

Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are neurodegenerative diseases characterized by the progressive loss of specific groups of neurons. Due to clinical, genetic and pathological overlap, both diseases are considered as the extremes of one disease spectrum and in a number of ALS and FTD patients, fused in sarcoma (FUS) aggregates are present. Even in families with a monogenetic disease cause, a striking variability is observed in disease presentation. This suggests the presence of important modifying genes. The identification of disease-modifying genes will contribute to defining clear therapeutic targets and to understanding the pathways involved in motor neuron death. In this study, we established a novel in vivo screening platform in which new modifying genes of FUS toxicity can be identified. Expression of human FUS induced the selective apoptosis of crustacean cardioactive peptide (CCAP) neurons from the ventral nerve cord of fruit flies. No defects in the development of these neurons were observed nor were the regulatory CCAP neurons from the brain affected. We used the number of CCAP neurons from the ventral nerve cord as an in vivo read-out for FUS toxicity in neurons. Via a targeted screen, we discovered a potent modifying role of proteins involved in nucleocytoplasmic transport. Downregulation of Nucleoporin 154 and Exportin1 (XPO1) prevented FUS-induced neurotoxicity. Moreover, we show that XPO1 interacted with FUS. Silencing XPO1 significantly reduced the propensity of FUS to form inclusions upon stress. Taken together, our findings point to an important role of nucleocytoplasmic transport proteins in FUS-induced ALS/FTD.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are adult-onset progressive neurodegenerative disorders, characterized by the aggregation of different proteins (1). ALS patients show selective loss of motor neurons in the brain and spinal cord, resulting in progressive muscle weakness and death within 2–5 years after symptom onset (2). The majority of ALS patients are classified as sporadic ALS (sALS), while about 10% of patients show a clear family history (fALS) (2). No effective therapy for ALS is available and the FDA-approved drugs riluzole and edaravone have only a limited impact on disease progression (3,4). FTD, the second most common form of early onset dementia, is characterized by the degeneration of cortical neurons in the brain (5) and is clinically subdivided into a behavioral and a language subtype (5).
Although ALS and FTD manifest in different ways, there is a significant overlap on clinical, genetic and neuropathological levels (6). The FUSopathies, a subgroup within this disease spectrum, are characterized by fused in sarcoma (FUS) inclusions in neurons and glial cells. With roles in the regulation of DNA and RNA metabolism, FUS is involved in many cellular processes and has more than 8000 RNA targets (7). Under normal physiological conditions, FUS is predominantly localized in the nucleus (8) but is able to shuttle between the nucleus and the cytoplasm through binding of Transportin (TNPO1) to its C-terminal nuclear localization signal (NLS) (9).
Mutations in FUS are responsible for a small but important subset of fALS and sALS cases, accounting for 4 and 1% of cases, respectively (10–13). Disease-causing mutations are mainly present in the N-terminal low-complexity domain and in the highly-conserved C-terminal NLS (7). This leads to the mislocalization of FUS and results in the formation of cytoplasmic FUS inclusions (14). In addition, mutations in the 3′ untranslated region of the FUS gene increase the expression levels of the FUS protein (15). In FTD patients, the FUS gene is not mutated, while FUS pathology is present in approximately 10% of these patients (6,16).
ALS patients show a striking variability in disease presentation, even within families with a monogenetic disease cause (2,17). Although median survival is only 3 years after symptom onset, approximately 10% of patients live longer than 10 years, without showing pathological differences (2,18). This suggests the presence of genetic modifiers. The identification of these genetic modifiers will have a great impact for predicting the disease course of patients, but also for understanding the pathways of motor neuron death and for the development of novel therapeutic strategies.
In this study, we developed a novel in vivo platform to screen for potential modifiers and identified factors involved in nucleocytoplasmic transport to play a role in FUS neurotoxicity. We created an ALS-FUS Drosophila model in which we observed the loss of a particular subset of neurons. We used this phenotype to screen for potent modifiers of FUS toxicity and first validated our model by confirming the modifying capacities of PINK1 and Parkin (PRKN), two known suppressors of FUS toxicity (19). Next, we studied the modifying role of proteins involved in nucleocytoplasmic transport. In line with the recent studies of C9orf72 and TDP-43-induced toxicity (20–24), we discovered that the nuclear pore protein, nucleoporin (Nup) 154 and the nuclear transport protein, Exportin1 (XPO1), suppressed FUS-induced neurotoxicity. In addition, we found evidence for an important role of XPO1 in the formation of FUS inclusions, suggesting XPO1 to be an important disease modifier of ALS/FTD pathogenesis.
Results
FUS overexpression in motor neurons induces pupal lethality with immature adult escapers
In order to create an ALS-FUS Drosophila model, we overexpressed human wild-type (WT) and mutant FUS (FUS p.R521H and FUS p.R521G, referred to as R521G hFUS and R521H hFUS) (25). To avoid any influence of the genomic environment on the transgene expression, the UAS-hFUS transgenes were targeted to the 35B landing site on chromosome 2 or to the VK31 landing site on chromosome 3. By comparing the effects of both fly lines, we excluded effects based upon transgene integration and have greater versatility to recombine the transgenes with different driver lines. Using the UAS-GAL4 expression system, spatial and temporal control of transgene expression was obtained.
To assess the effects of hFUS expression on neurons in vivo, we expressed hFUS transgenes in the motor neurons of Drosophila using the OK6-GAL4 driver. This resulted in partial pupal lethality as pharate adults were not able to hatch from their pupal case leading to significantly reduced eclosion rates (Fig. 1A). Although the pupal lethality was not 100%, all eclosing adults showed an immature phenotype characterized by unexpanded wings, disorganization of the scutellar bristles and defects in the sclerotization and melanization of the cuticle (Fig. 1A and B, middle panel).
Figure 1.
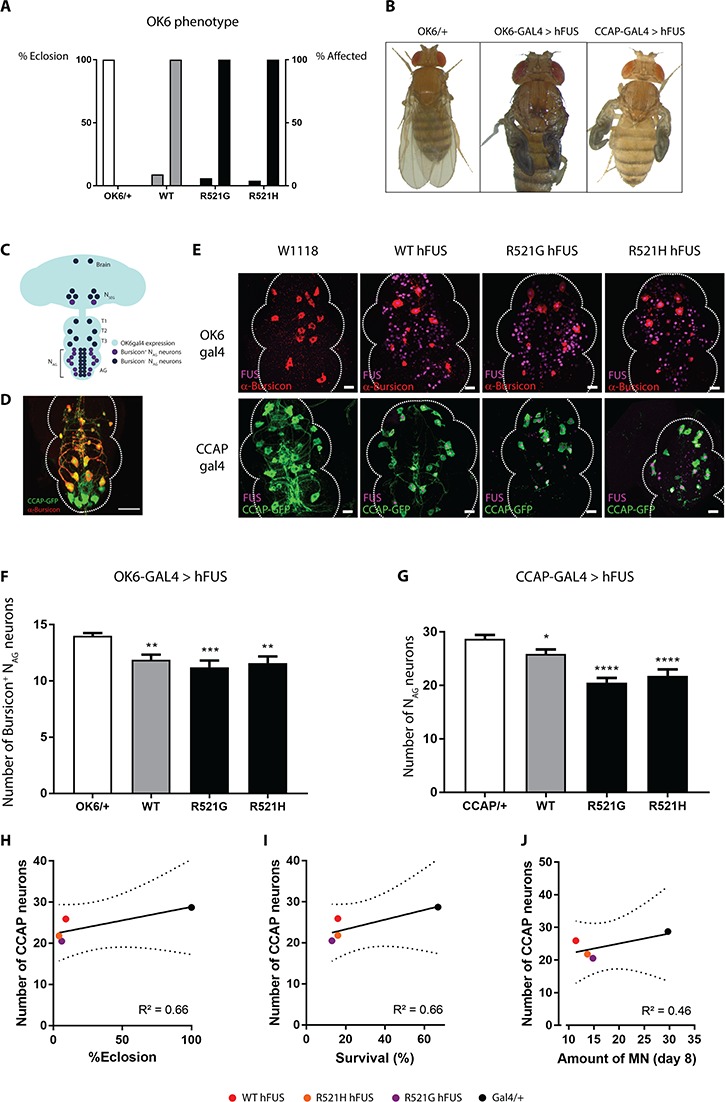
Motor neuronal expression of hFUS induces an immature fly phenotype with underlying NAG neuronal loss. (A) hFUS expression in motor neurons via OK6-GAL4 driver induced eclosion defects, as pharate adults are not able to hatch from their pupal case. All adult escapers (% affected, right bar) eclosed with immature defects. (B) Example of an affected fly showing cuticle hardening, tanning and wing inflation (middle panel) in comparison with control flies (left panel). Limiting hFUS expression to CCAP neurons recapitulates this immature phenotype (right panel). (C) OK6-GAL4 drives hFUS expression in motor neurons from the Drosophila central nervous system (light blue). CCAP-GAL4 drives expression in the CCAP neurons from the brain (NSEG) and the ventral nerve cord (T1, T2, T3 and NAG) in Bursicon+ NAG neurons (purple) and in Bursicon− NAG neurons (dark blue). (D) The CCAP;GFP-GAL4 line labeled the CCAP neurons with a GFP-tag (green) facilitating their visualization. The driver is used to drive expression of hFUS in all CCAP neurons (NSEG + T1-T3 + NAG). A subset of 15 CCAP neurons (red) secrete the neurohormone Bursicon (Bursicon+ NAG neurons). Scale bar = 20 μm. (E) Immunofluorescent confocal analysis revealed the loss of Bursicon+ NAG neurons (red) upon OK6-GAL4 driven hFUS expression (magenta, upper panel). Confocal analysis of the GFP-labeled CCAP neurons (green) that express hFUS (magenta) showed a loss of the NAG neurons (lower panel). The tip of the ventral nerve cord is oriented at the top. Scale bar = 20 μm. (F) Quantification of confocal images from upper panel of E. (G) Quantification of confocal images from lower panel of E. (H–J) Correlation between the number of CCAP neurons in young adult (CCAP-GAL4) and the eclosion phenotype upon motor neuronal expression of FUS (OK6-GAL4) (H), the survival of flies with motor neuronal FUS expression (D42-GAL4) (25) (I) and the amount of motor neurons present in aged flies (Day 8) with FUS expresssion in the motor neurons (D42-GAL4) (25) (J). For statistical testing one-way ANOVA and Holm-Sidak multiple comparison test was used. *P < 0.05, **P < 0.01, ****P < 0.0001. Data are presented as mean ± SEM, n = 15.
Loss of CCAP neurons underlies the immature phenotype
Wing expansion, sclerotization and melanization of the cuticle is regulated by Bursicon, a neuropeptide secreted by a subset of 15 ‘crustacean cardioactive peptide’ (CCAP) neurons in the ventral nerve cord (Bursicon+ NAG neurons) in the early post-eclosion phase (Fig. 1C and D) (26,27). In addition, these CCAP neurons regulate the different transitions in the fruit fly, inducing the eclosion motor program the fly needs to eclose from the pupal case (27). Given the phenotypes that we observed when overexpressing hFUS transgenes with OK6-GAL4, we investigated whether the observed defects were due to a defect in the CCAP neurons, as OK6-GAL4 also drives expression of hFUS transgenes in the CCAP neurons. Limiting human FUS expression to the CCAP neurons using the CCAP-GAL4 driver resulted in adult flies showing the same immature defects as observed when hFUS was expressed in all motor neurons (Fig. 1B, right panel). This strongly suggests that the CCAP neurons are indeed involved in the observed phenotype.
As expression of hFUS in CCAP neurons is capable of recapitulating the immature phenotype that we observed when hFUS was expressed in motor neurons, we reasoned that a defect in CCAP neurons underpins the immature phenotype in these flies. To investigate this, we visualized the Bursicon+ NAG neurons from the abdominal ganglia (AG) by immunohistochemical labeling of Bursicon (Fig. 1C and D). Mutant or WT human FUS expression in the motor neurons, using the OK6-GAL4 driver, induced a significant loss of Bursicon+ NAG neurons (Fig. 1E and F). In addition, CCAP-GAL4-driven hFUS expression induced the same defect in maturation as OK6-GAL4-driven expression. Therefore, we visualized the total population of CCAP neurons from the abdominal ganglia (NAG neurons) of the ventral nerve cord, by using a GFP-labeled CCAP-GAL4 line (CCAP;GFP-GAL4) (Fig. 1C–E). Analysis of the NAG neurons upon CCAP;GFP-GAL4-driven human WT or mutant FUS expression also showed a reduction in the total number of NAG neurons (i.e. sum of Bursicon+ NAG and Bursicon− NAG neurons) (Fig. 1E and G). When mutant hFUS was expressed in the CCAP neurons, this induced mainly the loss of the Bursicon+ NAG neurons. On the other hand, WT hFUS caused rather the loss of the Bursicon− NAG neurons (Supplementary Material, Fig. S1). As a consequence, our results strongly indicate that human FUS expression affects the maturation process of the adult fly due to the loss of the CCAP neurons.
Previously, we observed adult motor defects present in this ALS-FUS Drosophila model (25). To stress the relevance of these CCAP neurons for FUS toxicity, we correlated the loss of CCAP neurons to the adult phenotypes. The severe eclosion phenotype observed upon OK6-driven FUS expression correlated to the loss of the CCAP neurons in the young adult flies (Fig. 1H). Moreover, we correlated the FUS-induced loss of the CCAP neurons with the phenotypes that we observed in our Drosophila model when we studied the molecular determinants of FUS toxicity (25). Motor neuronal expression in adult flies resulted in a severely shortened lifespan, an age-dependent progressive motor performance defect and an age-dependent reduction of motor neurons. The loss of the CCAP neurons induced by FUS in young adult flies (2–4 h) correlated with the survival and motor function of these aged flies (in which FUS expression was induced later in life) (Fig. 1I–J).Therefore, we conclude that the FUS-induced loss of CCAP neurons is a valuable phenotype to study ALS-FUS toxicity.
Degeneration of NAG neurons is not due to defects in their development
To confirm that the loss of the NAG neurons was due to degeneration, rather than to developmental defects, we analyzed these neurons at different time points. We first determined the number of CCAP neurons from the ventral nerve cord in third instar larvae. Human WT or mutant FUS expression did not result in the loss of CCAP neurons at this stage (Fig. 2A). However, WT, R521G and R521H hFUS expression did induce a reduction in the number of NAG neurons at pharate adult stage and in newly eclosed adult flies (Fig. 2B and C). Because the NAG neurons were not affected at the third instar larval phase, we can exclude defects in the development of these NAG neurons as a cause of the reduced number of these neurons.
Figure 2.
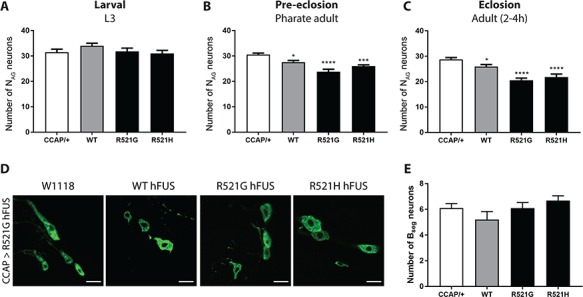
Loss of CCAP neurons is not due to developmental or regulatory defects. (A) No reduction in number of NAG neurons was observed in the ventral nerve cords of third instar larvae in comparison to control flies (CCAP/+). (B) A significant reduction of NAG neurons is observed in WT and mutant expressing pharate adults. (C) Newly eclosed flies (2–4 h old) showed progressive loss of NAG neurons induced by WT and mutant FUS expression. (D) Visualization of the regulatory CCAP neurons from the left subesophageal ganglion (NSEG) from the fly brain. (E) The number of regulatory NSEG neurons was not affected by human FUS expression in newly eclosed adult flies (2–4 h old). For statistical testing, one-way ANOVA and Holm–Sidak multiple comparison test was used. *P < 0.05, ***P < 0.001; ****P < 0.0001. Data are presented as mean ± SEM, n = 15, scale bar = 10 μm.
CCAP neurons from the subesophageal ganglia are not affected
Bursicon secretion from the Bursicon+ NAG neurons is regulated by the CCAP neurons located in the subesophageal ganglia of the fly brain (NSEG, Fig. 1C). To exclude that degeneration of the NAG neurons is the consequence of the death of CCAP neurons from the subesophageal ganglia (NSEG neurons), we analyzed the number of NSEG neurons. In newly eclosed flies, no significant reduction of these NSEG neurons was observed (Fig. 2D and E). This shows that the observed phenotype is not due to a regulatory defect of the NSEG neurons located in the subesophageal ganglia.
Prevention of apoptosis rescues the degeneration of NAG neurons
The CCAP neurons undergo programmed cell death within the first 24 h after eclosion, once the fly is fully mature. We observed a loss of the NAG neurons in very young adult flies when these neurons are still fully functional. An early initiation of this programmed cell death program could contribute to the degeneration of the NAG neurons. Therefore, we introduced apoptotic inhibitors in these neurons via CCAP;GFP-GAL4. It was reported before that expression of a versatile caspase inhibitor, p35, blocks programmed cell death in CCAP neurons and that expression of the apoptotic inhibitor, DIAP1, inhibits premature death of CCAP neurons (28). Co-expression of p35 or DIAP1 with R521G mutant human FUS expression in the NAG neurons (CCAP;GFP-GAL4) resulted in the full rescue of the degeneration of the NAG neurons from the ventral nerve cord (Fig. 3A and B). In contrast, expression of p35 or DIAP1 in NAG neurons (CCAP;GFP-GAL4) alone had no effect on the number of these NAG neurons (Fig. 3C). These results indicate that expression of human FUS in motor neurons resulted in enhanced apoptotic cell death of the NAG neurons. This neurodegenerative phenotype provides a suitable read-out to screen for potential suppressors of FUS toxicity.
Figure 3.
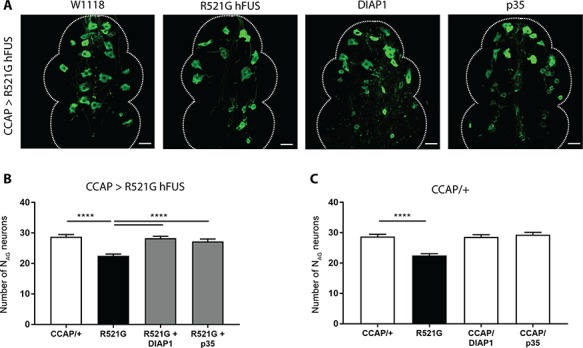
Selective overexpression of apoptosis inhibitors DIAP1 and p35 can fully rescue the NAG neuronal loss. (A) Confocal analysis of NAG neurons that co-express R521G hFUS and DIAP1 or p35 (CCAP > R521G hFUS). (B) Quantification of the number of NAG neurons from flies that co-express DIAP1 or p35 with R521G human FUS in CCAP neurons (CCAP;GFP-GAL4). (C) Quantification of control experiment where flies only express DIAP1 or p35 in their CCAP neurons (CCAP;GFP-GAL4). For statistical testing one-way ANOVA and Holm-Sidak multiple comparison test was used. ****P < 0.0001. Data are presented as mean ± SEM, n = 10, scale bar = 20 μm.
Downregulation of PRKN or PINK1 expression rescues FUS-induced neurodegeneration
To validate the potency of our novel in vivo screening system, we first tested its sensitivity to already known suppressors of FUS toxicity. Therefore, we used the hFUS transgenes targeted to the 35B landing site. We designed a fly line in which the CCAP;GFP-GAL4 driver and the R521G mutant human FUS were recombined on the second chromosome. The net effect of mutant FUS expression remained the same, which excludes an effect of the transgene integration site.
It was previously shown that the downregulation of PRKN or PINK1 resulted in the amelioration of WT and mutant FUS-induced toxicity (19). Therefore, as a proof of principle, we silenced the fly Pink1 or Parkin gene in CCAP neurons (CCAP;GFP-GAL4) by RNA interference (siRNAs) (Fig. 4B). siParkin and siPink1 fully rescued the degeneration of the NAG neurons (Fig. 4A and C), which we confirmed by using two independent siRNA lines (data not shown). In the absence of hFUS expression, no effect on the number of CCAP neurons was observed (Fig. 4D). These results confirm that downregulation of PRKN or PINK1 can suppress FUS-induced neurodegeneration. In addition, these results prove the effectiveness of our newly established in vivo model to screen for unknown suppressors of FUS-induced neurotoxicity.
Figure 4.
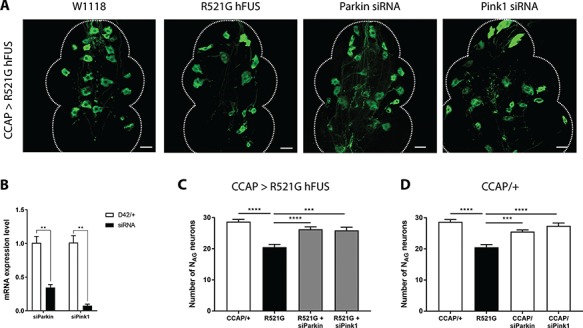
Silencing of PRKN and PINK1, known suppressors of FUS, rescues the loss of NAG neurons. (A) Confocal analysis of NAG neurons showed that siRNAs against Parkin and PINK1 mitigate degeneration of mutant FUS expressing CCAP neurons (CCAP;GFP-GAL4). (B) The downregulation of Parkin and Pink1 gene expression was verified by qRT-PCR. For statistical testing, we used an unpaired T-test. (C) Quantification of NAG neurons in R521G mutant flies. Suppression of PRKN and PINK1 expression rescued the number of R521G expressing NAG neurons. (D) Quantification of NAG neurons in control flies (no FUS expression). Suppression of PRKN and PINK1 expression in CCAP neurons without mutant hFUS expression did not influence the number of NAG neurons in comparison with the control CCAP/+. For statistical testing, one-way ANOVA and Dunnett’s multiple comparison test were used, unless other tests were mentioned. **P < 0.01, ****P < 0.0001. Data are presented as mean ± SEM, n = 10, scale bar = 20 μm.
Nucleocytoplasmic transport proteins play a role in FUS-induced neurodegeneration
Recent work in C9orf72 and TDP-43 ALS/FTD disease models suggest the pivotal role of the nucleocytoplasmic transport system in toxicity. In FUS proteinopathies, like ALS and FTD, FUS is depleted from the normal nuclear localization and aggregates in the cytoplasm (14). In order to investigate whether nucleocytoplasmic transport proteins play a role in FUS-induced toxicity, we performed a candidate-based screen. In this screen, we included suppressor genes for C9orf72-dependent neurodegeneration, previously reported by our group (20). We downregulated the expression of Nup107 and Nup154, two subunits of the nuclear pore complex, by RNA interference (Fig. 5B). In addition, we also tested the effect of downregulating two nuclear transport receptors, XPO1 (Emb in flies) and TNPO1 (Trn in flies) (Fig. 5B). Previously, TNPO1 was shown to be a molecular chaperone of FUS, able to suppress phase separation and stress granule association of FUS (29–32). Moreover, different studies in FUS Drosophila models identified TNPO1 as an enhancer of FUS toxicity (29,33). Therefore, TNPO1 can serve as a good control to test whether enhancers can be identified as well with our new in vivo screening tool. Silencing of Nup154 (Nup155 in humans) and XPO1 rescued the FUS-induced loss of NAG neurons (Fig. 5A and C), although neither of the siRNAs by themselves affected the number of NAG neurons (Fig. 5D). In contrast, downregulation of Nup107 or Trn expression did not ameliorate the human FUS-induced degeneration of NAG neurons (Supplementary Material, Fig. 5C). These results were confirmed by using two or three independent siRNA lines (Supplementary Material, Fig. S2). Downregulation of XPO1 or Nup154 did not result in decreased FUS expression levels (Supplementary Material, Fig. S2E–F), nor did silencing of Trn or Nup107 (data not shown). In conclusion, we identified Nup154 and XPO1 as potent suppressors of FUS-induced neurotoxicity.
Figure 5.
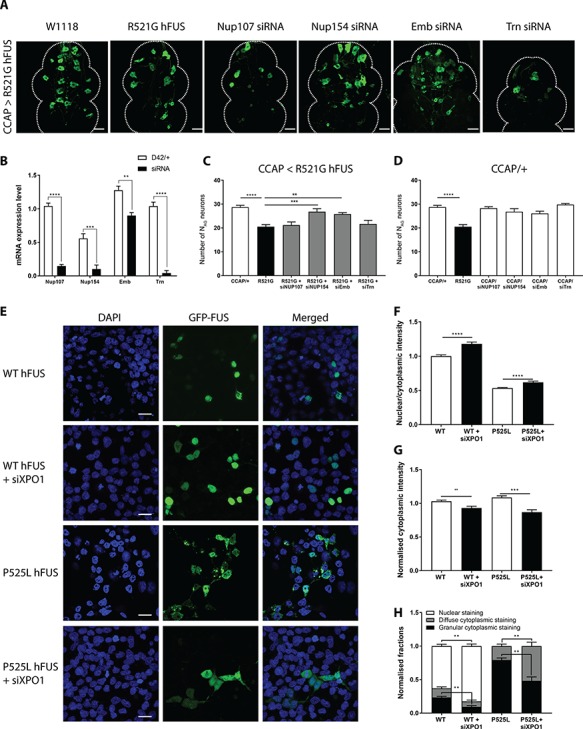
Nup154 and XPO1 are suppressors for FUS-induced neurotoxicity. (A) Confocal analysis of the NAG neurons. Knockdown of Nup154 (Nup155 in humans) and XPO1 (Emb) via siRNAs in R521G human FUS expressing CCAP neurons ameliorated the degeneration of NAG neurons. Downregulation of Nup107 or Trn does not rescue the loss of NAG neurons. (B) Downregulation of Nup107, Nup154, Emb and Trn gene expression was confirmed by qRT-PCR. For statistical testing, unpaired T-test was used. (C) Suppression of Nup154 and XPO1 expression rescued the number of R521G expressing NAG neurons. (D) Expression of Nup107, Nup154, XPO1 and Trn siRNAs by themselves did not affect the NAG neurons in comparison with the control CCAP/+. (E) Confocal imaging of HeLa cells cotransfected with GFP-labeled WT or P525L mutant human FUS transgenes and siXPO1. (F) siXPO1 increased the ratio of nuclear FUS intensity/cytoplasmic FUS intensity for both WT and mutant FUS. (G) siXPO1 decreased the total cytoplasmic WT and mutant FUS levels obtained by analysis of the cytoplasmic FUS intensities. (H) siXPO1 decreased the amount of cells with a granular and diffuse WT hFUS staining pattern and relocalized WT hFUS to the nucleus (see also confocal images in two top rows of E). siXPO1 induced a significant decrease of the amount of cells with a granular staining pattern upon mutant FUS transfection and a significant increase in the number of cells with diffuse cytoplasmic FUS staining (see also confocal images in two lower rows of E). Numbers were normalized to the total number of cells per picture. Ten pictures were analyzed per well. For statistical testing, one-way ANOVA and Dunnett’s or Tukey’s multiple comparison test were used, unless other tests were mentioned. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean ± SEM, n = 10 for the fly ventral nerve cords and n = 3 for the HeLa cells, scale bar = 20 μm.
Downregulation of XPO1 reduces the FUS propensity to form inclusions
The presence of cytoplasmic FUS inclusions is a hallmark of ALS-FUS and FTD-FUS pathology [11,29,30]. The identification of the nuclear transport receptor XPO1 as a suppressor of FUS neurotoxicity suggests that prevention of FUS nuclear export may be neuroprotective. To elucidate the effect of XPO1 silencing on the subcellular localization of the FUS protein, we transfected HeLa cells with WT and P525L GFP-labeled FUS together with siXPO1 (in order to silence XPO1 expression through RNA interference). We hypothesized that downregulation of XPO1 could influence the nuclear export of FUS, leading to the redistribution of FUS into the nucleus. We found that downregulation of XPO1 increased the nuclear/cytoplasmic ratio of the WT and mutant human FUS protein after stress induction with sodium arsenite (Fig. 5F). A decrease of the cytoplasmic FUS levels is underlying this increased nuclear/cytoplasmic ratio (Fig. 5G). Moreover, we observed a change in the FUS staining pattern (Fig. 5E and H). When XPO1 was not downregulated, WT FUS was mainly present in the nucleus although stress induction resulted in the mislocalization of FUS to the cytoplasm in a small amount of cells. In the cytoplasm of these stressed cells, WT FUS could be observed in a granular or diffuse staining pattern (Fig. 5E). NLS mutant FUS was almost completely depleted from the nucleus and mislocalized to cytoplasmic granules upon stress induction (Fig. 5E). Downregulation of XPO1 in WT human FUS expressing HeLa cells increased the amount of cells with FUS exclusively present in the nucleus (nuclear staining pattern) and decreased significantly the number of cells with a granular cytoplasmic staining pattern (i.e. granules are observed in the nucleus or cytoplasm; Fig. 5E and H). Moreover, knocking down XPO1 induced a significant shift of the NLS mutant FUS protein from a granular staining pattern to a diffuse staining pattern (Fig. 5E and H). Taken together, we showed that XPO1 contributes to the redistribution of FUS into inclusions, because silencing of XPO1 prevented FUS incorporation into these cytoplasmic granules. Since familial mutations in the NLS of the FUS protein lead to the formation of toxic cytoplasmic FUS aggregates (11,13) and FUS inclusions composed of WT protein are also observed in FTD cases (34), these results suggest an important role of XPO1 in the ALS/FTD pathogenesis.
To understand how XPO1 could influence the incorporation of FUS in cytoplasmic granules, we analyzed the localization of XPO1 in arsenite-treated HeLa cells. As shown in Figure 6A, XPO1 accumulated in FUS-positive cytoplasmic granules upon stress. This is in line with recent work that showed the localization of XPO1 in stress granules (35). Next, we determined whether XPO1 and FUS physically interact by performing Duolink proximity ligation assay (PLA). As shown in Figure 6B, the PLA dots (red) confirmed the interaction of WT and NLS mutant FUS with endogenous XPO1 in stressed conditions. Our data suggest that XPO1 can bind to FUS. In conclusion, XPO1 knockdown resulted in the less-efficient recruitment of FUS into cytoplasmic granules, suggesting that XPO1 stabilized FUS in the cytoplasmic granules (Fig. 6C). Therefore, we hypothesize that the downregulation of XPO1 can result in more soluble FUS. Hence, this could reduce the toxicity for the affected neuron. Thus, XPO1 can be an important therapeutic target to modify ALS/FTD pathogenesis.
Figure 6.
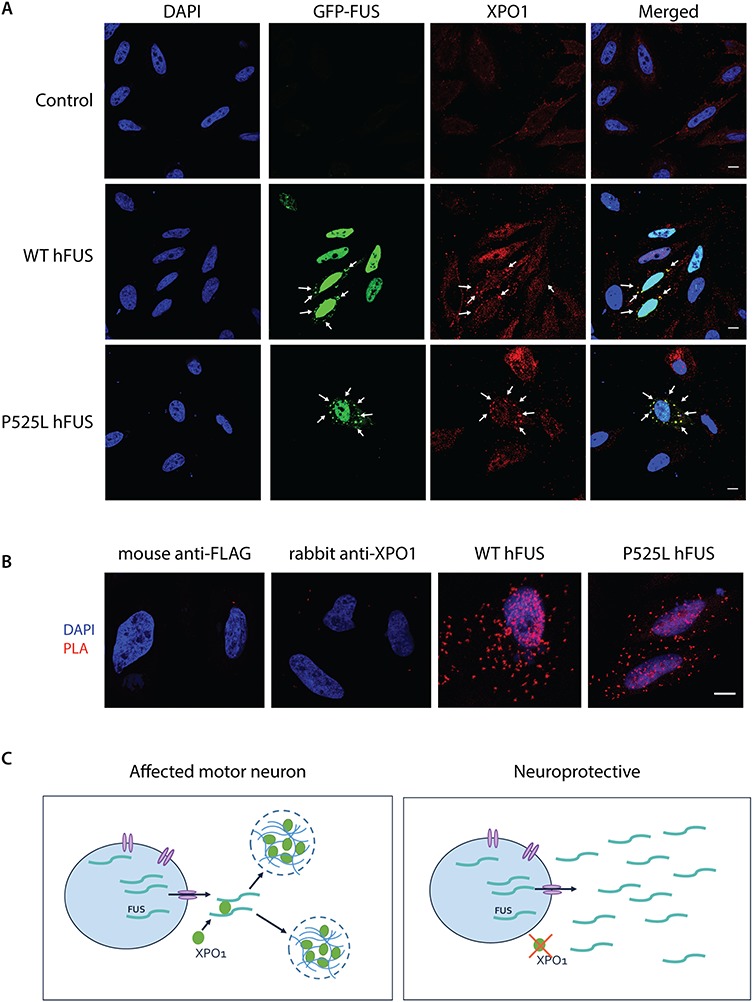
XPO1 interact with FUS in FUS-positive cytoplasmic granules. (A) Arsenite-treated HeLa cells (0.5 mm, 1 h) expressing GFP-tagged WT or P525L hFUS were stained with XPO1. White arrows indicate co-localization. (B) Duolink protein–protein interaction detection assay to investigate the physical interaction between FUS and XPO1 upon arsenite treatment. Staining with the primary antibodies (Mouse anti-FLAG or rabbit anti-XPO1) separately was negative. Hela cells expressing WT or P525L hFUS showed clear PLA signals (red dots) indicating that FUS interacted with XPO1 in stress conditions. Representative images. Scale bar = 10 μm. (C) Model illustrating our hypothesis. In stressed conditions, XPO1 binds to FUS and recruits FUS into stress granules present in the cytoplasm which could have negative effects (if there is too much FUS and/or if FUS is mutated). Silencing of XPO1 results in the recruitment of less FUS into the stress granules and can therefore be neuroprotective.
Discussion
In this study, we developed a novel in vivo screening platform, designed to identify novel genetic modifiers of FUS-induced toxicity. To achieve this, we created an ALS-FUS Drosophila model in which human FUS expression induced the early apoptotic loss of NAG neurons. We validated this model as an effective screening tool by confirming the role of PINK1 and PRKN as suppressors of FUS toxicity. In addition, via a candidate-based screen, we identified the nuclear pore protein Nup154 and the nuclear transport protein XPO1 as modifiers of FUS-induced neurotoxicity. Furthermore, XPO1 knockdown experiments in a cellular model suggest a role for XPO1 in the incorporation of FUS in cytoplasmic inclusions, which are the hallmarks of ALS-FUS and FTD-FUS. This hypothesis is strengthened by the observation that XPO1 and FUS co-localize in cytoplasmic FUS inclusions. In addition, a Duolink PLA confirmed the physical interaction between FUS and XPO1. The identification of XPO1 and Nup154 as suppressors of FUS toxicity offers new therapeutic targets in order to modify ALS/FTD.
Until now, the mechanism by which FUS causes neurotoxicity in ALS and FTD has not been fully elucidated. Mutant FUS is depleted from the nucleus and aggregates in the cytoplasm (14). This suggests a dual mechanism with both loss of nuclear function and gain of cytoplasmic toxic function. To gain better insights into the pathobiological mechanisms of FUS toxicity in ALS, we developed an ALS-FUS Drosophila model (25). We used the ΦC31 integrase-mediated cassette (36) and integrated the human FUS transgenes site-specifically in the VK31 and 35B landing sites to avoid any influence of the genomic environment on the transgene expression. The expression of human FUS in motor neurons induced pupal lethality phenotypes with the immature appearance of adult escapers. This is in line with the pupal lethality and motor performance defects induced by human FUS expression that was described previously in ALS-FUS Drosophila models (33,37). We observed similar phenotypes when hFUS transgenes were expressed in motor neurons using the D42-GAL4 driver (25), ruling out driver-dependent effects.
We identified a specific effect of hFUS expression during late metamorphosis and early adult maturation in Drosophila. Human FUS expression had a negative effect on the survival of NAG neurons. These neurons are known to regulate the pupal-to-adult transitions through a neuropeptide hormone cascade (38). The CCAP hormone is a key regulator of the pupal ecdysis motor program (27,39–41), with CCAP secretion stopping the pre-ecdysis program and turning on the ecdysis motor program (38). This signaling is needed to induce the motor behavior program, necessary for the fully developed fly to eclose. This, in turn, initiates the secretion of Bursicon from the NAG neurons (27). Subsequently, this neurohormone initiates post-eclosion processes to mature the adult fly. It is known that targeted ablation of CCAP neurons leads to pupal lethality with adult escapers showing immature cuticle and wing expansion phenotypes (41). We observed defects in pupal-to-adult transition with hFUS-induced apoptotic cell death of the NAG neurons. It is known that CCAP neurons undergo programmed cell death once the adult fly is fully mature (28). However, no defects in these neurons were observed at the third instar larval stage. Although these neurons undergo programmed cell death, the NAG neurons are still fully functional in very young adult flies at the time points (2 to 4 h) when we are examining these CCAP neurons (27,41). To prove the relevance for ALS toxicity, we correlated the loss of the NAG neurons to the eclosion defect that we observed upon motor neuronal FUS expression (Fig. 1A and H). Moreover, we correlated the FUS-induced loss of the CCAP neurons with the phenotypes that we observed in our Drosophila model when we studied the molecular determinants of FUS toxicity (25). Motor neuronal expression in adult flies resulted in a severely shortened lifespan, an age-dependent progressive motor performance defect and an age-dependent reduction of motor neurons (25). The loss of the CCAP neurons induced by FUS in young adult flies (2–4 h) correlated with the survival and motor function of these aged flies (in which FUS expression was induced later in life) (Fig. 1I–J). In addition, previous fly work on TDP-43, a protein similar in structure and function to FUS, supports the relevance of these neurons in ALS/FTD related toxicity. Vanden Broeck et al. showed that dTDP-43 overexpression resulted in the selective apoptosis of the Bursicon+ NAG neurons (42).
Although we investigated FUS in a small animal model, the role of FUS seems to be evolutionary conserved. FUS knockout models showed an increase in aneuploidy and chromosomal aberrations in affected cells, highlighting the importance of FUS in genomic maintenance and chromosomal stability (43). Our observations that FUS overexpression disrupted the pupal-to-adult transformation by inducing cell death of CCAP neurons, suggests an evolutionary conserved function of FUS in maintaining the post-developmental neuronal integrity. Furthermore, we observed that FUS disrupted a very specific subset of neurons in the Drosophila ventral nerve cord. The selectivity of human FUS vulnerability to this particular subset of neurons is reminiscent of the neuronal selectivity observed in ALS/FTD pathology (44,45). As a consequence, our ALS-FUS Drosophila model seems to be a suitable model to study FUS-induced neuronal toxicity.
As our aim was to establish a screening model to rapidly identify new disease modifiers of in vivo neuronal toxicity, we chose to use the small group of CCAP neurons which are GFP labeled and easy to count in the ventral nerve cord of the fly. Furthermore, the motor neuronal driver OK6 also drives FUS expression in CCAP neurons, suggesting that these neurons can be considered as motor neurons. One extra argument for this is that CCAP neurons induce the motor behavior which the fly needs to eclose (41). As explained previously, we found a nice correlation between the number of CCAP neurons and the hFUS-induced adult motor phenotypes. The CCAP neurons are also shown to develop ALS-related axonal transport defects in stages prior to the stages in which we observe the loss of these CCAP neurons (46). As axonal transport defects are believed to result in motor neuron death in ALS, CCAP/NAG neurons can be considered as mimics of motor neurons and are a relevant tool to study FUS-induced neurotoxicity.
Thanks to the CCAP;GFP-GAL4 line that labels the CCAP neurons with GFP, the visualization of these neurons is relatively easy. As a proof of principle, we confirmed the role of two known suppressors, PRKN and PINK1, before identifying new proteins that can suppress FUS-induced neurotoxicity. The rescue experiments with siParkin and siPINK1 lines, confirmed the potency of our model to screen for suppressors of FUS toxicity, whilst confirming their previous description as modifiers of FUS toxicity (19).
In a next step, we screened for new potent modifiers of FUS-induced toxicity. Downregulation of Nup154 and XPO1 suppressed FUS-induced toxicity, pointing to a pivotal role of nucleocytoplasmic transport in ALS/FTD pathogenesis. These results are in line with recent work on C9orf72 and TDP-43 providing evidence for a role of nucleocytoplasmic transport in ALS/FTD (20–24). In cell culture and Drosophila models, mutations in the FUS NLS induce aberrant binding of FUS to TNPO1, impairing nuclear import of FUS (14,33). This results in the accumulation of the protein in the cytoplasm (14,33). In addition, the nuclear/cytoplasmic ratio of mutant FUS in vitro conversely correlates with the age of disease onset in ALS-FUS patients (14,47). These results suggest that a proper nucleocytoplasmic transport machinery is important for neuronal health.
Recent in vitro and in vivo work on FUS toxicity showed that, beyond the regulation of nuclear import, TNPO1 has a molecular chaperone activity, suppressing phase separation and stress granule association of FUS (29–32). This and earlier work showed that downregulation of this nuclear transport protein enhanced observed eye and neurodegenerative phenotypes in FUS Drosophila models, identifying TNPO1 as an enhancer of FUS toxicity (29,33). Silencing of Trn in NAG neurons did not induce more pronounced loss of these neurons in comparison with R521G hFUS expression. One explanation for this could be that the loss of even more CCAP neurons resulted in the failure of the fly to eclose. This observation can be considered as a potential disadvantage. Nevertheless, the suppressor genes are the most relevant targets from the disease perspective. First, by using our model, we can identify modifiers improving neuronal health, as the read out of our screening model is the survival of the NAG neurons. These suppressors can be targeted by antisense oligonucleotides or inhibitors to influence disease onset and/or progression. Second, enhancers could be identified by assessing the effect of the candidate gene on the eclosion phenotype, although more processes can be involved. This could complicate the interpretation of the modifying mechanism of the candidate gene. Therefore, we focused in this study on the identification of suppressor genes, as these are relevant targets to develop new therapeutic strategies.
In contrast to Nup154, knockdown of Nup107 was not sufficient to rescue the loss of NAG neurons. This suggests that Nup154 is an indirect or general suppressor of ALS/FTD pathology, while Nup107 seems to be a selective suppressor for C9orf72 toxicity.
Previously, contradictory results were reported for the modifying capacity of XPO1 in Drosophila models of the C9orf72 repeat expansion. Our lab and others (20,22) reported that XPO1 was an enhancer of C9orf72-induced toxicity, while Zhang et al. (24) described XPO1 as a suppressor. In our ALS-FUS Drosophila model, XPO1 was a suppressor of FUS-induced neurotoxicity. Moreover, we discovered in HeLa cells a role of XPO1 in the propensity of FUS to form inclusions. Upon stress, FUS relocalizes to dynamic membraneless stress granules, and mutations in the NLS of FUS contribute to its increased recruitment into stress granules due to the accumulation of FUS in the cytoplasm (48). Consistent with this, expression of mutant FUS in HeLa cells promoted the recruitment of FUS into cytoplasmic granules. However, downregulation of XPO1 resulted in the redistribution of FUS diffusely in the cytoplasm. Moreover, our data showed that XPO1 and FUS co-localize in cytoplasmic granules and even physically interact, suggesting that XPO1 recruits and/or stabilizes FUS in cytoplasmic stress granules. This is in line with recent data demonstrating that XPO1 is a stress granule component (35). FUS was also shown to bind to XPO1 in a study that systematically mapped the relationship between cargoes and nuclear transport receptors in situ, by using proximity ligation coupled to mass spectrometry (49). Although our results show a clear interaction between XPO1 and FUS, it was recently reported that the export of FUS and TDP-43 is independent of XPO1 and most likely occurs through passive diffusion (50–52). NLS mutations of FUS induce defective nuclear import and result in the accumulation of FUS in the cytoplasm, leading to the toxic-gain-of-cytoplasmic-function of FUS. How XPO1 is recruited to the FUS-positive stress granules is still unclear. We showed that XPO1 binds to FUS. One possible explanation is that FUS recruits XPO1 and other nucleocytoplasmic transport factors via its NES or NLS to the stress granules (35). By silencing XPO1, we observed a reduction of the FUS-induced neurotoxicity and a decrease in the amount of cells with FUS inclusions. We hypothesize that XPO1 interacts with cytoplasmic FUS stimulating the formation of toxic cytoplasmic FUS inclusions (Fig. 6C). Further research is needed to confirm this hypothesis.
In conclusion, our newly developed in vivo screening platform is a suitable model system to identify suppressors of FUS toxicity. In this Drosophila model, human FUS expression caused neurodegeneration of a particular subset of CCAP neurons. Since WT and mutant FUS induced similar phenotypes, the identification of disease modifiers via our model could not only help to explain the pathogenesis in missense mutations, but also in FUS-proteinopathy patients with elevated FUS levels (15). Apart from providing new important mechanistic insights, these disease modifiers could be translated into new therapeutic strategies for these detrimental diseases.
Materials and Methods
Fly stocks
Drosophila melanogaster strains were maintained on standard yeast, cornmeal and agar-based medium (5% glucose, 5% yeast extract, 3.5% wheat flour, 0.8% agar) in a 12 h light/dark rhythm. Crosses were performed at 25°C. The w1118 (Canton-S10, BDSC Cat# 3605, RRID:BDSC_3605) line was used as WT control. FUS expressing transgenic flies were described previously (25). The CCAP-GAL4;UAS-mCD8-GFP stock was a kind gift of Dr Randall S. Hewes (University of Oklahoma, Norman, OK, USA). GAL4 driver lines P(GawB)OK6 (OK6-GAL4, BDSC Cat# 64199, RRID:BDSC_64199), P(CCAP-GAL4.P)16/CyO (CCAP-GAL4, BDSC Cat# 25685, RRID:BDSC_25685, P(UAS-DIAP1.H)3 (UAS-DIAP1, BDSC Cat# 6657, RRID:BDSC_6657), P(UAS-p35.H)BH1 (UAS-p35, BDSC Cat# 8651, RRID:BDSC_8651) were obtained from the Bloomington Stock center, as well as the deficiency kit and null mutant lines. RNAi lines were obtained from the Vienna Drosophila RNAi center (VDRC; Table 1).
Table 1.
RNAi lines obtained from VDRC
| Gene name | siRNA (VDRC) |
|---|---|
| PINK1 | 21860/GD, 109614/KK |
| PRKN | 47636/GD, 104363/KK |
| Nup107 | 22407/GD, 110759/KK |
| Nup154 | 21878/GD, 34710/GD, 106136/KK |
| XPO1 | 3347/GD, 31353/GD, 103767/KK |
| TNPO1 | 6543/GD, 6544/GD |
Eclosion test
Pharate adult flies were transferred to a petri dish and followed for 72 h. The percentage of eclosed flies was defined as ratio of the number of empty pupal cases to the number of total pupal cases. Eclosed flies were scored for immature phenotypes. If flies displayed signs of immaturity, they were scored affected. The signs of immaturity are as follows: unexpanded wings, disorganization of the scutellar bristles and absence of the sclerotization and melanization of the cuticle (mostly presented by a black dot on the back of the cuticle).
CCAP neuron counting
FUS variants were expressed in CCAP neurons using CCAP-GAL4;UAS-CD8-GFP (CCAP;GFP-GAL4) to drive expression. The brain and ventral nerve cord was dissected at different time points. A confocal image was taken every 2 μm using a Fluoview FV1000 confocal microscope (Olympus, Tokyo, Japan) or TCS SP8 confocal microscope (Leica, Wetzlar, Germany). The number of CCAP neurons was counted in the AG of the ventral nerve cord or the subesophageal ganglia of the brain.
Immunohistochemistry of fly central nervous system
Immunostaining on fly brain and ventral nerve cords was performed using standard techniques. The following primary antibodies were used: Rabbit anti-α-Bursicon (kindly provided by Benjamin White, Laboratory of Molecular Biology, National Institute of Mental Health, NIH, Bethesda, MD, USA) 1:4000 and mouse polyclonal anti-FUS (BD Biosciences Cat# 611385, RRID:AB_398907) 1:100. CCAP neurons were labeled with CD8-GFP tag in the plasma membrane. Alexa 555-conjugated secondary antibody (Bursicon) and Alexa 647-conjugated secondary antibody (FUS) were used at 1:500 (Invitrogen, Carlsbad, CA, USA; Cat# A-21202, RRID: AB_141607 and Cat#A-31571, RRID: AB_162542). Brains and ventral nerve cords were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA; Cat# H-1200, RRID:AB_2336790) and analyzed with a Fluoview FV1000 confocal microscope (Olympus, Tokyo, Japan) or TCS SP8 confocal microscope (Leica). CCAP neurons or Bursicon+ NAG neurons were counted. Images of multiple fluorescent-labeled brains and ventral nerve cords were obtained by sequential scanning of each channel at equal laser intensity. One way ANOVA was used to evaluate significant variation among genotypes and a post hoc Holm–Sidak test was used to control for multiple testing.
Transgene expression levels using qRT-PCR
Thirty larval central nervous systems (D42-GAL4-driven expression) were mixed with 300 μl TriPure reagent (Sigma Aldrich, St. Louis, MO, USA) and ground with a pestle. Total RNA was isolated by using standard procedures. cDNA was generated from 1 μg of RNA of each sample by using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer conditions. qRT-PCRs were performed on a StepOnePlus instrument (Applied Biosystems, Foster City, CA, USA) using TaqMan assays (Applied Biosystems) for PINK1, PRKN, TNPO1, XPO1, Nup107 and Nup154 and normalized to the housekeeping gene Rap2L. Assays were designed by the IDT PrimerQuest Tool (IDT, Coralville, IA, USA) and are stated in Table 2. Expression levels were normalized and analyzed using qBase+ (v.3.0, Biogazelle, Zwijnaarde, Belgium).
Table 2.
Sequences of the Taqman assays designed by the IDT PrimerQuest Tool
| Transgene | Forward primer (5′-3′) | Probe (5′-3′) | Reversed primer (5′-3′) |
|---|---|---|---|
| PINK1 | CGT GAT ATA CAC GCC AAC ATT T | TGA AGA CAA GAA GCA AGC GAG GCT | CAC TAC ATT GAC CAC CGA TTT G |
| PRKN | GAC TTC AGC CGG ATG ATC TAA A | AAG GAA CTA AGC GAT GCC ACG ACA | GAC CCA AGT CAC ATT GCT CTA T |
| TNPO1 | CGG ACA CAG CCA CTC AAA | TGG CCG TAC AGA TGA AAC TGG AGG | TTC AGT TTC GTC AGC ACA TAG A |
| XPO1 | TCT CTT GGA AGA ACC TGA ACA C | TGT GCT GGG CCA TTG GTT CCA TAT | GTT CGC ACA GGC CTA ATA GAT |
| Nup107 | GGG AGC ACA AGG TTA AGG AG | CAG GAG TTC GAT GGT TTG CTT GGC | CGA TGA ACG GAT CCA G |
| Nup154 | GGT GAC CAA GTC GGC TAT TC | ACG TGC CTG GCT GAC TAT CTA TTC | GTC TGA TTG AAC GTC CAG ATG TA |
Cell culture and transfection
HeLa cells (CLS Cat# 300194/p772_HeLa, RRID: CVCL_0030) were grown in DMEM/F12 high glucose GlutaMax (Gibco, ThermoFisher Scientific; Cat#31331028) supplemented with 10% standard fetal bovine serum (Gibco; Cat# 10500064) and 2% PenStrep (Invitrogen; Cat#15140–122) at 37°C in a humidified atmosphere with 5% CO2 for 24 h. HeLa cells were seeded in 96-well plates (TPP, Trasadingen, Switzerland) with a density of 7500 cells and transiently transfected using Lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions. HeLa cells were free of mycoplasm. GFP-labeled FUS constructs were kindly provided by Dr Simon Alberti (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany). The generation of the P525L mutant construct was previously described (25). siXPO1 was obtained from Dharmacon (Lafayette, CO, USA; Cat#J–003030–10).
Immunohistochemistry and microscopy of HeLa cells
Briefly, cells were fixed 24 or 48 h (when FUS is co-transfected with siXPO1) after transfection in 4% formaldehyde in phosphate buffered saline (PBS) and rinsed three times. XPO1 staining was performed as described previously (35). We blocked the cells in wash buffer (PBS, 0.1% Tween-20 and 2 mg/ml Heparin) supplemented with 3% donkey serum and 5% glycine for 1 h. Subsequently, cells were incubated with the rabbit anti-mouse CRM1 antibody (Novus Biologicals, Littleton, CO, USA; NBP2–16014), diluted 1:100, for 16 h at 4°C. After washing the cells four times with wash buffer, Alexa Fluor-647 conjugated secondary antibodies (Invitrogen) were applied to the cells for 3 h at RT. Secondary antibodies were diluted 1:1000 in wash buffer with 3% donkey serum. By confocal microscopy, we analyzed the localization of XPO1. In order to determine the recruitment of FUS into cytoplasmic granules, cells were analyzed using the CellInsight™ CX5 High Content Screening (HCS) Platform (ThermoFisher Scientific; Cat#CX51110). Nuclear and cytoplasmic intensities were analyzed with the HCS studio cell analysis software (ThermoFisher Scientific). Stress granules were induced by incubating the cells for 1 h with 0.5 mm NaAsO2 (Sigma).
Duolink assay
Duolink® proximity ligation assay (PLA®) (Sigma-Aldrich; DUO92101) was performed according to the manufacturer’s instructions. Briefly, samples were fixed with 4% paraformaldehyde in PBS for 20 min, washed three times and incubated for 1 h at 37°C in PBS containing 0.04% Triton X-100 and 5% normal donkey serum. Cells were incubated for 1 h at 37°C in 5% donkey serum containing the mouse monoclonal anti-FLAG primary antibody (Sigma-Aldrich, F3165). Subsequently, cells were washed with PBS and stained with the rabbit anti-CRM primary antibody (Novus Biologicals; NB100-79802) in PBS containing 0.04% Triton X-100 and 5% normal donkey serum for 1 h at RT. After washing the cells twice with 1× Duolink wash buffer A for 5 min, cells were incubated for 1 h in a pre-heated humidity chamber at 37°C with secondary antibodies conjugated with oligonucleotides (PLA probes anti-mouse MINUS and anti-rabbit PLUS). Next, a second wash step with Duolink washing buffer A was performed and cells were incubated with the Duolink ligation solution for 30 min in a pre-heated humidity chamber at 37°C. Cells were rinsed twice with Duolink washing buffer A for 5 min and subsequently incubated with the Duolink amplification-polymerase solution for 100 min at 37°C. The samples were then washed twice in 1× Duolink washing buffer B for 10 min at room temperature followed by a 1 min wash with 0.01× Duolink washing buffer B. The cells were then mounted using ProLong Gold Antifade Reagent with DAPI (Thermo Fisher Scientific; P36930). For confocal analysis of the cells, we used the Leica SP8 Confocal microscope. PLA signals were recognized as red fluorescent spots.
Western blot analysis
Fly heads were homogenized on ice in radioimmune precipitation buffer (RIPA, containing 50 mm Tris–HCl, 150 mm NaCl, 0.1% sodium deoxycholate [SDS],, 0.5% Na, 1% NP-40, [pH 8.0]) supplemented with protease inhibitor (Complete Protease inhibitor, Roche, Indianapolis, IN, USA). Homogenates were incubated on ice for 20 min and protein lysates were extracted by centrifugation (12000 rpm for 20 min at 4°C). Supernatants were used for immunoblotting. Protein concentrations were measured by Micro BCA assay kit (ThermoFisher Scientific; 23235). Western blotting was performed as described before (25). Briefly, 30 μg of protein was loaded on a 7.5% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. Membranes were blocked in 5% skimmed milk in PBS and probed with primary antibodies. Immunodetection was performed with specific secondary antibodies conjugated to horseradish peroxidase and the ECL-plus chemiluminescent detection system (Amersham Biosciences, Little Chalfont, UK) with bands quantified on a LAS-3000 Station (GE Healthcare, Chicago, IL, USA). The signal was normalized to the signal obtained from tubulin for quantification. Primary antibodies were used in a 1/500 dilution: rabbit anti-FUS (Bethyl Laboratories, A300-302A) and rabbit anti-tubulin (Cell signaling, #2125).
Statistical analysis
GraphPad Prism (GraphPad Software, La Jolla, CA, USA) was used to perform statistical analysis.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Lies Vanden Broeck, Dr Thomas Moens and Dr Maarten Jacquemyn for their support and kind suggestions. We thank Laura Fumagalli for her help with the Duolink PLA experiment. We thank Dr Dirk Daelemans (Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy, KU Leuven, Leuven, Belgium) for providing the CellInsight™ CX5 HCS Platform. We also thank Jana Hiers for maintaining the fly stocks.
Conflict of Interest statement. None declared.
Funding
KU Leuven (C1 and ‘Opening the Future’ Fund), the ‘Fund for Scientific Research Flanders’ (FWO-Vlaanderen, FWO G.0431.12 N); the Belgian Government (Interuniversity Attraction Poles Programme P7/16 initiated by the Belgian Federal Science Policy Office); the Thierry Latran Foundation; the ‘Association Belge contre les Maladies neuro-Musculaires—aide à la recherché ASBL’ (ABMM); the European Community’s Health Seventh Framework Programme (FP7/2007–2013; grant agreement n°259867); the EU Joint Programme—Neurodegenerative Disease Research (JPND) projects (STRENGTH and RiMod-FTD); the ALS Liga België (A Cure for ALS); the ALS Association (ID #2169); the Flemish government-initiated Flanders Impulse Program on Networks for Dementia Research (VIND); PhD fellowship from the Agency for Innovation by Science and Technology in Flanders (IWT; 131535 to J.S.); PhD fellowship from FWO-Vlaanderen (1197517 N to J.V.); E von Behring Chair for Neuromuscular and Neurodegenerative Disorders (to W.R.), Geneeskundige Stichting Koningin Elisabeth (GSKE) (to W.R.); the European Research Council under the European’s Seventh Framework Programme (FP7/2007–2013)/ERC (grant agreement no. 340429 to W.R.) and Laevers fund for ALS Research and the ‘Een Hart voor ALS’ fund (to W.R. and P.V.D.). Postdoctoral fellowship from FWO–Vlaanderen (to E.B.); Senior clinical investigatorship from FWO–Vlaanderen (to P.V.D.).
References
- 1. Nolan M., Talbot K. and Ansorge O. (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta. Neuropathol. Commun., 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Swinnen B. and Robberecht W. (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol., 10, 661–670. [DOI] [PubMed] [Google Scholar]
- 3. Bensimon G., Lacomblez L. and Meininger V. (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med., 330, 585–591. [DOI] [PubMed] [Google Scholar]
- 4. Writing Group on Behalf of The Edaravone Als 19 Study G. (2017) Open-label 24-week extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener., 18, 55–63. [DOI] [PubMed] [Google Scholar]
- 5. Young J.J., Lavakumar M., Tampi D., Balachandran S. and Tampi R.R. (2018) Frontotemporal dementia: latest evidence and clinical implications. Ther. Adv. Psychopharmacol., 8, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Radford R.A., Morsch M., Rayner S.L., Cole N.J., Pountney D.L. and Chung R.S. (2015) The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci., 9, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ling S.C., Polymenidou M. and Cleveland D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O. and Aman P. (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol., 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dormann D., Madl T., Valori C.F., Bentmann E., Tahirovic S., Abou-Ajram C., Kremmer E., Ansorge O., Mackenzie I.R., Neumann M. and Haass C. (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J., 31, 4258–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Damme P., Goris A., Race V., Hersmus N., Dubois B., Van Den Bosch L., Matthijs G. and Robberecht W. (2010) The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol., 17, 754–756. [DOI] [PubMed] [Google Scholar]
- 11. Kwiatkowski T.J., Bosco D.A., LeClerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T. et al. (2009) Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science, 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 12. Renton A.E., Chio A. and Traynor B.J. (2014) State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci., 17, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P. et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B. et al. (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J., 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sabatelli M., Moncada A., Conte A., Lattante S., Marangi G., Luigetti M., Lucchini M., Mirabella M., Romano A., Del Grande A. et al. (2013) Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet., 22, 4748–4755. [DOI] [PubMed] [Google Scholar]
- 16. Robberecht W. and Philips T. (2013) The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci., 14, 248–264. [DOI] [PubMed] [Google Scholar]
- 17. Al-Chalabi A., Hardiman O., Kiernan M.C., Chiò A., Rix-Brooks B. and Berg L.H. (2016) Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol., 15, 1182–1194. [DOI] [PubMed] [Google Scholar]
- 18. Pupillo E., Messina P., Logroscino G., Beghi E. and Group S (2014) Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann. Neurol., 75, 287–297. [DOI] [PubMed] [Google Scholar]
- 19. Chen Y., Deng J., Wang P., Yang M., Chen X., Zhu L., Liu J., Lu B., Shen Y., Fushimi K. et al. (2016) PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum. Mol. Genet., 25, 5059–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boeynaems S., Bogaert E., Michiels E., Gijselinck I., Sieben A., Jovicic A., De Baets G., Scheveneels W., Steyaert J., Cuijt I. et al. (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep., 6, 20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou C.C., Zhang Y., Umoh M.E., Vaughan S.W., Lorenzini I., Liu F., Sayegh M., Donlin-Asp P.G., Chen Y.H., Duong D.M. et al. (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci., 21, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Freibaum B.D., Lu Y., Lopez-Gonzalez R., Kim N.C., Almeida S., Lee K.H., Badders N., Valentine M., Miller B.L., Wong P.C. et al. (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature, 525, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jovicic A., Mertens J., Boeynaems S., Bogaert E., Chai N., Yamada S.B., Paul J.W. 3rd, Sun S., Herdy J.R., Bieri G. et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci., 18, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang K., Donnelly C.J., Haeusler A.R., Grima J.C., Machamer J.B., Steinwald P., Daley E.L., Miller S.J., Cunningham K.M., Vidensky S. et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature, 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bogaert E., Boeynaems S., Kato M., Steyaert J., Scheveneels W., Wilmans N., Haeck W., Schymkowitz J., Rousseau F., Callaerts P. et al. (2018) Molecular dissection of FUS points at synergistic effect of low complexity domains in toxicity. Cell Rep., 24, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peabody N.C., Diao F., Luan H., Wang H., Dewey E.M., Honegger H.W. and White B.H. (2008) Bursicon functions within the Drosophila CNS to modulate wing expansion behavior, hormone secretion, and cell death. J. Neurosci., 28, 14379–14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. White B.H. and Ewer J. (2014) Neural and hormonal control of postecdysial behaviors in insects. Annu. Rev. Entomol., 59, 363–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee G.G., Kikuno K., Nair S. and Park J.H. (2013) Mechanisms of postecdysis-associated programmed cell death of peptidergic neurons in Drosophila melanogaster. J. Comp. Neurol., 521, 3972–3991. [DOI] [PubMed] [Google Scholar]
- 29. Guo L., Kim H.J., Wang H., Monaghan J., Freyermuth F., Sung J.C., O'Donovan K., Fare C.M., Diaz Z., Singh N. et al. (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell, 173, e620, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.D., Simons M., Niessing D., Madl T. et al. (2018) Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell, 173, e713, 706–719. [DOI] [PubMed] [Google Scholar]
- 31. Yoshizawa T., Ali R., Jiou J., Fung H.Y.J., Burke K.A., Kim S.J., Lin Y., Peeples W.B., Saltzberg D., Soniat M. et al. (2018) Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell, 173, e622, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qamar S., Wang G., Randle S.J., Ruggeri F.S., Varela J.A., Lin J.Q., Phillips E.C., Miyashita A., Williams D., Strohl F. et al. (2018) FUS phase separation Is modulated by a molecular chaperone and methylation of arginine cation–pi interactions. Cell, 173, e715, 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackel S., Summerer A.K., Thommes C.M., Pan X., Voigt A., Schulz J.B., Rasse T.M., Dormann D., Haass C. and Kahle P.J. (2015) Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis., 74, 76–88. [DOI] [PubMed] [Google Scholar]
- 34. Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H.A. and Mackenzie I.R.A. (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain, 132, 2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang K., Daigle J.G., Cunningham K.M., Coyne A.N., Ruan K., Grima J.C., Bowen K.E., Wadhwa H., Yang P., Rigo F. et al. (2018) Stress granule assembly disrupts nucleocytoplasmic transport. Cell, 173, e917, 958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bateman J.R., Lee A.M. and Wu C.T. (2006) Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics, 173, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lanson N.A. Jr., Maltare A., King H., Smith R., Kim J.H., Taylor J.P., Lloyd T.E. and Pandey U.B. (2011) A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet., 20, 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davis M.M., O’Keefe S.L., Primrose D.A. and Hodgetts R.B. (2007) A neuropeptide hormone cascade controls the precise onset of post-eclosion cuticular tanning in Drosophila melanogaster. Development, 134, 4395–4404. [DOI] [PubMed] [Google Scholar]
- 39. Luan H., Lemon W.C., Peabody N.C., Pohl J.B., Zelensky P.K., Wang D., Nitabach M.N., Holmes T.C. and White B.H. (2006) Functional dissection of a neuronal network required for cuticle tanning and wing expansion in Drosophila. J. Neurosci., 26, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peabody N.C. and White B.H. (2013) Eclosion gates progression of the adult ecdysis sequence of Drosophila. J. Exp. Biol., 216, 4395–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park J.H., Schroeder A.J., Helfrich-Forster C., Jackson F.R. and Ewer J. (2003) Targeted ablation of CCAP neuropeptide-containing neurons of Drosophila causes specific defects in execution and circadian timing of ecdysis behavior. Development, 130, 2645–2656. [DOI] [PubMed] [Google Scholar]
- 42. Vanden Broeck L., Naval-Sanchez M., Adachi Y., Diaper D., Dourlen P., Chapuis J., Kleinberger G., Gistelinck M., Van Broeckhoven C., Lambert J.C. et al. (2013) TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep., 3, 160–172. [DOI] [PubMed] [Google Scholar]
- 43. Hicks G.G., Singh N., Nashabi A., Mai S., Bozek G., Klewes L., Arapovic D., White E.K., Koury M.J., Oltz E.M. et al. (2000) Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet., 24, 175–179. [DOI] [PubMed] [Google Scholar]
- 44. Van Damme P., Robberecht W. and Van Den Bosch L. (2017) Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech., 10, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ilieva H., Polymenidou M. and Cleveland D.W. (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol., 187, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baldwin K.R., Godena V.K., Hewitt V.L. and Whitworth A.J. (2016) Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet., 25, 2378–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boeynaems S., Bogaert E., Van Damme P. and Van Den Bosch L. (2016) Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol., 132, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aulas A. and Vande Velde C. (2015) Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front. Cell. Neurosci., 9, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mackmull M.T., Klaus B., Heinze I., Chokkalingam M., Beyer A., Russell R.B., Ori A. and Beck M. (2017) Landscape of nuclear transport receptor cargo specificity. Mol. Syst. Biol., 13, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ederle H., Funk C., Abou-Ajram C., Hutten S., Funk E.B.E., Kehlenbach R.H., Bailer S.M. and Dormann D. (2018) Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci. Rep., 8, 7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Archbold H.C., Jackson K.L., Arora A., Weskamp K., Tank E.M., Li X., Miguez R., Dayton R.D., Tamir S., Klein R.L. et al. (2018) TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep., 8, 4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pinarbasi E.S., Cagatay T., Fung H.Y.J., Li Y.C., Chook Y.M. and Thomas P.J. (2018) Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep., 8, 7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.