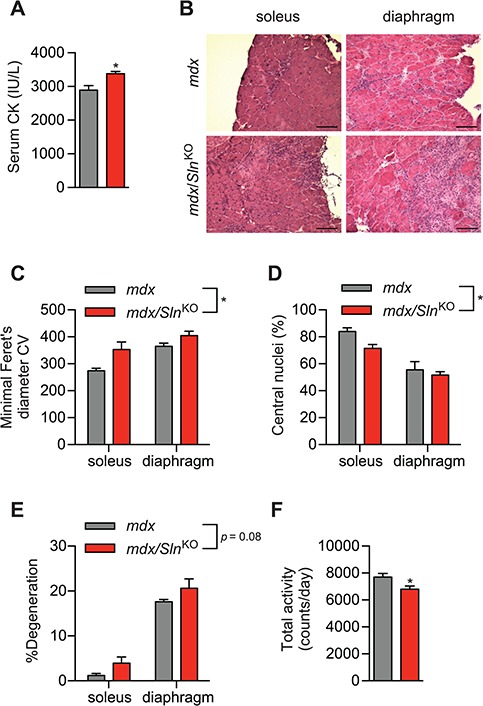
The mdx/SlnKO muscles display worsened dystrophic pathology compared with mdx muscles. (A) Serum CK levels from mdx/SlnKO and mdx mice. (B) H&E staining showing worsened dystrophic myopathy in mdx/SlnKO muscles with increased variability in the minimal Feret’s diameter, (C) lowered central nuclei counts (D) and increased muscle degeneration (% mononuclear cell and connective tissue infiltration; E). Total cage activity using CLAMS revealed a significant reduction in total activity in mdx/SlnKO mice compared with mdx. For (A) and (F), *denotes significance using a Student’s t-test comparing mdx versus mdx/SlnKO muscles, P ≤ 0.05; for (C-E), *denotes a significant main effect when using a two-way ANOVA, P ≤ 0.05. For CK and histological analyses, n = 3–4 per group; and for total activity, n = 8–13.
Discussion
In the present study, we examined the physiological significance of SLN in the mdx mouse model where SLN protein was previously found to be highly upregulated (Supplementary Material, Fig. S1 and 35). Based on our recent findings (33,34), we hypothesized that SLN would play a critical role in activating calcineurin and launching compensatory cellular signalling events that combat the dystrophic pathology in the mdx mouse. After generating the mdx/SlnKO mouse line we discovered that Sln deletion enhanced SERCA function, while impairing calcineurin signalling, leading to a slow-to-fast fibre-type shift, smaller myofibre CSA, worsened dystrophic pathology and reduced total cage activity.
Reductions in utrophin, stabilin-2 and calcineurin expression are altogether indicative of lowered calcineurin signalling in the mdx/SlnKO muscles compared with mdx. Along with calcineurin expression, we chose utrophin and stabilin-2 as our markers of calcineurin signalling in muscle since the expression of these proteins in muscle are largely controlled by calcineurin (13,21). More importantly, these proteins are critical in combating dystrophic pathology, and therefore serve as functional measures of calcineurin signalling that could explain why dystrophic pathology was worsened in the mdx/SlnKO mice. Utrophin is a dystrophin homolog that is upregulated in the mdx mouse to provide compensatory membrane stability, and mice lacking both dystrophin and utrophin display far worse dystrophic pathology when compared with mdx mice (14,16). Thus, the lowered utrophin expression not only indicates blunted calcineurin signalling, but also likely contributes to the augmented myofibre breakdown revealed with elevated serum CK levels and muscle degeneration in the mdx/SlnKO mice. Stabilin-2 is a phosphatidylserine receptor that is important for myoblast fusion, and genetic deletion of stabilin-2 leads to smaller myofibre CSA and impaired muscle regeneration (21). Therefore, our findings of lowered stabilin-2 expression are not only indicative of blunted calcineurin signalling, but could also explain the reductions in myofibre CSA and central nuclei counts, which mark muscle regeneration. Indeed, calcineurin signalling is critical for muscle regeneration (8,17–21) and we have recently uncovered a role for SLN in mediating muscle regeneration after acute cardiotoxin injury (37).
The slow-to-fast fibre-type shift observed in the mdx/SlnKO muscles could also be viewed as another indicator of impaired calcineurin signalling since calcineurin plays a critical role in activating the slow-oxidative phenotype (13,28,38–44). Correspondingly, we also found reduced COX IV and cytochrome c protein in the mdx/SlnKO muscles, which is consistent with calcineurin’s effect of enhancing mitochondrial content and respiratory function (45,46). It has been shown that promoting the oxidative phenotype can ameliorate muscular dystrophy (47) and it is well known that fast glycolytic fibres are more prone to dystrophic pathology compared with the slow-oxidative fibres (11,12). While glycolytic fibres generate more force thereby rendering them more susceptible to muscle damage in dystrophic conditions (12), it is also important to note that calcineurin signalling is much lower in these fibres compared with the slow-oxidative fibres (48). Indeed, utrophin expression has been highly linked to the slow-oxidative phenotype (13). Thus, our results also support the notion that faster fibres are more vulnerable to DMD or lack of dystrophin, as the slow-to-fast fibre-type shift observed in the mdx/SlnKO muscles was associated with worsened dystrophic pathology and reduced cage activity.
Since muscular dystrophy is characterized by extensive degeneration and regeneration cycles, an increase in fibre size variability serves as an accurate indicator of dystrophic pathology. Here, we show that mdx/SlnKO muscles have greater variation in the minimal Feret’s diameter, which is a robust measure of myofibre size. In addition to this, we also found a trending increase in muscle degeneration with the percent of areas occupied by mononuclear cell and connective tissue infiltration (6). Furthermore, we and others have found that mdx mice are ∼25% less active in their cages compared with wild-type (WT) mice likely as a result of overt muscle weakness (Supplementary Material, Fig. S2 and 49). Thus, our findings of reduced cage activity in the mdx/SlnKO mice compared with mdx mice further support the worsened dystrophic myopathy in these animals, and this may not be explained by a less active phenotype observed solely in the SlnKO mice since we have previously shown similar cage activity patterns between WT and SlnKO mice (50).
While there is some considerable debate regarding calcineurin’s role in regulating myofibre size (23–28), calcineurin’s stimulation of myoblast fusion (18–21) can increase myofibre CSA by increasing the relative amount of nuclei per fibre (20,51). We have recently published findings in other mouse models that support SLN’s role in regulating adaptive changes in myofibre size through calcineurin signalling (33,34). In another mouse model of muscle myopathy, we found that SLN was highly upregulated in the soleus muscles, and genetic deletion of Sln worsened the myopathic phenotype leading to smaller myofibres, as a result of impaired calcineurin signalling and lowered stabilin-2 (33). We also observed similar findings in soleus muscles subjected to tenotomy, which represents a model of soleus unloading (34). Conversely, in response to overloading stimuli, we recently found that plantaris muscles from SlnKO mice failed to hypertrophy as a result of impaired calcineurin signalling and lowered stabilin-2 expression (34). Altogether, our findings of smaller myofibre CSA in the mdx/SlnKO muscles compared with mdx are consistent with our previously published reports and further demonstrate the importance of SLN in maintaining muscle size across several myopathic/atrophic conditions.
Mechanistically, our study demonstrates that SLN activates calcineurin signalling and combats dystrophic pathology in mdx muscles by inhibiting SERCA function. Specifically, in the absence of SLN, soleus and diaphragm muscles from mdx/SlnKO mice displayed an increase in SERCA’s apparent affinity for Ca2+. Presumably, higher SERCA activity at sub-maximal Ca2+ concentrations would lead to lower cytosolic Ca2+ levels and less activation of calmodulin and thus calcineurin. The improvement in SERCA’s apparent affinity for Ca2+ is consistent with SLN’s well-known role as a SERCA modulator but was not associated with any changes in SERCA isoform expression. Although, inconsistent with the slow-to-fast fibre-type switch observed in the mdx/SlnKO muscles (52), this finding is in agreement with the notion that SERCA-myosin heavy chain (MHC) mismatch may occur under several myopathic conditions (53–55). Interestingly, previous studies have shown that improving SERCA function via SERCA overexpression can mitigate muscular dystrophy by reducing cytosolic Ca2+ overload and the ensuing muscle damage (56–58). We speculate that differences in the level of SERCA improvement could explain these discrepant results, as Sln deletion may not improve SERCA function to the extent of SERCA overexpression. Indeed, we only observed an improvement in SERCA’s apparent affinity for Ca2+ with no significant alterations in maximal activity, whereas SERCA1 overexpression was shown to enhance maximal SERCA activity 2–3-fold in mdx muscles (57). Thus, our findings suggest that the subtle improvement in SERCA function observed with Sln deletion may not overcome the Ca2+ overload in mdx muscles, but rather results in an exacerbated dystrophic phenotype by reducing calcineurin signalling. Similar findings were observed after overexpressing parvalbumin in the mdx soleus, where increasing the content of this calcium buffering protein worsened the dystrophic pathology by impairing calcineurin activation (10).
While our study was under review, another study was published demonstrating that genetic deletion of Sln alleviated the dystrophic pathology and enhanced muscle function in the mdx/utrophin double knockout mouse (59). We believe that these discrepant findings can be explained by the fact that muscles obtained from mdx/utrophin double knockout mice display greater SLN upregulation when compared with mdx mice (35). Given that our findings show a critical role for SLN in mediating utrophin expression via calcineurin signalling in the mdx mouse, the greater SLN expression in the mdx/utrophin double knockout mouse likely represents a compensatory event attempting to increase utrophin protein. However, in the absence of the utrophin gene, the greater upregulation of SLN likely becomes a maladaptive response that further impairs SERCA function and Ca2+ handling and thus contributing to the worsened dystrophic pathology in the mdx/utrophin double knockout mouse. Thus, our results show that when utrophin expression remains intact, as with mdx mice and DMD patients, SLN upregulation is an adaptive response to increase utrophin expression and combat muscular dystrophy.
To date, there is no cure for DMD, and therapeutic strategies are aimed towards improving quality of life while slowing disease trajectory, and the results presented here reveal SLN as a potential therapeutic target. In this respect, treatment of mdx mice with corticosteroids, which is one of the therapies currently being used in the management of DMD, has been shown to alleviate dystrophic myofibre pathology via calcineurin activation and increased utrophin expression (60). Since corticosteroids also increase Sln expression (61), it stands to reason that the activation of calcineurin signalling in corticosteroid-treated skeletal muscle may be dependent on upregulation of SLN expression. Similarly, high-fat feeding has been shown to improve muscle function in mdx mice (62), and we and others have shown that high-fat feeding increases SLN expression by 3–5-fold (63,64). Thus, to further demonstrate SLN’s therapeutic potential in the treatment of muscular dystrophy, we are currently investigating whether the benefits provided to mdx mice from corticosteroid treatment and high-fat feeding are mediated by SLN and its downstream effects on calcineurin signalling.
In our study we questioned whether the mdx soleus and diaphragm muscles would be affected differently with Sln deletion, and although we did not detect any significant interactions with our two-way analyses of variance (ANOVAs) we found that the average reduction in myofibre CSA was significantly greater in the mdx/SlnKO soleus compared with diaphragm (−48 ± 6% versus −17 ± 3%, P = 0.0004). Similarly, we found that the increase in fibre size variability (soleus, 1.28 ± 0.10-fold versus diaphragm, 1.11 ± 0.04-fold, P = 0.17) and the reduction in central nuclei (soleus, −15 ± 3% versus diaphragm, −7 ± 4%, P = 0.21) was greater in the soleus compared with diaphragm; however, neither were statistically significant. Taken together, these data suggest that Sln deletion may impact the soleus muscle more than the diaphragm. Although purely speculative, this could be due to the fact that the disease phenotype is more severe in the mdx diaphragm relative to the soleus.
One limitation to our study is that we only assessed muscle pathology in mice at 3–6 months of age. This age was selected in order to bypass the initial necrosis and regeneration stage observed within the first 4 weeks of the mdx mouse (65,66). Thus, to fully comprehend the physiological role of SLN in the mdx mouse, future studies should examine the effects of Sln deletion in the mdx mouse across all ages, particularly in older mdx mice (20 months old) where the phenotype more closely resembles DMD pathology (67). Nevertheless, our present results show the importance of SLN protein in combatting dystrophic pathology in young adult mdx mice.
In summary, our study demonstrates for the first time that upregulation of SLN in the mdx mouse represents an adaptive response set out to enhance calcineurin signalling. Altogether, our findings suggest that SLN could be a novel therapeutic target in the management of muscular dystrophy, while also adding further evidence in support of calcineurin’s therapeutic potential for muscle disease. With SLN’s critical role in combatting muscle pathology in mdx mice, we propose that further increasing SLN protein may alleviate the dystrophic pathology in mdx mice and perhaps in DMD patients, and this will be examined further with future studies.
Materials and Methods
Animals
Sln KO mice were described previously (68) and were a kind gift from Dr Muthu Periasamy. To generate the mdx/SlnKO and mdx colonies, homozygous mdx females (XmdxXmdx) were purchased from the Jackson Laboratory (001801, Bar Harbor, ME, USA) and crossed with homozygous SlnKO males. The resulting F1 progeny were hemizygous mdx males (XmdxY) and heterozygous mdx females (XmdxX) that were all heterozygous for Sln deletion. Experimental mdx and mdx/SlnKO (50:50, C57BL6/J:C57BL10) mice were obtained after crossing the hemizygous mdx males with heterozygous mdx females from F1. All animals used in the study were adult mice ranging from 3 to 6 months of age. Animals were housed in an environmentally controlled room with a standard 12:12 hour light–dark cycle and allowed access to food and water ad libitum. All animal procedures were reviewed and approved by the Animal Care Committee of the University of Waterloo and are consistent with the guidelines established by the Canadian Council on Animal Care.
Tissue collection
All mice were sacrificed by cervical dislocation and the diaphragm and soleus muscles were isolated and either homogenized in homogenizing buffer (250 mm sucrose, 5 mm HEPES, 0.2 mm PMSF, 0.2% [w/v] NaN3) or embedded in optimal cutting temperature (O.C.T.) compound (Tissue-Tek, Sakura Finetek, Torrance, CA, USA) frozen in liquid nitrogen-cooled isopentane and then stored at −80°C until further analysis.
SERCA activity assay
SERCA activity was measured in muscle homogenates over Ca2+ concentrations ranging from pCa 7.2 to 5.4 in the presence of the Ca2+ ionophore A23187 (C7522, Sigma Aldrich, St. Louis, MO, USA) using a Ca2+-dependent, enzyme-linked spectrophotometric plate reader assay that has been described previously (69). Maximal SERCA activity was taken from the raw data, whereas SERCA’s apparent affinity for Ca2+ was measured with the pCa50. The pCa50 is the negative logarithm of the [Ca2+] required to elicit half-maximal SERCA activity and was obtained from SERCA activity-pCa curves. The SERCA activity-pCa curves were generated with GraphPad PrismTM (Graphpad Software, La Jolla, CA, USA) by non-linear regression curve fitting, using an equation for a general cooperative model for substrate activation.
Western blotting
Western blotting was performed to determine expression levels of SLN, SERCA1a, SERCA2a, utrophin, calcineurin, stabilin-2, COX IV and cytochrome c in soleus and diaphragm muscles. The primary antibody directed against SLN was generated by Lampire Biological Laboratories (Everett, PA, USA) (70). The SERCA1a antibody (A52) was a kind gift from Dr David MacLennan (University of Toronto), whereas the SERCA2a antibody (MA3-919) was purchased from ThermoFisher Scientific (Waltham, MA, USA). The utrophin (610896) antibody was purchased from BD Biosciences (Franklin Lakes, NJ, USA), and the primary antibodies for calcineurin and stabilin-2 were obtained from Milliopore (Burlington, MA, USA) (07-1491) and Biorbyt (orb158499), respectively. Both primary antibodies for COX IV (sc-69630) and cytochrome c (sc-13156) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Quantitation of optical densities was performed using GeneTools (Syngene, Frederick, MD, USA) and values were normalized to total protein after ponceau staining.
Histochemical and immunofluorescent staining
Soleus and diaphragm muscles embedded in O.C.T. compound were cut into 10 μm thick cryosections with a cryostat (Thermo Fisher Scientific) maintained at −20°C. H&E staining was performed and images were acquired with a brightfield Nikon (Melville, NY, USA) microscope linked to a PixeLink digital camera (Navitar, Rochester, NY, USA). ImageJ software was used to examine central nuclei, muscle degeneration (percent of area occupied by mononuclear and connective tissue infiltration (6)) and the minimal Feret’s diameter, which is the minimum distance of parallel tangents at opposing borders of the muscle fibre (36).
Immunofluorescence analysis of MHC expression was performed as previously described (71) with primary antibodies against MHCI, MHCIIa and MHCIIb to assess fibre-type distribution. Slides were visualized with an Axio Observer Z1 fluorescent microscope equipped with standard red, green, blue filters, an AxioCam HRm camera and AxioVision software (Carl Zeiss, Jena, Germany). Quantification of fibre distribution and CSA was performed using ImageJ (National Institutes of Health, Bethesda, MD, USA) software.
Serum CK analysis
Mice were anaesthetized using somnotol (0.65 mg/kg body weight) and blood from the left ventricle was drawn into a syringe. Blood was centrifuged at 5000g for 8 min and the serum was decanted and stored at −80°C until analysis. CK activity was measured using a kinetic fluorometric assay as previously described (72).
Cage activity
Daily cage activity was measured using CLAMS as previously described (50). This system is equipped with X- and Z-axes infrared photocell detectors that allow monitoring total cage activity. Mice were placed in the CLAMS for a 3 day period for three separate trials and had free access to food and water.
Statistics
All values are presented as standard error of the mean. Comparisons between mdx and mdx/SlnKO mice were performed using either a Student’s t-test or two-way ANOVA. Since, dystrophic pathology differs across soleus and diaphragm muscles from mdx mice with the diaphragm presenting with a more severe phenotype, we also examined the potential interaction effect between genotype and muscle type with a two-way ANOVA. Statistical significance was set to P ≤ 0.05.
Supplementary Material
Acknowledgements
We thank Dawn McCutcheon and Jean Flanagan for their expertise in generating mouse breeding colonies and the Animal Health Technicians at the University of Waterloo for their help in maintaining them. We thank Dr Muthu Periasamy (Sanford, Bernam, Orlando, USA) for providing the SlnKO mice. A doctoral award from CIHR supported V.A.F.
Conflict of Interest statement. None declared.
Funding
Canadian Institutes of Health Research (MOP 86618 and MOP 47296 to A.R.T.).
References
- 1. Emery A.E. (2002) The muscular dystrophies. Lancet, 359, 687–695. [DOI] [PubMed] [Google Scholar]
- 2. Bulfield G., Siller W.G., Wight P.A. and Moore K.J. (1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA, 81, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Angus L.M., Chakkalakal J.V., Mejat A., Eibl J.K., Belanger G., Megeney L.A., Chin E.R., Schaeffer L., Michel R.N. and Jasmin B.J. (2005) Calcineurin-NFAT signaling, together with GABP and peroxisome PGC-1{alpha}, drives utrophin gene expression at the neuromuscular junction. Am. J. Physiol. Cell Physiol., 289, C908–C917. [DOI] [PubMed] [Google Scholar]
- 4. Chakkalakal J.V., Harrison M.A., Carbonetto S., Chin E., Michel R.N. and Jasmin B.J. (2004) Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum. Mol. Genet., 13, 379–388. [DOI] [PubMed] [Google Scholar]
- 5. Sakuma K. and Yamaguchi A. (2010) The functional role of calcineurin in hypertrophy, regeneration, and disorders of skeletal muscle. J. Biomed. Biotechnol., 2010, 721219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stupka N., Gregorevic P., Plant D.R. and Lynch G.S. (2004) The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol., 107, 299–310. [DOI] [PubMed] [Google Scholar]
- 7. Stupka N., Michell B.J., Kemp B.E. and Lynch G.S. (2006) Differential calcineurin signalling activity and regeneration efficacy in diaphragm and limb muscles of dystrophic mdx mice. Neuromuscul. Disord., 16, 337–346. [DOI] [PubMed] [Google Scholar]
- 8. Stupka N., Schertzer J.D., Bassel-Duby R., Olson E.N. and Lynch G.S. (2008) Stimulation of calcineurin Aalpha activity attenuates muscle pathophysiology in mdx dystrophic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol., 294, R983–R992. [DOI] [PubMed] [Google Scholar]
- 9. Chakkalakal J.V., Michel S.A., Chin E.R., Michel R.N. and Jasmin B.J. (2006) Targeted inhibition of Ca2+ /calmodulin signaling exacerbates the dystrophic phenotype in mdx mouse muscle. Hum. Mol. Genet., 15, 1423–1435. [DOI] [PubMed] [Google Scholar]
- 10. Zein M.A., Chin E.R., Jasmin B.J. and Michel R.N. (2012) Targeted expression of the non-native Ca+2-buffering protein parvalbumin exacerbates the dystrophic phenotype in mdx mouse slow muscle fibers. FASEB J., 26, 1086.1023. Published abstract.. [Google Scholar]
- 11. Webster C., Silberstein L., Hays A.P. and Blau H.M. (1988) Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell, 52, 503–513. [DOI] [PubMed] [Google Scholar]
- 12. Consolino C.M. and Brooks S.V. (2004) Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J. Appl. Physiol. (1985), 96, 633–638. [DOI] [PubMed] [Google Scholar]
- 13. Chakkalakal J.V., Stocksley M.A., Harrison M.A., Angus L.M., Deschenes-Furry J., St-Pierre S., Megeney L.A., Chin E.R., Michel R.N. and Jasmin B.J. (2003) Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc. Natl. Acad. Sci. USA, 100, 7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M. and Davies K.E. (1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell, 90, 717–727. [DOI] [PubMed] [Google Scholar]
- 15. Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S. and Sanes J.R. (1997) Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell, 90, 729–738. [DOI] [PubMed] [Google Scholar]
- 16. Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M. and Davies K. (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med., 4, 1441–1444. [DOI] [PubMed] [Google Scholar]
- 17. Chen H.H., Chen W.P., Yan W.L., Huang Y.C., Chang S.W., Fu W.M., Su M.J., Yu I.S., Tsai T.C., Yan Y.T. et al. (2015) NRIP is newly identified as a Z-disc protein, activating calmodulin signaling for skeletal muscle contraction and regeneration. J. Cell Sci., 128, 4196–4209. [DOI] [PubMed] [Google Scholar]
- 18. Horsley V., Friday B.B., Matteson S., Kegley K.M., Gephart J. and Pavlath G.K. (2001) Regulation of the growth of multinucleated muscle cells by an NFATC2-dependent pathway. J. Cell Biol., 153, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horsley V., Jansen K.M., Mills S.T. and Pavlath G.K. (2003) IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell, 113, 483–494. [DOI] [PubMed] [Google Scholar]
- 20. Horsley V. and Pavlath G.K. (2003) Prostaglandin F2(alpha) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J. Cell Biol., 161, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park S.Y., Yun Y., Lim J.S., Kim M.J., Kim S.Y., Kim J.E. and Kim I.S. (2016) Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat. Commun., 7, 10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hudson M.B. and Price S.R. (2013) Calcineurin: a poorly understood regulator of muscle mass. Int. J. Biochem. Cell Biol., 45, 2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bodine S.C., Stitt T.N., Gonzalez M., Kline W.O., Stover G.L., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J.C., Glass D.J. et al. (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol., 3, 1014–1019. [DOI] [PubMed] [Google Scholar]
- 24. Serrano A.L., Murgia M., Pallafacchina G., Calabria E., Coniglio P., Lomo T. and Schiaffino S. (2001) Calcineurin controls nerve activity-dependent specification of slow skeletal muscle fibers but not muscle growth. Proc. Natl. Acad. Sci. USA., 98, 13108–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parsons S.A., Millay D.P., Wilkins B.J., Bueno O.F., Tsika G.L., Neilson J.R., Liberatore C.M., Yutzey K.E., Crabtree G.R., Tsika R.W. et al. (2004) Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J. Biol. Chem., 279, 26192–26200. [DOI] [PubMed] [Google Scholar]
- 26. Parsons S.A., Wilkins B.J., Bueno O.F. and Molkentin J.D. (2003) Altered skeletal muscle phenotypes in calcineurin Aalpha and Abeta gene-targeted mice. Mol. Cell. Biol., 23, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dunn S.E., Chin E.R. and Michel R.N. (2000) Matching of calcineurin activity to upstream effectors is critical for skeletal muscle fiber growth. J. Cell Biol., 151, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naya F.J., Mercer B., Shelton J., Richardson J.A., Williams R.S. and Olson E.N. (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J. Biol. Chem., 275, 4545–4548. [DOI] [PubMed] [Google Scholar]
- 29. Asahi M., Kurzydlowski K., Tada M. and MacLennan D.H. (2002) Sarcolipin inhibits polymerization of phospholamban to induce superinhibition of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs). J. Biol. Chem., 277, 26725–26728. [DOI] [PubMed] [Google Scholar]
- 30. Odermatt A., Becker S., Khanna V.K., Kurzydlowski K., Leisner E., Pette D. and MacLennan D.H. (1998) Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem., 273, 12360–12369. [DOI] [PubMed] [Google Scholar]
- 31. Gamu D., Bombardier E., Smith I.C., Fajardo V.A. and Tupling A.R. (2014) Sarcolipin provides a novel muscle-based mechanism for adaptive thermogenesis. Exerc. Sport Sci. Rev., 42, 136–142. [DOI] [PubMed] [Google Scholar]
- 32. Gorski P.A., Glaves J.P., Vangheluwe P. and Young H.S. (2013) Sarco(endo)plasmic reticulum calcium ATPase (SERCA) inhibition by sarcolipin is encoded in its luminal tail. J. Biol. Chem., 288, 8456–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fajardo V.A., Gamu D., Mitchell A., Bloemberg D., Bombardier E., Chambers P.J., Bellissimo C., Quadrilatero J. and Tupling A.R. (2017) Sarcolipin deletion exacerbates soleus muscle atrophy and weakness in phospholamban overexpressing mice. PLoS One, 12, e0173708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fajardo V.A., Rietze B.A., Chambers P.J., Bellissimo C., Bombardier E., Quadrilatero J. and Tupling A.R. (2017) Effects of sarcolipin deletion on skeletal muscle adaptive responses to functional overload and unload. Am. J. Physiol. Cell Physiol, 313, C154--C161. [DOI] [PubMed] [Google Scholar]
- 35. Schneider J.S., Shanmugam M., Gonzalez J.P., Lopez H., Gordan R., Fraidenraich D. and Babu G.J. (2013) Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil., 34, 349–356. [DOI] [PubMed] [Google Scholar]
- 36. Briguet A., Courdier-Fruh I., Foster M., Meier T. and Magyar J.P. (2004) Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord., 14, 675–682. [DOI] [PubMed] [Google Scholar]
- 37. Bellissimo C., Bombardier E., Chambers P., Fajardo V.A. and Tupling A.R. (2016) Sarcolipin ablation impairs muscle regeneration after acute injury. FASEB J., 30, 1224.1228. Published abstract. [Google Scholar]
- 38. Berchtold M.W., Brinkmeier H. and Muntener M. (2000) Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev., 80, 1215–1265. [DOI] [PubMed] [Google Scholar]
- 39. Bigard X., Sanchez H., Zoll J., Mateo P., Rousseau V., Veksler V. and Ventura-Clapier R. (2000) Calcineurin co-regulates contractile and metabolic components of slow muscle phenotype. J. Biol. Chem., 275, 19653–19660. [DOI] [PubMed] [Google Scholar]
- 40. Chin E.R., Olson E.N., Richardson J.A., Yang Q., Humphries C., Shelton J.M., Wu H., Zhu W., Bassel-Duby R. and Williams R.S. (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev., 12, 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Long Y.C., Glund S., Garcia-Roves P.M. and Zierath J.R. (2007) Calcineurin regulates skeletal muscle metabolism via coordinated changes in gene expression. J. Biol. Chem., 282, 1607–1614. [DOI] [PubMed] [Google Scholar]
- 42. McCullagh K.J., Calabria E., Pallafacchina G., Ciciliot S., Serrano A.L., Argentini C., Kalhovde J.M., Lomo T. and Schiaffino S. (2004) NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. USA, 101, 10590–10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schiaffino S. and Serrano A. (2002) Calcineurin signaling and neural control of skeletal muscle fiber type and size. Trends Pharmacol. Sci., 23, 569–575. [DOI] [PubMed] [Google Scholar]
- 44. Swoap S.J., Hunter R.B., Stevenson E.J., Felton H.M., Kansagra N.V., Lang J.M., Esser K.A. and Kandarian S.C. (2000) The calcineurin-NFAT pathway and muscle fiber-type gene expression. Am. J. Physiol. Cell Physiol., 279, C915–C924. [DOI] [PubMed] [Google Scholar]
- 45. Ryder J.W., Bassel-Duby R., Olson E.N. and Zierath J.R. (2003) Skeletal muscle reprogramming by activation of calcineurin improves insulin action on metabolic pathways. J. Biol. Chem., 278, 44298–44304. [DOI] [PubMed] [Google Scholar]
- 46. Jiang L.Q., Garcia-Roves P.M., Castro Barbosa T. and Zierath J.R. (2010) Constitutively active calcineurin in skeletal muscle increases endurance performance and mitochondrial respiratory capacity. Am. J. Physiol. Endocrinol. Metab., 298, E8–E16. [DOI] [PubMed] [Google Scholar]
- 47. Selsby J.T., Morine K.J., Pendrak K., Barton E.R. and Sweeney H.L. (2012) Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS One, 7, e30063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schiaffino S. and Reggiani C. (2011) Fiber types in mammalian skeletal muscles. Physiol. Rev., 91, 1447–1531. [DOI] [PubMed] [Google Scholar]
- 49. Tatem K.S., Quinn J.L., Phadke A., Yu Q., Gordish-Dressman H. and Nagaraju K. (2014) Behavioral and locomotor measurements using an open field activity monitoring system for skeletal muscle diseases. J. Vis. Exp., 91, 51785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bombardier E., Smith I.C., Gamu D., Fajardo V.A., Vigna C., Sayer R.A., Gupta S.C., Bal N.C., Periasamy M. and Tupling A.R. (2013) Sarcolipin trumps beta-adrenergic receptor signaling as the favored mechanism for muscle-based diet-induced thermogenesis. FASEB J., 27, 3871–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Allen D.L., Roy R.R. and Edgerton V.R. (1999) Myonuclear domains in muscle adaptation and disease. Muscle Nerve, 22, 1350–1360. [DOI] [PubMed] [Google Scholar]
- 52. Tupling A.R. (2004) The sarcoplasmic reticulum in muscle fatigue and disease: role of the sarco(endo)plasmic reticulum Ca2+-ATPase. Can. J. Appl. Physiol., 29, 308–329. [DOI] [PubMed] [Google Scholar]
- 53. Hamalainen N. and Pette D. (2001) Myosin and SERCA isoform expression in denervated slow-twitch muscle of euthyroid and hyperthyroid rabbits. J. Muscle Res. Cell Motil., 22, 453–457. [DOI] [PubMed] [Google Scholar]
- 54. Salanova M., Schiffl G. and Blottner D. (2009) Atypical fast SERCA1a protein expression in slow myofibers and differential S-nitrosylation prevented by exercise during long term bed rest. Histochem. Cell Biol., 132, 383–394. [DOI] [PubMed] [Google Scholar]
- 55. Talmadge R.J., Roy R.R., Bodine-Fowler S.C., Pierotti D.J. and Edgerton V.R. (1995) Adaptations in myosin heavy chain profile in chronically unloaded muscles. Basic Appl Myol, 5, 117–137. [PubMed] [Google Scholar]
- 56. Goonasekera S.A., Lam C.K., Millay D.P., Sargent M.A., Hajjar R.J., Kranias E.G. and Molkentin J.D. (2011) Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest., 121, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mazala D.A., Pratt S.J., Chen D., Molkentin J.D., Lovering R.M. and Chin E.R. (2015) SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol., 308, C699–C709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morine K.J., Sleeper M.M., Barton E.R. and Sweeney H.L. (2010) Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum. Gene Ther., 21, 1735–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Voit A., Patel V., Pachon R., Shah V., Bakhutma M., Kohlbrenner E., McArdle J.J., Dell'Italia L.J., Mendell J.R., Xie L.H. et al. (2017) Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun., 8, 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. St-Pierre S.J., Chakkalakal J.V., Kolodziejczyk S.M., Knudson J.C., Jasmin B.J. and Megeney L.A. (2004) Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J., 18, 1937–1939. [DOI] [PubMed] [Google Scholar]
- 61. Gayan-Ramirez G., Vanzeir L., Wuytack F. and Decramer M. (2000) Corticosteroids decrease mRNA levels of SERCA pumps, whereas they increase sarcolipin mRNA in the rat diaphragm. J. Physiol., 524(Pt 2), 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Radley-Crabb H.G., Fiorotto M.L. and Grounds M.D. (2011) The different impact of a high fat diet on dystrophic mdx and control C57Bl/10 mice. PLoS currents, 3, RRN1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bal N.C., Maurya S.K., Sopariwala D.H., Sahoo S.K., Gupta S.C., Shaikh S.A., Pant M., Rowland L.A., Bombardier E., Goonasekera S.A. et al. (2012) Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med., 18, 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bombardier E., Smith I.C., Vigna C., Fajardo V.A. and Tupling A.R. (2013) Ablation of sarcolipin decreases the energy requirements for Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases in resting skeletal muscle. FEBS Lett., 587, 1687–1692. [DOI] [PubMed] [Google Scholar]
- 65. Carnwath J.W. and Shotton D.M. (1987) Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci., 80, 39–54. [DOI] [PubMed] [Google Scholar]
- 66. Dangain J. and Vrbova G. (1984) Muscle development in mdx mutant mice. Muscle Nerve, 7, 700–704. [DOI] [PubMed] [Google Scholar]
- 67. Lefaucheur J.P., Pastoret C. and Sebille A. (1995) Phenotype of dystrophinopathy in old mdx mice. Anat. Rec., 242, 70–76. [DOI] [PubMed] [Google Scholar]
- 68. Babu G.J., Bhupathy P., Timofeyev V., Petrashevskaya N.N., Reiser P.J., Chiamvimonvat N. and Periasamy M. (2007) Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc. Natl. Acad. Sci. USA, 104, 17867–17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duhamel T.A., Green H.J., Stewart R.D., Foley K.P., Smith I.C. and Ouyang J. (2007) Muscle metabolic, SR Ca(2+) -cycling responses to prolonged cycling, with and without glucose supplementation. J. Appl. Physiol. (1985), 103, 1986–1998. [DOI] [PubMed] [Google Scholar]
- 70. Fajardo V.A., Bombardier E., Vigna C., Devji T., Bloemberg D., Gamu D., Gramolini A.O., Quadrilatero J. and Tupling A.R. (2013) Co-expression of SERCA isoforms, phospholamban and sarcolipin in human skeletal muscle fibers. PLoS One, 8, e84304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bloemberg D. and Quadrilatero J. (2012) Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One, 7, e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Szasz G., Gruber W. and Bernt E. (1976) Creatine kinase in serum: 1. Determination of optimum reaction conditions. Clin. Chem, 22, 650–656. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.