

Abstract
Double homeobox 4 (DUX4), the causative gene of facioscapulohumeral muscular dystrophy (FSHD), is ectopically expressed in the skeletal muscle cells of FSHD patients because of chromatin relaxation at 4q35. The diminished heterochromatic state at 4q35 is caused by either large genome contractions [FSHD type 1 (FSHD1)] or mutations in genes encoding chromatin regulators, such as SMCHD1 [FSHD type 2 (FSHD2)]. However, the mechanism by which DUX4 expression is regulated remains largely unknown. Here, using a myocyte model developed from patient-derived induced pluripotent stem cells, we determined that DUX4 expression was increased by oxidative stress (OS), a common environmental stress in skeletal muscle, in both FSHD1 and FSHD2 myocytes. We generated FSHD2-derived isogenic control clones with SMCHD1 mutation corrected by clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR associated 9 (Cas9) and homologous recombination and found in the myocytes obtained from these clones that DUX4 basal expression and the OS-induced upregulation were markedly suppressed due to an increase in the heterochromatic state at 4q35. We further found that DNA damage response (DDR) was involved in OS-induced DUX4 increase and identified ataxia-telangiectasia mutated, a DDR regulator, as a mediator of this effect. Our results suggest that the relaxed chromatin state in FSHD muscle cells permits aberrant access of OS-induced DDR signaling, thus increasing DUX4 expression. These results suggest OS could represent an environmental risk factor that promotes FSHD progression.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is a genetically inherited disease that causes progressive weakness of the skeletal muscle. FSHD type 1 (FSHD1) is caused by reduced D4Z4 repeats (1–10 repeats) combined with permissive polymorphisms containing a polyadenylation (poly(A)) signal (PAS) at the sub-telomeric region 4q35. Similarly, FSHD type 2 (FSHD2) is caused by reduced D4Z4 repeats combined with permissive polymorphisms containing a PAS at 4q35, but the repeats exceed 10, and mutations in chromatin regulators, including SMCHD1 (structural maintenance of chromosomes flexible hinge domain containing 1), are also present (1–3). The genomic mutations in both FSHD1 and FSHD2 result in chromatin relaxation at 4q35, which is characterized by DNA hypomethylation and a reduction in the levels of histone 3 lysine 9 trimethylation (H3K9me3) and heterochromatin protein 1γ (HP1γ). Thus, PAS stabilizes double homeobox 4 (DUX4) transcripts to permit ectopic expression of the gene (3–6). The aberrant DUX4 expression exerts toxic effects in skeletal muscle cells through its transcriptional activity (7–9). FSHD1 and FSHD2 patients show similar clinical phenotypes, suggesting an identical molecular pathology (10).
FSHD shows unique clinical characteristics compared to other types of muscular dystrophies including relatively late onset of the disease phenotypes (typically during the second decade), asymmetric patterns of muscle weakness and large variations in disease progression among patients (11). This variability in symptoms is partially explained by the approximately inverse correlation between the number of D4Z4 repeats and clinical severity (12), but this explanation remains incomplete because clinical variability is found even among patients harboring the same number of D4Z4 repeats (13). Based on a series of clinical and genetic studies, the current consensus is that endogenous DUX4 expression plays a causative role in FSHD pathogenesis; the varying clinical characteristics of the disease would strongly support the existence of exogenous factors that modulate the clinical phenotype by affecting events upstream or downstream of DUX4 expression. Accordingly, a recent study showed that estrogens could function in mediating the sex-related differences in the disease by antagonizing DUX4 downstream events without altering DUX4 expression, thus protecting against the impaired differentiation of patient-derived myoblasts (14). Regarding events upstream of DUX4, previous pharmacological studies have identified several upstream regulators of DUX4 (15–18), but no extracellular factor that increases DUX4 expression has been reported so far. Moreover, the low-level expression of DUX4 raises questions regarding its functional impact; only extremely few cultured cells (1/1000 cells) show detectable DUX4 expression at the translational level (19). Furthermore, to date, no evidence has been reported of DUX4 protein expression in FSHD patient biopsies. However, endogenous DUX4 expression was shown to be sufficient for inducing cellular toxicity during the differentiation of myoblasts into myotubes or for impairing the differentiation of pluripotent stem cells into cells of skeletal muscle lineage (20,21). Thus, low but substantial DUX4 expression in cultured cells derived from distinct patients with FSHD appears to reflect differences in clonal conditions and disease progression of the patients (19). In addition, a recently reported rodent FSHD model showed that the muscular pathological phenotype depends on DUX4 transgene expression levels (22). A consideration of these findings led us to hypothesize that an external factor modulates disease onset and progression in FSHD patients through the transcriptional regulation of DUX4.
Oxidative stress (OS) is one of the major stresses affecting skeletal muscle function in the context of both homeostasis and pathology (23,24). The involvement of OS in FSHD pathology is supported by certain clinical and experimental studies. Clinical studies have revealed that OS markers are elevated in FSHD muscle as compared with the level in healthy muscle (25) and that oral supplementation of antioxidants partially improves muscle function in FSHD patients (26). In addition, the results of in vitro experimental studies have shown that FSHD myoblasts are vulnerable to H2O2 stimulation, a model of OS, and that DUX4-induced endogenous OS contributes to aberrant differentiation (27–29). Moreover, a series of transcriptomic studies revealed that DUX4 altered the transcription of OS-response genes (30–32). Thus, the findings obtained to date have positioned OS downstream of DUX4 in FSHD pathology, but the possibility that OS affects DUX4 expression by acting as an upstream factor has not been investigated.
In the present study, using myocytes differentiated from induced pluripotent stem cells (iPSCs) derived from both FSHD1 and FSHD2 patients, we demonstrated that OS increases DUX4 expression specifically in FSHD muscle cells. By using genome-editing technology, we generated isogenic controls of FSHD2 iPSCs by correcting the SMCHD1 mutation; myocytes derived from these isogenic controls showed suppressed basal DUX4 expression and partially recovered heterochromatic markers at 4q35. Notably, the OS-induced DUX4 increase was also suppressed in these gene-corrected myocytes. Lastly, we attributed the DUX4 increase by OS to aberrant access of DNA damage response (DDR) signaling to the opened FSHD 4q35 region and further identified ataxia-telangiectasia-mutated (ATM) kinase as a mediator of this effect. These results suggest that OS could represent an environmental risk factor that promotes FSHD progression through the regulation of DUX4.
Results
Generation of iPStet-MyoD clones from FSHD1 and FSHD2 patients and a healthy control donor
We prepared iPSCs from one FSHD1 patient harboring three D4Z4 repeats (F1), one FSHD2 patient harboring an SMCHD1 mutation (F2) and one healthy donor with no FSHD symptoms (HC) by using a previously established method, in which episomal vectors carrying the reprogramming factors Oct3/4, Sox2, Klf4 and L-Myc were transfected into skin fibroblasts of HC and F1 and blood cells of F2 (Supplementary Material, Table S1). To obtain iPSCs possessing the potential to efficiently differentiate into myocytes, we used a tetracycline-inducible MyoD piggyBac vector (tet-MyoD) that was constructed previously; this enabled the transfected iPSCs (iPStet-MyoD) to express MyoD, a master regulator gene for skeletal muscle differentiation, and subsequently to differentiate into myosin heavy chain (MyHC)-positive myocytes only when Dox was added to the medium (Fig. 1A) (33). The expression of stage-specific embryonic antigen (SSEA)-4 and tumor-related antigen (TRA)-1–60, which are indicators of undifferentiated pluripotent stem cells, was detected by immunocytochemistry, demonstrating that the iPStet-MyoD clones retained undifferentiated pluripotent characteristics (Fig. 1B; Supplementary Material, Fig. S1A). To model myocytes, myogenic differentiation was performed on these iPStet-MyoD clones through Dox-inducible MyoD overexpression. On day 8, >80% of the nuclei were positioned inside MyHC-positive cells in each clone, indicating that comparable myocyte lineage was induced from two iPStet-MyoD clones from each donor (HC#1, HC#2, F1#1, F1#2, F2#1 and F2#2) (Fig. 1C and D; Supplementary Material, Fig. S1B and C). The results of the reverse-transcription quantitative polymerase chain reaction (RT-qPCR) analysis for pluripotency markers (OCT3/4, NANOG and SOX2) and muscle-lineage markers (MYOG, MYH3 and CKM) confirmed the efficient myogenic differentiation of each cell line (Fig. 1E and F; Supplementary Material, Fig. S1D and E). Moreover, RT-qPCR analysis for DUX4 and its reported downstream targets (ZSCAN4, TRIM43 and MBD3L2) revealed considerably higher expression of these molecules in the differentiated state than in the undifferentiated state in the F1 and F2 clones but not in the HC clone (Fig. 1G; Supplementary Material, Fig. S1F). The expression of ACTB, which we used as another reference gene, was almost comparable between the undifferentiated and differentiated states (Fig. 1H; Supplementary Material, Fig. S1G). In the case of the F1#1 clone, DNA methylation analysis specific for the allele harboring short D4Z4 repeats revealed that DNA methylation was stably low during the reprogramming from fibroblasts to iPSCs or differentiation from iPSCs to myocytes as compared to the healthy level (> 26%) reported previously by using the same methods (Fig. S2) (34). These data suggested that the myocytes differentiated from the human iPSC clones were suitable for investigating FSHD-related DUX4 expression.
Figure 1.
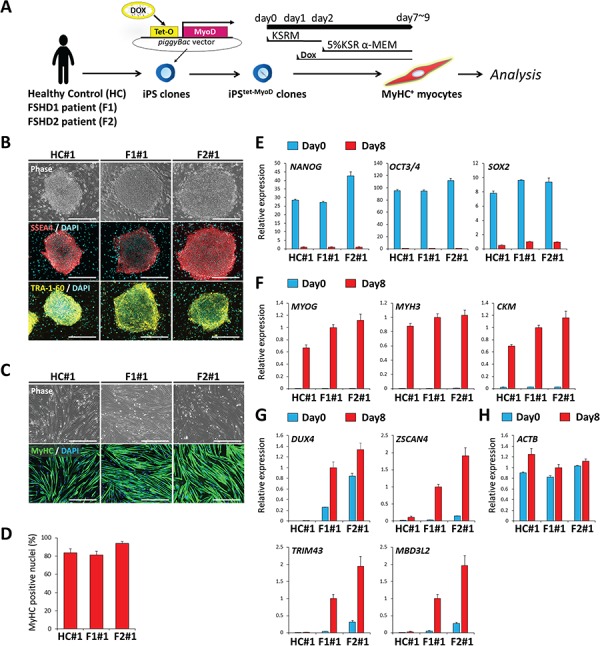
iPStet-MyoD clones were generated from a healthy control and from FSHD1 and FSHD2 patients. (A) Scheme of the differentiation of iPSC clones to myocytes. (B) Representative images of immunofluorescence staining of SSEA4 (red) and TRA-1-60 (yellow) in undifferentiated iPSCtet-MyoD clones. Scale bar: 500 μm. (C) Representative images of immunofluorescence staining of MyHC (green) in differentiated myocytes at day 8. Scale bar: 500 μm. (D) Differentiation efficiency was quantified by calculating the percentage of MyHC positive nuclei in total nuclei at day 8 (n = 4). (E–H) RT-qPCR analysis of all clones at day 0 (undifferentiated) and day 8 (differentiated) (n = 3) for (E) pluripotency markers, (F) myogenic markers, (G) DUX4 and its downstream targets and (H) ACTB as a reference gene. Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to F1#1 at day 8 (differentiated). Data information: in (D–H), data are represented as mean ± standard error of the mean.
OS increased DUX4 expression in both FSHD1 and FSHD2 myocytes
To investigate whether OS increases DUX4 expression, we stimulated myocytes with H2O2, which served here as an OS model. RT-qPCR analysis of DUX4 mRNA expression in myocytes showed that H2O2 stimulation for 24 h, which is physiologically mild to allow cell survival (for more than 80%), significantly increased the transcription of DUX4 and its targets, a phenomenon not observed in the healthy control (Fig. 2A; Supplementary Material, Fig. S3). The H2O2 stimulation did not increase the MYH3 level, indicating that the DUX4 increase was not because of accelerated myogenic differentiation, as DUX4 expression is known to increase after myogenic differentiation (Fig. 2A). F1#1 and F2#1 myocytes showed an almost dose-dependent increase of DUX4 expression and transcriptional activity (Fig. 2B). Conversely, treatment with NAC, an antioxidant, attenuated the DUX4 increase induced by H2O2, indicating that the OS caused by H2O2 stimulation was responsible for the DUX4 increase (Fig. 2C). Time-course analysis further revealed that the increase of DUX4 transcription started between 6 and 12 h after H2O2 stimulation, suggesting that an early response to OS mediates the increase in transcription (Fig. 2D). The increase of ZSCAN4 occurred later than the increase of DUX4, suggesting that the DUX4 increase was because of an increase in the amount of transcription per cell, not because of selection for the population with higher DUX4 expression (Fig. 2D). In contrast, F1 fibroblasts did not express DUX4 regardless of H2O2 stimulation (Supplementary Material, Fig. S4). These data demonstrated that OS increased DUX4 expression in FSHD muscle cells.
Figure 2.
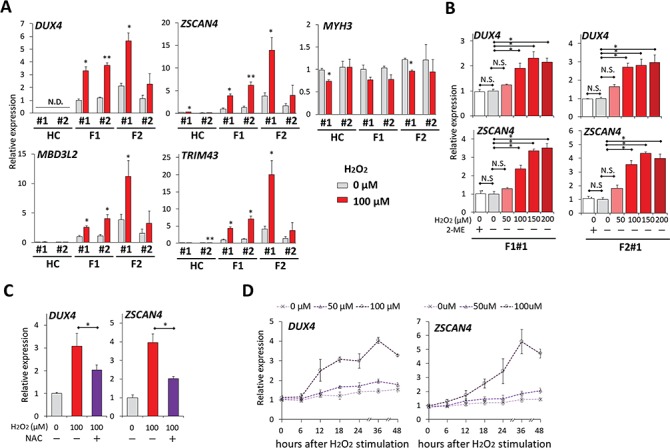
OS increased DUX4 expression in FSHD myocytes. (A) RT-qPCR was performed for DUX4, its downstream targets (ZSCAN4, TRIM43 and MBD3L2) and MYH3 in HC#1, HC#2, F1#1, F1#2, F2#1 and F2#2 myocytes incubated with or without 100 μM H2O2 for 24 h at day 8 after differentiation (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to F1#1 without H2O2. (B) RT-qPCR was performed for DUX4 and ZSCAN4 in F1#1 and F2#1 myocytes incubated with 2-mercaptoethanol (2-ME) or with H2O2 at various concentrations for 24 h at day 8 after differentiation (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition with 0 μM H2O2 without 2-ME in each clone. (C) NAC was pretreated 1 h before H2O2 stimulation to F1#1 myocytes at day 8. 24 h after H2O2 addition, RT-qPCR was performed for DUX4 and ZSCAN4 (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition without H2O2 or NAC. (D) Time-course analysis of DUX4 and ZSCAN4 mRNA expression by RT-qPCR after H2O2 stimulation (n = 3 per each time point). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition with 0 μM H2O2. Data information: all data are represented as mean ± SEM. *P ≤ 0.05, *P ≤ 0.01 [Student’s t-test in (A) and (C), one way ANOVA followed by Dunnett’s Multiple Comparison Test in (B)].
DUX4 expression in FSHD2 isogenic control clones showed basal suppression and resistance to OS
To ascertain whether the OS-induced DUX4 increase was a disease-specific event, we generated isogenic control cell lines by correcting the SMCHD1 mutation in F2#1 iPStet-MyoD cells by using the CRISPR/Cas9 system together with donor template vectors for homology-directed repair (Fig. 3A). Patient F2 carried a heterozygous mutation with a 15 bp deletion and 1 bp substitution in exon 32 of SMCHD1, which was modified to generate two isogenic clones (IC#1 and IC#2) in which the mutations were replaced with the wild-type sequence (Fig. 3A and B). These isogenic clones retained the expression of pluripotency markers and exhibited comparable ability to differentiate into MyHC-positive myocytes (Fig. 3C–E and G–H). SMCHD1 protein expression was increased in isogenic control myocytes compared to parental F2#1 myocytes, indicating haploinsufficiency of SMCHD1 in the parental clone myocytes (Fig. 3F). In IC#1 and IC#2 myocytes, DUX4 and its downstream targets were expressed at substantially lower levels than in parental clone F2#1 myocytes, indicating that the SMCHD1 mutation was responsible for the basal DUX4 transcription in these F2 myocytes (Fig. 3I). Comparable ACTB expression confirmed these observations (Fig. 3J). Moreover, the myocytes differentiated from the isogenic control clones showed only a slight increase in DUX4 expression following H2O2 stimulation, and the expression level in the isogenic control clones treated with the highest concentration of H2O2 was still lower than that in the parental clone without stimulation (Fig. 3K; Supplementary Material, Fig. S5A). Together, these data indicate that the DUX4 expression level and its marked OS-induced increase were dependent on the disease-causing SMCHD1 mutation in FSHD2.
Figure 3.
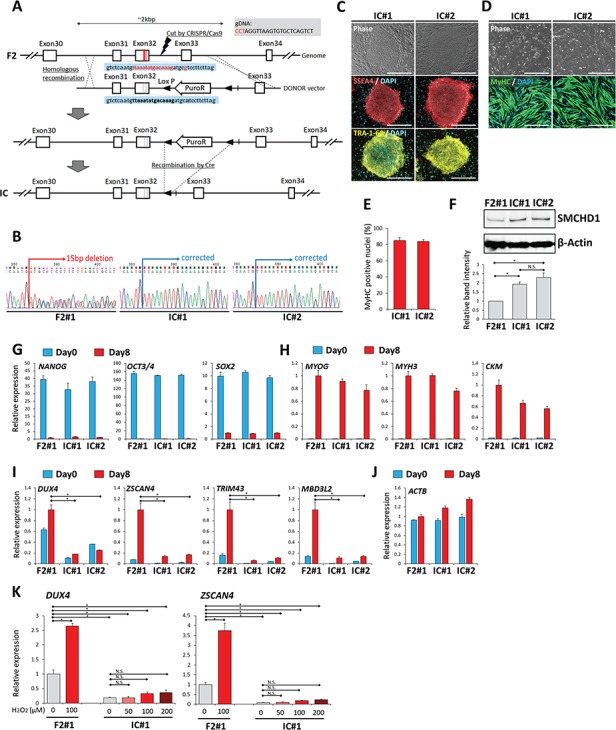
Basal DUX4 expression and increased DUX4 expression by OS were suppressed by gene modification in FSHD2 myocytes. (A) Scheme of the gene correction of the SMCHD1 mutation (shown in red letters) in F2 clone. The expected sequence (equal to wild-type sequence) after correction is shown in bold black letters. (B) The sequence around the mutation site was checked. F2#1 clone has a heterozygous 15 bp deletion followed by one point mutation (A), resulting in an ambiguous signal pattern, but genetically modified IC#1 and IC#2 clones showed a single pattern consistent with the expected sequence shown in (A), confirming the right correction. (C) Representative images of immunofluorescence staining of SSEA4 (red) and TRA-1-60 (yellow) in undifferentiated iPSCtet-MyoD clones of IC#1 and IC#2. Scale bar: 500 μm. (D) Representative images of immunofluorescence staining of MyHC (green) in differentiated myocytes at day 8 of IC#1 and IC#2. Scale bar: 500 μm. (E) Differentiation efficiency was quantified by calculating the percentage of MyHC positive nuclei in total nuclei at day 8 (n = 4). (F) Representative result of western blotting analysis for SMCHD1 (upper panel) and β-Actin (lower panel) in the myocytes of each clone at day 8. The lowest graph shows quantification of relative band intensity. Band intensity of SMCHD1 was normalized to that of β-Actin and subsequently to F2#1 (n = 3). (G–J). RT-qPCR analysis of all clones on day 0 (undifferentiated) and day 8 (differentiated) (n = 3) for (G) pluripotency markers, (H) myogenic markers, (I) DUX4 and its downstream targets and (J) ACTB as a reference gene. Relative expression levels were normalized to RPLP0 as an internal control in each sample and subsequently to F2#1 at day8 (differentiated). (K) Differentiated myocytes at day 8 after induction from F2#1 and IC#1 clones were incubated with or without H2O2 for 24 h, and RT-qPCR was performed for DUX4 and ZSCAN4 (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to F2#1 without H2O2. Data information: in (E–K) data are represented as mean ± SEM. *P ≤ 0.05, N. S. not significant [one-way ANOVA followed by Dunnett’s Multiple Comparison Test in (I) and by Tukey‘s test in (F) and (K)].
Heterochromatic status at 4q35 was partially increased in F2 isogenic control myocytes
FSHD 4q35 is characterized by chromatin relaxation through DNA hypomethylation and reduced levels of H3K9me3 and HP1γ (4,5). Unexpectedly, DNA methylation in the 4qA allele was not significantly increased after the gene correction (Fig. 4A; Supplementary Material, Fig. S5B). However, chromatin immunoprecipitation (ChIP) followed by RT-qPCR analysis revealed that the accumulation of H3K9me3 and HP1γ had increased in the D4Z4 regions in isogenic control myocytes, whereas H3K4me2, which we used as a transcriptional activation marker, was not significantly altered (Fig. 4B; Supplementary Material, Fig. S5C). Moreover, SMCHD1 accumulation was also increased in isogenic control myocytes, indicating SMCHD1 haploinsufficiency at the D4Z4 region in the F2 clone (Fig. 4B; Supplementary Material, Fig. S5C). These data indicated that a reduction of heterochromatic histone modification underlays the DUX4 basal expression and increase by OS in FSHD myocytes.
Figure 4.

The 4q35 genome in FSHD2 isogenic control myocytes showed a more heterochromatic state at the protein level. (A) DNA methylation analysis on the 4q35 region by bisulfite sequencing. White and black circles mark unmethylated and methylated CpG, respectively. The lower quartile (Q1) and range of the percent methylation are shown above each column. (B) ChIP RT-qPCR was performed on 4q35 for H3K9me3, H3K4me2 (as a negative control), HP1ɤ and SMCHD1 (n = 4). Relative % input was normalized to F2#1. Data information: all data are represented as mean ± SEM. *P < 0.05, **P < 0.01 (Student’s t test, unpaired).
DDR occurred before DUX4 increase under OS
Because H2O2 stimulation exerts a broad range of effects on cells including DNA damage (23,35), we next investigated whether DDR was involved in the OS-induced increase of DUX4. At 6 h after H2O2 stimulation, when DUX4 expression was not yet affected by H2O2 in FSHD myocytes (Fig. 2D), the expression of γ-H2AX, an indicator of DDR, was increased in each clone similarly (Fig. 5A and B). For F1#1 myocytes, the expression of γ-H2AX showed an H2O2 dose-dependent increase (Fig. 5C and D). These data indicate that the DDR by OS occurs prior to DUX4 increase in FSHD myocytes.
Figure 5.
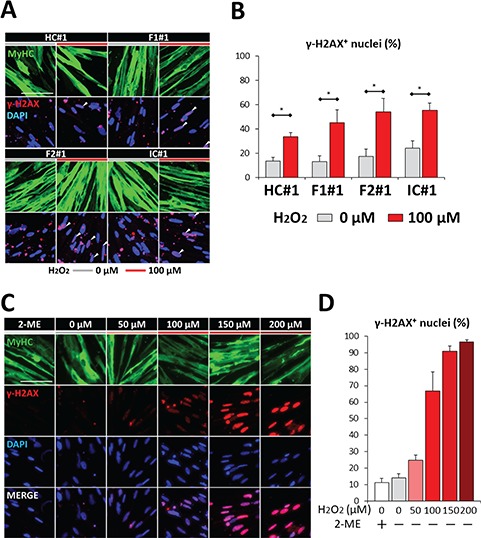
DDR occurred after H2O2 stimulation but before DUX4 expression increased. (A) Representative images of immunofluorescence staining of MyHC (green) and ɤ-H2AX (red) in HC#1, F1#1, F2#1 and IC#1 myocytes at day 8 in the presence or absence of H2O2. Cells were fixed 6 h after H2O2 addition. Scale bar: 100 μm. (B) The percentages of ɤ-H2AX positive nuclei in total nuclei in (A) (n = 3). (C) Representative images of immunofluorescence staining of MyHC (green) and ɤ-H2AX (red) in F1#1 myocytes at day 8 incubated with 2-ME or with H2O2 at various concentrations for 6 h. Scale bar: 100 μm. (D) The percentages of ɤ-H2AX positive nuclei in total nuclei in (C) (n = 3). Data information: in (B) and (D), data are represented as mean ± SEM. *P ≤ 0.05 [Student’s t-test in (B)].
Other genotoxic stresses increased DUX4 expression in FSHD myocytes
Moreover, because ultraviolet C (UV-C) exposure induces DDR, we examined its effect and found that a short exposure of myocytes to UV-C (such that most cells survived for 24 h) also increased the expression of DUX4 and its targets, but not of MYH3 (Supplementary Material, Fig. S6A–C). The expression of γ-H2AX was similarly increased in each clone before UV-C affected DUX4 transcription (Supplementary Material, Fig. S6D and E). We examined whether DNA damage alone is sufficient to induce the DUX4 increase using mitomycin C (MMC), a DNA damage inducer. Myocytes treated with MMC also showed an increased expression of DUX4 and its targets, but not of MYH3 (Fig. 6A and B). F1#1 myocytes showed an MMC dose-dependent increase of DUX4 expression and transcriptional activity (Fig. 6C). The expression of γ-H2AX was increased in each clone similarly before MMC affected DUX4 transcription (Fig. 6D and E). These data indicated that genotoxic stresses could increase DUX4 expression in FSHD myocytes.
Figure 6.
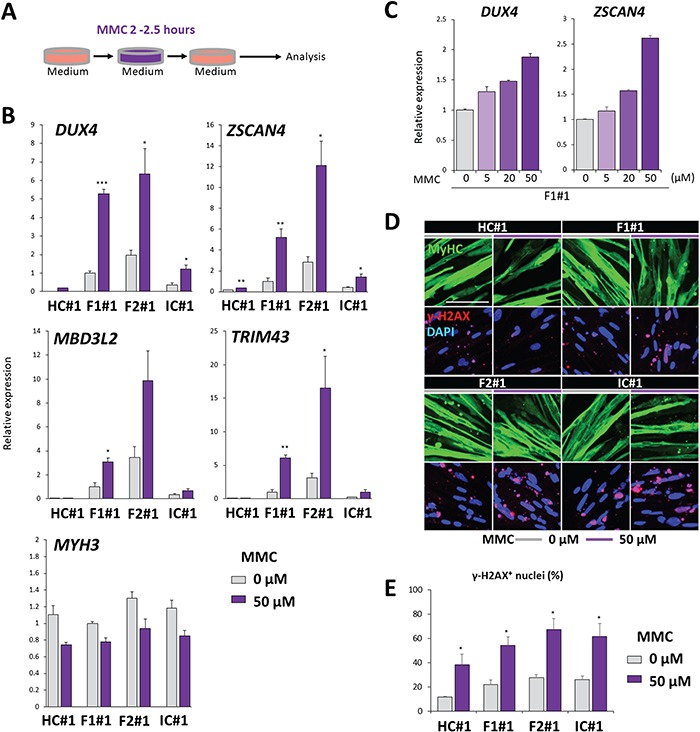
MMC exposure increased DUX4 expression. (A) Scheme of the MMC exposure. Differentiated myocytes at day 8 after induction were exposed to MMC for 2–2.5 h, incubated for 9 or 24 h and analyzed. (B) RT-qPCR was performed for DUX4, its downstream targets (ZSCAN4, TRIM43 and MBD3L2), and MYH3 in HC#1, F1#1, F2#1 and IC#1 myocytes exposed to MMC for 2.5 h and subsequently incubated for 24 h (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to F1#1 without UV-C exposure. (C) RT-qPCR was performed for DUX4 and ZSCAN4 in F1#1 myocytes exposed to MMC at various concentrations for 2 h and subsequently incubated for 24 h (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition without MMC exposure. (D) Representative images of immunofluorescence staining of MyHC (green) and ɤ-H2AX (red) in HC#1, F1#1, F2#1 and IC#1 myocytes at day 8 exposed to MMC for 2.5 h. Cells were fixed for 9 h after the initiation of MMC exposure. Scale bar: 100 μm. (E) The percentages of ɤ-H2AX positive nuclei in total nuclei in (C) (n = 3). Data information: in (B), (C) and (E), data are represented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 [Student’s t-test in (B) and (E)].
OS indirectly increased DUX4 through ATM-mediated DDR
DDR is primarily controlled by three phosphoinositide 3-kinase-related kinases: ATM, ATM and Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK) (36,37). To identify the kinase that is responsible for OS-induced DUX4 increase, F1#1 myocytes were stimulated with H2O2 in the presence or absence of specific kinase inhibitors. Whereas the inhibition of ATR or DNA-PK did not markedly affect DUX4 expression, the inhibition of ATM attenuated the DUX4 increase induced by H2O2 in a concentration-dependent manner (Fig. 7A). Moreover, treatment with the ATM inhibitor caused a decrease in the number of γ-H2AX-positive nuclei under the OS condition in F1 myocytes, which indicated that the DDR triggered by H2O2 stimulation was prevented by ATM inhibition (Fig. 7B and C). Although OS can directly activate members of the stress-induced MAP kinase family to regulate gene expression (23,38), the inhibition of MEK, JNK or p38 MAPK did not suppress the OS-induced DUX4 increase; therefore, this increase appeared not to be a direct outcome of stress responses (Supplementary Material, Fig. S7A). Lastly, the DUX4 increase in F2#1 myocytes under the OS condition was also attenuated by the ATM inhibitor (Fig. 7D) and inhibition of ATM did not decrease expression of DUX4 and ZSCAN4 in FSHD myocytes in the absence of OS (Supplementary Material, Fig. S7B and S7C). Overall, these data indicated that DDR mediates the OS-induced DUX4 increase through ATM activation in FSHD myocytes.
Figure 7.
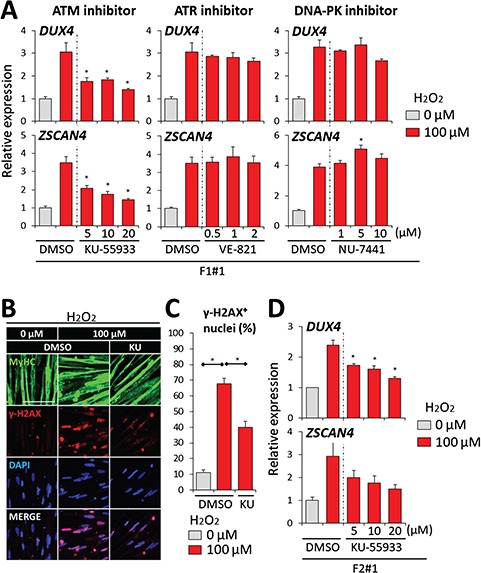
ATM inhibition suppressed DUX4 increase by OS. (A) RT-qPCR was performed for DUX4 and ZSCAN4 in F1#1 myocytes under H2O2 stimulation treated with KU-55933, VE-821 or NU-7441 (inhibitors for ATM, ATR or DNA-PK, respectively) (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition with 0 μM H2O2. Dimethyl sulfoxide (DMSO) was added to each sample to keep its final concentration comparable among all conditions. (B) Representative images of immunofluorescence staining of MyHC (green) and ɤ-H2AX (red) in F1#1 myocytes in the absence or presence of 100 μM H2O2 and 20 μM KU-55933 (KU). DMSO was added to each sample to keep its final concentration comparable among all the conditions. (C) The percentages of ɤ-H2AX positive nuclei in total nuclei in (B) (n = 3). (D) RT-qPCR was performed for DUX4 and ZSCAN4 in F2#1 myocytes under H2O2 stimulation and treated with KU-55933 (n = 3 for each condition). Relative expression levels were normalized to RPLP0 as an internal control in each sample and then to the condition with 0 μM H2O2. DMSO was added to each sample to keep its final concentration comparable among all conditions. Data information: in (A), (C) and (D), data are represented as mean ± SEM. *P ≤ 0.05 [one-way ANOVA followed by Dunnett’s Multiple Comparison Test in (A), (C) and (D)].
Discussion
FSHD is caused by genetic disorders but shows variations in disease progression among patients, suggesting the existence of certain external factors that modulate the pathological condition. Using patient iPSCs, this study has demonstrated, to our knowledge for the first time, that the expression of DUX4, the gene primarily responsible for FSHD pathology, is increased by external cellular stress. Furthermore, we have shown that the DUX4 increase induced by OS is mediated by the DDR signaling pathway, which suggests that diverse types of genotoxic stress can increase DUX4 expression and could therefore represent risk factors for FSHD onset or progression.
The involvement of OS in FSHD pathology has been previously reported by several groups. DUX4-expressing muscle cells showed transcriptome enrichment in OS-related gene expression (30–32). Moreover, exogenous DUX4 expression in mouse myoblasts or human immortalized myoblasts resulted in vulnerability to OS (28,39). Endogenous DUX4 expression in immortalized FSHD myoblasts also led to vulnerability to OS and provoked the production of endogenous reactive oxygen species (ROS) followed by DDR to cause partial failure in differentiation (29). However, these findings regarding the association of OS with FSHD provide insights only into the downstream consequences of DUX4 expression. In contrast, our results suggest OS and DNA damage could act as upstream inducers of DUX4 expression in FSHD. Collectively, these findings indicate that a positive-feedback loop in signaling cascades could be formed among OS, DNA damage and endogenous DUX4 expression. During myogenic differentiation in mouse and human myoblasts, transient DNA strand breakage is followed by the formation of γ-H2AX foci (40,41). This effect might underlie the ‘burst’ of DUX4 expression in a limited population of FSHD myoblasts during differentiation (20). In the context of differentiated muscle cells, which are recognized to be resistant to diverse stressors (42,43), including H2O2 and DNA damage inducers, DNA damage can be accumulated without cell death by suppressing the p53-mediated apoptosis pathway (42,43). This effect might contribute to the increase in DUX4 expression induced in muscle fibers by OS and other genotoxic stresses. Thus, OS and DNA damage are likely to influence FSHD pathology through the regulation of DUX4 expression.
ATM performs multiple functions in DDR signaling at DNA lesion sites, including transcriptional regulation and DNA repair. We propose that the diminished heterochromatic state at the 4q35 region, characterized by a reduction of H3K9me3 and HP1γ, increases the region’s sensitivity to DNA damage-induced ATM activity, which results in DUX4 increase. A considerable amount of evidence has implicated chromatin compaction in the regulation of ATM activity. HDAC inhibition in human fibroblasts permits an ATM-mediated transcriptional upregulation of MCL1 and Gadd45α, which are ATM targets, through the recruitment of E2F1 transcription factor (44). In the context of DNA repair, heterochromatin serves as a barrier to the expansion of ATM-dependent γ-H2AX (45,46). Moreover, a decrease in chromatin compaction, which can be induced by reducing the level of linker histone H1, enhances resistance to DNA damage through an increased accessibility of DDR factors to the lesion sites (47). Thus, we suggest that non-FSHD 4q35 is transcriptionally silenced and is less sensitive to the DNA damage-induced ATM-mediated transcriptional machinery, whereas FSHD 4q35 permits the ATM-mediated machinery to access the lesion site (Supplementary Material, Fig. S8).
Notably, in our FSHD models, activity of DUX4 was dependent on myogenic differentiation in both types of FSHD and was associated with a disease-related mutation on SMCHD1 in FSHD2 (Fig. 1G and 3I), which indicates that our models are useful for further FSHD pathological studies. Isogenic controls generated for the FSHD2 clone showed robust suppression of endogenous DUX4 expression, which agrees with previous genetic studies (6). Intriguingly, SMCHD1 was found to recover the heterochromatic state according to the level of a protein marker (although the level was still lower than that in the healthy control; data not shown), but not in terms of the DNA methylation level (Fig. 4). This observation indicates that the DNA methylation status, once determined, remains stable and is resistant to post-developmental modification by epigenetic factors, whereas alterations of heterochromatic protein markers are sufficient to suppress DUX4 expression at 4q35. This finding supports the modification of histone markers as a therapeutic strategy for FSHD.
Although the expression of DUX4 in FSHD myocytes upregulated downstream transcriptional targets, we did not observe marked phenotypic changes, including apoptosis or impaired differentiation, which apparently disagrees with previous reports (15,21). We propose two possible reasons for this phenotypic absence. The first is that the survival duration of our FSHD myocyte models might be too short to allow for notable phenotypic changes under the mild OS conditions used in this study. A higher H2O2 concentration therefore may be recommended but at the same time risks cell death independently of DUX4 expression (Supplementary Material, Fig. S3). Instead, a more mature muscle lineage showing longer survival would be preferred. The second is that the myocytes induced in our model system, which was shown to mimic the embryonic myogenic lineage, might maintain resistance to DUX4 activity. Given that DUX4 expression is detected even in the FSHD embryo and that the disease phenotypes manifest long after birth in most FSHD cases (48), embryonic myocytes might be capable of tolerating DUX4 expression. To investigate this possibility, a more mature state of muscle cells could be useful.
In accordance with previous studies, our findings indicate that increased OS is a risk factor for FSHD patients. The elevated OS could potentially be attenuated through supplementation with ROS scavengers or even by including anti-oxidative nutrients in the daily diet. Overall, our study has revealed, by using patient-derived iPSCs, a previously unrecognized mechanism by which DUX4 expression is regulated in FSHD muscle cells. This finding could provide a basis for drug development and discovery of therapeutic targets for FSHD.
Materials and Methods
Ethical approval
This study was approved by the Ethics Committee of the Graduate School of Medicine, Kyoto University and the Kyoto University Hospital (Approval numbers #R0091 and #G259) and was conducted according to the guidelines of the Declaration of Helsinki. To protect confidentiality, all patient information were kept anonymous, and a written informed consent was obtained from the study participants.
Cell line and cell culture
Human dermal fibroblasts were donated by one Japanese male FSHD1 patient and one healthy donor. Human blood cells were obtained from one Japanese female FSHD2 patient (34). Donor clinical information is listed in Supplementary Table S1. All human iPSCs used in this study were established by overexpressing four transcription factors by using an episomal vector and were maintained on inactivated mouse feeder cells, as previously described (33), in primate embryonic stem (ES) cell medium (ReproCELL, Japan) supplemented with 4 ng/mL recombinant human basic fibroblast growth factor (Oriental Yeast, Japan).
Generation of iPStet-MyoD clones
To prepare iPStet-MyoD clones (iPSCs that express MyoD in response to doxycycline), the MyoD element was cloned into the PB-TAC-ERN vector by using Gateway cloning to generate PB-MyoD as previously described (33). iPSCs were transfected with plasmids, including PBase and PB-MyoD, by using a NEPA21 electroporator (Nepagene, Japan) and plated on mouse feeder cells for clone selection and culture maintenance as previously described (49).
Myocyte differentiation
iPStet-MyoD cells were treated with accutase (Nacalai Tesque, Japan) to separate them into single cells and plated at a density of 3–5 × 105 cells/well in 6-well dishes coated with Matrigel (BD Biosciences, USA) in primate ES cell medium supplemented with 100 μg/mL neomycin sulfate (Nacalai Tesque) and 10 μM Y-27632 (Nacalai Tesque). On the following day, the culture medium was replaced with primate ES cell medium containing 1–2.5 μg/mL doxycycline (Dox; LKT Laboratories, USA). Induction was performed in 5% KSR/α-MEM containing Dox and 2-mercaptoethanol (2-ME) supplement for 5–7 days. Dox was removed at least 1 day before harvest for gene expression study.
Correction of SMCHD1 mutation in iPSCs
The mutation in SMCHD1 was corrected through homologous recombination (HR)-mediated knock-in using the CRISPR/Cas9 system (CRISPR and Cas9 endonuclease system) as described in (50). Briefly, the single-guide RNA (sgRNA) sequence was designed for an intron site near the mutation site by using CRISPRdirect online software (http://crispr.dbcls.jp/) (51) and cloned into pHL-H1-ccdB-mEF1a-RiH (Addgene ID: 60601). For the HR template, bilateral ~1 kb regions interposing the sgRNA target site with the mutation in exon 32 replaced by the wild-type sequence were cloned and inserted before and after the puromycin-resistance gene element with two loxP sites in pENTR-Donor-MCS2. The sgRNA vector, the HR vector and pHL-EF1a-SphcCas9-iP-A (Addgene ID: 60599) were cotransfected using a NEPA21 electroporator into F2 iPStet-MyoD cells. Subcloning was performed after drug selection for 1 week. Among the obtained clones, those harboring the homozygous HR-derived sequence were selected and transfected with a Cre recombinase construct, and subcloning was again performed to select the clones that had lost the puromycin resistance. Lastly, the sequence was confirmed to contain only one loxP site in each allele. The primers used in these procedures are listed in Supplementary Table S2.
Immunofluorescence analysis
Cultured cells were fixed with 2% formaldehyde in phosphate buffered saline (PBS), washed in PBS, refixed with 100% methanol and blocked with Blocking One solution (Nacalai Tesque) at 4 °C. Fixed samples were incubated overnight at 4 °C with primary antibodies diluted in 10% Blocking One/PBST [PBS containing 0.2% Triton X-100 (Santa Cruz, USA)], washed repeatedly with PBST and incubated for 1 h with secondary antibodies (diluted in 10% Blocking One/PBST) and the nuclear stain DAPI (Sigma; 1:5000). Samples were examined, and images were captured using a BZ9000 system (Keyence, Japan) at 200× and 400× magnification. The antibodies used in this study are listed in Supplementary Table S3. Myocyte differentiation efficiency is shown as the percentage of nuclei within MyHC-positive areas in total nuclei per field.
RNA extraction and real-time RT-qPCR
Total RNA was extracted using a ReliaPrep RNA Miniprep System (Promega) as per the manufacturer’s instructions. cDNA was synthesized from the extracted RNA by using ReverTra Ace Master Mix with gDNA Remover (TOYOBO, Japan). Real-time RT-qPCR was performed using SYBR Green probe sets (Applied Biosystems, USA) and a Step One Plus thermal cycler (Applied Biosystems), and a standard curve was prepared for each target. RPLP0, which encodes a ribosomal protein, was used as the internal control in all assay. The primer sets used in this study are listed in Supplementary Table S4.
Protein extraction and western blotting
Cultured cells were washed with PBS and collected in tubes as pellets. Cell pellets were lysed in radio-immunoprecipitation assay buffer (Nacalai Tesque, Kyoto, Japan) containing 1% (v/v) Protease Inhibitor Cocktail (Nacalai Tesque) and then sonicated with a Bioruptor (BM EQUIPMENT, Japan) for 9 cycles (high intensity, 15/15-s on/off per cycle). Protein concentrations of the lysates were measured using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. Protein lysate (5 and 1 μg, for SMCHD1 and β-Actin) were mixed with NuPAGE LDS Sample Buffer and NuPAGE Sample Reducing Agent (Thermo Fisher Scientific) and heated at 70°C for 10 min. Samples were then separated on a 3–8% NuPAGE Novex tris-acetate mini gel (Thermo Fisher Scientific) at 150 V for 60 min with the addition of NuPAGE antioxidant (Thermo Fisher Scientific) during electrophoresis. The fractionated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using an iBlot system (Thermo Fisher Scientific) with the program, P0, 9 min. The membrane was blocked with Blocking One (Nacalai Tesque) and incubated with primary antibody against SMCHD1 (Sigma-Aldrich, USA) at 4°C overnight, otherwise the membrane was blocked overnight for β-Actin. After three washes with PBST (containing 0.1% Tween-20), the membrane was incubated for 1 h at room temperature with peroxidase labeled anti-rabbit IgG antibody (Vector Laboratories, USA) as a secondary antibody for SMCHD1 or with anti-β-Actin−Peroxidase antibody (Sigma-Aldrich, USA). Detection was carried out with SuperSignal West Femto substrate (Thermo Fisher Scientific). Visualization of images was performed by ImageQuant LAS 4000 system (GE Healthcare Life Science, UK). Images were analyzed with ImageJ (https://imagej.nih.gov/ij/) to calculate relative band intensity.
ChIP and ChIP qPCR analysis
ChIP analysis was performed by using an EpiScope ChIP Kit (TAKARA, Japan) according to the manufacturer’s instructions with partial modifications. Briefly, 2–5 × 106 cells of each iPSC clone were plated and differentiated in 10 cm dishes to obtain equally confluent myocytes. The cells were cross-linked for 5 min at room temperature with 1% formaldehyde and lysed, and the lysates were sonicated with a Bioruptor for 12 cycles (high intensity, 30/30-s on/off per cycle). Fragmented lysates were incubated with antibodies conjugated to mouse IgG magnetic beads (TAKARA) or Dynabeads Protein G (Thermo Fisher Scientific) overnight. The beads were precipitated and washed in a series of buffers supplied in the kit and with DynaMag (Thermo Fisher Scientific). Immunoprecipitated beads and 10% of the input chromatin was purified by reversing the crosslink and treating with RNase A and Proteinase K according to the manufacturer’s instructions. RT-qPCR followed by the ΔΔCt method was used to quantify the D4Z4 elements. Relative % input was calculated by dividing the amount of immunoprecipitated chromatin by that of the input and then normalizing relative to the F2 sample. The following primer pair was used for D4Z4 elements: 5′-CCGCGTCCGTCCGTGAAA-3′ and 5′-TCCGTCGCCGTCCTCGTC-3′ (4).
DNA methylation analysis
Genomic DNA was extracted using a GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, USA) as per the manufacturer’s protocol, and 500 ng–1 μg of DNA was treated with bisulfite by using an EpiTect DNA bisulfite kit (QIAGEN, Germany) according to the manufacturer’s guidelines. For the F1 clone, the methylation level of the FSHD allele was quantified using the pyrosequencing technique as described in (34). Briefly, PCR was performed using a PyroMark PCR Kit (QIAGEN), and 10 μL of the biotinylated PCR product was affinity purified using Streptavidin Sepharose High Performance (GE Healthcare Life Science) and PyroMark Q24 Advanced CpG Reagents (QIAGEN). For the F2 isogenic controls IC#1 and IC#2, PCR and DNA methylation analysis of the 4qA distal region were performed according to the protocol of the 4qA BSS assay as described in (52) for QUMA online software (http://quma.cdb.riken.jp/index_j.html) (53); the analysis was performed using the default parameters.
Reagents
Hydrogen peroxide mixed with a stabilizer (H2O2, Sigma) was added at various concentrations by diluting a 1 mM stock immediately before use on day 8 of myocyte differentiation after the depletion of 2ME for 1 day. N-acetylcysteine (NAC) was dissolved in water, and the pH was adjusted to 7.0. NAC was added to the cells 1 h before H2O2 stimulation. KU-55933 (Selleck Chemicals, USA), VE-821 (AdooQ BioScience, USA), NU-7441 (Selleck Chemicals), PD0325901 (Cayman Chemical, USA), SP600125 (Selleck Chemicals) and SB203580 (Sigma) were dissolved in DMSO (Sigma) and added 1 h before H2O2 stimulation. MMC was added at various concentration for 2 or 2 and a half h and then washed once and replaced with medium.
UV-C irradiation
For the light source of UV-C, the lamps inside the laminar flow cabinet were used. Myocytes that were differentiated on 6 cm dishes were washed once, and medium was replaced with PBS. Myocytes were then positioned on a fixed position inside the laminar flow cabinet, exposed to UV-C, and washed once. Finally, PBS was replaced with medium.
Supplementary Material
Acknowledgements
We are grateful to the donors who gave their cells for the establishment of iPSCs. We thank Dr Kohei Hamanaka and Dr Hiroaki Mitsuhashi for constructive discussion and Dr Takashi Ikeda and Dr Masaki Yagi for technical instruction. Editage (www.editage.jp) proofread an earlier draft of this paper. We also thank Dr Peter Karagiannis for reading the manuscript. M.S.-H. appreciates all the members of his laboratory for kind support because of his physical handicap. M.S.-H. especially appreciates his parents and Mrs. Nao Sasaki, his wife, for generous and unwavering supports.
Conflict of Interest statement. None declared.
Funding
Acceleration Program for Intractable Diseases Research utilizing Disease-specific iPS cells, which were provided by the Japan Agency for Medical Research and Development, AMED (to H.S. and A.H.); the Japan Society for the Promotion of Science KAKENHI (17 J04509 to M.S.-H. and 15H05581 to A.H.); The Intramural Research Grant [29-4] for Neurological and Psychiatric Disorders of National Center of Neurology and Psychiatry and the Fugaku Trust for Medical Research (to R.M.).
References
- 1. Tawil R., Maarel S.M. and Tapscott S.J. (2014) Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet. Muscle, 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daxinger L., Tapscott S.J. and Maarel S.M. (2015) Genetic and epigenetic contributors to FSHD. Curr. Opin. Genet. Dev., 33, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lemmers R.J.L.F., Vliet P.J., Klooster R., Sacconi S., Dauwerse J.G., Snider L., Straasheijm K.R., Van Ommen G.J., Padberg G.W., Miller D.G. et al. (2010) A unifying genetic model for facioscapulohumeral muscular dystrophy. Science, 329, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zeng W., De Greef J.C., Chen Y.Y., Chien R., Kong X., Gregson H.C., Winokur S.T., Pyle A., Robertson K.D., Schmiesing J.A. et al. (2009) Specific loss of histone H3 lysine 9 trimethylation and HP1γ/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet., 5, e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lemmers R.J.L.F., Goeman J.J., Van der Vliet P.J., Van Nieuwenhuizen M.P., Balog J., Vos-Versteeg M., Camano P., Ramos Arroyo M.A., Jerico I., Rogers M.T. et al. (2015) Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet., 24, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lemmers R.J.L.F., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W.E., Amell A.M., Van Der Vliet P.J., Almomani R., Straasheijm K.R. et al. (2012) Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet., 44, 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., Van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R. et al. (2012) DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell, 22, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shadle S.C., Zhong J.W., Campbell A.E., Conerly M.L., Jagannathan S., Wong C.J., Morello T.D., Maarel S.M. and Tapscott S.J. (2017) DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet., 13, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whiddon J.L., Langford A.T., Wong C.-J., Zhong J.W. and Tapscott S.J. (2017) Conservation and innovation in the DUX4-family gene network. Nat. Genet., 49, 935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Greef J.C., Lemmers R.J.L.F., Camaño P., Day J.W., Sacconi S., Dunand M., Van Engelen B.G.M., Kiuru-Enari S., Padberg G.W., Rosa A.L. et al. (2010) Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology, 75, 1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ricci G., Zatz M. and Tupler R. (2014) Facioscapulohumeral muscular dystrophy: more complex than it appears. Curr. Mol. Med., 14, 1052–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ricci E., Galluzzi G., Deidda G., Cacurri S., Colantoni L., Merico B., Piazzo N., Servidei S., Vigneti E., Pasceri V. et al. (1999) Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann. Neurol., 45, 751–757. [DOI] [PubMed] [Google Scholar]
- 13. Statland J., Donlin-Smith C., Tapscott S., Van Der Maarel S. and Tawil A. (2015) Milder phenotype in facioscapulohumeral muscular dystrophy patients with the largest residual D4Z4 fragments. Neurology, 84, 2147–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Teveroni E., Pellegrino M., Sacconi S., Calandra P., Cascino I., Farioli-Vecchioli S., Puma A., Garibaldi M., Morosetti R., Tasca G. et al. (2017) Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Invest., 127, 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Block G.J., Narayanan D., Amell A.M., Petek L.M., Davidson K.C., Bird T.D., Tawil R., Moon R.T. and Miller D.G. (2013) Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet., 22, 4661–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huichalaf C., Micheloni S., Ferri G., Caccia R. and Gabellini D. (2014) DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PLoS One, 9, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma V., Pandey S.N., Khawaja H., Brown K.J., Hathout Y. and Chen Y.-W. (2016) PARP1 differentially interacts with promoter region of DUX4 gene in FSHD myoblasts. 7, 95–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campbell A.E., Oliva J., Yates M.P., Zhong J.W., Shadle S.C., Snider L., Singh N., Tai S., Hiramuki Y., Tawil R. et al. (2017) BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle, 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Snider L., Geng L.N., Lemmers R.J.L.F., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., Maarel S.M., Tapscott S.J. et al. (2010) Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet., 6, e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rickard A.M., Petek L.M. and Miller D.G. (2015) Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet., 24, 5901–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caron L., Kher D., Lee K.L., McKernan R., Dumevska B., Hidalgo A., Li J., Yang H., Main H., Ferri G. et al. (2016) A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy-affected skeletal muscles. Stem Cells Transl. Med., 5, 1145–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bosnakovski D., Chan S.S.K., Recht O.O., Hartweck L.M., Gustafson C.J., Athman L.L., Lowe D.A. and Kyba M. (2017) Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun., 8, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mason S. and Wadley G.D. (2014) Skeletal muscle reactive oxygen species: a target of good cop/bad cop for exercise and disease. Redox Rep., 19, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kozakowska M., Pietraszek-Gremplewicz K., Jozkowicz A. and Dulak J. (2015) The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes. J. Muscle Res. Cell Motil., 36, 377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turki A., Hayot M., Carnac G., Pillard F., Passerieux E., Bommart S., De Mauverger E.R., Hugon G., Pincemail J., Pietri S. et al. (2012) Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med., 53, 1068–1079. [DOI] [PubMed] [Google Scholar]
- 26. Passerieux E., Hayot M., Jaussent A., Carnac G., Gouzi F., Pillard F., Picot M.C., Böcker K., Hugon G., Pincemail J. et al. (2015) Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: a double-blind randomized controlled clinical trial. Free Radic. Biol. Med., 81, 158–169. [DOI] [PubMed] [Google Scholar]
- 27. Winokur S.T., Barrett K., Martin J.H., Forrester J.R., Simon M., Tawil R., Chung S.A., Masny P.S. and Figlewicz D.A. (2003) Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord., 13, 322–333. [DOI] [PubMed] [Google Scholar]
- 28. Bou Saada Y., Dib C., Dmitriev P., Hamade A., Carnac G., Laoudj-Chenivesse D., Lipinski M. and Vassetzky Y.S. (2016) Facioscapulohumeral dystrophy myoblasts efficiently repair moderate levels of oxidative DNA damage. Histochem. Cell Biol., 145, 475–483. [DOI] [PubMed] [Google Scholar]
- 29. Dmitriev P., Bou Saada Y., Dib C., Ansseau E., Barat A., Hamade A., Dessen P., Robert T., Lazar V., Louzada R.A.N. et al. (2016) DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med., 99, 244–258. [DOI] [PubMed] [Google Scholar]
- 30. Winokur S.T., Chen Y.W., Masny P.S., Martin J.H., Ehmsen J.T., Tapscott S.J., Maarel S.M., Hayashi Y. and Flanigan K.M. (2003) Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet., 12, 2895–2907. [DOI] [PubMed] [Google Scholar]
- 31. Celegato B., Capitanio D., Pescatori M., Romualdi C., Pacchioni B., Cagnin S., Viganò A., Colantoni L., Begum S., Ricci E. et al. (2006) Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics, 6, 5303–5321. [DOI] [PubMed] [Google Scholar]
- 32. Tsumagari K., Chang S.-C., Lacey M., Baribault C., Chittur S.V., Sowden J., Tawil R., Crawford G.E. and Ehrlich M. (2011) Gene expression during normal and FSHD myogenesis. BMC Med. Genomics, 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanaka A., Woltjen K., Miyake K., Hotta A., Ikeya M., Yamamoto T., Nishino T., Shoji E., Sehara-Fujisawa A., Manabe Y. et al. (2013) Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling miyoshi myopathy in vitro. PLoS One, 8, e61540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamanaka K., Goto K., Arai M., Nagao K., Obuse C., Noguchi S., Hayashi Y.K., Mitsuhashi S. and Nishino I. (2016) Clinical, muscle pathological, and genetic features of Japanese facioscapulohumeral muscular dystrophy 2 (FSHD2) patients with SMCHD1 mutations. Neuromuscul. Disord., 26, 472. [DOI] [PubMed] [Google Scholar]
- 35. Bou Saada Y., Zakharova V., Chernyak B., Dib C., Carnac G., Dokudovskaya S. and Vassetzky Y.S. (2017) Control of DNA integrity in skeletal muscle under physiological and pathological conditions. Cell. Mol. Life Sci., 74, 3439–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blackford A.N. and Jackson S.P. (2017) ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell, 66, 801–817. [DOI] [PubMed] [Google Scholar]
- 37. Christmann M. and Kaina B. (2013) Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res., 41, 8403–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Torres M. and Forman H.J. (2003) Redox signaling and the MAP kinase pathways. Biofactors, 17, 287–296. [DOI] [PubMed] [Google Scholar]
- 39. Bosnakovski D., Xu Z., Gang E.J., Galindo C.L., Liu M., Simsek T., Garner H.R., Agha-Mohammadi S., Tassin A., Coppée F. et al. (2008) An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J., 27, 2766–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larsen B.D., Rampalli S., Burns L.E., Brunette S., Dilworth F.J. and Megeney L.A. (2010) Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci., 107, 4230–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer U., Ludwig N., Raslan A., Meier C. and Meese E. (2016) Gene amplification during myogenic differentiation. Oncotarget, 7, 6864–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Latella L., Lukas J., Simone C., Lorenzo P., Puri P.L. and Bartek J. (2004) Differentiation-induced radioresistance in muscle cells differentiation-induced radioresistance in muscle cells. Mol. Cell. Biol., 24, 6350–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fortini P., Ferretti C., Pascucci B., Narciso L., Pajalunga D., Puggioni E.M.R., Castino R., Isidoro C., Crescenzi M. and Dogliotti E. (2012) DNA damage response by single-strand breaks in terminally differentiated muscle cells and the control of muscle integrity. Cell Death Differ., 19, 1741–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang E.R., Choi J.D., Park M.A., Jeong G., Cho H. and Lee J.-S. (2010) ATM modulates transcription in response to histone deacetylase inhibition as part of its DNA damage response. Exp. Mol. Med., 42, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cowell I.G., Sunter N.J., Singh P.B., Austin C.A., Durkacz B.W. and Tilby M.J. (2007) γH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS One, 2, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim J.A., Kruhlak M., Dotiwala F., Nussenzweig A. and Haber J.E. (2007) Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol., 178, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murga M., Jaco I., Fan Y., Soria R., Martinez-Pastor B., Cuadrado M., Yang S.M., Blasco M.A., Skoultchi A.I. and Fernandez-Capetillo O. (2007) Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol., 178, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ferreboeuf M., Mariot V., Bessières B., Vasiljevic A., Attié-Bitach T., Collardeau S., Morere J., Roche S., Magdinier F., Robin-Ducellier J. et al. (2014) DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum. Mol. Genet., 23, 171–181. [DOI] [PubMed] [Google Scholar]
- 49. Shoji E., Sakurai H., Nishino T., Nakahata T., Heike T., Awaya T., Fujii N., Manabe Y., Matsuo M. and Sehara-Fujisawa A. (2015) Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep., 5, 12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H.L., Gee P., Ishida K. and Hotta A. (2016) Efficient genomic correction methods in human iPS cells using CRISPR-Cas9 system. Methods, 101, 27–35. [DOI] [PubMed] [Google Scholar]
- 51. Naito Y., Hino K., Bono H. and Ui-Tei K. (2015) CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics, 31, 1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jones T.I., Yan C., Sapp P.C., McKenna-Yasek D., Kang P.B., Quinn C., Salameh J.S., King O.D. and Jones P.L. (2014) Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenetics, 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumaki Y., Oda M. and Okano M. (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res., 36, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.