

Abstract
Retinitis pigmentosa (RP) is an inheritable retina degenerative disease leading to blindness. Despite the identification of 70 genes associated with RP, the genetic cause of ∼40% of RP patients remains to be elucidated. Whole-exome sequencing was applied on the probands of a RP cohort of 68 unsolved cases to identify candidate genetic mutations. A homozygous missense variant (c.173C > T, p.T58 M) was found in HKDC1 in two unrelated families presenting late-onset retinal degeneration. This variant affects highly conserved amino acid residue and is very rare in several databases and absent in 4000 ethnic-matched controls. Mutant HKDC1 protein partially lost hexokinase activity. Hkdc1 is expressed in the mouse retina and localized to photoreceptor inner segments. To elucidate the in vivo roles of Hkdc1 in the retina, we generated Hkdc1 knockout (KO) mouse models using CRISPR/Cas9 technique. Two independent alleles were identified and backcrossed to C57BL/6 J for 6 generations. Absence of HKDC1 expression in the Hkdc1 KO retina was confirmed by western blot and immunostaning using HKDC1 antibody. Hkdc1 KO mice exhibited reduced scotopic electroretinogram response and thinner outer nuclear layer, similar to some of the human patient phenotypes. Loss of Hkdc1 led to mislocalization of rhodopsin to the inner segments and cell bodies of rods in some regions in the retina. Taken together, our data demonstrated that HKDC1 is associated with autosomal recessively inherited RP.
Introduction
Retinitis pigmentosa (RP, MIM 268000) is a collection of inherited retinal degenerative disorders that mainly affects rod cells and has an estimated worldwide prevalence of 1:4000 (1–3). RP is a clinically heterogeneous disorder; some affected individuals develop severe vision loss in childhood, whereas mildly affected patients remain asymptomatic until mid-adulthood (3). RP patients usually exhibit night blindness at the early stage of the disease, followed by progressive peripheral vision loss, tunnel vision and eventual central vision loss (4–6). Progressive death of photoreceptors (including both rods and cones) is the main underlying cause for this progressive vision loss. In RP course, cone death usually occurs after rod death because survival of cones is dependent on regular functions of rods.
As a hereditary disease, RP can be inherited in autosomal-dominant (30–40%), autosomal-recessive (50–60%) or X-linked (5–15%) manners. Non-Mendelian inheritance patterns, including digenic and maternal inheritances, have also been reported to account for a small proportion of cases (3,7,8). To date, more than 70 genes associated with RP have been reported (9), but these known genes only explain ∼60% of RP cases (3).
Whole-exome sequencing (WES) has been demonstrated as a powerful tool for investigating causative genes of retinal disease (10–15). In the present study, we investigated potential mutations in two autosomal recessive retinitis pigmentosa (arRP) families using the WES method and identified a missense variant (c.173C > T, p.T58 M) in HKDC1. Further functional studies demonstrated that mutant HKDC1 partially lost its hexokinase activity and mice lacking Hkdc1 gene showed retinal degeneration features: decreased scotopic electroretinogram (ERG) response, abnormal rhodopsin protein localization and photoreceptor cell death. These results demonstrate that HKDC1 is a candidate gene for RP disease.
Results
WES identified a homozygous variant in HKDC1 in two RP families
In a first-cousin consanguineous family 1 (Fig. 1A), proband IV:2 is a 71-year-old male who developed night blindness at the age of 54. Vision loss was evident after this age, and he only had hand-motion visual acuity at age 65 (Supplementary Material, Table S1). Fundus examination revealed pigment deposits in the retina (Fig. 2A), while his sister IV:5 showed normal fundus (Fig. 2B). In family 2 (Fig. 1B), proband II:1 is a 39-year-old male who complained to have night blindness at age 29. He had a 2-year history of reduced vision in both eyes, with visual acuity of /40 Oculus Dexter (OD) and 20/50 [Oculus Sinister (OS] (Supplementary Material, Table S1). Fundus examination revealed irregular pigmentation patterns, mainly in the posterior pole and along the arcade vessels of both eyes (Fig. 2C). Visual field testing revealed peripheral vision defects in both eyes (Fig. 2D). Optical coherence tomography (OCT) examination showed discontinuous ellipsoid and interdigitation zones in both eyes and a disrupted Retinal Pigment Epithelium (RPE) layer in both eyes (Fig. 2E). Full-field ERG examination showed severely reduced rod responses and oscillatory potentials in both eyes (Fig. S1). However, combined and cone responses were only mildly reduced in both eyes (Fig. S1).
Figure 1.
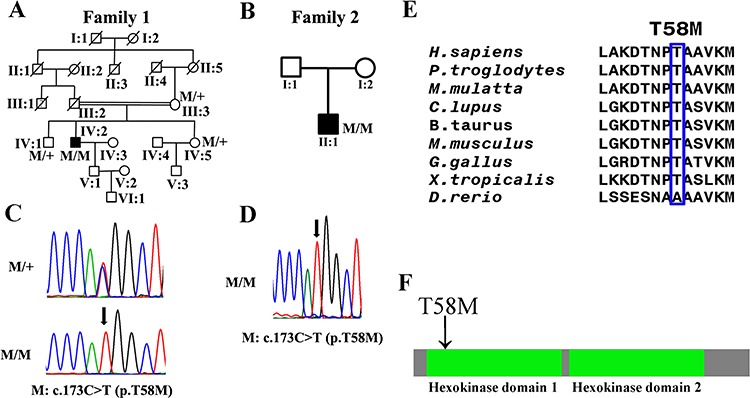
Genetic findings in this study. (A, B) The pedigree maps of two RP families with HKDC1 mutations. (C, D) Sanger sequencing confirmed the segregation of the homozygous mutation, c.173C > T, p.T58 M of the HKDC1 gene in families 1 and 2. Arrows indicate the position of altered nucleotides. (E) Protein sequence alignment of amino sequences surrounding the HKDC1 mutation with its orthologues from H. sapiens to Xenopus tropicalis. Note that the amino acid sequences surrounding the affected amino acid residues are highly conserved. (F) The mutant amino acid locates at the first hexokinase domain in HKDC1 protein.
Figure 2.
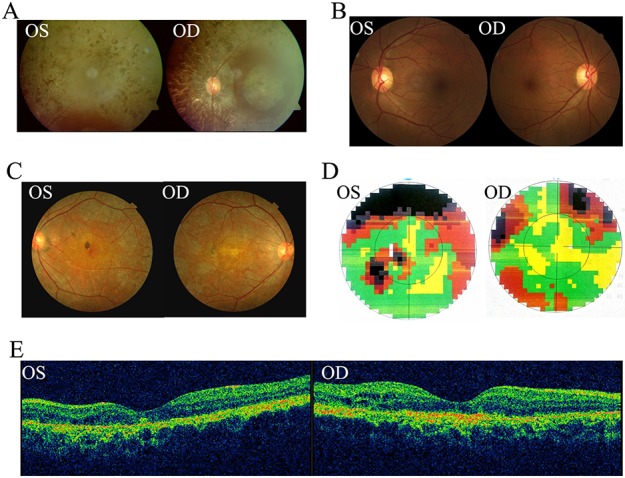
Clinical images of affected patients with RP. (A) Fundus photography shows pigment deposits of patient IV:2 in family 1. (B) Fundus images shown normal retina of IV:5. (C) Fundus images show irregular pigmentation patterns, mainly in the posterior pole and along the arcade vessels of both eyes of patient II:1 in family 2. (D) Visual field examination reveals peripheral defects in both eyes and paracentral scotoma loss in the left eye. (E) OCT examination shows a discontinuous ellipsoid zone and interdigitation zone in both eyes, and a disrupted RPE layer in both eyes.
To reveal the genetic causes for these two RP families, we performed WES on the genomic DNA of the two affected probands. For the proband of family 1, subject IV:2, 70320 Single-nucleotide polymorphisms (SNPs) and 3567 Indels were identified (Supplementary Material, Table S2). No causal mutations were found in known retinal disease genes. Candidate variants presented with frequencies of <0.1% in dbSNP database, 1000 Genomes, NHLBI Exome Sequencing Project, the ExAC Browser (Beta) and Genome Aggregation Database (gnomAD) were selected. This resulted in 218 rare protein-altering variants (Supplementary Material, Table S3). Filtering using 2117 in-house non-RP controls further narrowed the list to five candidate variants (Supplementary Material, Table S4). These five biallelic variants were subjected to candidate disease gene prioritization strategy to exclude or prioritize each gene for further functional testing. We first checked if the gene is expressed in the human retina as determined by human retinal RNA-seq data (16). We then checked if mutations in the gene are already associated with a human phenotype as determined by Online Mendelian Inheritance in Man (OMIM) and knockout (KO) animal models of each gene were available in Zebrafish Information Network or Mouse Genome Informatics (MGI). Potential deleteriousness of non-synonymous variants was determined by 12 in silico prediction scores included in dbNSFP. A variant was considered pathogenic if it was predicted damaging by 10/12 of the scores (17). A homozygous variant, c.173C > T, p.T58 M, in the HKDC1 gene was deemed the most likely candidate for the cause of RP disease in family A. This variant was predicted to be damaging or pathologic by 11 of the 12 scoring (Supplementary Material, Table S4). Sanger sequencing was performed to validate the authenticity (Fig. 1A, C). It was also found homozygous in an unrelated patient II:1 of family 2 (variants in each step of the analysis were listed in Supplementary Material, Table S5–7) from North China (Fig. 1B, D).
HKDC1 encodes 917 amino acids and contains 2 hexokinase domains. The variant occurs in the second exon2 of HKDC1 and results in a Threonine to Methionine substitution. It is unique in our in-house database of 2117 geographically matched individuals analyzed by WES and was not observed in 4 000 ethnic-matched control individuals (Supplementary Material, Table S8). Its frequency in 277 202 alleles from the gnomAD is 0.02886% (80/277 202), and no homozygous status was presented. Three additional different changes involving this codon (Thr 58 to Ala, Ser and Lys) have been reported in the gnomAD. In Asian population, the frequency of p.T58 M is 0.003445. All of these variants are rare and none is in the homozygous state. The amino acid residue affected by the p.T58 M variant is evolutionarily highly conserved from Homo sapiens to X. tropicalis (Fig. 1E) and was located at the first hexokinase domain in HKDC1 protein (Fig. 1F).
Segregation analysis with SNPs surrounding the c.173C > T, p.T58 M variant was performed to determine the haplotype in these two families (Supplementary Material, Table S9). rs3740598, which was 13 468 bp to the variant, was only found in patient 2. A second SNP, rs3740600, which was 22 835 bp to the variant, was only found in patient 1 (Supplementary Material, Table S9). This data suggested that c.173C > T, p.T58 M variant in these two patients might be more likely due to independent event rather than founder mutation.
The missense variant negatively impact HKDC1 function
Further bioinformatics analysis revealed that HKDC1 was evolutionarily conserved and shared ∼70% sequence homology with HK1 (Fig. S2), which is expressed in the retina (Fig. S3) and was reported to be associated with autosomal dominant retinitis pigmentosa (adRP) (18,19). HKDC1 encodes a kinase that phosphorylates glucose to generate glucose-6-P (19). To assess the impacts of the variant on expression, cellular localization and activity of HKDC1 protein, we introduced the p.T58 M change into the HKDC1 coding sequence and constructed mutant HKDC1 expression plasmid. Western blotting (WB) experiments revealed that unique immune band at ∼110 kDa position can be detected when hybridized with HKDC1 antibody or FLAG-tag antibody (which indicated the specificity of HKDC1 antibody), and the expression of the mutant HKDC1 protein was comparable to that of wild-type (WT) protein (Fig. 3A). Immunofluorescence staining indicated that both the WT and mutant HKDC1 proteins were localized to cytoplasm in NIH3T3 cells (Fig. 3B). In order to evaluate the effect of p.T58 M on the hexokinase activity of HKDC1, we performed hexokinase activity assay on WT and mutant HKDC1 protein. Compared with WT control, mutant HKDC1 partially lost (∼30%) hexokinase activity (Fig. 3C, D). Taken together, these data suggested that HKDC1 is a candidate gene for these two RP families.
Figure 3.
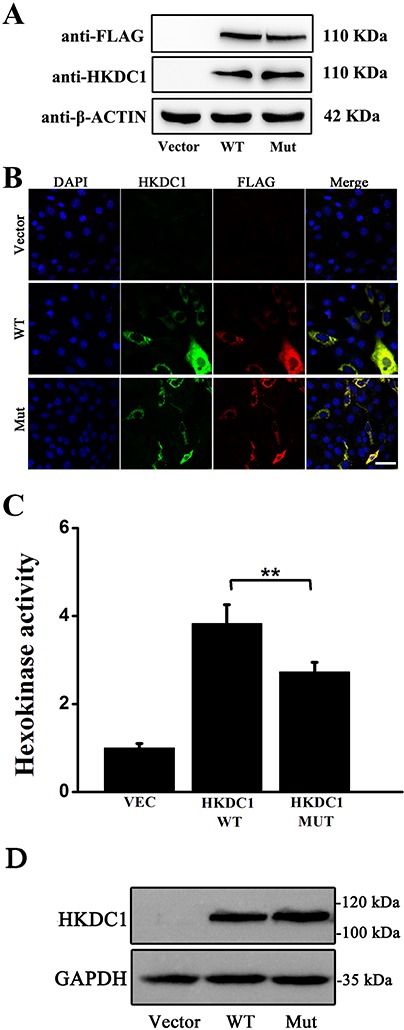
Expression, localization and functional analysis of mutant HKDC1 protein. (A) Expression of WT and mutant HKDC1 proteins in 293 T cells. WB analysis showed mutant HKDC1 protein was stable. Vector served as the negative control. Flag-tag was used to verify the specificity of HKDC1 antibody and β-actin was used as the loading control. Uncropped gel pictures were shown in Figure S7. (B) Expression and localization of WT and mutant HKDC1 proteins in NIH3T3 cells. Immunocytochemistry study using anti-HKDC1 antibody indicated that both WT and mutant HKDC1 proteins were localized in the cytoplasm. (C) A hexokinase assay revealed partial loss of kinase activity in the T58 M mutation. Vec designated the empty pCMV expression vector as a negative control. HK1 was used as a positive control. Sample size: n = 4. A representative result of three independent experiments was shown for A–C. ***, P-value < 0.001. (D) Protein expression analysis of WT and mutant HKDC1 revealed by WB. WT and mutant HKDC1 were expressed at similar level.
Hkdc1 is expressed in the mouse retina and localized to the inner segment of photoreceptors
Reverse transcription-polymerase chain reaction (RT-PCR) showed that Hkdc1 mRNA expression was present in mouse retina, brain, cerebellum, liver, lung, kidney, spleen, pancreas and intestine (Fig. 4A). The precise localization of HKDC1 in the retina is unknown. We preformed immunohistochemistry on sections of mouse retinas using an anti-HKDC1 antibody and CNG1A1. In the mouse retina, HKDC1 did not co-localize with CNG1A1 (photoreceptor OS marker, detected by mouse monoclonal antibody against CNG1A1, clone 1D1), indicating that HKDC1 protein mainly localizes to the inner segment (Fig. 4B), similar to the localization of HK1 in the inner segment (Fig. S3).
Figure 4.
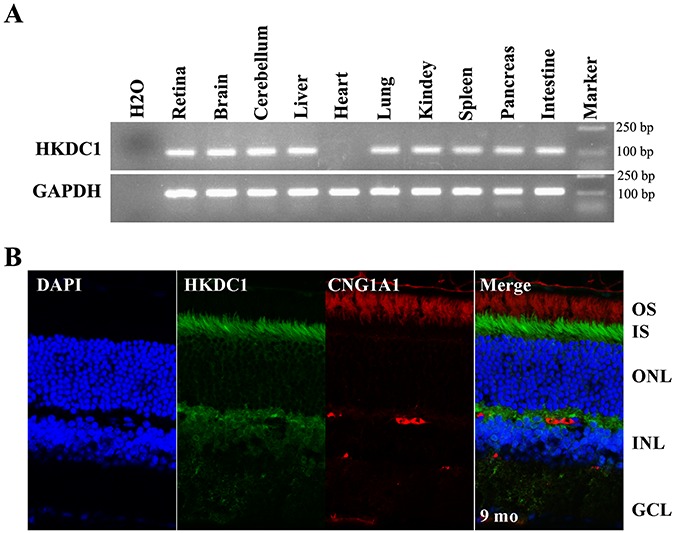
Expression and localization of HKDC1 in the mouse retina. (A) Reverse transcription PCR analysis of expression of Hkdc1 in the mouse retina, retina, brain, cerebellum, liver, heart, lung, kidney, spleen, pancreas and intestine. GAPDH was used as loading control. Hkdc1 is widely expressed. Uncropped gel pictures were shown in Figure S7. (B) Immunofluorescence labeling of retina cryosections from C57BL/6 J mice at age of 9 months using rabbit anti-HKDC1 polyclonal antibody. HKDC1 antibody was co-stained with mouse monoclonal antibody for CNG1A1 (1D1). DAPI was used to counterstain the nuclei. IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. A representative result of three independent experiments was shown. Scale bar, 20 μm.
Generation of Hkdc1 KO mice
HKDC1 has been reported as a hexokinase associated with high glucose conditions in pregnancy (21), while its in vivo roles remain unclear. To better illustrate the in vivo functions of Hkdc1, we generated Hkdc1 KO mice using CRISPR/Cas9 technology (Fig. 5A). Target site with the sequence of CCTGTATCACATGCGGCTCTCGG in exon 2 of Hkdc1 was selected. Two independent frameshift deletion mutant alleles were generated from two independent founder mice: Mut1, a 7-bp deletion: NM_145419.1, c.93_99delCTCGGATG; Mut2, an 8-bp deletion: NM_145419.1, c.87_94delGCGGCTCT (Fig. 5B). It should be noted that C57BL/6 J strain from the Jackson Laboratory was used to generate Hkdc1 KO alleles, which do not harbor the rd8 mutation in Crb1 gene (Fig. S4). We backcrossed both Hkdc1 Mut1 and Mut2 to C57BL/6 J for 6 generations and generated mutant mice with the genotypes of Mut1/Mut1, Mut2/Mut2 and Mut1/Mut2. Hkdc1 mutant mice were born at Mendelian ratio. To confirm the effect of Hkdc1 KO, we performed WB and immunohistochemistry studies using the anti-HKDC1 antibody, which recognizes a region near the C-terminal. HKDC1 protein was absent in the Mut1/Mut1, Mut2/Mut2 and Mut1/Mut2 mice retinas (Fig. 5C). In the Hkdc1Mut1/Mut2 KO mutant retina, no HKDC1 labeling was visible in photoreceptors (Fig. 5D). In addition, we performed RNA-Seq analysis on Hkdc1 KO retina to assess the expression of mutant Hkdc1 mRNA. The long isoform is the major transcript and the mutant mRNA with deletion was present in the retina transcriptome (Fig. S5). This observation was verified by RT-PCR with primers cover different regions along the transcript (Fig. S6). These findings demonstrated that HKDC1 protein was absent in Hkdc1 KO mice, and the KO alleles were null alleles. We used Mut1/Mut2 mice for further study and defined them as KO mouse.
Figure 5.
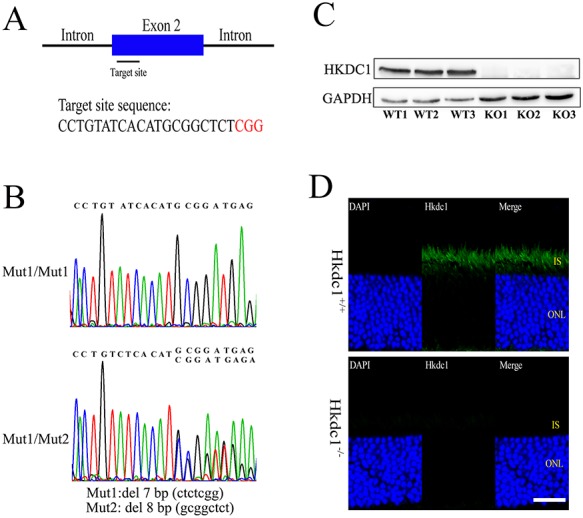
Generation of Hkdc1 KO mice using CRISPR/Cas9 technique. (A) Illustration describing the position of the target site and its sequence in the Hkdc1 gene in mouse. (B) Representative Sanger sequence results of the Hkdc1 KO mice used in this study. Mut1: NM_145419.1, c.93_99delCTCGGATG. Mut2: NM_145419.1, c.87_94delGCGGCTCT. (C) WB analysis of HKDC1 protein expression in retinas from WT or KO mice. KO1, KO2 and KO3 represented Mut1/Mut1, Mut2/Mut2 and Mut1/Mut2 genotypes, respectively. β-Actin was used as a loading control. Sample size: n = 3. Uncropped gel pictures were shown in Figure S7. (D) Immunofluorescence staining of mouse retinas from WT and KO mice at 9 months of age. DAPI was used to stain the nuclei. IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar, 20 μm. For C and D, a representative result of three independent experiments was shown.
Retinal degeneration phenotype in Hkdc1 KO mice
To test the overall function of Hkdc1 KO retinas, we performed ERG examination on Hkdc1 KO mice and littermate controls. All mutant mice at 9 months of age with genotypes Mut1/Mut1, Mut2/Mut2 or Mut1/Mut2 exhibited reduced scotopic ERG response (Figs. 6A1–A3, S7). The mean a-wave amplitude was reduced by ∼50% in KO mice. However, no obvious difference was observed in photopic responses (Fig. S8). The heterozygous mice did not show any abnormality in the ERG test (Fig. S9). To determine the pathological changes underlying the abnormal ERG results, we utilized immunohistochemistry to study retinal cryosections from WT and KO mice. In agreement with the ERG results, immunostaining revealed mislocalization of rhodopsin protein in some areas of the KO retina at 9 months of age (Figs. 6B1, B2 and S10). Rhodopsin is a critical OS protein essential for normal visual function, and the biosynthesis and transport of this protein are tightly regulated. Mislocalized rhodopsin is cytotoxic and leads to rod death (22). We also stained cryosections with markers of the Na-K pump (Fig. 6C1, C2) and cone opsin, as well as 594-conjugated peanut agglutinin. However, there was no significant difference between WT and KO retinas, indicating that cone function was not disturbed at 9 months of age (Figs. 6D1, D2 and S8).
Figure 6.
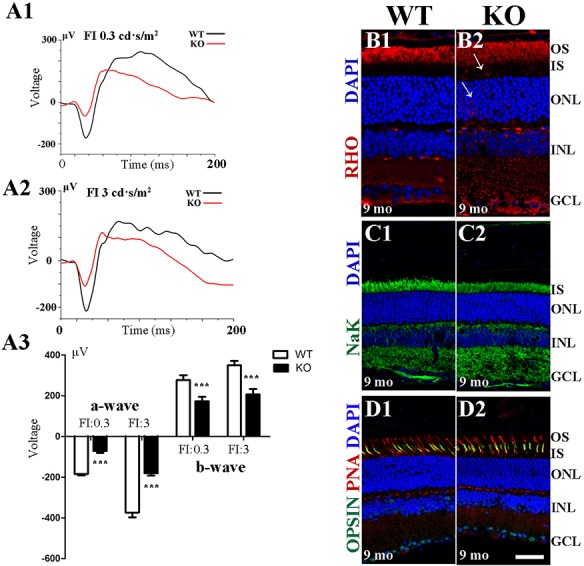
ERG examination and immunofluorescence staining analysis of Hkdc1 KO retinas. (A1–A3) KO mice at 9 month of ages show reduced scotopic ERG responses for both the a-wave and b-wave at the light intensities of 0.3 and 3 cd·s/m2. Sample size: n = 4. (B1–D2) Immunofluorescence staining of mouse retinas from WT and KO mice at 9 months of age. DAPI was used to counterstain the nuclei. Rhodopsin mislocalized to the inner segment and cell body in KO retinas. IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar, 20 μm. A representative result of three independent experiments was shown.
We next applied Hematoxylin and Eosin (H&E) staining to assess whether the retinal outer nuclear layer (ONL) thickness was changed in 9-month-old, 11-month-old and 17-month-old KO mouse retinas. The results showed that 9-month-old and 11-month-old retinal ONL thickness did not change significantly (Fig. 7A–F), but the 17-month-old retinal thickness was significantly reduced in the Hkcd1 KO retinas (Fig. 7G–I). Inner nuclear layer of 17-month-old KO retinas was also thinner (Fig. 7G–I).
Figure 7.
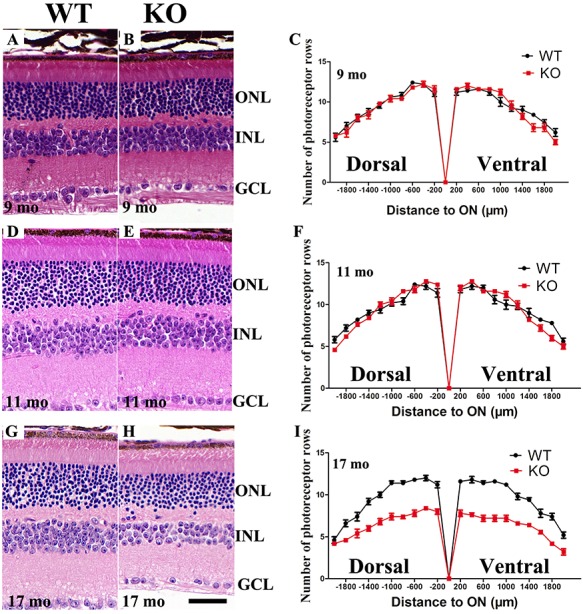
Retinal degeneration of Hkdc1 KO mice. (A–C) At 9 months of age, H&E staining of retina sections revealed that KO mice did not exhibit visible cell loss. (D–F) At 11 months of age, H&E staining of retina sections revealed that KO retina did not exhibit visible cell loss. (G–I) At 17 months of age, KO mice exhibited reduced number of cells per row. Number of cells in the INL was also reduced. Sample size: n = 4 per group.
Discussion
In this study, by using WES and genetic KO mouse models, we identified a missense variant in HKDC1 that linked to arRP in two families, highlighting the importance of HKDC1 in the retina. Functional studies indicated partial loss of hexokinase activity in the p.T58 M protein. Genetic KO mouse models exhibited reduced scotopic light response and degeneration of photoreceptors. In some area of the KO retina, rhodopsin was mislocalized to the cell bodies or inner segment. Rhoodopsin is an important protein for the proper development and function of photoreceptors. Mutations in the rhodopsin gene (RHO) are a leading cause of adRP and account for ∼25% of adRP cases (3). Alloway et al. reported that accumulation of rhodopsin-arrestin complex led to retina degeneration (22). Our genetic analysis combined with functional studies demonstrated that the missense variant in HKDC1 is associated with RP.
In both patients studied in this research and in our KO mouse models, the phenotypes of retinal degeneration were rather mild. Patient from family 1 did not exhibit vision loss and night blindness until middle age. Patient from family 2 only showed moderate peripheral vision loss at the age of 39. In agreement with these data, Hkdc1 KO mice did not exhibit visible retinal phenotypes until 9 months of age. The mild phenotype might be due to existence of other hexokinases in the retina. Further investigations are required to uncover the molecular functions of HKDC1 in the retina.
In a previous report, Ludvik et al. reported that homozygous Hkdc1 KO mice were embryonic lethal (20). The Hkdc1 allele they used was a KO first allele from Knockout Mouse Project (KOMP). The KO first allele was designed to be a KO by splicing of the cDNA of targeted gene to the LacZ cassette, which was inserted upstream of a critical exon(s). This allele can generate a null allele of the gene selected. The genetic background of the KOMP ES cells is C57BL/6 N. However, the exact background of the Hkdc1tm1a was not clear (20). In our study, two Hkdc1 frameshift deletion alleles were generated on C57BL6/J genetic background using CRISPR/Cas9 method. Homozygous mutants of both alleles and the compound heterozygous mutation of Mut1/Mut2 were all viable with no visible abnormality. Mutant mice exhibited retinal degeneration phenotypes at late stage (Figs. 6, 7). Saleheen et al. recently described a homozygous HKDC1 nonsense in a cohort with a high rate of consanguinity (22). Unfortunately, no clinical phenotype information was available for this individual. Other genetic modifiers might contribute to survival of this HKDC1 KO subject. The difference between our KO lines and the KO first line reported previously (20) warrant further investigation.
HKDC1 is a new hexokinase gene, which is homologous to HK1–4 in the hexokinase gene family involving in glucose metabolism (23,24). Based on sequence homology, HKDC1 is most similar to HK1, and these genes are in close genetic proximity on Chromosome 10 in humans. Despite this high homology, the enzyme activity of HKDC1 is much lower than that of HK1 (20). It is possible that functions of HKDC1 are related but become distinct when HKDC1 evolved from HK1 during evolution. HKDC1 might arise through a tandem duplication event either by translocation or transposition. The newly formed hexokinase gene might gain tissue-specific activity in the retina. In addition, HKDC1 was described to have a putative role in phosphorylation of glucose during maternal metabolism during pregnancy (20). Genetic variants in the first intron of HKDC1 were associated with gestational hyperglycemia and alleles associated with higher maternal 2-h glucose levels decrease HKDC1 expression. Guo et al. demonstrated that HKDC1 protein had hexokinase activity using purified HKDC1 protein and its activity was ∼20% of the specific activity of HK1 under the same condition (23). Such low hexokinase activity may have prevented earlier detection of HKDC1 as a hexokinase by less sensitive biochemical screening. Based on these finding, they proposed to name HKDC1 as HK5. In our study, we also detected hexokinase activity using 293 T cells transient expressing HKDC1 and T58 M mutant. T58 M mutant protein lost 30% of hexokinase activity under the same condition (Fig. 3). We tested fasting glucose level and glucose intolerance in both male and pregnant female Hkdc1 KO mice. Compared to littermate controls, no obvious difference was observed in KO mice (Fig. S11).
HK1 mutations cause severe non-spherocytic hemolytic anemia and hereditary motor and sensory neuropathy (25–28). Previous reports have shown that mutations in HK1 result in adRP (18,19). However, the identified HK1 mutation did not affect hexokinase activity, suggesting an alternative unknown mechanism involved in this adRP condition. It has been established that HK1 plays essential roles in both glucose sensing and glucose metabolism (27). Previous studies have showed that HK1 (named HXK1 in Arabidopsis) mutation lacking catalytic activity were able to initiate various signaling functions in gene regulation, cell proliferation, growth and senescence, demonstrating the uncoupling of glucose signaling from glucose metabolism (28). Similar to HXK1 in the Arabidopsis, HKDC1 might function as a glucose sensor in the mammalian retina. In addition, neither patient exhibited non-spherocytic hemolytic anemia or motor and sensory neuropath.
In summary, by using WES analysis and genetic KO mouse model, we demonstrate that HKDC1 is a candidate gene associated with arRP.
Materials and Methods
Subjects and clinical evaluation
This study adhered to the Declaration of Helsinki. All study protocols were approved by institutional ethics committees of Sichuan Provincial People’s Hospital, Peking Union Medical College Hospital, Peking Union Medical College and Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine. Written informed consents were obtained from all subjects who participated in this study. All participants were diagnosed by a clinical ophthalmologist, geneticist and pediatrician based mainly on fundus photographic and angiographic changes.
Genomic DNA preparation
Peripheral blood from members of the two RP families and control individuals were collected into EDTA anticoagulant tubes and then genomic DNA were extracted using a blood DNA extract kit according to the manufacture’s protocol (Qiagen, Germantown, Maryland, USA). DNA was stored at −20°C for subsequent analysis.
Library preparation and WES
Approximately 1 μg of the genomic DNA sample was sheared into fragments of 200–500 bp in length. Then the sheared fragments were end repaired and Klenow exonuclease was used to add an ‘A’ base to the 3′ end. Illumina index adaptors were ligated to the repaired ends and then eight cycles of PCR amplification were applied to each sample. A total of 50 pre-capture DNA libraries were pooled together in each capture reaction. The targeted DNA was captured by NimbleGenSeqCap EZ Hybridization and Wash kit (NimblegenSeqCap EZ Human Exome Library v.2.0, Roche, Redwood City, CA, USA) following the manufacturer’s protocols for WES. Captured libraries were then sequenced on Illumina HiSeq 2500 (Illumina, San Diego, CA, USA).
Bioinformatics analysis
Sequencing data was analyzed by the software of NextGene V2.3.4 and reads were compared to the hg19 human reference sequence from The Universoty of California, Santa Cruz (UCSC) (http://genome.ucsc.edu/) by burrows-wheeler aligner (BWA) (http://bio-bwa.sourceforge.net/). Variants were called using Atlas2 and noted by ANNOtation of genetic VARiant (ANNOVAR). Variant frequency data were obtained from the dbSNP database, 1000 Genomes (http://browser.1000genomes.org/index.html), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/, ESP6500), the ExAC Browser (Beta) (http://exac.broadinstitute.org/), gnomAD browser beta (http://gnomad.broadinstitute.org/) and 2805 in-house non-RP controls. The strategies for prioritization and determination of pathogenicity variants were: (1) variants with total read depth >5X with the snp quality score >50; (2) minor allele frequency <0.1% in all the six variant databases for recessive genes; (3) variants were SNVs (stoploss, stopgain, non-synonymous, non-frameshift substitution, non-frameshift insertion, non-frameshift deletion, frameshift insertion, frameshift deletion) or splice site variants (splicing within 7-bp of a splicing junction); (4) variants were consistent with the pattern of inheritance models (homozygous/compound heterozygous); (5) the gene is expressed in the human retina as determined by human retinal RNA-seq data (16); (6) variants were assessed for the potential deleteriousness as determined by 12 in silico prediction scores included in dbNSFP. If the variant wasn’t predicted damaging by 10/12 of the scores the variant was considered likely benign. Sanger sequencing and segregation analysis were performed for variant confirmation in the whole family members.
Variant validation
To confirm the variant identified by WES, primer sequences were designed to amplify genomic DNA fragment containing the candidate locus. The primer sequences were as follows: HKDC1-F, 5′-TAGGGAGGCTTTGGGAATCC-3′ and HKDC1-R, 5′-GCAGAGAGAGGGCTAGAGAG-3′. DNA samples from the two families and 4000 control individuals were subjected to Sanger sequencing.
mRNA analysis by RT-PCR
Total RNA was extracted from male C57BL/6 J mice tissues including retina, brain, cerebellum, liver, heart, lung, kidney, spleen, pancreas and intestine. The cDNA was synthesized by Superscript cDNA Synthesis Kit (Invitrogen, Waltham, MA, USA) and then used as template for PCR with Hkdc1 (Hkdc1-ms-F: 5′-CCACTGCCTCTGTGAAGATG-3′, Hkdc1-ms-R: 5′-GACCCGAAACTTGGATCCTC-3′) and Gapdh (Gapdh-ms-F: 5′-CGTCCCGTAGACAAAATGGT-3′, Gapdh-ms-R: 5′-TTGATGGCAACAATCTCCAC-3′) primers. The PCR products were subjected to DNA electrophoresis on a 3% agarose gel.
Site-direct mutagenesis
C-terminal Flag-tagged HKDC1 expression vectors were purchased from Origene Inc. (Rockville, MD, USA). Point mutation was introduced into the WT HKDC1 cDNA by site-directed mutagenesis using a QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). The recombinant plasmids containing HKDC1-Flag fusion constructs were verified by DNA sequencing.
Immunocytochemistry
NIH3T3 cells (American Type Culture Collection, ATCC, Manassas, VA, USA) were seeded on coverslips and transiently cultured in DMEM medium with high glucose (HyClone) supplemented with 10% foetal bovine serum and 1% (vol/vol) penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. Cells were seeded in 24-well plates (Corning, NY, USA) and transfected at 70% confluency with 600 ng of pCMV6-HKDC1 or empty vectors using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions. Cells were harvested after 48 h and fixed in 4% paraformaldehyde for 15 min at room temperature. After blocking with 1 × PBS containing 5% normal goat serum and 0.2% Triton X-100, cells were incubated with anti-Flag antibody (Cat# F1804, Sigma, St Louis, MO, USA, 1:500 dilution) and HKDC1 antibody (Cat# 209846, Abcam, Cambridge, MA, USA, 1:200 dilution) at 4°C overnight. Alexa-Fluor 594/488-conjugated goat anti-mouse/rabbit secondary antibody (Cat# A11005 and A11008, Invitrogen, Waltham, MA, USA, 1:500 dilution) was applied and nuclei were counter stained with DAPI (Cat# D8417, Sigma, St Louis, MO, USA).
Hexokinase activity assay
Hexokinase activity of crude cytosolic fractions was determined using a Hexokinase colorimetric assay kit (Cat# K789–100, BioVision, Milpitas, CA, USA), following the manufacturer’s instructions. Briefly, cells were transfected with 1 μg WT and mutant HKDC1 expression vectors using Lipofactamine 3000 and harvested and washed twice. A total of 1 mL of exaction solution was added to every 5 × 106 cells. Cells were extracted via ultrasonication and then centrifuged for 10 min at 4°C at 8000 g to remove any insoluble material. A total of 10 μl cell extract, 10 μl reagent 3 and 180 μl reagent 2 were added into 96-well plate and mixed, and then OD 340 nm A1 at 20s and A2 at 320 s was measured. Hexokinase activity was calculated as ∆A = A2-A1 and presented as relative activity, as compared to the value of empty vector.
Generation of Hkdc1 KO mice by the CRISPR/Cas9 system
All animal research followed National Institutional of Health guideline. All animal experiment study protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Sichuan Provincial People’s Hospital. Cervical dislocation was used for mouse euthanasia.
We generated Hkdc1 KO mouse on the C57/BL6J (The Jackson Laboratory, Bar Harbor, Maine, USA, stock number 000664) strain using CRISPR/Cas9 method. The sgRNA sequence 5′-CCTGTATCACATGCGGCTC TCGG-3′ is located in the second exon of Hkdc1 gene. Two independent mutant alleles were generated: deletion of 7 base-pair in exon 2 (Mut1) and deletion of 8 base-pairs in exon 2 (Mut2). Both of these two mutations led to frameshift change in Hkdc1 transcript, resulting in truncation of the HKDC1 protein. Both mutant stains were backcrossed to C57BL/6 J for 5 generations before intercross to remove potential off-target variants that might be introduced by CRISPR/Cas9. Genotypes were confirmed by Sanger sequencing with the following primers: Hkdc1-Forward, 5′-TGGTTAGGAACACATAGAACAAA-3′ and Hkdc1-Reverse, 5′-CTAAGGGGCTGGTATGGGAAT-3′.
WB analysis
293 T cells or retinas from the WT or Hkdc1 KO mice were lysed in SDS lysis buffer (2% SDS and 62.5 mM Tris–HCl, pH 6.8, containing protease inhibitors cocktail tablets ordered from Roche Inc.) and sonicated three times for 5 s. Equal amount of protein (20 μg) were loaded to a 10% polyacrylamide gel and analyzed by immunoblotting. The antibodies used for WB are anti-Flag (Cat# F1804, Sigma, St Louis, MO, USA, 1:3000 dilution), anti-HKDC1 (Cat# ab150764, Abcam, Cambridge, MA, USA) and anti-β-Actin (Cat# 20536–1-AP, Proteintech, Chicago, IL, USA). Horseradish peroxidase (HRP) conjugated goat anti-rabbit secondary antibody (Cat#7074, Cell Signaling Technology, USA) was used for immunoblotting.
Immunohistochemistry analysis
For immunohistochemistry analysis, eyes from the WT and KO mice at the age of 9 months were enucleated and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 2 h, soaked in 30% sucrose in PBS for 2 h for dehydration. After dehydration, the lenses were removed and the eyes were embedded into OCT compound. 10 μm cryosections were then collected for the latter immunofluorescent analysis. The following primary antibodies were used: HKDC1 (Cat# ab150764, Abcam, Cambridge, MA, USA, 1:100 dilution), rhodopsin (mouse monoclonal antibody, 1D4, Cat# MA1–722, Thermo Fischer,USA, 1:500 dilution), Na-K pump (Monoclonal Na-K antibody, Thermo Fisher Scientific, Waltham, MA, U.S.A, Cat # MA3–928, 1:500 dilution), opsin (Red/Green, Cat# AB5405, Millipore, Burlington, MA, USA, 1:500), 594-conjugated peanut agglutinin (PNA, Cat# RL1072, Vector laboratories, Burlingame, CA, USA), Alexa Fluor 488- and Alexa Fluor 594-tagged secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI, Cat# D8417, Sigma, St Louis, MO, USA, 1:2000) were also used in the experiments. Eye sections were viewed under a confocal microscope (LSM800, Carl Zeiss, Jena, Germany).
ERGs in mice
Mice at the age of 9 months were dark-adapted overnight and all the subsequent procedures were performed under dim red light. The mice were anesthetized using a combination of ketamine (16 mg/kg body weight) and xylazine (80 mg/kg body weight) in normal saline. After their pupils were dilated, dark-adapted ERGs were recorded on with the responses to short-wavelength flashes over 4.0 log units to the maximum intensity with the Espion Visual Electrophysiology System from Diagnosis, LLC (Littleton, MA, USA). Cone-mediated ERGs were recorded with white flashes after 20 min of complete light adaptation.
Measurement of retina outer nuclear layer
Eyes from WT or KO mice were removed, marked on the nasal side for orientation and were fixed overnight in 1.22% glutaraldehyde and 0.8% paraformaldehyde in 0.08 M phosphate buffer, embedded in paraffin and cut into 5-μm sections. H&E-stained retinas were counted for the rows of photoreceptors in the outer nuclear layer. Three measurement of the outer nuclear layer were taken every 200 μm from the optic nerve and averaged. The optic nerve was designated as 0 μm.
Supplementary Material
Acknowledgements
The authors want to thank all affected individuals and their family members for their participation. The funders had no role in study design, data collection and analysis or preparation of the manuscript.
Conflict of Interest statement. None declared.
Electronic Database Information
GenBank, http://www.ncbi.nlm.nih.gov/genbank/; 1000 Genomes, http://www.internationalgenome.org/; ExAC Browser, http://exac.broadinstitute.org/; GeneMatcher, https://genematcher.org/; GnomAD Browser, http://gnomad.broadinstitute.org/; HGMD, http://www.hgmd.cf.ac.uk/; Mouse Genome Informatics, http://www.informatics.jax.org/; NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/; OMIM, http://www.omim.org/; Optimized CRISPR Design, http://crispr.mit.edu/; RetNet - Retinal Information Network, https://sph.uth.edu/retnet/; Protein Databank, http://www.rcsb.org/pdb/; Zebrafish Information Network,http://zfin.org/; https://komp.org/alleles.php.
Funding
National Natural Science Foundation of China (http://www.nsfc.gov.cn/; 81770950, 21561142003, 81470668 to X.Z.; 81700876 to L.Z.; 81790643, 81430008 to Z.Y.; 81770964, 81470642 to P.Z; 81525006, 81670864 to C.Z.; 81470669 to R.S.; 81670895 to L.H.; 31471196 to H.W.); National Key Scientific Research Program (www.most.gov.cn; 2015CB554100 to X.Z.; 2016YFC0905200 to Z.Y.); Department of Science and Technology of Sichuan Province (www.scst.gov.cn; 2016TD0009, 2014JQ0023, 2017TJPT0010 to X.Z.); Chinese Postdoctoral Science Foundation (res.chinapostdoctor.org.cn; 2016M600734; 2108YSZH0020 to L.Z.); Foundation Fighting Blindness (www.blindness.org; CD-CL-0808-0470-PUMCH, CD-CL-0214-0631-PUMCH to R.S.; BR-GE-0613-0618-BCM to R.C.); Beijing Natural Science Foundation (www.bjnsf.org; 7152116 to R.S.); National Key Basic Research Program of China (www.most.gov.cn; 2013CB967500 to C.Z.); National Eye Institute (www.nih.gov; R01EY022356, R01EY018571, EY002520 to R.C.), NIH Shared Instrument Grant (1S10RR026550 to R.C.); National Key Research and Development Program of China (2016YFC0902100 to H.W.).
References
- 1. Bunker C.H., Berson E.L., Bromley W.C., Hayes R.P. and Roderick T.H. (1984) Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol., 97, 357–365. [DOI] [PubMed] [Google Scholar]
- 2. Grøndahl J. (1987) Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin. Genet., 31, 255–264. [DOI] [PubMed] [Google Scholar]
- 3. Hartong D.T., Berson E.L. and Dryja T.P. (2006) Retinitis pigmentosa. Lancet, 368, 1795–1809. [DOI] [PubMed] [Google Scholar]
- 4. Daiger S., Sullivan L. and Bowne S. (2013) Genes and mutations causing retinitis pigmentosa. Clin. Genet., 84, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hong D.-H., Pawlyk B.S., Shang J., Sandberg M.A., Berson E.L. and Li T. (2000) A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl. Acad. Sci., 97, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Namburi P., Ratnapriya R., Khateb S., Lazar C.H., Kinarty Y., Obolensky A., Erdinest I., Marks-Ohana D., Pras E., Ben-Yosef T. et al. (2016) Bi-allelic truncating mutations in CEP78, encoding centrosomal protein 78, cause cone-rod degeneration with sensorineural hearing loss. Am. J. Hum. Genet., 99, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mansergh F.C., Millington-Ward S., Kennan A., Kiang A.-S., Humphries M., Farrar G.J., Humphries P. and Kenna P.F. (1999) Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am. J. Hum. Genet., 64, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kajiwara K., Berson E.L. and Dryja T.P. (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264, 1604–1608. [DOI] [PubMed] [Google Scholar]
- 9. Daiger S.P., Sullivan L.S., Bowne S.J., Koboldt D.C., Blanton S.H., Wheaton D.K., Avery C.E., Cadena E.D., Koenekoop R.K. and Fulton R.S. (2016) Identification of a Novel Gene on 10q22.1 Causing Autosomal Dominant Retinitis Pigmentosa (adRP). Adv. Exp. Med. Biol., 854, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin Z.B., Huang X.F., Lv J.N., Xiang L., Li D.Q., Chen J., Huang C., Wu J., Lu F. and Qu J. (2014) SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat. Commun., 5, 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El Shamieh S., Neuille M., Terray A., Orhan E., Condroyer C., Demontant V., Michiels C., Antonio A., Boyard F., Lancelot M.E. et al. (2014) Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am. J. Hum. Genet., 94, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeitz C., Jacobson S.G., Hamel C.P., Bujakowska K., Neuille M., Orhan E., Zanlonghi X., Lancelot M.E., Michiels C., Schwartz S.B. et al. (2013) Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet., 92, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu M., Xie Y.A., Abouzeid H., Gordon C.T., Fiorentino A., Sun Z., Lehman A., Osman I.S., Dharmat R., Riveiro-Alvarez R. et al. (2017) Mutations in the spliceosome component CWC27 cause retinal degeneration with or without additional developmental anomalies. Am. J. Hum. Genet., 100, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coppieters F., Ascari G., Dannhausen K., Nikopoulos K., Peelman F., Karlstetter M., Xu M., Brachet C., Meunier I., Tsilimbaris M.K. et al. (2016) Isolated and syndromic retinal dystrophy caused by biallelic mutations in RCBTB1, a gene implicated in ubiquitination. Am. J. Hum. Genet., 99, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nikopoulos K., Farinelli P., Giangreco B., Tsika C., Royer-Bertrand B., Mbefo M.K., Bedoni N., Kjellstrom U., El Zaoui I., Di Gioia S.A. et al. (2016) Mutations in CEP78 cause cone-rod dystrophy and hearing loss associated with primary-cilia defects. Am. J. Hum. Genet., 99, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farkas M.H., Grant G.R., White J.A., Sousa M.E., Consugar M.B. and Pierce E.A. (2013) Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics, 14, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soens Z.T., Li Y., Zhao L., Eblimit A., Dharmat R., Li Y., Chen Y., Naqqeb M., Fajardo N., Lopez, I. et al. (2015) Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet. Med., 18, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sullivan L.S., Koboldt D.C., Bowne S.J., Lang S., Blanton S.H., Cadena E., Avery C.E., Lewis R.A., Webb-Jones K., Wheaton D.H. et al. (2014) A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest Ophthalmol. Vis. Sci., 55, 7147–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang F., Wang Y., Zhang B., Zhao L., Lyubasyuk V., Wang K., Xu M., Li Y., Wu F. and Wen C. (2014) A missense mutation in HK1 leads to autosomal dominant retinitis pigmentos. Invest. Ophthalmol. Vis. Sci., 55, 7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ludvik A.E., Pusec C.M., Priyadarshini M., Angueira A.R., Guo C., Lo A., Hershenhouse K.S., Yang G.Y., Ding X., Reddy T.E. et al. (2016) HKDC1 is a novel hexokinase involved in whole-body glucose use. Endocrinology, 157, 3452–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo C., Ludvik A.E., Arlotto M.E., Hayes M.G., Armstrong L.L., Scholtens D.M., Brown C.D., Newgard C.B., Becker T.C., Layden B.T. et al. (2015) Coordinated regulatory variation associated with gestational hyperglycaemia regulates expression of the novel hexokinase HKDC1. Nat. Commun., 6, 6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alloway P.G., Howard L. and Dolph P.J. (2000) The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron, 28, 129–138. [DOI] [PubMed] [Google Scholar]
- 23. Saleheen D., Natarajan P., Armean I.M., Zhao W., Rasheed A., Khetarpal S.A., Won H.H., Karczewski K.J., O'Donnell-Luria A.H., Samocha K.E. (2017) at al, Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature., 544, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Irwin D.M. and Tan H. (2008) Molecular evolution of the vertebrate hexokinase gene family: Identification of a conserved fifth vertebrate hexokinase gene. Comparative biochemistry and physiology. Part D, Genomics Proteomics, 3, 96–107. [DOI] [PubMed] [Google Scholar]
- 25. Wijk R., Rijksen G., Huizinga E.G., Nieuwenhuis H.K. and Solinge W.W. (2003) HK Utrecht: missense mutation in the active site of human hexokinase associated with hexokinase deficiency and severe nonspherocytic hemolytic anemia. Blood, 101, 345–347. [DOI] [PubMed] [Google Scholar]
- 26. Bianchi M. and Magnani M. (1995) Hexokinase mutations that produce nonspherocytic hemolytic anemia. Blood cells Mol. Dis., 21, 2–8. [DOI] [PubMed] [Google Scholar]
- 27. Hantke J., Chandler D., King R., Wanders R.J., Angelicheva D., Tournev I., McNamara E., Kwa M., Guergueltcheva V., Kaneva R. et al. (2009) A mutation in an alternative untranslated exon of hexokinase 1 associated with hereditary motor and sensory neuropathy -- Russe (HMSNR). Eur. J. Hum. Genet., 17, 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore B., Zhou L., Rolland F., Hall Q., Cheng W.H., Liu Y.X., Hwang I., Jones T. and Sheen J. (2003) Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science, 300, 332–336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.