

Abstract
The human chr15q11-q13 imprinted cluster is linked to several disorders, including Prader–Willi (PWS) and Angelman (AS) syndromes. Recently, disease modeling approaches based on induced pluripotent stem cells (iPSCs) have been used to study these syndromes. A concern regarding the use of these cells for imprinted disease modeling is the numerous imprinting defects found in many iPSCs. Here, by reprogramming skin fibroblasts from a control and AS individuals, we generated several iPSC lines and addressed the stability of imprinting status across the PWS/AS domain. We focused on three important regulatory DNA elements which are all differentially methylated regions (DMRs), methylated on the maternal allele: the PWS imprinting center (PWS-IC), which is a germline DMR and the somatic NDN and MKRN3 DMRs, hierarchically controlled by PWS-IC. Normal PWS-IC methylation pattern was maintained in most iPSC lines; however, loss of maternal methylation in one out of five control iPSC lines resulted in a monoallelic to biallelic switch for many imprinted genes in this domain. Surprisingly, MKRN3 DMR was found aberrantly hypermethylated in all control and AS iPSCs, regardless of the methylation status of the PWS-IC master regulator. This suggests a loss of hierarchical control of imprinting at PWS/AS region. We confirmed these results in established iPSC lines derived using different reprogramming procedures. Overall, we show that hierarchy of imprinting control in donor cells might not apply to iPSCs, accounting for their spectrum of imprinting alterations. Such differences in imprinting regulation should be taken into consideration for the use of iPSCs in disease modeling.
Introduction
Genomic imprinting is a mammalian epigenetic phenomenon that causes a subset of genes to be monoallelically expressed according to their parental origin. These genes typically cluster together in 20–25 chromosomal regions in the mammalian genome containing genes exclusively expressed from the maternally or the paternally inherited alleles. Regulation of imprinting at these clusters relies on a cis-acting element known as imprinting center (IC), which exhibits parental-specific differences in CpG methylation inherited from the germline (reviewed in 1,2). Apart from the IC, other differentially methylated regions (DMRs) are also important for regulating imprinted expression of certain genes in the cluster. These DMRs, known as secondary DMRs, acquire differential methylation after the implantation stages of development and their methylation status and action is dependent hierarchically from the IC. That is, if the IC shows aberrant loss or gain of methylation, secondary DMRs show inappropriate loss or gain of methylation in the same parental allele (1,3). The molecular mechanisms underlying hierarchical regulation between germline and secondary DMRs remain to be deciphered.
(Epi)genetic alterations at these imprinted regions cause congenital syndromes known as imprinting disorders. There are at least 12 of these syndromes that typically affect growth, development, metabolism and brain function (4). The human chr15q11-q13 (Fig. 1A) harbors an imprinted cluster of genes associated with two classical imprinted neurodevelopmental disorders, Angelman Syndrome (AS) (OMIM#105830) and Prader–Willi Syndrome (PWS) (OMIM#176270). Patients with AS are characterized by severe intellectual disability, developmental delay, seizure, ataxia and happy demeanor (5). AS is caused by the loss of the maternally inherited copy of the E3 ubiquitin ligase UBE3A gene, which is expressed from both alleles in the majority of tissues, but exclusively expressed from the maternal allele in neurons (6,7). Patients with PWS suffer from neonatal hypotonia and poor feeding initially, but children eventually develop hyperphagia-induced obesity and typically exhibit obsessive-compulsive behaviors. PWS is believed to be caused by the loss of two or more paternally expressed imprinted genes within the segment from MKRN3 to the small nucleolar RNA (snoRNA) genes (8) (Fig. 1A). More recently, other diseases have been linked to this imprinted region, such as 15q11-q13 duplication syndrome (OMIM#608636), characterized by intellectual disability and autistic spectrum disorder (9), Schaaf-Yang syndrome (caused by paternally inherited MAGEL2 mutations) (OMIM#615547), which is phenotypically related to PWS and autism (10) and central precocious puberty 2 (caused by paternally inherited MKRN3 mutations) (OMIM#615346) (11).
Figure 1.
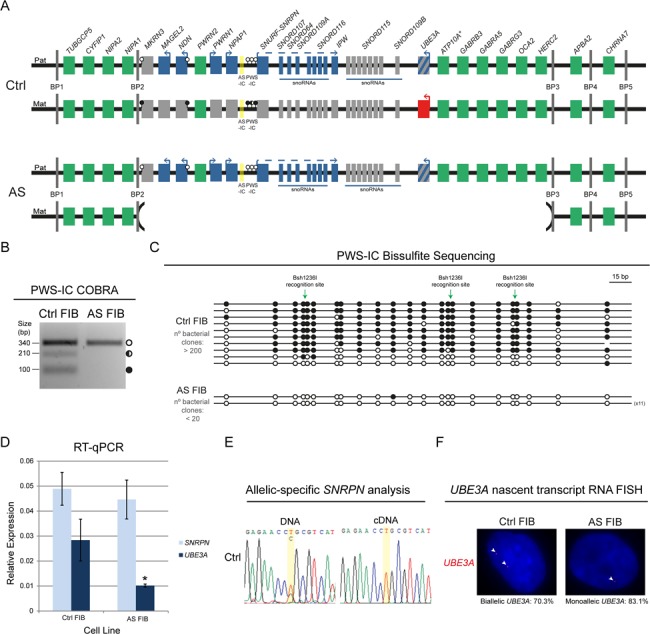
Stable imprinting of the chr15q11-q13 region in Ctrl and AS FIBs. (A) Scheme of the predictive epigenetic and transcriptional profile of the human chr15q11-q13 region in Ctrl and AS FIBs. Green rectangles, non-imprinted genes (* indicates that ATP10A imprinting status is uncertain); blue rectangles, imprinted genes expressed from the paternal allele; red rectangles, imprinted genes expressed from the maternal allele; gray rectangles, non-expressed imprinted genes; the rectangle with blue and gray stripes represents neuronal-specific imprinted expression of the paternal UBE3A gene; AS-IC is highlighted in yellow; white lollipops, unmethylated DNA; black lollipops, fully methylated DNA; half black lollipops, partially methylated DNA; Pat, paternal allele; Mat, maternal allele; BP1 to 5, breaking points 1 to 5. (B) PWS-IC COBRA for Ctrl and AS FIBs. White circle, unmethylated band; half black circle, partially methylated band; black circle, fully methylated band. (C) Bisulfite sequencing analysis for PWS-IC in Ctrl and AS FIBs. Each line represents the methylation profile of an independent PCR amplicon as determined by Sanger sequencing and BiQ Analyser. White dots, unmethylated CpGs; black dots, methylated CpGs; Bsh1236I recognition sites used for PWS-IC COBRA are indicated; the numbers in brackets represent the number of times the same amplicon was sequenced; estimated number of bacterial clones obtained in the Luria-Bertani (LB) agar plates is indicated. (D) RT-qPCR analysis for SNRPN and UBE3A in Ctrl and AS FIBs. Relative expression has been normalized to the GAPDH housekeeping gene; data are the mean ± standard error of the mean (SEM) of at least three independent experiments; significant differences between Ctrl and AS FIBs are indicated with an asterisk as P-value < 0.05, unpaired Student’s t-test. (E) Allelic-specific SNRPN genomic and expression analysis in Ctrl FIBs assayed by Sanger sequencing. Yellow stripe indicates the rs705 SNP. (F) UBE3A nascent-transcript RNA FISH in Ctrl and AS FIBs. Percentage of biallelic/monoallelic UBE3A FISH signals was calculated by counting a minimum of 64 cells per sample.
Imprinting regulation within the chr15q11-q13 region is controlled by a bipartite IC composed of two elements: AS imprinting center (AS-IC) and PWS imprinting center (PWS-IC; reviewed in 6). AS-IC is an 880 bp region located 35 kb upstream of the binary SNURF-SNRPN promoter. AS-IC is postulated to drive transcription from upstream exons of the SNURF-SNRPN gene in the maternal germline to establish a CpG methylation imprint at the PWS-IC (12). PWS-IC is situated on the promoter and exon 1 of the SNURF-SNRPN gene (Fig. 1A). This region is methylated on the maternal allele and unmethylated on the paternal allele. The unmethylated paternal allele drives the expression of a polycistronic transcript which includes the SNURF-SNRPN pair, the non-coding transcript IPW and several snoRNAs on the distal part of the cluster (Fig. 1A). In neurons, transcription continues further to produce the UBE3A anti-sense (UBE3A-ATS) long non-coding RNA which is believed to silence the paternal UBE3A allele (6,13). The proximal part of the cluster is composed by several intronless genes exclusively expressed from the paternal chromosome, including the MKRN3 and NDN genes. These two genes have differentially methylated CpG islands at their respective promoters which, like the PWS-IC, are unmethylated on the paternally inherited allele and methylated on the silent maternal allele (14,15). Mutations or epimutations (changes in DNA methylation pattern) of the PWS-IC alter the epigenetic profile, including of the NDN and MKRN3 DMRs, and allelic expression of the whole region (16,17), confirming PWS-IC as the master regulator of imprinting of the PWS/AS region in somatic tissues. An exception to this was observed in two PWS patients with an unusual milder phenotype. These PWS patients presenting maternal uniparental disomy with methylated PWS-IC on both alleles have unexpected expression of a few imprinted genes commonly silenced in PWS, including MKRN3. This suggest that some relaxation on imprinting regulation governed by PWS-IC can occur at this 6 Mb imprinted cluster, at least, in a disease context (18) suggesting that PWS/AS locus can be susceptible to independent epigenetic changes throughout the cluster.
In recent years, the emergence of the induced Pluripotent Stem Cell (iPSC) technology gave the unprecedented possibility to reprogram patient-derived somatic cells into stem-like cells to study a wide variety of human disorders (19). In a disease context, patient-derived iPSCs can be differentiated into the affected cell-type or tissue to function as a disease cellular model. iPSCs can and have been also used for modeling imprinted diseases (20). However, an important issue is whether ICs conserve their DMR status during the global epigenetic rewiring accompanying somatic reprogramming into iPSCs. Indeed, several reports have clearly pointed to the existence of multiple, but variable, imprinting defects in iPSCs (21–24), although the molecular mechanisms causing these defects remain largely unknown. For instance, whether ICs remain as the elements that hierarchically control the epigenome of an entire imprinted cluster in iPSCs, as they normally do in somatic cells, has not been addressed.
iPSCs have been derived from patients with imprinted syndromes associated with the 15q11-q13 region (25–29) to start unravelling molecular and cellular features associated with these syndromes (30,31). Interestingly, PWS-IC methylation status is reported to be generally stable in these AS and PWS-derived iPSCs (25,27,30). However, other studies reported a tendency for a certain degree of hypomethylation of the PWS-IC in normal or PWS-derived cells (23,32), though the functional consequences of PWS-IC hypomethylation, if any, have not been addressed. Interestingly, genome-wide studies suggest that the promoter of MKRN3 has a tendency to gain methylation in normal iPSCs (22,23). Whether this is dependent or independent of the PWS-IC methylation status remains unknown.
By reprogramming skin fibroblasts with the Yamanaka factors from healthy and AS individuals, we generated two groups of isogenic iPSC lines (five lines from the healthy control individual and five lines from an AS patient). Interestingly, we could detect a maternal to paternal epigenotype switch at the PWS-IC in one of our control iPSCs. Such alteration has widespread consequences for the imprinting regulation of the locus. Interestingly, we also found that the PWS-IC loses its ability to influence epigenetic status of the MKRN3 DMR, not only in our derived lines, but also in reported control and AS iPSCs. Our results indicate an alteration of the hierarchical influence of regulatory regions within imprinted loci in iPSCs which should be taken into consideration for disease modeling.
Results
Correct imprinting status of the chr15q11-q13 in skin fibroblasts derived from a healthy donor and a patient with AS
Fibroblasts from a 30-year-old female patient with AS and an age- and sex-matched healthy individual were derived from a punch skin biopsy and established in culture using standard conditions (33). The patient with AS harbors a 4.8 Mb deletion of the maternal 15q11-q13 locus, between breaking points 2 and 3 (from MKRN3 to HERC2 genes, including UBE3A). Megadeletions, as this one, are the most common defects in AS, accounting for 70–80% of the cases (5) (Fig. 1A). To understand whether imprinting status was correctly maintained in the cultured fibroblasts, we analyzed the methylation status of the PWS-IC, the master regulator of imprinting at the 15q11-q13 region by combined bisulfite restriction analysis (COBRA) and bisulfite sequencing. For COBRA, genomic DNA was initially treated with sodium bisulfite to distinguish methylated from unmethylated CpGs. After amplification using a nested Polymerase Chain Reaction (PCR) approach (33), a restriction enzyme was used to recognize three methylation-dependent retained sites within the PCR product (Supplementary Material, Table S1). AS fibroblasts (AS FIBs), as expected, only have an unmethylated allele, which was not cleaved by the restriction enzyme and corresponds to the single paternal allele. In contrast, in control fibroblasts (Ctrl FIBs) from the healthy donor, we could identify several bands that correspond to both fully methylated (three bands of similar size: 93, 110 and 115 bp), partially methylated (∼230 bp) and unmethylated (340 bp) alleles (Fig. 1B). Presumably, the unmethylated band corresponds to the paternal allele, while both fully and partially methylated bands correspond to the maternal allele. These results were confirmed by bisulfite sequencing, a technique whereby post-PCR single amplicons are ligated, cloned and then sequenced to retrieve methylation information of several CpG dinucleotides within the PCR product. After ligation and cloning, there was a reduced number of bacteria colonies for AS FIBs compared to Ctrl FIBs in replicate experiments (AS FIBs < 20 and Ctrl FIBs > 200) (Fig. 1C). Despite the clear cloning bias toward methylated amplicons in this assay, both unmethylated and methylated amplicons were observed for Ctrl FIBs, while only unmethylated amplicons were observed for AS FIBs (Fig. 1C). All in all, these results suggest that cultured fibroblasts maintained the expected methylation status of PWS-IC.
We then explored the expression and imprinting profile of the paternally expressed SNRPN imprinted gene and the biallelically expressed UBE3A gene, both of which are absent from the maternal chromosome in AS FIBs (Fig. 1A). The SNRPN levels were found to be equivalent between AS and Ctrl FIBs, while the levels of UBE3A were found to be at least 2-fold decreased in AS FIBs compared to Ctrl FIBs (Fig. 1D), suggesting that absence of the expressing maternal copy results in around 50% decrease in UBE3A expression. To address the imprinted expression of SNRPN in Ctrl FIBs, we were fortunate to find the single nucleotide polymorphism (SNP) rs705 in the 5′UTR of SNRPN gene, allowing for allelic-specific expression analysis. While both nucleotides (T/C) could be detected at the deoxyribonucleic acid (DNA) level, only one of them (T) was detected at the complementary DNA (cDNA) level indicating that SNRPN is, as expected, monoallelically expressed (Fig. 1E). In contrast to SNRPN, no SNP could be found in UBE3A exons (data not shown). Therefore, we developed a Stellaris™ UBE3A RNA Flourescent In Situ Hybridyzation (RNA FISH) (35) using a probe detecting the UBE3A nascent transcript. As expected most of Ctrl FIBs (70.3%) have two fluorescent dots for UBE3A, while the vast majority of AS FIBs has only one dot (83.1%) (Fig. 1F). These results suggest that the two UBE3A alleles in Ctrl FIBs and the single paternal copy of UBE3A in AS FIBs are expressed as expected, since UBE3A imprinting is only reported in neurons (6,7). In conclusion, a profile consistent with the expected epigenetic and transcriptional landscape was maintained in cultured Ctrl and AS FIBs.
Creation of iPSCs from a healthy donor and a patient with AS
To study the stability of imprinting at the chr15q11-q13 region in normal and in disease context, we reprogrammed both Ctrl and AS FIBs to obtain distinct iPSC lines. The creation of two set of isogenic lines, one from Ctrl FIBs and other from AS FIBs, greatly limits the influence of genetic factors on putative imprinting defects. Yamanaka reprogramming was performed using a lentiviral vector expressing the OCT4, SOX2, KLF4, C-MYC (OSKM) cassette plus a fluorescent tractable marker (dTomato) using a previously established protocol (36). At the beginning of reprogramming, activation of the OSKM cassette could be followed with the dTomato fluorescent trackable marker (data not shown), which became silenced 3 weeks later as previously shown (36). Five independent iPSC clones from both Ctrl and AS FIBs were isolated. They were initially cultured in feeder-dependent conditions, but adapted to feeder-free mTeSR1 conditions around passage 4, displaying embryonic stem cell (ESC)-like morphology and self-renewal capacities (data not shown). Karyotyping was done for the majority of the iPSC clones and found to be normal with the exception for Ctrl C and AS B iPSCs, which displayed a few chromosomal aberrations (Supplementary Material, Fig. S1A). iPSCs were then characterized for expression of stem cell factors by both immunofluorescence (IF) and Reverse Transcriptase-quantitative Polymerase Chain Reaction (RT-qPCR) analysis with more detailed analyses performed for the Ctrl E, Ctrl B and AS E iPSC lines. As expected, NANOG and SOX2 markers strongly stained all iPSC lines checked by IF (Fig. 2A). Likewise, stem-cell markers (OCT4, SOX2, NANOG and REX1), but not the fibroblast marker COL1A1, were expressed as assessed by RT-qPCR in the same cells (Fig. 2B). These results were confirmed by RT-qPCR analysis for the full panel of Ctrl and AS iPSCs and compared to the published AG1-0 iPSC line (Supplementary Material, Fig. S1B) which harbors a similar megadeletion to our AS iPSC lines (24). These results confirmed that we successfully reprogrammed healthy and AS FIBs into five independent stem-like clones of cells.
Figure 5.

Hypermethylation of the MKRN3 DMR is a frequent event in iPSCs and escapes control by the PWS-IC. (A) Summary table of the features of the five established iPSC lines used. (B) PWS-IC and MKRN3 DMR COBRA in the five iPSC lines (and also in Ctrl FIBs for MKRN3 DMR). White circle, unmethylated band; half black circle, partially methylated band; black circle, fully methylated band. (C) Scheme of the expected and the observed epigenetic and transcriptional profile of the human chr15q11-q13 region in Ctrl and AS iPSC lines. Blue rectangles, imprinted genes expressed from the paternal allele; red rectangles, imprinted genes expressed from the maternal allele; gray rectangles, silenced imprinted genes; white lollipops, unmethylated DNA; black lollipops, methylated DNA; Pat, paternal allele; Mat, maternal allele; green arrowheads highlight the epigenetic errors found; iPSCs lines in black correspond to the lines generated in this study; iPSCs in red correspond to the previously published IPSC lines.
Figure 2.
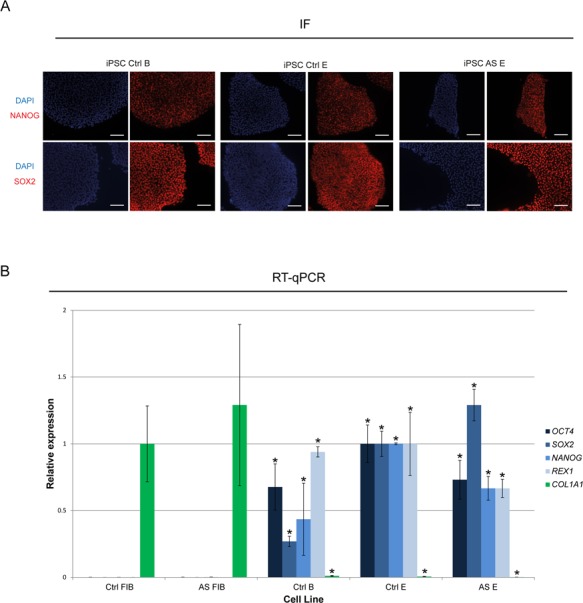
Stem-cell markers are expressed in Ctrl and AS iPSCs. (A) IF for SOX2 and NANOG (in red) in Ctrl B, Ctrl E and AS E iPSCs. Nuclei are marked with DAPI in blue; bar scale = 100 μm. (B) RT-qPCR analyses for the fibroblast marker (COL1A1) and the stem cell markers (OCT4, SOX2, NANOG and REX1) in Ctrl FIB, AS FIB, Ctrl B, Ctrl E and AS E iPSCs. Relative expression has been normalized to the GAPDH housekeeping gene; data are the mean ± SEM of at least two independent experiments; COL1A1 RT-qPCR values were normalized to 1 for the Ctrl FIBs sample, while OCT4, SOX2, NANOG and REX1 RT-qPCR values were normalized to 1 for the Ctrl E iPSC sample; significant differences toward the Ctrl FIBs sample are indicated with an asterisk as P-value < 0.05, unpaired Student’s t-test.
Reprogramming-induced methylation changes in PWS-IC cause loss of imprinting at the chr15q11-q13
To assess the stability of imprinting status of the chr15q11-q13 locus in normal and in disease context, we first analyzed whether the methylation status of the PWS-IC inherited from the initial Ctrl and AS FIBs was maintained upon reprogramming. For that, we performed COBRA for PWS-IC in the generated iPSC lines. All AS iPSC lines presented only the unmethylated paternal allele, preserving the methylation status inherited from AS FIBs (Fig. 3A and Supplementary Material, Fig. S2A). Four of the Ctrl iPSC lines (Ctrl A, C, D & E) also maintained the inherited methylation pattern from Ctrl FIBs, presenting both an unmethylated band, presumably corresponding to the paternal allele, and partially and fully methylated bands, presumably corresponding to the maternal allele (Fig. 3A and Supplementary Material, Fig. S2A). These results agree with previous studies demonstrating that methylation pattern at PWS-IC remains stable (25,27,30). Interestingly, in contrast to the other Ctrl iPSC lines, Ctrl B iPSCs exhibit only the unmethylated band (Fig. 3A). Since the nearby rs705 SNP of the SNRPN gene was still present in Ctrl B iPSC line (Fig. 3C), a deletion of the PWS-IC was unlikely to explain this result. Instead, this data suggests a maternal-to-paternal epigenotype switch of the PWS-IC. Bisulfite sequencing confirmed the COBRA results: Ctrl B iPSC, like AS A iPSC, only had unmethylated amplicons, while Ctrl A iPSC had both methylated and unmethylated alleles (overrepresentation of methylated amplicons due to cloning bias as also found for Ctrl FIBs) (Fig. 3B). To rule out that the putative loss of methylation on the maternal PWS-IC was not due to a global methylation loss, we decided to interrogate the methylation status of the IC from another imprinted region, the InterGenic-DMR (IG-DMR) of the DLK1-MEG3 domain by COBRA. IG-DMR is known to be frequently hypermethylated in iPSCs (22,23). We observed that in Ctrl B iPSC line, IG-DMR has the expected methylation pattern with both the presence of the unmethylated and methylated bands, ruling out an overall methylation defect in this line. Interestingly, a bias toward the methylation band (AS A, B, C & D), or complete reversal to a single methylated band was observed for the IG-DMR (Ctrl C & D) (Fig. 3A and Supplementary Material, Fig. S3A), as previously found in iPSCs (22,23).
Figure 3.
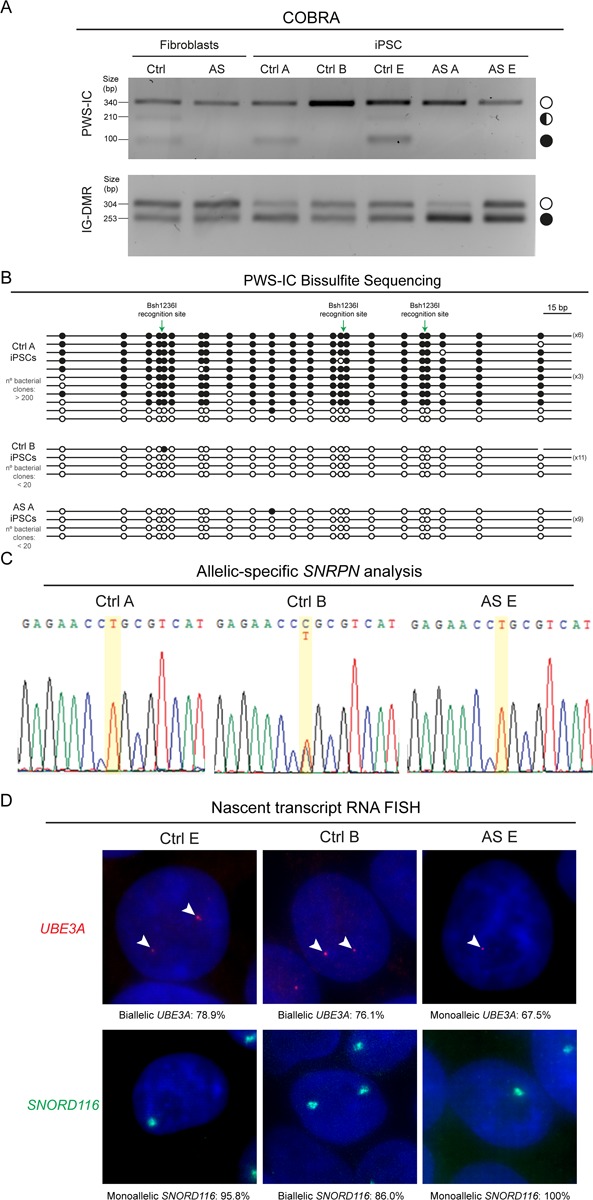
Loss of methylation at the maternal PWS-IC in Ctrl B iPSC results in loss of imprinting of genes at the chr15q11-q13. (A) PWS-IC and IG-DMR COBRA for Ctrl and AS FIBs and several iPSCs (Ctrl A, Ctrl B, Ctrl E; AS A and AS E). White circle, unmethylated band; half black circle, partially methylated band; black circle, fully methylated band. (B) Bisulfite sequencing analysis for PWS-IC in Ctrl A, Ctrl B and AS A iPSCs. Each line represents the methylation profile of an independent PCR amplicon as determined by Sanger sequencing and BiQ Analyser; white dots, unmethylated CpGs; black dots, methylated CpGs; Bsh1236I recognition sites used for PWS-IC COBRA are indicated; the numbers in brackets represent the number of times the same amplicon was sequenced; estimated number of bacterial clones obtained in the LB agar plate is indicated. (C) Allelic-specific SNRPN expression analysis assayed by Sanger sequencing for the Ctrl A, Ctrl B and AS E iPSCs. Yellow stripe indicates the rs705 SNP. (D) Nascent-transcript RNA FISH for UBE3A and SNORD116 in Ctrl E, Ctrl B and AS E iPSCs. Percentage of biallelic/monoallelic UBE3A and SNORD116 FISH signals was calculated by counting a minimum of 100 cells per sample.
The putative imprinting defect in Ctrl B iPSCs highlights the existence of methylation alterations at the level of PWS-IC in iPSCs. A tendency for decreased methylation at PWS-IC in certain iPSCs has been previously suggested (23,32), however the functional consequences of such an alteration has never been explored. Taking advantage of the known exonic SNP (rs705) in the SNRPN gene, we asked whether loss of methylation at the maternal PWS-IC in Ctrl B iPSCs resulted in loss of imprinting, i.e. biallelic expression of the SNRPN gene. Indeed, allelic-specific Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) showed that SNRPN, which is paternally expressed in the normal Ctrl A, Ctrl D, AS D and AS E iPSCs, becomes biallelically expressed in Ctrl B iPSCs (Fig. 3C; Supplementary Material, Fig. S3B). Moreover, nascent transcript RNA FISH for the SNORD116 snoRNA cluster showed a switch from the monoallelic to the biallelic state in Ctrl B iPSCs, but not in normal Ctrl E or AS E iPSCs (Fig. 3D). As expected expression of the UBE3A gene which is not imprinted in iPSCs remains biallelic in the Ctrl B and Ctrl E lines, and expressed from the sole paternal allele in the AS E line (Fig. 3D). Importantly, the reported imprinting status in Ctrl B or in any other iPSCs was not altered in the range of cell passages we studied (passages 7–19). In conclusion, we found that one of the generated iPSCs lost methylation of the maternal PWS-IC and, as a consequence, displayed biallelic expression of the polycistronic RNA initiated from the promoter region where PWS-IC is situated.
PWS-IC fails to fully control the epigenetic landscape across the chr15q11-q13 in iPSCs
Imprinting dysregulation is a known side-effect of current iPSC reprogramming protocols, with many ICs switching their methylation patterns and causing loss of imprinting of the neighbouring genes (21–24). However, whether ICs remain the imprinting master regulator within a cluster in iPSCs has never been functionally dissected. Taking advantage of our isogenic Ctrl iPSC lines with normal (Ctrl A, C, D & E) and abnormal (Ctrl B) methylation patterns at PWS-IC (Fig. 3A and B; Supplementary Material, Fig. S3A), we decided to explore the impact of these changes on the methylation profile of the two most-studied somatic DMRs (NDN and MKRN3 DMRs) at the proximal part of the chr15q11-q13 region by COBRA. For the NDN DMR, we verified that the normal Ctrl E iPSC line exhibits the same band pattern as the Ctrl FIBs sample, with both a strong unmethylated and a weak methylated band (Fig. 4A). In contrast, Ctrl B iPSCs only have an unmethylated band (Fig. 4A), suggesting that the maternal allele fully lost its normal methylation. Through the presence of an exonic SNP at the NDN gene (rs2192206), we verified that NDN became biallelically expressed in Ctrl B iPSCs, remaining monoallelically expressed in Ctrl E iPSCs, as in the original Ctrl FIBs sample (Fig. 4B). These results suggest a maternal-to-paternal epigenotype switch on the NDN DMR, similar to the changes at the level of the PWS-IC in Ctrl B iPSCs. Therefore, NDN DMR seems to be hierarchically dependent on the PWS-IC in iPSCs.
Figure 4.
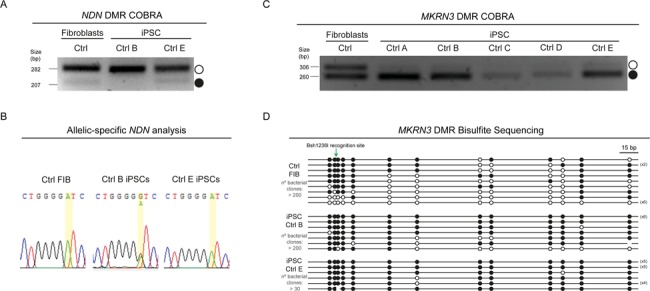
MKRN3 DMR, but not NDN DMR, is hypermethylated in Ctrl iPSCs. (A) NDN COBRA for Ctrl FIBs, Ctrl B and Ctrl E iPSCs. White circle, unmethylated band; black circle, methylated band. (B) Allelic-specific NDN expression analysis assayed by Sanger sequencing for Ctrl FIBs, Ctrl B and Ctrl E iPSCs. Yellow stripe indicates the rs2192206 SNP. (C) MKRN3 COBRA for Ctrl FIBs and all the isogenic Ctrl iPSCs. White circle, unmethylated band; black circle, methylated band. (D) Bisulfite sequencing analysis for MKRN3 DMR in Ctrl FIBs, Ctrl B and Ctrl E iPSCs. Each line represents the methylation profile of an independent PCR amplicon as determined by Sanger sequencing and BiQ Analyser; white dots, unmethylated CpGs; black dots, methylated CpGs; Bsh1236I recognition site used for MKRN3 DMR COBRA is indicated; the numbers in brackets represent the number of times the same amplicon was sequenced; estimated number of bacterial clones obtained in the LB agar plates is indicated.
We then investigated the methylation pattern at the MKRN3 DMR. While both a methylated and an unmethylated band were observed for Ctrl FIBs, we verified that, surprisingly, all the Ctrl iPSCs displayed a tendency for hypermethylation at the MKRN3 DMR by COBRA (Fig. 4C). This occurred independently of the methylation pattern at the PWS-IC, since the Ctrl B iPSC line also exhibits the same hypermethylation pattern as the other Ctrl iPSCs. This result was confirmed by bisulfite sequencing: while unmethylated and methylated amplicons were sequenced in the Ctrl FIBs sample, both Ctrl B and Ctrl E iPSCs showed a clear accumulation of hypermethylated amplicons (Fig. 4D). Moreover, all the AS iPSCs lines also showed a tendency for MKRN3 to become hypermethylated (Supplementary Material, Fig. S3A). All together, these results suggest that MKRN3 DMR no longer responds to the PWS-IC methylation status and tends to become hypermethylated during iPSC reprogramming. This finding showed that iPSCs, in contrast to the donor somatic cells, exhibit a loss of hierarchical control of imprinting by PWS-IC in the chr15q11-q13 region. The reasons why some regions, such as the MKRN3 DMR, but not others (e.g. NDN DMR) gain ectopic methylation during iPSC derivation need to be further explored. An interesting perspective is that the lack of the neuronal-specific MKRN3 expression, but not of NDN (or MAGEL2) during reprogramming (Supplementary Material, Fig. S3B), might explain the aberrant gain of methylation on the paternal allele.
MKRN3 DMR hypermethylation, independent of PWS-IC regulation, is a frequent event in iPSCs
To corroborate the results obtained with our iPSC lines, we examined the methylation profile of both the PWS-IC and MKRN3 DMR in five independent iPSC lines previously generated using different reprogramming methods (Fig. 5A) (25,37–39) (Silva et al., in preparation). This includes the AG1-0 line derived from an AS patient with a similar megadeletion to our AS iPSCs (25). Using COBRA, we found the normal methylation pattern at the PWS-IC, with both unmethylated (corresponding to the paternal allele) and partially and fully methylated bands (corresponding to the maternal allele) in three out of the five previously generated iPSC lines (Fig. 5B). As expected, the AG1-0 AS iPSC line only showed the sole unmethylated paternal allele, as previously found in all our derived AS iPSCs (Fig. 3A and Supplementary Material, Fig. S3A). Interestingly, the iUC-DAND5_455/10 (iUC-DAND5 for short) iPSC line, with no genetic defects in chr15q11-q13, seems to have lost maternal methylation at the PWS-IC (Fig. 5B), similar to the Ctrl B IPSC line (Fig. 3A). This highlights for the fact that PWS-IC hypomethylation defects in iPSCs are more recurrent than initially appreciated.
We then verified the methylation pattern of the MKRN3 DMR in these iPSCs. In four out of the five iPSCs, including AG1-0, a clear tendency for hypermethylation at the level of the MKRN3 DMR is observed. This demonstrated that MKRN3 DMR hypermethylation is indeed frequent in iPSCs regardless of the reprogramming procedure. As described for the Ctrl B iPSC line, hypomethylation of the maternal PWS-IC in the iUC-DAND5 iPSC did not impact the gain of methylation at the MKRN3 DMR (Fig. 5B). Only the iPSC6.2 line was found to have both the unmethylated and methylated alleles (with some bias toward the unmethylated allele). Overall, by using established iPSC lines, we confirmed the results from our isogenic system. Indeed, we confirmed that MKRN3 DMR hypermethylation and loss of hierarchical control of the IC of the Prader–Willi/Angelman imprinted cluster are frequent events occurring upon reprogramming into iPSCs (summarized in Fig. 5C).
Discussion
Imprinting defects are known to accumulate in iPSCs (21–24); however, the frequency and causes underlying these defects remain to be understood. In particular, it is not known whether imprinting dysregulation is driven only by epigenetic changes in the ICs or whether epigenetic alterations in other regions could also account for imprinting errors accumulated during reprogramming. In this study, we evaluated the impact of Yamanaka reprogramming in the imprinting regulation of the PWS/AS region on chr15q11-q13. We found that PWS-IC, the master regulator of chr15q11-q13 imprinting, is mostly resistant to changes in its parental-inherited methylation profile. However, loss of maternal methylation was found in one derived and one published iPSC lines. Loss of maternal methylation at the PWS-IC caused loss of imprinting of many genes within the cluster. Interestingly, PWS-IC lost the ability to control differential methylation at the MKRN3 DMR, which showed a propensity to gain aberrant methylation in iPSCs. These results highlight, for the first time, a change in the hierarchy of imprinting regulation within imprinted domains in iPSCs.
Skin fibroblasts in culture seem to maintain their methylation profiles intact across the chr15q11-q13 in both Ctrl and AS FIBs (Fig. 1). We cannot completely rule out for undetectable imprinting defects in a minority of fibroblasts, since we have not performed single-cell analysis (except for UBE3A nascent-transcript RNA FISH). In any case, these results are in agreement with previous publications, suggesting stable imprinting in cultured skin fibroblasts (25,27). To generate isogenic Ctrl and AS iPSC lines, we used a standardized lentiviral approach (36) for somatic reprogramming, which has been previously used to generate AS iPSCs carrying UBE3A mutations (27). We confirmed that our five isogenic AS and five isogenic Ctrl iPSCs have ESC-like morphology, self-renewal capacity, express stem cell markers and mostly have a normal karyotype (Fig. 2 and Supplementary Material, Fig. S2). The use of independent but genetically identical iPSCs lines provided a good starting point to study the epigenetic stability of the PWS/AS imprinting cluster in both healthy background and disease context.
We began by addressing the status of PWS-IC in the different iPSCs. We detected that the karyotypically normal Ctrl B iPSC line lost methylation on the maternal allele, while the paternal allele remained unmethylated in both Ctrl and AS iPSCs (Fig. 3A and Supplementary Material, Fig. S2A). Loss of methylation at the PWS-IC was also seen in the IUC-DAND5 iPSC line (Fig. 5B), suggesting that the maternal PWS-IC has some propensity to lose methylation during reprogramming, while the paternal PWS-IC seems to remain unmethylated and inert to methylation aberrations. This data contrasts with previous studies reporting a stable parental-inherited methylation pattern in healthy, AS and PWS iPSCs for PWS-IC (25,27,30). However, our results corroborate other findings reporting partial hypomethylation of the PWS-IC in normal or PWS-derived iPSCs (23,32). A direct implication of this is that AS iPSCs derived from patients with megabase deletions (65–70% of patients with AS) (5) with only the unmethylated paternal PWS-IC are less likely to accumulate imprinting errors at this element and, thus, might provide a good cellular system to model AS. In contrast, PWS iPSCs derived from patients with megabase deletions (70–80% of patients with PWS) (8) containing only the methylated maternal PWS-IC will be prone to hypomethylation defects with possible consequences in imprinting regulation of the locus. In any case, independently of acquiring reprogramming-induced imprinting defects or not, the methylation profile of iPSCs remains thereafter stable through cell passage and cell differentiation (data not shown) (23,30). Nevertheless, our results suggest that recurrent screening for methylation errors at the level of PWS-IC should be advised in iPSCs used to study diseases affecting the chr15q11-q13 region.
Given the observed maternal-to-paternal epigenotype switch in the PWS-IC in Ctrl B iPSCs (Fig. 3A), we explored the extent to which imprinting of the chr15q11-q13 region was affected. We observed that genes within the polycistronic transcript initiated at the PWS-IC, such as SNRPN and SNORD116 genes, became biallelically expressed (Fig. 3C and D). Loss of imprinting was also seen for the NDN gene, which lies on the proximal part of the domain. This was accompanied by the loss of maternal methylation at the NDN DMR (Fig. 4A and B). Overall, we report, for the first time in iPSCs, that loss of maternal PWS-IC has widespread effects on imprinting regulation of the chr15q11-q13 region. Interestingly, around 3% of AS cases arise from similar imprinting defects resulting in loss of maternal methylation of the PWS-IC (5). In principle, such a reprogramming-induced imprinting defect can be used to model the cases of AS caused by loss of maternal methylation at the PWS-IC, a distinct molecular mechanism causing the disease from the megabase deletions or mutations in the UBE3A gene.
A surprising finding from our study was the fact that in contrast to the NDN DMR, the MKRN3 DMR does not respond to alterations in the PWS-IC methylation status (Fig. 5B). Moreover, a clear tendency to become hypermethylated was seen, not only in all our healthy and AS-derived iPSCs, but also on previously established iPSCs, with only one exception (the iPSC6.2 line) (Fig. 4C and D; Fig. 5B; Supplementary Material, Fig. S3A). These results confirm unexplored reports on MKRN3 DMR hypermethylation in iPSCs previously gathered from genome-wide studies (22,23). To our knowledge, MKRN3 DMR hypermethylation has never been observed in any other tissues or cell types. Furthermore, our findings clearly point toward a loss of the hierarchical regulation of imprinting at the chr15q11-q13, a situation only reported once before in two PWS patients with milder phenotype (18). Indeed, PWS-IC no longer exerts its master imprinting regulator role as it normally does in somatic cells (16,17). The reasons why changes in the hierarchy of imprinting regulation for the chr 15q11-q13 occur in iPSCs remains to be understood. It is also unclear why MKRN3 DMR gains methylation, while the same is not observed for NDN DMR. A possible explanation centers on the fact that MKRN3, in contrast to NDN, is silent from both alleles and presumably becomes prone to acquire methylation on the unmethylated, albeit silenced, paternal allele. This likely occurs during the reprogramming process since the MKRN3 DMR remains differentially methylated in fibroblasts, despite the fact that MKRN3 gene is also not expressed in this cell type. Further studies will be necessary to understand the role of the initial cell source (the only iPSC line with no MKRN3 hypermethylation was derived from CD34+ progenitors) (Fig. 5A and B) or of reprogramming methods on the aberrant hypermethylation of the MKRN3 DMR in many iPSCs.
By exploring the mechanisms of imprinting regulation at the PWS/AS imprinted cluster in depth, we documented several anomalies on methylation pattern and unravelled changes in the hierarchy of imprinting regulation in iPSCs. Whether imprinting defects at other clusters (e.g. DLK1-DIO3 cluster is also affected in some of our iPSC lines) might also lead to changes in the imprinting regulatory principles documented for normal somatic cells will need to be explored in the future. In any case, our study clearly suggests that imprinting defects in iPSCs might also occur in somatic or secondary DMRs, regardless of epigenetic alterations at the respective IC. This increases the current spectrum of imprinting defects associated to this cell type likely to cause loss of imprinting for certain gene(s) within an imprinted cluster. This also highlights for the need to screen for epigenetic defects in all imprinting regulatory regions within a cluster and not only at the IC. Interestingly, by presenting such flexibility of imprinting regulation, iPSCs might become a good cellular model to study the relationships between regulatory elements within an imprinted domain to achieve long-range transcriptional regulation. Finally, the impact these hierarchical changes of imprinting regulation will have on iPSC differentiation toward committed lineages will certainly be an important issue to explore in the future in order to improve the use of iPSCs for disease modeling.
Materials and Methods
Patient-derived fibroblasts
Fibroblast punch biopsies from both the patient with AS and the age- and sex-matched individual were collected in Hospital de Santa Maria (Centro Hospitalar Lisboa Norte) in accordance with European and National ethical regulation and approved by the Ethical Committee of Instituto de Medicina Molecular João Lobo Antunes (iMM) and Hospital de Santa Maria. For the Ctrl individual, written informed consent was obtained. For the patient with AS, written informed consent was obtained from the patient’s legal guardians. Isolation and maintenance of fibroblasts were performed according to a standardized protocol (33). For AS FIBs, a 4.8 Mb deletion of the 15q11-q13 between breaking points 2 and 3 (from MKRN3 to HERC2 genes) was confirmed using a chromosomal array CGH 180K (performed by Serviço de Genética, Departamento de Patologia, Faculdade de Medicina, Universidade do Porto). Human fibroblasts were maintained in DMEM supplemented with 10% fetal bovine serum, 1 mm L-Glutamine, and 100 units/ml penicillin, 100 μg/ml streptomycin using products from Life Technologies in a humid incubator at 37°C with 5% (vol/vol) CO2.
Yamanaka reprogramming
iPSC reprogramming of AS and Ctrl FIBs was performed using a previously published protocol (36). Briefly, pluripotency of adult human AS and Ctrl FIBs was induced with a polycistronic lentiviral vector encoding for the reprogramming OSKM cassette and a red fluorescent protein (dTomato) under the spleen focus-forming virus promoter (36). Transduced fibroblasts were harvested via trypsinization and transferred onto mouse embryonic fibroblasts (MEFs) 3 days after transduction and thereafter cultured in human embryonic stem cell (hES) medium composed by DMEM F-12 (Gibco, Invitrogen), 15% Knock-out serum replacement, 1 mm L-glutamine (Gibco, Invitrogen), 0.1 mm nonessential amino acids (Sigma-Aldrich), 100 μM β-mercaptoethanol (Sigma-Aldrich), 100 units/ml penicillin, 100 μg/ml streptomycin (Gibco, Invitrogen) and 10 ng/ml of basic Fibroblast Growth Factor (bFGF; Peprotech). From day 21 of reprogramming, the first ESC-like colonies appear and single colonies were picked based on morphology and further expanded on a MEF feeder layer. A total of five iPSC lines derived from AS and Ctrl FIBs each were obtained.
iPSC culture
iPSCs were initially expanded in hES medium under MEF feeder-dependent conditions. When colonies reached 50–75% confluency, cells were passed with a splitting ratio from 1:2 to 1:4 using 0.5 mm EthyleneDiamineTetraacetic Acid (EDTA) in Phosphatase Buffered Saline (PBS), avoiding single-cell suspensions. Medium was changed every day. After two/three passages without or with minimal spontaneous differentiation in feeder-dependent conditions, cells were transferred to Matrigel (Corning)-coated plates in mTeSR1 (STEMCELL Technologies) medium supplemented with 50 units/ml penicillin, 50 μg/ml streptomycin (Life Technologies), using the same enzyme-free method. Medium was changed every day. Cells were passaged at least twice in feeder-free conditions before any experiment was performed.
DNA methylation analysis
Genomic DNA was purified using conventional phenol:chloroform:isoamyl alcohol extraction. Bisulfite treatment was performed using the EZ DNA Methylation Gold Kit (Zymo Research) following manufacturer’s guidelines. Bisulfite treated DNA was amplified by PCR (MKRN3 DMR and NDN DMR) or nested PCR (PWS-IC and IG-DMR) using the primers and conditions summarized in Supplementary Material, Table S1. For COBRA, PCR products were extracted from the agarose gel using the NZYGelpure kit (NZYTech) and then submitted to a restriction digest with BstUI/Bsh1236I (Thermo Fisher) to distinguish methylated from unmethylated alleles based on the original status of the CGCG sequence. For bisulfite sequencing of the PWS-IC and the MKRN3 DMR, PCR products were cloned into the pGEM-T Easy vector (Promega) and at least 10 clones from each sample were sequenced. Methylation analysis was performed using BiQ Analyser (40).
RT-qPCR
Total RNA was isolated from fibroblasts, iPSCs and brain cortex using NYZol (NZYtech) and then DNAse I treated (Roche) to remove contaminating DNA. RNA template was reverse transcribed using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche), according to the manufacturer’s instructions for RT-qPCR. cDNA was subjected to qPCR with iTaqTM SYBR® Green Supermix (Bio-Rad) in a ViiA7 Applied Biosystems machine, according to the manufacturer’s instructions using the primers in Supplementary Material, Table S2.
RT-PCR
cDNA was subjected to PCR to study MKRN3, NDN, MAGEL2 and GAPDH expression. The primer pairs and PCR conditions are described in Supplementary Material, Table S2.
Allelic-specific expression analysis
Primers were designed to amplify genomic regions around frequent exonic SNPs in the human population for several genes within chr15q11-q13 imprinted cluster (data not shown). SNPs for SNRPN (rs705) and NDN (rs2192206) genes were found in the Ctrl samples. cDNA collected from Ctrl FIBs and Ctrl A, Ctrl B, Ctrl E and/or AS E iPSC samples were then amplified by PCR and the product was sequenced to determine the monoallelic or biallelic status of these genes. Primers for PCR and sequencing and respective PCR conditions are in Supplementary Material, Table S2.
RNA FISH
In order to analyze UBE3A allelic-specific expression in Ctrl FIBs, AS FIBs and Ctrl B, Ctrl E and AS E iPSCs, custom Stellaris™ RNA FISH probes designed using the Stellaris™ Probe Designer software (Biosearch Technologies). The dye used was Quasar® 570 and the region used for the design of the probe was Chr15:25353077-25360996 (GRCh38.p7). Probe sequences are in Supplementary Material, Table S3. Cells were cultured to 75% confluency and hybridization conditions for RNA FISH were followed according to Stellaris™ guidelines using a final concentration of 125 nM of probe per coverslip.
For SNORD116 RNA FISH probe, a RP11-186C7 bacterial artificial chromosome (BAC; BACPAC Resources Center) was prepared using the Nick Translation Kit (Abbot) with Green dUTP (Enzo Life Sciences). RNA FISH was done accordingly to established protocols (41) in Ctrl B, Ctrl E and AS E iPSCs. After the RNA FISH procedure for both probes, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich), diluted 1:5000 in 2× Saline Sodium Citrate (SSC) buffer for 5 min at room temperature (RT) and mounted with Vectashield Mounting Medium (Vectorlabs). Cells were observed with the widefield fluorescence microscope Zeiss Axio Observer (Carl Zeiss MicroImaging) with 63× oil objective using the filter sets FS43HE, FS38HE and FS49. Digital images were analyzed with the FIJI platform (42). UBE3A and SNORD116 fluorescent signals were counted in at least 64 cells of FIBs and at least 100 cells of iPSCs.
IF
iPSCs were cultured in Matrigel-coated glass coverslips to 75% confluency. Cells were washed with PBS and fixed with 3% ParaFormAldehyde (PFA) for 10 min at RT, permeabilized with 0.5% Triton 100× (Sigma-Aldrich) in PBS on ice for 5 min and incubated for 15 min at RT in blocking solution containing 1% Bovine Serum Albumin (Sigma-Aldrich) in PBS. The cells were stained with the primary antibody diluted in blocking solution according to Supplementary Material, Table S4 for 45 min in a humid chamber. Cells were then incubated with the secondary antibody Cy™3 AffiniPure F(ab')2 Fragment Goat Anti-Mouse IgG (H+L) (dilution 1:200; Jackson ImmunoResearch Laboratories Inc.) for 45 min. DAPI (0.2 mg/ml) was used to stain the DNA and mark the nuclei by incubating at RT for 2 min. Mounting and imaging was performed as stated for RNA FISH.
Karyotyping
iPSCs were expanded until they have reached the logarithmic phase (70% confluency). Colcemid was added to the cell medium at 10 μl/ml and cells were incubated in a humid incubator at 37°C with 5% (vol/vol) CO2 for 4 h. Cells were detached with accutase, centrifuged at 300 g for 3 min and washed with PBS three times. Cells were then incubated with 55 mm of KCl at 37°C for 30 min. After centrifugation for 15 min at 400 g, most of the supernatant was removed and only 1 ml of supernatant was left for resuspending the cells. Fixative solution (3:1 ratio of methanol:glacial acetic acid) was added dropwise and the mixture was incubated on ice for 10 min. After centrifugation at 400 g, most of the supernatant was removed and only 1 ml of supernatant was left for resuspending the cells. The steps of adding fixative solution, incubation on ice and centrifugation were repeated three more times and the nuclei were stored at −20°C before G-banding karyotype (performed by GenoMed, Diagnósticos de Medicina Molecular SA).
Supplementary Material
Acknowledgements
We would like to thank Natasha Zamudio (University of Melbourne) and Rachel Duffie (Columbia University) for critical reading of the manuscript and Vanessa Borges Pires (Instituto de Medicina Molecular João Lobo Antunes-iMM JLA) for technical help. We also thank the following people for providing genomic DNA samples from established iPSC lines: Marta Ribeiro and Rodrigo Gonçalves (iMM JLA) for the DF6-9T line, José M. Inácio (Chronic Diseases Research Center - CEDOC) for the iUC-DAND5 line and Teresa P. Silva and Evguenia Beckman (iMM JLA and Institute for Bioengineering and Biosciences - iBB) for the F002.1A.13 and iPSC6.2 lines.
Conflict of Interest statement. None declared.
Funding
Fundação para a Ciência e Tecnologia (PTDC/BEX-BCM/2612/2014 and IF/00242/2014), Fundação Amélia de Mello (Pedro José de Mello Costa Duarte/2015 fellowship) and LISBOA-01-0145-FEDER-007391, project confunded by FEDER, through POR Lisboa 2020-Programa Operacional Regional de Lisboa, PORTUGAL 2020 and Fundação para a Ciência e Tecnologia.
References
- 1. Barlow D.P. and Bartolomei M.S. (2014) Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol., 6, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pólvora Brandão D. and Rocha S.T. (2018) Genomic imprinting at the transcriptional level. eLS. John Wiley & Sons, Ltd, Chichester. [Google Scholar]
- 3. Xie W., Barr C.L., Kim A., Yue F., Lee A.Y., Eubanks J., Dempster E.L. and Ren B. (2012) Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell, 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soellner L., Begemann M., Mackay D.J., Gronskov K., Tumer Z., Maher E.R., Temple I.K., Monk D., Riccio A., Linglart A. et al. (2017) Recent advances in imprinting disorders. Clin. Genet., 91, 3–13. [DOI] [PubMed] [Google Scholar]
- 5. Margolis S.S., Sell G.L., Zbinden M.A. and Bird L.M. (2015) Angelman syndrome. Neurotherapeutics, 12, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chamberlain S.J. (2013) RNAs of the human chromosome 15q11-q13 imprinted region. Wiley Interdiscip. Rev. RNA, 4, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rougeulle C., Glatt H. and Lalande M. (1997) The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet., 17, 14–15. [DOI] [PubMed] [Google Scholar]
- 8. Cassidy S.B., Schwartz S., Miller J.L. and Driscoll D.J. (2012) Prader–Willi syndrome. Genet. Med., 14, 10–26. [DOI] [PubMed] [Google Scholar]
- 9. Finucane B.M., Lusk L., Arkilo D., Chamberlain S., Devinsky O., Dindot S., Jeste S.S., LaSalle J.M., Reiter L.T., Schanen N.C. et al. (2016) 15q Duplication Syndrome and Related Disorders GeneReviews®. Seattle: University of Washington, Seattle, : 1993–2018. [Google Scholar]
- 10. Schaaf C.P., Gonzalez-Garay M.L., Xia F., Potocki L., Gripp K.W., Zhang B., Peters B.A., McElwain M.A., Drmanac R., Beaudet A.L. et al. (2013) Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat. Genet., 45, 1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abreu A.P., Dauber A., Macedo D.B., Noel S.D., Brito V.N., Gill J.C., Cukier P., Thompson I.R., Navarro V.M., Gagliardi P.C. et al. (2013) Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med., 368, 2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith E.Y., Futtner C.R., Chamberlain S.J., Johnstone K.A. and Resnick J.L. (2011) Transcription is required to establish maternal imprinting at the Prader–Willi syndrome and Angelman syndrome locus. PLoS Genet., 7, e1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rougeulle C., Cardoso C., Fontes M., Colleaux L. and Lalande M. (1998) An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet., 19, 15–16. [DOI] [PubMed] [Google Scholar]
- 14. Jay P., Rougeulle C., Massacrier A., Moncla A., Mattei M.G., Malzac P., Roeckel N., Taviaux S., Lefranc J.L., Cau P. et al. (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader–Willi syndrome chromosomal region. Nat. Genet., 17, 357–361. [DOI] [PubMed] [Google Scholar]
- 15. Jong M.T., Gray T.A., Ji Y., Glenn C.C., Saitoh S., Driscoll D.J. and Nicholls R.D. (1999) A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum. Mol. Genet., 8, 783–793. [DOI] [PubMed] [Google Scholar]
- 16. Bielinska B., Blaydes S.M., Buiting K., Yang T., Krajewska-Walasek M., Horsthemke B. and Brannan C.I. (2000) De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat. Genet., 25, 74–78. [DOI] [PubMed] [Google Scholar]
- 17. Ohta T., Gray T.A., Rogan P.K., Buiting K., Gabriel J.M., Saitoh S., Muralidhar B., Bilienska B., Krajewska-Walasek M., Driscoll D.J. et al. (1999) Imprinting-mutation mechanisms in Prader–Willi syndrome. Am. J. Hum. Genet., 64, 397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rogan P.K., Seip J.R., White L.M., Wenger S.L., Steele M.W., Sperling M.A., Menon R. and Knoll J.H. (1998) Relaxation of imprinting in Prader-Willi syndrome. Hum Genet, 103, 694–701. [DOI] [PubMed] [Google Scholar]
- 19. Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K. and Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 20. Chamberlain S.J., Germain N.D., Chen P.F., Hsiao J.S. and Glatt-Deeley H. (2016) Modeling genomic imprinting disorders using induced pluripotent stem cells. Methods Mol. Biol., 1353, 45–64. [DOI] [PubMed] [Google Scholar]
- 21. Bar S., Schachter M., Eldar-Geva T. and Benvenisty N. (2017) Large-scale analysis of loss of imprinting in human pluripotent stem cells. Cell Rep., 19, 957–968. [DOI] [PubMed] [Google Scholar]
- 22. Ma H., Morey R., O'Neil R.C., He Y., Daughtry B., Schultz M.D., Hariharan M., Nery J.R., Castanon R., Sabatini K. et al. (2014) Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature, 511, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Muller F.J., Wang Y.C., Boscolo F.S. et al. (2012) Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell, 10, 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pick M., Stelzer Y., Bar-Nur O., Mayshar Y., Eden A. and Benvenisty N. (2009) Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells, 27, 2686–2690. [DOI] [PubMed] [Google Scholar]
- 25. Chamberlain S.J., Chen P.F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S. and Lalande M. (2010) Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl. Acad. Sci USA, 107, 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Germain N.D., Chen P.F., Plocik A.M., Glatt-Deeley H., Brown J., Fink J.J., Bolduc K.A., Robinson T.M., Levine E.S., Reiter L.T. et al. (2014) Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism, 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stanurova J., Neureiter A., Hiber M., Oliveira Kessler H., Stolp K., Goetzke R., Klein D., Bankfalvi A., Klump H. and Steenpass L. (2016) Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep., 6, 30792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stelzer Y., Sagi I., Yanuka O., Eiges R. and Benvenisty N. (2014) The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader–Willi syndrome. Nat. Genet., 46, 551–557. [DOI] [PubMed] [Google Scholar]
- 29. Yang J., Cai J., Zhang Y., Wang X., Li W., Xu J., Li F., Guo X., Deng K., Zhong M. et al. (2010) Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader–Willi syndrome. J. Biol. Chem., 285, 40303–40311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burnett L.C., LeDuc C.A., Sulsona C.R., Paull D., Eddiry S., Levy B., Salles J.P., Tauber M., Driscoll D.J., Egli D. et al. (2016) Induced pluripotent stem cells (iPSC) created from skin fibroblasts of patients with Prader–Willi syndrome (PWS) retain the molecular signature of PWS. Stem Cell Res., 17, 526–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fink J.J., Robinson T.M., Germain N.D., Sirois C.L., Bolduc K.A., Ward A.J., Rigo F., Chamberlain S.J. and Levine E.S. (2017) Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun., 8, 15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okuno H., Nakabayashi K., Abe K., Ando T., Sanosaka T., Kohyama J., Akamatsu W., Ohyama M., Takahashi T., Kosaki K. et al. (2017) Changeability of the fully methylated status of the 15q11.2 region in induced pluripotent stem cells derived from a patient with Prader–Willi syndrome. Congenit. Anom. (Kyoto), 57, 96–103. [DOI] [PubMed] [Google Scholar]
- 33. Takashima A. (1998) Establishment of fibroblast cultures. Current Protocols in Cell Biology. 00, 2.1.1--2.1.12, doi: 10.1002/0471143030.cb0201s00. [DOI] [PubMed] [Google Scholar]
- 34. Judson H., Hayward B.E., Sheridan E. and Bonthron D.T. (2002) A global disorder of imprinting in the human female germ line. Nature, 416, 539–542. [DOI] [PubMed] [Google Scholar]
- 35. Orjalo A., Johansson H.E. and Ruth J.L. (2011) StellarisTM fluorescence in situ hybridization (FISH) probes: a powerful tool for mRNA detection. Nat. Meth., 8, i--ii. [Google Scholar]
- 36. Warlich E., Kuehle J., Cantz T., Brugman M.H., Maetzig T., Galla M., Filipczyk A.A., Halle S., Klump H., Scholer H.R. et al. (2011) Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol. Ther., 19, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burridge P.W., Thompson S., Millrod M.A., Weinberg S., Yuan X., Peters A., Mahairaki V., Koliatsos V.E., Tung L. and Zambidis E.T. (2011) A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One, 6, e18293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cristo F., Inacio J.M., Rosas G., Carreira I.M., Melo J.B., Almeida L.P., Mendes P., Martins D.S., Maio J., Anjos R. et al. (2017) Generation of human iPSC line from a patient with laterality defects and associated congenital heart anomalies carrying a DAND5 missense alteration. Stem Cell Res., 25, 152–156. [DOI] [PubMed] [Google Scholar]
- 39. Yu J., Hu K., Smuga-Otto K., Tian S., Stewart R., Slukvin II. and Thomson J.A. (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science, 324, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bock C., Reither S., Mikeska T., Paulsen M., Walter J. and Lengauer T. (2005) BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics, 21, 4067–4068. [DOI] [PubMed] [Google Scholar]
- 41. Chaumeil J., Augui S., Chow J.C. and Heard E. (2008) Combined immunofluorescence, RNA fluorescent in situ hybridization, and DNA fluorescent in situ hybridization to study chromatin changes, transcriptional activity, nuclear organization, and X-chromosome inactivation. Methods Mol. Biol., 463, 297–308. [DOI] [PubMed] [Google Scholar]
- 42. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.