

Abstract
Prader–Willi syndrome (PWS), an imprinted neurodevelopmental disorder characterized by metabolic, sleep and neuropsychiatric features, is caused by the loss of paternal SNORD116, containing only non-coding RNAs (ncRNAs). The primary SNORD116 transcript is processed into small nucleolar RNAs (snoRNAs), which localize to nucleoli, and their spliced host gene 116HG, which is retained at its site of transcription. While functional complementation of the SNORD116 ncRNAs is a desirable goal for treating PWS, the mechanistic requirements of SNORD116 RNA processing are poorly understood. Here we developed and tested a novel transgenic mouse which ubiquitously expresses Snord116 on both a wild-type and a Snord116 paternal deletion (Snord116+/–) background. Interestingly, while the Snord116 transgene was ubiquitously expressed in multiple tissues, splicing of the transgene and production of snoRNAs was limited to brain tissues. Knockdown of Rbfox3, encoding neuron-specific splicing factor neuronal nuclei (NeuN) in Snord116+/–-derived neurons, reduced splicing of the transgene in neurons. RNA fluorescence in situ hybridization for 116HG revealed a single significantly larger signal in transgenic mice, demonstrating colocalization of transgenic and endogenous 116HG RNAs. Similarly, significantly increased snoRNA levels were detected in transgenic neuronal nucleoli, indicating that transgenic Snord116 snoRNAs were effectively processed and localized. In contrast, neither transgenic 116HG nor snoRNAs were detectable in either non-neuronal tissues or Snord116+/- neurons. Together, these results demonstrate that exogenous expression and neuron-specific splicing of the Snord116 locus are insufficient to rescue the genetic deficiency of Snord116 paternal deletion. Elucidating the mechanisms regulating Snord116 processing and localization is essential to develop effective gene replacement therapies for PWS.
Introduction
Prader–Willi syndrome (PWS) is a neurodevelopmental disorder characterized by a broad range of symptoms including hypotonia and failure to thrive in infancy followed by the onset of hyperhagia, intellectual impairment, obsessive-compulsive tendencies and sleep abnormalities including shorter sleep duration and daytime sleepiness (1). PWS is caused by paternal deficiency of the maternally imprinted 15q11-q13 locus, which encodes a long neuron-specific transcript (Fig. 1). Expression of this locus originates at the PWS imprinting control region (IC) at the 5ʹ end of SNRPN, extends through small nucleolar RNA (snoRNA) clusters SNORD116 (27 copies in human, >40 copies in mouse) and SNORD115 (47 copies in human, 37 copies in mouse) and terminates antisense to the maternally expressed UBE3A (UBE3A-ATS) (2–5). PWS is considered a contiguous gene disorder, as most patients carry large paternal deletions of this locus; however, analyses of PWS patients have revealed that small deletions of the SNORD116 cluster of snoRNAs are also sufficient to cause PWS (6–9). Snord116 is processed to form two non-coding RNAs (ncRNAs): Snord116 snoRNAs and the Snord116 host gene (116HG). Snord116 snoRNAs are intronically embedded within the non-coding Snord116 primary transcript, and although they currently have no known target sequence, they localize to the nucleolus in mature neurons (10–12), detectable by RNA fluorescence in situ hybridization (FISH) (Fig. 1). It has also been proposed that these orphan snoRNAs are further processed, generating short fragments with non-canonical functions. Generation of these processed snoRNAs (psnoRNAs) is not well understood; however, it has been suggested that they may arise from the same host intron and depend on stability and protein interactions (13). The nucleolar accumulation of Snord116 snoRNAs coincides with increased transcription of the locus, an increase in nucleolar size during early postnatal development and chromatin decondensation of the paternally expressed Snrpn-Ube3a allele, detected by DNA FISH (10).
Figure 1.
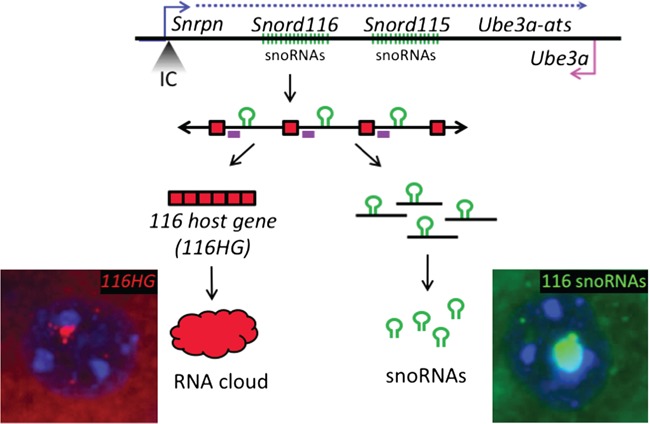
The PWS locus encodes a long ∼1 Mb transcript containing the Snord116 and Snord115 snoRNA clusters. The imprinting control (IC) region is located at the 5ʹ end of Snrpn, with paternal expression of this locus extending through the Snord116 and Snord115 snoRNA clusters and the antisense to the maternally expressed Ube3a in neurons. The long paternally expressed transcript is shown in blue and maternally expressed Ube3a is shown in pink. The primary Snord116 transcript is comprised of a repeating array of exons (red) flanking intron-embedded snoRNAs (green). Processing of this primary transcript produces snoRNAs from the introns of Snord116 and the 116HG from the spliced exons of Snord116. Localization of the 116HG (red) and Snord116 snoRNAs (green) are shown by RNA FISH (blue = DAPI). Location of Rbfox3 binding motifs indicated on primary transcript as purple boxes.
116HG is a long ncRNA (lncRNA) generated from the spliced exons of the Snord116 primary transcript. 116HG is retained within the nucleus and localizes to its paternally decondensed site of transcription, forming an ‘RNA cloud’, which associates with the Snord116 locus as well as other genes with epigenetic, metabolic and circadian functions regulating their expression in a time-of-day-dependent manner (11). The 116HG cloud is significantly larger during light hours, corresponding to the downregulated expression of its gene targets during sleep (14). Colocalized nuclear accumulation of the unspliced transcript and the spliced 116HG indicate that splicing occurs locally at the site of paternal expression (11). Although splicing is required for 116HG formation and Snord116 snoRNA biogenesis, the mechanism of this process and its specificity for neurons is not well understood.
In an effort to determine phenotypes associated with genes within the PWS region, mouse models carrying deletions of nearly every paternally expressed gene in the human 15q11-q13 locus, either individually or as part of a large deletion, have been created and characterized (15). Although some models exhibit a subset of PWS-like phenotypes, issues of lethality complicate phenotypic analyses of adult mice in some models, and no current mouse model of PWS exhibits consistency in the late onset obesity characteristic of PWS (15–17). Currently, two mouse models (PWScrm+/p– and Snord116+/–) carry deletions of the minimal Snord116 PWS critical region, and display postnatal growth deficiency characteristic of the early failure to thrive phenotype exhibited by PWS patients as well as some hyperphagic behavior (18,19). Activation of maternal Snord116 expression rescued the growth retardation and postnatal lethality phenotypes of the PWScrm+/p– small deletion model, supporting Snord116 as the PWS-critical region (20). Although the snoRNAs have previously been the main focus of studies of the Snord116 locus, transgenic expression of a single copy of Snord116 snoRNA failed to rescue the phenotype of a Snrpn-Ube3a deletion mouse model, suggesting either that the Snord116 functional unit is not restricted solely to the snoRNAs or that multiple copies are required (19,21). Reintroduction of multiple copies of Snord116 snoRNAs expressed from the introns of another host gene failed to rescue the growth retardation phenotype of PWScrm+/p– mice, highlighting the functional significance of the 116HG (20). Importantly, the regulation of circadian and metabolic gene expression by 116HG leads to dysregulated energy expenditure in mice deficient for paternal Snord116, suggesting that the lncRNA 116HG may play a role in the pathogenesis of PWS (14). Finally, the introns of the Snord116 primary transcript may play a role in the regulation ofUbe3a-ats progression by regulating chromatin compaction through the formation of R-loops (22). These studies have illustrated the genetic complexity of the Snord116 locus and the potential functional capacity of multiple elements within this lncRNA.
We designed a novel transgenic Snord116 mouse to characterize the processing and formation of these Snord116 RNA products, and define the mechanism regulating the brain specificity of these processes. By driving expression of Snord116 with a ubiquitous promoter, we investigated the requirements for Snord116 processing in multiple tissues and the potential of our transgene to compensate for the molecular deficits observed in Snord116+/- mice. We show that Snord116 expression is not sufficient for the production of snoRNAs or the 116HG, and that the formation of these products is blocked at the level of tissue-specific splicing. Our data demonstrate a requirement for Rbfox3 in the neuron-specific splicing of Snord116, and an active paternal Snord116 allele in the localization of its processed RNA components, providing a better understanding of the requirements of Snord116 function in the development of potential future therapies for PWS.
Results
Transgenic Snord116 integrates into the genome and is expressed in many tissues
To reintroduce Snord116 in a highly expressed, ubiquitous manner to paternally Snord116-deficient mice, we engineered a novel ‘complete’ Snord116 transgene (Ctg) containing three complete Snord116 repeats under the control of a cytomegalovirus (CMV) promoter (Fig. 2A). Each unit of the Snord116 repeat contained an intronically encoded snoRNA flanked by 116HG exons, as organized in the genomic DNA. This construct was randomly inserted into the genome by pronuclear injection of fertilized oocytes to create a transgenic mouse carrying nine copies of the construct, representing 27 copies of the Snord116repeat unit (Fig. 2B). Mice were tested for transgene insertion by polymerase chain reaction (PCR) using primers specific to the transgene and inverse PCR was performed to identify the genomic location of the transgene integration at 7qE3, approximately 47 Mb from the endogenous Snord116 locus (Fig. 2C). This falls outside of the 15q11-q13 imprinted locus and is therefore not expected to be co-regulated with endogenous Snord116. Transgenic Snord116 insertion overlaps with the poorly characterized gene Gm1966, which has been reported as both an unprocessed pseudogene as well as a protein-coding gene in different databases. Overall, no anatomical differences were observed between the brains of the wild-type and transgenic mice.
Figure 2.
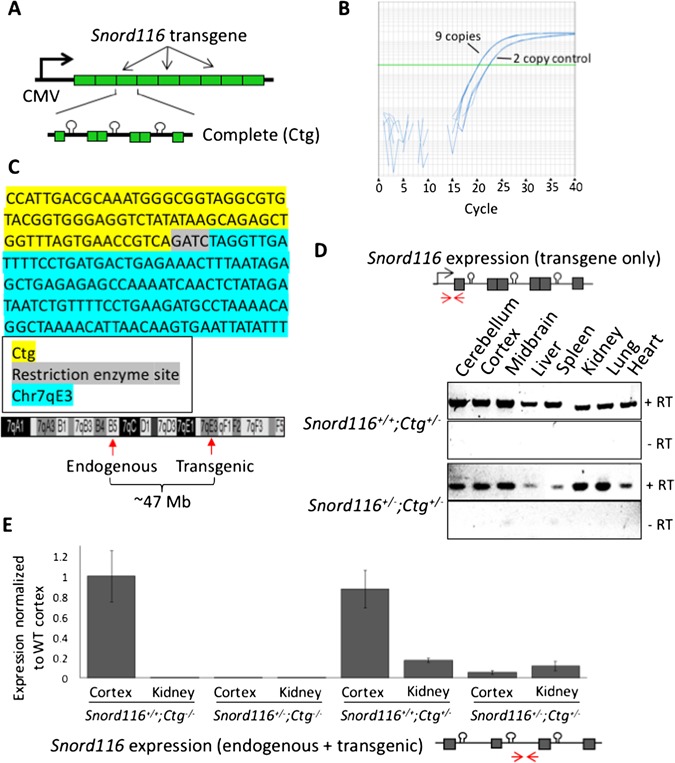
The Snord116 transgene contains 27 copies of the Snord116 repeat unit and is ubiquitously expressed. (A) Schematic of the Snord116 transgene construct design containing nine copies of the 3-copy Snord116 repeat unit (Ctg). (B) Copy number analysis indicates insertion of the Snord116 transgene nine times in Snord116+/+;Ctg+/– transgenic mice. (C) The location of Snord116 transgene insertion was identified at chromosome 7qE3 by inverse PCR. The sequence amplified at the ligation between the 5ʹ end of the transgene and its site of insertion mapped to chromosome 7 is shown. Diagram of chromosome 7 indicating the locations of the endogenous and transgenic loci ∼47 Mb apart. (D) The Snord116 transgene shows ubiquitous expression in all tissues tested using transgene-specific primers on both WT and Snord116+/– backgrounds. The locations of the transgene-specific primers are shown as red arrows in the construct diagram. (E) Quantification of combined endogenous and transgenic Snord116 primary transcript expression for all genotypes in cortex and kidney indicates the relative contribution of the Snord116 transgene both in the presence and absence of endogenous Snord116. Primer locations are shown as red arrows in the transcript diagram. Statistical significance testing by analysis of variance (ANOVA) with Tukey correction is provided in Supplementary Table 1.
The Snord116+/– PWS mouse model carries a ∼150 kb deletion of Snord116, representing the smallest region of overlap for human PWS deletions (6,7,9,19). Snord116+/– mice recapitulate the neonatal failure to thrive exhibited by PWS patients and exhibit altered energy expenditure (14,19). We crossed Snord116+/+;Ctg+/– transgenic females to Snord116+/– males to produce mice deficient for endogenous paternal Snord116 but carrying the transgene (Snord116+/–;Ctg+/–) and littermate controls (Snord116+/+;Ctg+/–, Snord116+/+;Ctg–/– and Snord116+/–;Ctg–/–). To ensure that the Snord116 transgene was not transcriptionally silenced, we tested the expression of the transgene in both WT (Snord116+/+;Ctg+/–) and PWS (Snord116+/–;Ctg+/–) backgrounds, confirming expression in several tissues, including those in which Snord116 is not endogenously expressed (12) (Fig. 2D). Quantification of combined endogenous and transgenic Snord116 expression indicates that the expression levels of the transgenic primary transcript may be impacted by its chromosomal context and processing (Fig. 2E, Supplementary Material, Table S1).
Transgenic Snord116 snoRNAs localize with endogenous snoRNAs specifically in neuronal nucleoli in wild-type but not Snord116+/– mice
Endogenous Snord116 expression is restricted to neurons in mice; however, we sought to determine if transgenic Snord116 was sufficient to recruit the required processing factors for the generation of functional snoRNAs. The ubiquitous expression pattern of the Snord116 transgene allowed us to examine the processing and localization of Snord116 snoRNAs outside of the neuronal lineage. We examined Snord116 snoRNA localization by RNA FISH in coronal brain sections using probes that detect both the endogenous and transgenic Snord116 RNAs (Fig. 3A, Supplementary Material, Figs S1 and 2). Despite ubiquitous expression of the Snord116 transcript in multiple tissues, detection of snoRNA nucleolar localization was limited to neurons in the brain, as it was not observed in non-neuronal nuclei. Snord116 snoRNAs were clearly detected in WT nucleoli of Purkinje neurons in the cerebellum and localized to the single large nucleolus, which we have previously characterized by α-nucleolin staining (10). As expected, snoRNAs were not observed in Snord116+/– neurons due to the lack of paternal Snord116 expression. In Snord116+/+;Ctg+/– mice, the intensity of the nucleolar snoRNA RNA FISH signalwas significantly greater than in WT nucleoli (Snord116+/+;Ctg–/–) (Fig. 3B), indicating that transgenic Snord116 potentially contributes to increasing the snoRNA population. SnoRNAs were also detected in the cortex of Snord116+/+;Ctg–/– and Snord116+/+;Ctg+/– mice. However, in the absence of endogenous Snord116 (Snord116+/–;Ctg+/–), expression of the transgene was not sufficient to produce detectable snoRNA accumulation within neuronal nucleoli in any brain region or in non-neuronal tissues (Supplementary Material, Figs. S1 and 2). These results indicate that although transgenic Snord116 is able to contribute to endogenous snoRNAs, transcription of the primary transcript is not sufficient for the processing or localization of these RNAs in non-neuronal and Snord116-deficient neuronal tissues.
Figure 3.
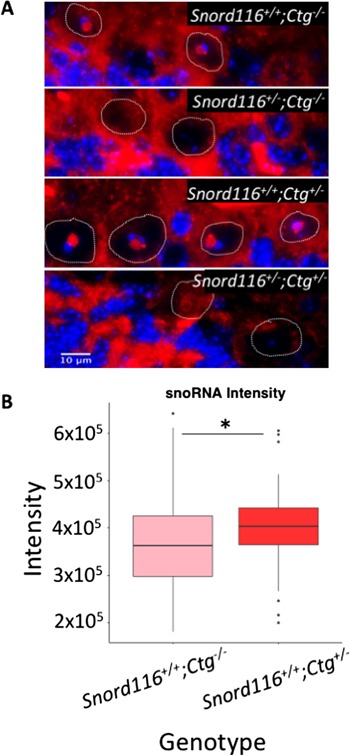
Transgenic Snord116 colocalizes with endogenous Snord116, but does not localize to nucleoli in neurons lacking endogenous Snord116 expression. (A) RNA FISH for Snord116 snoRNAs in Purkinje neurons. Nuclei are stained with DAPI (blue) and the boundaries of Purkinje nuclei are denoted by white dotted lines. Snord116 snoRNAs are shown in red localized within nucleoli of Purkinje neurons. (B) Quantification of RNA FISH signal shows significantly stronger Snord116 snoRNA signal in Snord116+/+;Ctg+/– Purkinje nucleoli compared to Snord116+/+;Ctg–/–. *P = 0.006 by t-test.
Neuron-specific splicing of transgenic Snord116 requires Rbfox3/NeuN
The discrepancy between expression of the primary transcript and production of functional snoRNA products from the Snord116 transgene led us to determine if splicing was the limiting factor in snoRNA processing. Using transgene-specific primers, we assessed splicing of transgenic Snord116 in several neuronal and non-neuronal tissues of Snord116+/+;Ctg+/– and Snord116+/–;Ctg+/– mice. Reverse transcription-PCR (RT-PCR) analysis demonstrated that splicing of transgenic Snord116 was restricted to neuronal tissues of both Snord116+/+;Ctg+/–and Snord116+/–;Ctg+/–mice (Fig. 4A) even though primary transcripts were detected in multiple tissue types (Fig. 2D). These results demonstrate that splicing may explain the tissue-specific differences between transcript expression and snoRNA localization, but not the deficiency of snoRNA processing in neurons of Snord116+/–;Ctg+/– mice.
Figure 4.
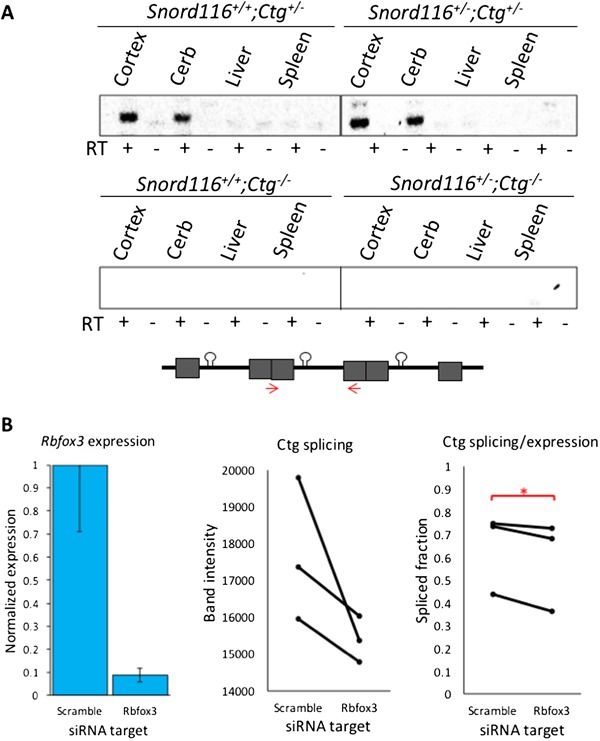
Splicing of transgenic Snord116 is restricted to neuronal tissues and is reduced by Rbfox3 knockdown. (A) Splicing of transgenic Snord116 is unique to neuronal tissues in both Snord116+/+;Ctg+/– and Snord116+/–;Ctg+/– mice. Bands indicate the spliced product of the Ctg transgene. RT (–) lanes indicate negative reverse transcriptase controls. The locations of splice primers are shown as red arrows in the construct diagram. (B) Knockdown of Rbfox3 expression in Snord116+/–;Ctg+/– NPC-derived neurons reduces splicing of Snord116. Rbfox3 levels are shown in the left panel, normalized to Gapdh. Knockdown levels are graphed relative to levels detected in the negative scramble siRNA condition. Absolute intensity of the spliced Ctg is shown for each condition in the middle panel. The spliced fraction of the expressed transgene (splicing/expression) is significantly lower after Rbfox3 knockdown compared to a scramble siRNA control (right panel). *P<0.05 by paired t-test.
NeuN is commonly used in immunofluorescence staining as a neuron-specific marker, and has now been identified as the neuron-specific splicing factor, Rbfox3 (23). To test the hypothesis that Rbfox3/NeuN regulates neuron-specific splicing of the Snord116 transgene, we performed an siRNA knockdown of Rbfox3 in neurons derived from Snord116+/–;Ctg+/– neural progenitor cells (NPCs), reducing Rbfox3 expression to about 9% of levels detected in scramble siRNA-treated NPCs (Fig. 4B and Supplementary Material, Fig. S3). The expression of the transgenic Snord116 primary transcript was unaffected following Rbfox3 siRNA knockdown; however, the proportion of the primary transcript that was spliced (splicing/expression) was significantly reduced (Fig. 4B and Supplementary Material, Fig. S3). To further examine a role for Rbfox3 in Snord116 splicing, we utilized a published dataset of RNA sequencing from Rbfox3 knockout mouse cerebral cortex (24). Visualization of exon junctions within the Snord116 locus by sashimi plot using the Integrative Genomics Viewer (IGV) browser demonstrated that loss of Rbfox3 leads to pronounced dysregulation of exon splicing between Snord116 and Ube3a (Supplementary Material, Fig. S4). Together, these results demonstrate that Rbfox3 levels affect neuronal Snord116 splicing, and suggest that Rbfox3/NeuN is essential for the processing of intron-embedded snoRNAs.
The transgenic host gene 116HG RNA cloud localizes to the endogenous, but not the transgenic Snord116 locus, only in wild-type neurons
In addition to the nucleolar snoRNAs, the spliced exons of the Snord116 locus are retained as an RNA cloud that localizes to the site of transcription on the active paternal allele in wild-type neurons (22) (Fig. 1). We therefore asked whether the transgenically-encoded 116HG localized to its own site of transcription using RNA FISH with a probe designed to detect the spliced junctions of both endogenous and transgenic 116HG. Similarly to the Snord116 snoRNA localization, 116HG FISH signals were observed as a single nuclear cloud in both Snord116+/+;Ctg–/– and Snord116+/+;Ctg+/– neurons, but not Snord116+/–;Ctg–/– or Snord116+/–;Ctg+/– neurons (Fig. 5A and Figs. S5 and 6). In mice of two different ages (5.5 months and1 year), the single 116HG RNA FISH signal was significantly larger in Snord116+/+;Ctg+/– compared to Snord116+/+;Ctg–/– neurons (Fig. 5B), suggesting that the transgenic spliced 116HG localized and contributed to the 116HG RNA cloud on the endogenous paternal allele. By DNA FISH, distinct nuclear locations of the three alleles (endogenous maternal and paternal, plus transgene) were observed (Fig. 5C), demonstrating the absence of colocalization of the transgenic allele with the active paternal allele at the chromosomal level. In addition to our molecular analysis, we measured body weight for all genotypes from weaning until postnatal week 11. It has previously been reported that Snord116+/– mice weigh significantly less than their WT littermates within this time frame (14,19), therefore we sought to determine whether the Snord116 transgene would rescue the deletion phenotype. Concordant with the lack of Snord116 molecular rescue of the 116HG RNA cloud and the Snord116 snoRNAs, Snord116+/–;Ctg+/– mice had a significantly lower body weight than Snord116+/+;Ctg–/– mice, similar to the weights observed in Snord116+/–;Ctg–/– mice (Fig. 6A and Supplementary Material, Table S2) (14). Interestingly, the additive effect of endogenous and transgenic Snord116 (Snord116+/+;Ctg+/–) also resulted in decreased body weight compared to expression of the endogenous locus alone (Snord116+/+;Ctg–/–), reaching significance at postnatal week 11, suggesting that expression in tissues outside of the brain as in Snord116+/+;Ctg+/– and Snord116+/–;Ctg+/– mice may have a significant impact on metabolism.
Figure 5.
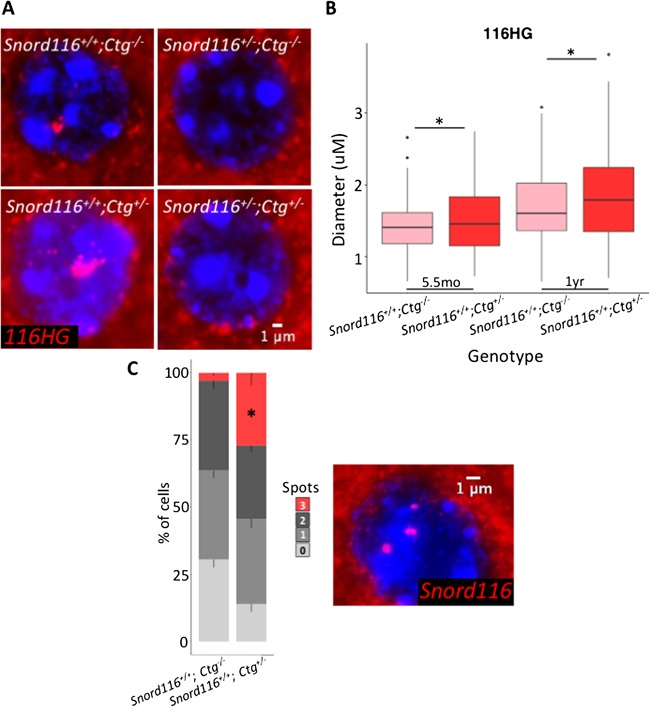
Transgenic 116HG colocalizes with the endogenous 116HG RNA cloud, but is not detected in the nuclei of neurons lacking endogenous Snord116 expression. (A) RNA FISH for 116HG in a cortical neuronal nucleus. Nuclei are stained with DAPI (blue) and the 116HG RNA FISH signal is shown in red. (B) Quantification of RNA FISH signal shows a single significantly larger 116HG RNA cloud in Snord116+/+;Ctg+/– neuronal nuclei compared to Snord116+/+;Ctg–/–. T-test, P = 0.002. (C) DNA FISH of Snord116 in neuronal nuclei shows three detectable Snord116 loci (red) in a significant proportion of Snord116+/+;Ctg+/– neuronal nuclei (blue), indicating a lack of colocalization. *P<0.0001 by t-test.
Figure 6.
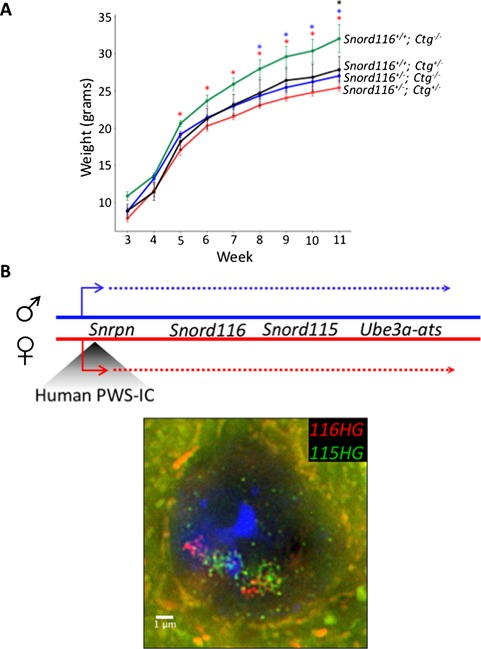
Transgenic Snord116 does not rescue the weight phenotype observed in Snord116+/– mice. (A) Weight curves of all genotypes from Snord116+/–;Ctg–/– X Snord116+/+;Ctg+/– cross. Mice carrying the Snord116 transgene, either on a WT or Snord116+/– background weigh significantly less than WT mice, and are phenotypically more similar to Snord116+/– mice. N = 4 Snord116+/+;Ctg–/– (WT), 4 Snord116+/+;Ctg+/– (Ctg/WT), 13 Snord116+/–;Ctg–/– (Prad), 13 Snord116+/–;Ctg+/– (Ctg/Prad). *P<0.05 by repeated measures ANOVA, Benjamini–Hochberg post-hoc correction (results in Supplementary Material, Table S2). (B) Map of the 15q11-q13 locus in PWS-ICHs/+ mice with the paternal allele indicated in blue and the maternal allele indicated in red. RNA FISH for 116HG (red) and 115HG (green) in a PWS-ICHs/+ neuronal nucleus, with two decondensed alleles, shows the formation of two distinct RNA clouds for each cluster, indicating the requirement for a decondensed endogenous locus for the localization of the spliced 116HG.
To test the hypothesis that a second requirement for 116HG complementation is an active endogenous Snord116 locus, we utilized another transgenic mouse model, in which the maternal mouse PWS-IC has been replaced by the human PWS-IC (PWS-ICHs/+) (25). Because the human PWS-IC does not become imprinted in mice, the normally silent maternal allele undergoes chromatin decondensation and expresses Snrpn, snoRNAs and Ube3a-ats in neurons, similar to the paternal allele (Fig. 6B) (10). In neuronal nuclei of PWS-ICHs/+ mice, two distinct 116HG RNA clouds were observed by RNA FISH, demonstrating that the requirement for 116HG RNA localization is an active endogenous Snord116 allele, not simply transcription of Snord116. Together, these results demonstrate that complementation by transgenic Snord116 requires additional factors both in trans (Rbfox3/NeuN) and in cis (chromatin decondensation).
Discussion
This study utilized a novel 27-copy Snord116 transgenic mouse to characterize the requirements for Snord116 processing and assess the molecular functional capacity of transgenic Snord116 in the absence of endogenous paternal expression. We demonstrated that transgenic Snord116 colocalizes with endogenous Snord116 snoRNAs and 116HG in the brain; however, transgenic Snord116 expression alone is not sufficient for the formation of functional RNAs from this locus, as they are not detected in tissues outside of the brain. Additionally, trafficking of transgenic RNAs is impaired in the absence of endogenous paternal Snord116; therefore, nuclear RNA cloud formation is observed only in the presence of endogenous Snord116 expression. Further, splicing of the Snord116 transgene was restricted to the brain, despite expression in many tissues. This process is partially blocked by the knockdown of Rbfox3 in NPC-derived neurons, suggesting a role for this neuron-specific splicing factor in the processing of Snord116. The introduction of transgenic Snord116 also fails to rescue the weight phenotype observed in Snord116+/– mice.
Splicing represents a critical step in the regulation of gene expression in all tissues; however, the brain exhibits the highest levels of tissue-specific alternative splicing (26–28). Such tissue-specific splicing reflects the intricate cellular connections and functional diversity within the brain and exemplifies the complex expression and regulatory networks observed in the brain and during development. Brain-specific splicing factors provide an important mechanism for the co- and post-transcriptional regulation involved in processes such as neurogenesis and synapse formation through the spatiotemporal control of RNA expression, processing and localization (29,30). The Rbfox family of splicing factors is important in the regulation of brain development, with Rbfox3 expression uniquely confined to mature postmitotic neurons (31). Neuronal co-expression and the presence of the (U)GCAUG-binding motif (32,33) within the introns of the Snord116 transcript support the role for Rbfox3 in the processing of Snord116 in the brain. In addition, a recent study modeling the Rbfox3 regulatory network by crosslinking and immunoprecipitation followed by high-throughput sequencing identified significant peaks within the introns of Ipw, validating our Snord116-specific analysis of Rbfox3 activity (33). Interestingly, RBFOX3 has been shown to be expressed at low levels in some immortalized human cell lines, where levels may be sufficient for the production of SNORD116 RNA products, or may be compensated for by the expression of other splicing factors including RBFOX1 or RBFOX2.
Ube3a, another gene within the 15q11-q13 locus, is paternally imprinted in neurons, in which the paternal expression of the Snrpn-Ube3a transcript extends through the anti-sense to Ube3a, blocking transcription of paternal Ube3a and leading to maternal-specific expression (34–36). Interestingly, in Rbfox3/NeuN-negative cells of the suprachiasmatic nucleus (SCN), paternal Ube3a expression is observed (37). Due to its high level of GC skew, transcription through Snord116 results in the formation of R-loops, modulating the balance between chromatin state and transcription elongation. Stabilization of these R-loops by topotecan treatment stalls RNA polymerase II progression, blocking transcription through Ube3a-ats and resulting in biallelic Ube3a expression (22). Conversely, a study of genome stability demonstrated the ability of the ASF/SF2 splicing factor to interact with R-loops, repressing their formation through interaction with the nascent RNA (38). The Rbfox3-dependent silencing of paternal Ube3a in the SCN suggests that neuron-specific splicing of Snord116 by Rbfox3 may be important for maintaining the balance ofR-loop formation, promoting transcription through Ube3a-ats and paternal imprinting of Ube3a in neurons.
It is important to note that the chromatin context of the transgene likely contributes to differences in the expression and processing of the primary transcript, which are lower than WT levels. It is therefore possible that the level of Snord116 expression produced by the transgene alone in Snord116+/–; Ctg+/– mice may not be sufficient to compensate for the loss of endogenous Snord116. Despite low levels of transgenic primary transcript however, in the presence of splicing by Rbfox3 and the endogenous chromatin for localization, this transgenic pri-mary transcript contributes significantly to the formation of mature RNA products in the brain. This indicates a potential multi-level deficit in the production of functional RNAs from transgenic Snord116 in Snord116+/–;Ctg+/– neurons and in all non-neuronal tissues. Paternal Snord116 is GC skewed, resulting in R-loop formation, histone displacement and chromatin decondensation upon neuronal transcription and the specific localization of the 116HG to its site of transcription at the decondensed paternal allele suggests a chromatin-state-dependent accumulation of 116HG (22). The formation of two 116HG RNA clouds in PWS-ICHs/+ neuronal nuclei demonstrates the requirement for a decondensed endogenous allele for the localization of Snord116, suggesting that chromatin decondensation may mediate RNA–DNA interactions necessary to anchor the 116HG to its proper subnuclear domain. The lack of phenotypic rescue by the Snord116 transgene is in agreement with the lack of rescue at the molecular level; however, the negative combinatorial effect of endogenous and transgenic Snord116 may reflect the aberrant expression of Snord116 in tissues outside of the brain important for metabolism. Further study of the mechanisms of Snord116 localization would benefit our understanding of the requirements for Snord116 function and enable the development of more effective therapies in the future.
Materials and Methods
Mouse husbandry
C57BL/6J (WT) and B6(Cg)-Snord116tm1.1Uta/J (Snord116+/–) mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Ctg+/– mice were generated by the Mouse Biology Program (UC Davis). All mice were housed in a 12 h light:12 h dark temperature-controlled room and fed a standard diet of PicoLab mouse chow 20 (PMI International, St Louis, MO, USA). Heterozygous deletion male mice (Snord116+/–) were bred with heterozygous Ctg transgenic females (Ctg+/–) to generate littermates of each of the following genotypes: Snord116+/+;Ctg–/– (WT), Snord116+/–;Ctg–/– (Prad), Snord116+/+;Ctg+/– (Ctg/WT), Snord116+/–;Ctg+/– (Ctg/Prad). All mice used for this study were male and tissues were collected during light hours (ZT0-ZT12).
RNA FISH and DNA FISH
Snord116 BAC RP23-358G20 was ordered from BACPAC Resources (Children’s Hospital Oakland Research Institute). Nick translation of DIG-labeled probes and DNA FISH was performed as described previously (10). RNA FISH was performed as described previously (10).
116HG probes = AATGCAACCCTTTTAACTCAG (Exiqon), pooled probes (Agilent).
snoRNA probe = TTCCGATGAGAGTGGCGGTACAGA (Exiqon).
Microscopy
Slides were imaged on an Axioplan 2 fluorescence microscope (Carl Zeiss, Inc., NY, USA), equipped with a Qimaging Retiga EXi high-speed uncooled digital camera and analyzed using iVision Software (BioVision Technologies, Inc., Exton, PA, USA). Images were captured using a 100× oil objective and 1× camera zoom. 116HG RNA cloud measurements were taken as two perpendicular cross sections through the center of the RNA cloud and averaged for RNA cloud size. snoRNA intensity was measured as the sum of the intensity of each pixel divided by the area measured. All measurements were converted from pixel counts to microns according to objective and zoom (1px = 0.069 μm). All measurements and spot counting were blinded to minimize bias.
Reverse transcriptase PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR)
RNA was isolated with the RNeasy mini kit (Qiagen, Hilden, Germany) and complementary DNA (cDNA) was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen). For qRT-PCR, cDNA was amplified using the SensiFAST SYBR Lo-Rox Kit (Bioline, London, UK). Custom primers were designed using Primer3 software.
Ctg expression (transgene specific):
F – taagcagagctggtttagtgaacc; R – aacagttcgatggagactcagttgg
Ctg splicing (transgene specific):
F – cctgagttaaaaggcggccg; R – gccatttcctctgcatgttt
Rbfox3 expression:
F – aattttcccgaattgcccgaac; R – atgaagcagcacagacagacaa
Snord116 expression (endogenous + transgenic):
F – atgcaggctgctggtagagt; R – cctccaaggcttagctcctt
Gapdh expression (39):
F – caaggagtaagaaaccctggacc; R – cgagttgggatagggcctct
NPC and neuron culture
Embryos were dissected and cortices removed at E15. NPCs were isolated as described previously (40) and cultured in NEP Complete media supplemented with GlutaMAX. To differentiate, neurospheres were dissociated with Accutase (Invitrogen) and filtered for a single-cell suspension. Plates were coated with Poly-D-lysine (Sigma, St. Louis, MO, USA) and laminin (Invitrogen) and media was changed to Neurobasal with retinoic acid and brain-derived neurotrophic factor.
siRNA knockdown
Rbfox3 was knocked down using a pool of three Stealth RNAi siRNA or a negative control siRNA at 55 pmol RNAi per 60 mm dish (Life Technologies, Carlsbad, CA, USA). Neurons were differentiated for 7 days followed by 3 days of knockdown. RNA was then extracted and evaluated for knockdown efficiency by qRT-PCR and expression/splicing by RT-PCR.
Inverse PCR
Genomic DNA was isolated from tail clipping using the Gentra Puregene Kit (Qiagen). DNA was digested with DpnII and circularized by T4 ligation (Promega, Madison, WI, USA). Primers were designed to the known transgene sequence to amplify the unknown flanking genomic region. The PCR product was gel purified and sequenced by Sanger sequencing. Sequences flanking the DpnII restriction site were mapped to the transgene and the genome flanking.
Inverse PCR primers:
F – gatttccaagtctccaccccat; R – ggctatgaactaatgaccccgt
RNA-sequencing analysis
SRA files were downloaded from GEO (GSE84786) (24) and converted to fastq files using fastq-dump, splitting files for paired-end reads. Reads were trimmed for adapters and quality using the following parameters: ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:8:T LEADING:15 TRAILING:15 SLIDINGWINDOW:4:15. An insert size of 200 bp was used based on Picard metrics and reads were aligned to mm10 using TopHat2. Bam index files were built using Picard Tools and stranded bed files were created and used to create bedGraph and bigwig files for visualization as custom tracks on the UCSC genome browser. Raw bam files were loaded into the IGV browser to create sashimi plots (MISO framework) (41,42).
Supplementary Material
Funding
National Institute of Health (R01 NS076263, R56 NS076263-06 to J.M.L., T32 GM007377 to R.L.C.); Intellectual and Developmental Disabilities Research Center (U54 HD079125) and the Foundation for Prader–Willi Research.
Conflict of Interest statement. None declared.
References
- 1. Cassidy S.B., Schwartz S., Miller J.L. and Driscoll D.J. (2012) Prader–Willi syndrome. Genet. Med., 14, 1–10. [DOI] [PubMed] [Google Scholar]
- 2. Runte M., Hüttenhofer A., Groß S., Kiefmann M., Horsthemke B. and Buiting K. (2001) The IC-SNURF–SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet., 10, 2687–2700. [DOI] [PubMed] [Google Scholar]
- 3. Landers M., Bancescu D.L., Le Meur E., Rougeulle C., Glatt-Deeley H., Brannan C., Muscatelli F. and Lalande M. (2004) Regulation of the large (∼1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res., 32, 3480–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sutcliffe J.S., Nakao M., Christian S., Orstavik K.H., Tommerup N., Ledbetter D.H. and Beaudet A.L. (1994) Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet., 8, 52–58. [DOI] [PubMed] [Google Scholar]
- 5. Buiting K., Saitoh S., Gross S., Dittrich B., Schwartz S., Nicholls R.D. and Horsthemke B. (1995) Inherited microdeletions in the Angelman and Prader–Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet., 9, 395–400. [DOI] [PubMed] [Google Scholar]
- 6. Duker A.L., Ballif B.C., Bawle E.V., Person R.E., Mahadevan S., Alliman S., Thompson R., Traylor R., Bejjani B.A., Shaffer L.G. et al. (2010) Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader–Willi syndrome. Eur. J. Hum. Genet., 18, 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith A.J., Purmann C., Walters R.G., Ellis R.J., Holder S.E., Van Haelst M.M., Brady A.F., Fairbrother U.L., Dattani M., Keogh J.M. et al. (2009) A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet., 18, 3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sahoo T., Del Gaudio D., German J.R., Shinawi M., Peters S.U., Person R.E., Garnica A., Cheung S.W. and Beaudet A.L. (2008) Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet., 40, 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bieth E., Eddiry S., Gaston V., Lorenzini F., Buffet A., Auriol F.C., Molinas C., Cailley D., Rooryck C., Arveiler B. et al. (2014) Highly restricted deletion of the SNORD116 region is implicated in Prader–Willi Syndrome. Eur. J. Hum. Genet., 23, 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leung K.N., Vallero R.O., Dubose A.J., Resnick J.L. and Lasalle J.M. (2009) Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet., 18, 4227–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vitali P., Royo H., Marty V., Bortolin-Cavaille M.-L. and Cavaille J. (2010) Long nuclear-retained non-coding RNAs and allele-specific higher-order chromatin organization at imprinted snoRNA gene arrays. J. Cell Sci., 123, 70–83. [DOI] [PubMed] [Google Scholar]
- 12. Cavaillé J., Buiting K., Kiefmann M., Lalande M., Brannan C.I., Horsthemke B., Bachellerie J.P., Brosius J. and Hüttenhofer A. (2000) Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. U.S.A., 97, 14311–14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falaleeva M. and Stamm S. (2013) Processing of snoRNAs as a new source of regulatory non-coding RNAs: SnoRNA fragments form a new class of functional RNAs. Bioessays, 35, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Powell W.T., Coulson R.L., Crary F.K., Wong S.S., Ach R.A., Tsang P., Yamada N.A., Yasui D.H. and LaSalle J.M. (2013) A Prader–Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum. Mol. Genet., 22, 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bervini S. and Herzog H. (2013) Mouse models of Prader–Willi Syndrome: a systematic review. Front. Neuroendocrinol., 34, 107–119. [DOI] [PubMed] [Google Scholar]
- 16. Purtell L., Qi Y., Campbell L., Sainsbury A. and Herzog H. (2017) Adult-onset deletion of the Prader–Willi syndrome susceptibility gene Snord116 in mice results in reduced feeding and increased fat mass. Transl. Pediatr., 6, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polex-Wolf J., Lam B.Y.H., Larder R., Tadross J., Rimmington D., Bosch F., Jiménez Cenzano V., Ayuso E., Ma M.K.L., Rainbow K. et al. (2018) Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader–Willi syndrome. J. Clin. Invest., 128, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skryabin B.V., Gubar L.V., Seeger B., Pfeiffer J., Handel S., Robeck T., Karpova E., Rozhdestvensky T.S. and Brosius J. (2007) Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet., 3, e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ding F., Li H.H., Zhang S., Solomon N.M., Camper S.A., Cohen P. and Francke U. (2008) SnoRNA Snord116 (Pwcr1/MBll-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One, 3, e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rozhdestvensky T.S., Robeck T., Galiveti C.R. and Raabe C.A. (2016) Maternal transcription of non-protein coding RNAs from the PWS-critical region rescues growth retardation in mice. Scientific Reports, 6, DOI: 10.1038/srep20398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsai T.F., Jiang Y.H., Bressler J., Armstrong D. and Beaudet A.L. (1999) Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader–Willi syndrome. Hum. Mol. Genet., 8, 1357–1364. [DOI] [PubMed] [Google Scholar]
- 22. Powell W.T., Coulson R.L., Gonzales M.L., Crary F.K., Wong S.S., Adams S., Ach R.A., Tsang P., Yamada N.A., Yasui D.H. et al. (2013) R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. U.S.A., 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim K.K., Adelstein R.S. and Kawamoto S. (2009) Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J. Biol. Chem., 284, 31052–31061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang H.-Y., Hsieh P.-F., Huang D.-F., Chin P.-S., Chou C.-H., Tung C.-C., Chen S.-Y., Lee L.-J., Shur-Fen Gau S. and Huang H.-S. (2015) RBFOX3/NeuN is required for hippocampal circuit balance and function. Scientific Reports, 5, DOI: 10.1038/srep17383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnstone K.A., DuBose A.J., Futtner C.R., Elmore M.D., Brannan C.I. and Resnick J.L. (2006) A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum. Mol. Genet., 15, 393–404. [DOI] [PubMed] [Google Scholar]
- 26. Grange P., Gratadou L., Delord M., Dutertre M. and Auboeuf D. (2010) Splicing factor and exon profiling across human tissues. Nucleic Acids Res., 38, 2825–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu Q., Modrek B. and Lee C. (2002) Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res., 30, 3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yeo G., Holste D., Kreiman G. and Burge C.B. (2004) Variation in alternative splicing across human tissues. Genome Biol., 5, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng S. and Black D.L. (2014) Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet, 29, 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chih B., Gollan L. and Scheiffele P. (2006) Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron, 51, 171–178. [DOI] [PubMed] [Google Scholar]
- 31. Duan W., Zhang Y.-P., Hou Z., Huang C., Zhu H., Zhang C.-Q. and Yin Q. (2015) Novel insights into NeuN: from neuronal marker to splicing regulator. Mol. Neurobiol., 53, 1637–1647. [DOI] [PubMed] [Google Scholar]
- 32. Minovitsky S., Gee S.L., Schokrpur S., Dubchak I. andConboy J.G. (2005) The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res., 33, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weyn-vanhentenryck S.M., Mele A., Yan Q., Sun S., Farny N., Zhang Z., Xue C., Herre M., Silver P.A. and Michael Q. (2014) HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep., 6, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rougeulle C., Cardoso C., Fontes M., Colleaux L. and Lalande M. (1998) An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet., 19, 15–16. [DOI] [PubMed] [Google Scholar]
- 35. Meng L., Person R.E. and Beaudet A.L. (2012) Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet., 21, 3001–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meng L., Person R.E., Huang W., Zhu P.J., Costa-Mattioli M. and Beaudet A.L. (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet., 9, e1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones K.A., Han J.E., DeBruyne J.P. and Philpot B.D. (2016) Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci. Rep., 6, 28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aguilera A. and Garcia-Muse T. (2012) R loops: from transcription byproducts to threats to genome stability. Mol. Cell, 46, 115–124. [DOI] [PubMed] [Google Scholar]
- 39. Siepka S.M., Yoo S.H., Park J., Song W., Kumar V., Hu Y., Lee C. and Takahashi J.S. (2007) Circadian mutant overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell, 129, 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hutton S.R. and Pevny L.H. (2008) Isolation, culture, and differentiation of progenitor cells from the central nervous system. Cold Spring Harb. Protoc., 3, DOI: 10.1101/pdb.prot5077. [DOI] [PubMed] [Google Scholar]
- 41. Katz Y., Wang E.T., Airoldi E.M. and Burge C.B. (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods, 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Katz Y., Wang E.T., Silterra J., Schwartz S., Wong B.,Thorvaldsdóttir H., Robinson J.T., Mesirov J.P., Airoldi E.M. and Burge C.B. (2015) Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics, 31, 2400–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.