

Abstract
Background
Cancer cachexia is a metabolic wasting syndrome that is strongly associated with a poor prognosis. The initiating factors causing fat and muscle loss are largely unknown. Previously, we found that leukaemia inhibitory factor (LIF) secreted by C26 colon carcinoma cells was responsible for atrophy in treated myotubes. In the present study, we tested whether C26 tumour‐derived LIF is required for cancer cachexia in mice by knockout of Lif in C26 cells.
Methods
A C26 Lif null tumour cell line was made using CRISPR‐Cas9. Measurements of cachexia were compared in mice inoculated with C26 vs. C26Lif−/− tumour cells, and atrophy was compared in myotubes treated with medium from C26 vs. C26Lif−/− tumour cells. Levels of 25 cytokines/chemokines were compared in serum of mice bearing C26 vs. C26Lif−/− tumours and in the medium from these tumour cell lines.
Results
At study endpoint, C26 mice showed outward signs of sickness while mice with C26Lif−/− tumours appeared healthy. Mice with C26Lif−/− tumours showed a 55–75% amelioration of body weight loss, muscle loss, fat loss, and splenomegaly compared with mice with C26 tumours (P < 0.05). The heart was not affected by LIF levels because the loss of cardiac mass was the same in C26 and C26Lif−/− tumour‐bearing mice. LIF levels in mouse serum was entirely dependent on secretion from the tumour cells. Serum levels of interleukin‐6 and G‐CSF were increased by 79‐fold and 68‐fold, respectively, in C26 mice but only by five‐fold and two‐fold, respectively, in C26Lif−/− mice, suggesting that interleukin‐6 and G‐CSF increases are dependent on tumour‐derived LIF.
Conclusions
This study shows the first use of CRISPR‐Cas9 knockout of a candidate cachexia factor in tumour cells. The results provide direct evidence for LIF as a major cachexia initiating factor for the C26 tumour in vivo. Tumour‐derived LIF was also a regulator of multiple cytokines in C26 tumour cells and in C26 tumour‐bearing mice. The identification of tumour‐derived factors such as LIF that initiate the cachectic process is immediately applicable to the development of therapeutics to treat cachexia. This is a proof of principle for studies that when carried out in human cells, will make possible an understanding of the factors causing cachexia in a patient‐specific manner.
Keywords: Skeletal muscle, Atrophy, Wasting, CRISPR, Tumour, Cachexia, LIF
Introduction
Cachexia is a devastating complication of cancer. It causes weakness, metabolic dysfunction, and intolerance to cancer treatment.1 The loss of skeletal muscle is a clinically significant feature of cachexia because it is a strong prognosticator of mortality.1, 2, 3, 4 Therefore, the alleviation of muscle wasting would reduce morbidity and allow for continued cancer treatment. Reviews of the literature indicate that triggering factors include blood‐borne proteins secreted from the tumour and from the patient's response to the tumour.5, 6, 7, 8, 9, 10, 11 Tumour‐derived factors that initiate cachexia must exist in the circulation, and their identities are just beginning to be investigated.
Pro‐inflammatory cytokines and other factors are elevated in mice and humans with cancer cachexia.6, 12, 13, 14 The most highly studied of these include interleukin‐6 (IL‐6) and tumour necrosis factor‐alpha (TNFα). However, antibodies administered to mice or humans against these molecules have had mixed effectiveness at improving health.6, 12, 14, 15, 16 More recently, multiplex immunoassays have made possible the measurement of many circulating factors simultaneously in humans and in preclinical mouse models of cancer cachexia, providing a broader picture of the growth and immune‐related factors that may play a role in cachexia.12, 17, 18 A difficulty, however, is linking the increases in circulating factors to a causal role in muscle wasting. In addition, different tumour types and different animal species appear to cause wasting by different mechanisms.14, 19, 20, 21, 22
Recently, we developed an approach to systematically identify the blood‐borne factors causing cancer cachexia by reasoning that proteins secreted by tumours activate muscle signalling pathways and their associated transcription factors, which in turn regulate muscle target genes. To test this principle, we carried out a study using a medium conditioned by tumour cells from the C26 colon carcinoma [C26 conditioned medium (CM)],23 the most commonly used mouse model of cancer cachexia. We screened for activated signalling pathways in myotubes treated with C26 CM and found robust activation of Stat‐dependent transcription but no activation of other commonly studied pathways in muscle. Furthermore, genetic and pharmacological inhibition showed that Jak2/Stat3 activation was required for C26 CM‐induced myotube atrophy. Moreover, we and others have shown that genetic inhibition of Stat3 in skeletal muscle of C26 mice blocks fibre atrophy.23, 24 Based on these combined findings, we assayed C26 CM for ligands belonging to the IL‐6 protein family, because this family has established activators of Jak/Stat signalling. We subsequently performed antibody neutralization studies to find that leukaemia inhibitory factor (LIF), but not other IL‐6 family members, was entirely responsible for C26 CM‐induced myotube atrophy.23 Thus, although high levels of IL‐6 induce Stat3 and a significant elevation in circulating IL‐6 is a common finding in mice with C26 tumours,6, 25 it is not required for C26 CM‐induced myotube atrophy.
Leukaemia inhibitory factor is well documented as a survival factor for mouse embryonic stem cells.26 Earlier work on the molecular action of LIF on muscle cells showed that it inhibits myogenesis.27, 28 The anti‐myogenic effect might be related more to ERK induction than to STAT3,28 although we showed that both STAT3 and ERK are required for the wasting effect of LIF on muscle.23
Interestingly, in previous C26 experiments, we found increased circulating levels of LIF at 19 and 25 days post‐inoculation, the latter of which was our cachexia endpoint. This preceded the increase in circulating IL‐6, which was increased at 25 days, but not at 19 days.23 Furthermore, neutralizing LIF in C26 CM‐treated myotubes blocked the majority of IL‐6 production by cultured myotubes. These data suggested a role for LIF in regulating cytokines such as IL‐6, found in the blood of C26 tumour‐bearing mice.
Therefore, in the present study, we directly tested whether LIF secretion by C26 tumour cells is necessary to induce cachexia in vivo, in mice carrying the C26 tumour. To do this, we employed C26Lif−/− cancer cells, made using CRISPR/Cas9, and inoculated mice with these cells compared with C26 tumour cells. To identify circulating factors that might be dependent on LIF, we measured levels of 23 different cytokines or chemokines in mice with C26 or C26Lif−/− tumours. Our findings from this study clearly demonstrate that LIF secreted by C26 tumour cells is required for the majority of muscle atrophy and for cachexia‐related changes in the C26 tumour‐bearing mouse. In addition, we identified several elevated cytokines in the C26 mouse that are regulated by C26 tumour‐derived LIF.
Methods
Genetic knockout of leukaemia inhibitory factor from C26 tumour cells
In order to remove LIF protein from the C26 tumour cell line, we submitted C26 cells to Applied Biological Materials (ABM, Vancouver, CA) to knock out the Lif gene using ABM's method of lentivirus‐delivered CRISPR‐Cas9. The mouse Lif gene was targeted in a unique part of the second exon, and a bi‐allelic knockout was produced. The DNA sequencing results provided by ABM verified the two mutant alleles in the knockout clone. The mutant alleles each had sequence deletions, one of 2 bp and the other of 4 bp in the target site in exon 2. We analysed the sequences against the known mouse LIF gene data online using MacVector, in order to predict the sequence of the shorter peptides made in the C26Lif−/− cells.
Cell culture and conditioned medium
C2C12 mouse myoblasts (American Type Culture Collection, Manassas, VA) were maintained in growth medium (GM) containing Dulbecco's modified Eagle medium (DMEM) high glucose, supplemented with penicillin–streptomycin (Invitrogen) and L‐glutamine (Invitrogen) plus 10% fetal bovine serum (Invitrogen) and incubated at 37°C in 5% CO2. For myoblast differentiation, cells were grown in differentiation medium (DM) containing DMEM (plus penicillin–streptomycin and L‐glutamine) with 2% horse serum (Invitrogen) at 37°C in 5% CO2. C26 adenocarcinoma cells (National Cancer Institute, Frederick, MD) were plated and maintained at 37°C and 5% CO2, in GM.
Conditioned medium for C26 cells was made using the methods we have published23 with modifications.7 C26 and C26Lif−/− cells were grown in GM at 37°C in 5% CO2. When the plates reached 90% of confluence, the GM was removed, and the cells were washed twice with sterile phosphate buffered saline (PBS) and three times with DMEM with no serum plus antibiotics and glutamine. DM was added to the cells, and after 24 h, the medium was collected and centrifuged in 50 mL Falcon tubes at 3645 g for 15 min at 4°C. Aliquots of the medium were stored at −80°C for up to a year. CM treatment of myotubes was 33% CM in DM. Treatment for control myotubes was DM. Recombinant mouse LIF (R&D Systems) was added to C26Lif−/− CM at the same concentration (1500 pg/mL) found in C26 CM and used to treat myotubes.
Measurement of C26 and C26Lif−/− cell viability and doubling time in culture
C26 and C26Lif−/− cells were plated in 12‐well tissue culture plates at a density of 2 × 104 cells per well in GM on day 0. Each day for 7 days, three individual wells were harvested, and the live cell count for each was determined using the EVE Automated Cell Counter (NanoEnTek, Waltham, MA). Trypan blue was used to distinguish live and dead cells at each time point. Counts were averaged for each day and plotted on a logarithmic scale. Doubling time (DT) for each cell line was calculated from the exponential phase of each growth curve (between days 2 and 4) using the formula:
where t = 48 h, log2 = 0.3, log = log base 10, C1 = first cell count within exponential growth phase (at day 2), C2 = second cell count within exponential growth phase 48 h after C1.
Measurement of myotube diameter in cell culture
For all variables measured, three independent wells were used to calculate mean values for control and treated myotubes. Cells were grown in a six‐well plate overnight before they were switched to DM. After 4 days of differentiation, myotubes were photographed under phase at 20× magnification at 0, 24, 48, and 72 h using a Nikon TS‐500 inverted fluorescent microscope. DM was refreshed each day. Six to 10 randomly selected fields per well (four wells per group) were photographed by a Spot RT camera and Spot Software (Diagnostic Instruments). At least 100 diameters were measured per group using MetaMorph Imaging software (Universal Imaging). The area of a box that was traced along a 100 μm length of each myotube was sampled to measure average myotube diameter.
Immunocytochemistry of myotubes
For immunocytochemistry, myotubes were fixed in 1.5% formaldehyde in HBSS for 30 min, washed in PBS, permeabilized in 1% Triton X‐100, washed in PBS‐Tween, and then blocked in 3% BSA in PBS‐Tween. Myotubes were incubated with mouse monoclonal anti‐myosin MF20 (Developmental Studies Hybridoma Bank, Iowa City, IA) antibody, washed, and then incubated with goat anti‐mouse fluorescein‐conjugated Alexa Fluor 488. Myotubes were visualized through an FITC‐HYQ filter, and images were taken as described previously.
C26 inoculation and characterization in mice
All animal studies were performed with approval from the University of Florida Institutional Animal Care and Use Committee. Prior to inoculation into mice, C26 wild‐type and knockout cells were trypsinized and washed twice with PBS and then re‐suspended in sterile 1X PBS. Tumours were produced by subcutaneous injection of both flanks with 5 × 105 cancer cells in 100 μL of PBS (106 cells total per mouse) into 8‐week‐old male CDF1 mice purchased from Charles River Laboratories (Wilmington, MA). Control groups consisted of equal volumes of 1X PBS injected into age‐matched mice. Mice were anaesthetized using inhaled isoflurane during the procedure and administered two doses of buprenorphine immediately and 12 h postoperatively. Animals were maintained on a 12:12 L/D cycle, were housed individually, and were given access to food and water ad libitum. Mouse weights and tumour diameters were measured and recorded throughout the duration of the study. Flank tumours were allowed to reach an endpoint of 2 cm in maximum diameter. At this time, mice were anaesthetized using inhaled isoflurane, a laparotomy performed, and a 27.5‐gauge needle inserted into the abdominal aorta and ~500 μL of blood withdrawn. Blood was expressed into a gold top BD Microtainer® blood collection tube, containing clot activator/serum separator tube gel, inverted five times and incubated at room temperature for 30–60 min. Samples were then centrifuged at 2500 g for 10 min at 4°C, and separated serum removed and stored at −80°C. One tibialis anterior (TA) and both gastrocnemius muscles were harvested, weighed, flash frozen, and stored at −80°C. The other TA muscle was harvested, weighed, embedded in OCT, and immediately frozen in isopentane cooled in liquid nitrogen prior to storage at −80°C. The heart, spleen, tumour, and epididymal fat were harvested and weighed. The experiment was repeated, so there was a total of 12 mice in each of the three groups. Not all measurements were made on all mice except for body mass, muscle mass, and tumour mass (see figure legends).
Haematoxylin and eosin staining
Tibialis anterior muscles were equilibrated at −20°C for 1 h prior to sectioning. A microtome cryostat was used to cut 10‐μm‐thick serial transverse sections, which were transferred to positively charged glass slides. The slides were then sequentially submerged in 100% ethanol for 1 min, 70% ethanol for 1 min, dH20 for 2 min, and Gill's Haematoxylin for 2 min. Sections were then washed thoroughly in dH20 followed by sequential submersions in the following solutions: Scott's Solution for 15 s, dH20 for 2 s, 70% ethanol for 1 min, Eosin for 2 min, 95% ethanol with gentle shaking for 1 min, 100% ethanol for 30 s, and Xylene for 3 min. Slides were allowed to dry for 30 min and then mounted with glass cover‐slips using Permount. All sections were visualized and images captured using a Leica DM5000B microscope (Leica Microsystems Bannockburn, IL) and the Leica Application Suite, version 3.5.0 software. This software was also used to trace and measure muscle fibre cross‐sectional area (CSA).
Measurement of cell culture and in vivo cytokine and chemokine levels
Soluble analytes in mouse serum and cell culture conditioned media were assayed using the Milliplex® Premixed 25‐Plex Mouse Immunology Multiplex Assay (Merck Millipore, Darmstadt, Germany) according to the manufacturer's protocol, as described before.17 Briefly, serum or CM was incubated in multiscreen filter microtiter plates (EMD Millipore, San Jose, CA) with beads coated with primary antibodies overnight at 4°C. After washing, PE‐conjugated anti‐cytokine antibodies were added and incubated for an additional 2 h at room temperature. Following washing, data were acquired on a Luminex 200® system running xPONENT® 3.1 software (Luminex, Austin, TX) and analysed using a 5‐paramater logistic spline‐curve fitting method using Milliplex® Analyst V5.1 software (Vigene Tech, Carlisle, MA). Cytokines were quantified using a standard curve and 5‐parameter logistics to determine pg/mL. The analytes tested were GCSF, GMCSF, IFNγ, IL1α, IL1β, IL2, IL4, IL5, IL6, IL7, IL‐9, IL10, IL12 (p40), IL12 (p70), IL‐13, IL15, IL17, IP‐10, KC, MCP1, MIP1α, MIP1β, MIP2, RANTES, and TNFα. IL‐9 and IL‐17 data did not pass internal quality control standards and are eliminated from our results.
In addition to the Milliplex assay, we used two independent ELISA assays for LIF (R&D Quantikine MLF00, Minneapolis) and IL‐6 (eBioscience, Ready‐Set‐Go mouse, San Diego) carried out according to the manufacturers' protocols.
Statistics
A one‐way ANOVA was used to test for overall group differences using GraphPad Prism 5.03 (GraphPad Software, La Jolla, CA). Tukey's post‐doc test was used to identify the statistical differences between control, C26 and C26Lif−/− groups (P < 0.05). In cases where there was unequal variance or unequal n among the three groups, a non‐parametric post hoc test (Dunn's) was used (P < 0.05). This is indicated in the figure legends.
Results
Bi‐allelic knockout of the mouse Lif gene in C26 tumour cells
One clone of C26 called 6a had a sequence deletion mutant on each allele: one allele had a 2 bp deletion and the other allele had a 4 bp deletion in exon 2 (Figure 1A). This produced a frame shift at the 47th and 46th amino acid, respectively, of the full‐length mouse LIF protein (the 24th and 23rd amino acid of the mature secreted form of LIF) (Figure 1B). Additionally, these frameshift mutants in exon 2 produced early terminations.
Figure 1.

Location of allelic deletion mutants targeted to mouse Lif exon2. (A) The LIF nucleotide (n.t.) sequence begins in exon 2 at nucleotide 103 of the full‐length DNA sequence, excluding introns. For the clone used (‘6a’), one allele is ‘6a mutant2’, missing n.t. 138–139. The other allele is ‘6a mutant4’, missing n.t. 137–140. (B) The deletion mutations caused frameshifts at amino acid 47 and 46 of the full‐length protein, respectively, and early terminations denoted by *. Amino acid sequence of wild type (mLIF), 6a mutant2, and 6a mutant 4 are shown for exon 1 and 2, and part of exon 3. The full‐length wild‐type mouse LIF peptide is 203 amino acids (a.a.).
C26 and C26Lif−/− cell viability and doubling time in culture
Visually, the C26Lif−/− tumour cells had the same morphology and growth characteristics as wild‐type C26 cells. However, counting the viable C26 and C26Lif−/− cells over a 7 day period the DT of viable C26 cells was 15 h during the log phase of growth compared with 20 h for C26Lif−/− cells (see Figure S1 ). Cell viability of C26 cells at days 3, 4, and 5 in culture was on average 76%, whereas C26Lif−/− cell viability was 61% during this same period of time (not shown). Cell viability during the lag (days 1 and 2) and stationary (days 6 and 7) phases of growth was the same among the two tumour types.
Cytokine/chemokine levels in medium conditioned by C26 and C26Lif−/− tumour cells
The levels of secreted LIF in CM from C26 and C26Lif−/− cells were 1450 pg/mL and undetectable, respectively, as measured by ELISA (Figure 2A). To determine if other secreted factors made by C26 cells were affected by knockout of the Lif gene, selected cytokine/chemokines were measured using multiplex analyte profiling. Medium from C26 and C26Lif−/− cells was assayed and compared with culture medium without cells. In addition to LIF, cytokines that were secreted at high levels (>1000 pg/mL) in C26 medium were IP‐10 (CXCL10), MCP‐1, and KC (CXCL1) (Figure 2B). Cytokines that were secreted in moderate amounts (100–200 pg/mL) in C26 medium were RANTES, MIP‐2, and GM‐CSF, and cytokines secreted at low levels (10–50 pg/mL) in C26 medium were MIP‐1α, MIP‐1β, and IL‐6 (Figure 2C). Each of these cytokines, except GM‐CSF and KC, were much lower or not detected in medium from C26Lif−/− cells. The remainder of the cytokines measured in CM had very low or not detectable levels (Table 1).
Figure 2.
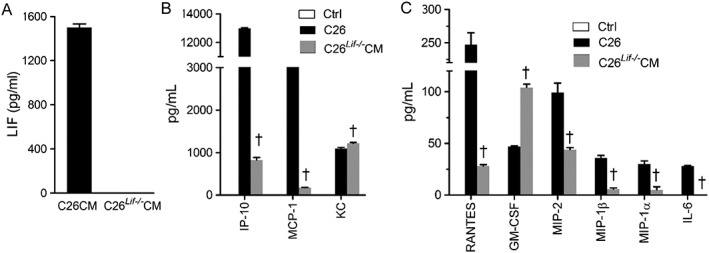
Cytokine concentrations in conditioned medium (CM) from C26 and C26Lif−/− cells. (A) LIF. (B) IP‐10, MCP‐1, and KC concentration. (C) RANTES, GM‐CSF, MIP‐2, MIP‐1β, MIP‐1α, and IL‐6 concentration. ctrl = control. All values plotted (black and grey bars) are significantly greater than control values measured in DM (not detected). The CM made was used for all experiments, and cytokine levels were measured in triplicate in all cases. †Significantly different from C26 value (P < 0.05).
Table 1.
Cytokine concentrations in differentiation medium (control) and in medium from C26 and C26Lif−/− cells
| Control | C26 | C26Lif−/− | |
|---|---|---|---|
| Mean (SEM) | Mean (SEM) | Mean (SEM) | |
| Cytokine | n = 3 | n = 3 | n = 3 |
| IP‐10 | n.d. | 12984 (61) | 824 (60)a |
| MCP‐1 | n.d. | 9370 (38) | 178 (2.4)a |
| KC | n.d. | 1092 (29) | 1216 (23)a |
| RANTES | n.d. | 247 (18) | 28 (1.4)a |
| GM‐CSF | n.d. | 47 (0.7) | 104 (3.3)a |
| MIP‐2 | n.d. | 99 (9.2) | 44 (1.9)a |
| MIP‐1α | n.d. | 30 (3) | 5 (3)a |
| MIP‐1β | n.d. | 36 (2.4) | 5.7 (1.3)a |
| IL‐6 | n.d. | 28 (0.6) | n.d.a |
| IL‐5 | n.d. | 16 (1.1) | n.d.a |
| G‐CSF | n.d. | 8.7 (0.3) | 3.4 (0.3)a |
| IL‐1α | n.d. | 9.3 (1) | n.d.a |
| IL‐1β | n.d. | 4.8 (0.8) | n.d.a |
| IL‐15 | n.d. | 7.6 (2.4) | n.d.a |
| IL‐10 | n.d. | n.d. | n.d. |
| IL‐4 | n.d. | n.d. | n.d. |
| IL‐7 | n.d. | n.d. | n.d. |
| IL‐13 | n.d. | n.d. | n.d. |
| IL‐2 | n.d. | n.d. | n.d. |
| IL‐12(p40) | n.d. | n.d. | n.d. |
| IL‐12(p70) | n.d. | n.d. | n.d. |
| IFNγ | n.d. | n.d. | n.d. |
| TNFα | n.d. | n.d. | n.d. |
All values are pg/mL. SEM, standard error of the mean; n.d., not detected.
Different from C26 CM value (P < 0.05).
The effect of CM from C26 vs. leukaemia inhibitory factor knockout (C26Lif−/−) tumour cells on C2C12 myotube diameter
We previously reported, using antibody neutralization, that LIF in the C26 CM is required for myotube atrophy.23 To further support these findings, using a genetic approach, we compared C26 and C26Lif−/− CM on myotube diameter. Following 24, 48, and 72 h of treatment with C26 CM, myotubes had a significantly smaller diameter. In contrast, myotubes did not atrophy, at any time point, in response to treatment with C26Lif−/− CM (Figure 3A–C). When LIF was added to C26Lif−/− CM at the same concentration as found in the C26 CM (1500 pg/mL), the atrophy‐inducing ability of the C26Lif−/− CM was restored (see Figure S2 ). These data confirm our previous study23 and provide further support that C26 tumour cell‐derived LIF is indispensable for myotube atrophy.
Figure 3.
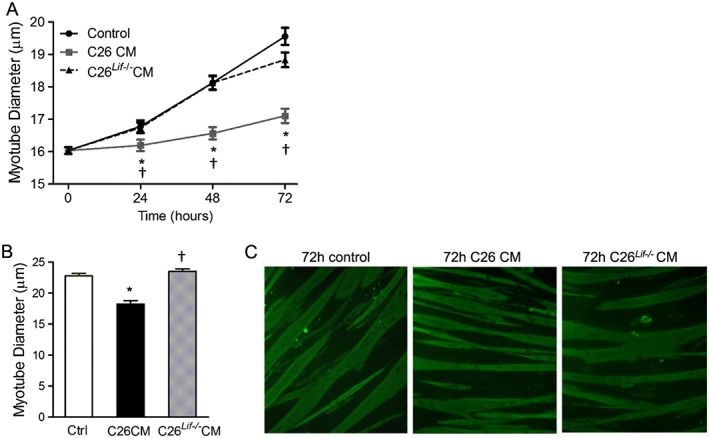
Diameter of control (ctrl), C26 CM, and C26Lif−/− CM‐treated myotubes. (A) Measurements made at 0 (day 4), and 24, 48, and 72 h of treatment with C26 CM or C26Lif−/− CM. At least 100 myotubes were measured per group. (B) Measurement of fluorescent myotubes after immunocytochemistry at 72 h treatment. (C) Representative images of fluorescent myotubes from 72 h control, C26 CM, and C26Lif−/− CM‐treated groups. *Significantly different from control value at same time point (P < 0.05). †Significantly different from C26 value (P < 0.05).
C26 and C26Lif−/− tumours in mice: effects on tumour size, body mass, morbidity, and spleen size
In order to determine the role of tumour‐derived LIF in vivo, we inoculated mice with C26 or C26Lif−/− cells and monitored tumour growth and cachexia progression. The tumours in mice inoculated with C26Lif−/− cells grew more slowly than mice with C26 cells, such that for a tumour endpoint diameter of 1.5–2 cm, C26 tumour‐bearing mice were sacrificed at 27 days post‐inoculation, while C26Lif−/− tumour‐bearing mice were sacrificed at 34 to 46 days post‐inoculation. The C26Lif−/− mice lost less body weight over the course of tumour growth compared with C26 mice (Figure 4A). Importantly, C26 mice were visibly sick at study endpoint as evidenced by hunched posture, reduced activity and grooming behaviour, and piloerection as commonly seen in these mice,29 but C26Lif−/− mice did not exhibit these characteristics (Figure 4B). Despite the tumours from C26 and C26Lif−/− mice showing the same mass at sacrifice (Figure 4C), C26 mice had a 35% lower tumour‐free body mass than control mice, while C26Lif−/− mice had an 18% lower tumour‐free body mass (Figure 4D). Therefore, the C26Lif−/− mice showed a significant 55% attenuation of cachexia compared with C26 mice. Consistent with the notion that mice bearing C26Lif−/− tumours did not show outward signs of severe sickness compared with those bearing C26 tumours, splenomegaly, as measured by spleen mass, was increased by 153% in the C26 tumour animals, whereas mice with C26Lif−/− tumours had a 50% increase in spleen mass (Figure 4E). This represents a significant 65% attenuation of splenomegaly in C26Lif−/− mice.
Figure 4.
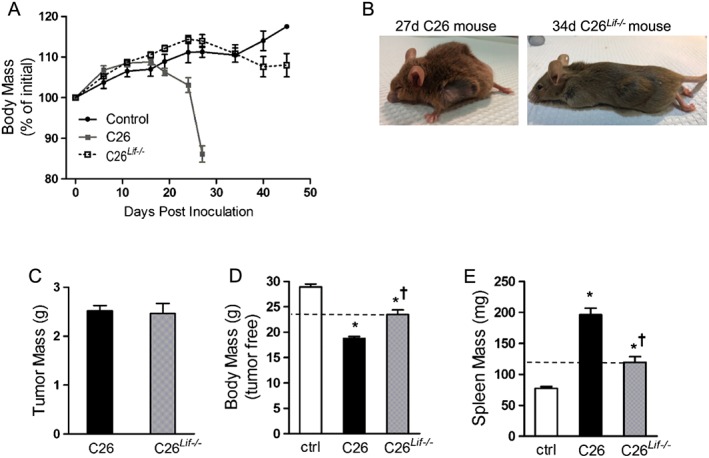
Growth characteristics of control (ctrl), C26, and C26Lif−/− tumour‐bearing mice (n = 12 per group). (A) Body mass during the weeks following tumour cell inoculation. (B) Photographs of tumour‐bearing mice immediately prior to sacrifice. Sacrifice was at 27 days post‐inoculation for C26 mice and 34 to 46 days post‐inoculation for C26Lif−/− mice. (C) Tumour weight at sacrifice. (D) Tumour‐free body mass at sacrifice. (E) Spleen mass at sacrifice. *Significantly different compared with control value (P < 0.05). †Significantly different from C26 value (P < 0.05). A Dunn's post hoc test was used for data in (D) and (E).
C26 and C26Lif−/− tumours in mice: effects on muscle mass, muscle fibre size, and fat mass
Gastrocnemius muscle mass from C26 mice was 31% lower than control mice while mass from C26Lif−/− mice was 12% lower (Figure 5A). This represents a significant 60% attenuation of muscle atrophy in the C26Lif−/− mice compared with C26 mice. Similar results were seen for TA muscle mass in tumour‐bearing mice (Figure 5B). C26 mice had 33% lower TA mass compared with control mice, while C26Lif−/− mice had a 14% lower TA mass, representing a significant 55% attenuation of atrophy. Measurement of TA muscle fibre CSA confirmed significant myofiber atrophy in C26 mice, with a 33% decrease in mean TA muscle fibre CSA (Figure 5C–E). In C26Lif−/− mice, TA fibre atrophy was significantly attenuated by 76% compared with C26 mice. The TA fibre size frequency distribution showed that in muscle of C26 mice, there were more small fibres and less fibre size heterogeneity than muscle from control or C26Lif−/− mice. Fibre areas from C26Lif−/− mice had a very similar pattern of fibre size distribution compared with control mice. Mean epididymal fat mass was 650 mg in control mice and decreased to less than 50 mg in C26 mice (Figure 5F). However, fat mass in C26 Lif−/− mice was not statistically different from controls, although they were more variable and had a mean value of 400 mg. Interestingly, heart mass was decreased by 27% and 21% in C26 and C26Lif−/− mice, respectively (Figure 5G), indicating that LIF did not significantly contribute to heart atrophy in C26 mice.
Figure 5.
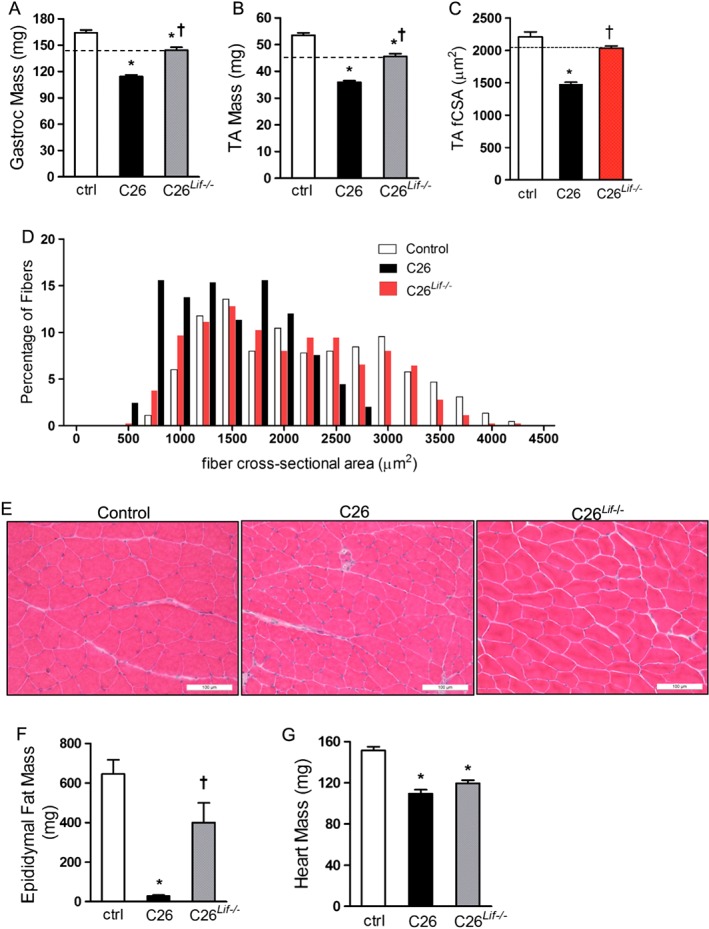
Muscle and fat mass in control (ctrl), C26, and C26Lif−/− tumour‐bearing mice. (A) Gastrocnemius muscle mass. (B) Tibialis anterior (TA) muscle mass. For (A) and (B), n = 24 per group. (C) TA fibre cross‐sectional areas from four muscles per group (300 fibres measured per muscle). (D) Fibre size frequency distribution of fibre areas from TA muscle. (E) Representative photomicrograph of haematoxylin and eosin stained sections of TA muscle from each group. (F) Epididymal fat mass. (G) Heart muscle mass. For G and H (control n = 6, C26 n = 6, C26Lif−/− n = 8). *Significantly different compared with control value (P < 0.05). †Significantly different from C26 value (P < 0.05). A Dunn's post hoc test was used for data in (F).
C26 and C26Lif−/− tumours in mice: effects on circulating cytokine and chemokine levels
Of the 24 proteins measured in the serum of tumour‐bearing mice, only IL‐12 (p40) and IL‐13 were not detected by the multiplex immunoassay. LIF was measured by ELISA. Cytokines/chemokines that were significantly elevated in C26 mice compared with controls were LIF, IL‐6, TNFα, G‐CSF, IL‐1β, IL‐10, IL‐2, and IP‐10 (Figure 6A–H). LIF levels were extremely low in C26Lif−/− mice indicating that neither the tumour nor the host made LIF (Figure 6A). Of the elevated proteins in C26 mice, IL‐6, TNFα, G‐CFS, IL‐1β, and IL‐10 may be dependent on the secretion of tumour‐derived LIF because these proteins were not significantly elevated in C26Lif−/− mice (Figure 6B–F). In contrast, the three‐fold increase of IP‐10 in C26 mice was further increased in C26Lif−/− mice (Figure 6H). In two cases, protein levels were unchanged in C26 mice but were significantly elevated in C26Lif−/− mice (Figure 6I, J). These proteins were MCP‐1, and most remarkably KC (Cxcl‐1). In two cases (RANTES and MIP‐1β), cytokines were lower in C26 serum but were not different in C26Lif−/− mice compared with controls (Table 2). Circulating levels of the remaining cytokines measured, IL‐7, IFNγ, IL‐4, IL‐1α, IL‐5, IL‐12(p70), IL‐15, MIP‐1α, MIP‐2, and GM‐CSF were not statistically different in tumour‐bearing mice compared with controls (Table 2).
Figure 6.
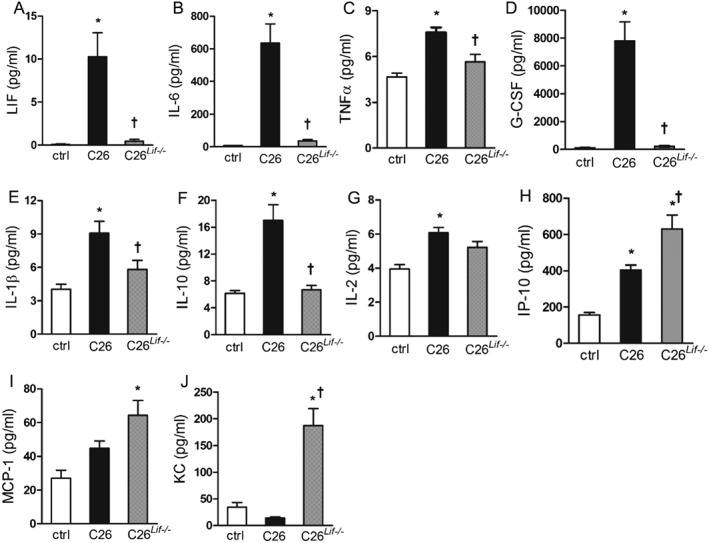
Cytokine concentration in serum of control (ctrl), C26, and C26Lif−/− tumour‐bearing mice at time of sacrifice. All values are pg/mL. (A) LIF, (B) IL‐6, (C) TNFα, (D) G‐CSF, (E) IL‐1β, (F) IL‐10, (G) IL‐2, (H) IP‐10, (I) MCP‐1, and (J) KC. *Significantly different compared with control value (P < 0.05). †Significantly different from C26 value (P < 0.05). For all cytokine measurements, the number of mice used were control (n = 6), C26 (n = 11), and C26Lif−/− (n = 7). A Dunn's post hoc test was used in all cases.
Table 2.
Serum cytokine concentration from mice at the study endpoint.
| Control | C26 | C26Lif−/− | |
|---|---|---|---|
| Mean (SEM) | Mean (SEM) | Mean (SEM) | |
| Cytokine | n = 6 | n = 11 | n = 7 |
| LIF | n.d. | 10.3 (2.8)a | 0.45 (0.23)b |
| IL‐6 | 8.1 (0.65) | 635 (117.9)a | 36.2 (8.7)b |
| TNFα | 4.7 (0.24) | 7.6 (0.33)a | 5.6 (0.51)b |
| G‐CSF | 114.0 (24.1) | 7793 (1363)a | 222 (56.1)b |
| IL‐1β | 4.0 (0.46) | 9.1 (1.1)a | 5.8 (0.80)b |
| IL‐10 | 6.2 (0.41) | 17.0 (2.3)a | 6.7 (0.65)b |
| IL‐2 | 3.9 (0.25) | 6.1 (0.30)a | 5.2 (0.35) |
| IP‐10 | 156.6 (12.6) | 404 (27.2)a | 631 (76.4)a , |
| MCP‐1 | 27.0 (4.8) | 44.7 (4.4) | 64.2 (8.8)a |
| KC | 34.3 (8.9) | 13.9 (2.1) | 187 (31.6)a , b |
| GM‐CSF | 9.9 (2.2) | 14.0 (1.8) | 7.3 (0.95)b |
| MIP‐1β | 14.9 (1.1) | 10.3 (0.75)a | 16.6 (1.6)b |
| RANTES | 7.7 (1.7) | 5.1 (0.18)a | 7.6 (0.56)b |
| IL‐1α | 312 (15.2) | 505 (95.7) | 503 (87.7) |
| IL‐5 | 11.7 (1.2) | 7.4 (1.1) | 11.1 (1.7) |
| IL‐4 | 3.9 (0.10) | 4.4 (0.44) | 3.9 (0.07) |
| IFNγ | 7.1 (0.92) | 5.8 (0.45) | 6.7 (0.64) |
| IL‐7 | 8.3 (1.2) | 7.7 (0.92) | 8.3 (1.7) |
| IL‐15 | 26.2 (6.5) | 18.2 (3.4) | 22.5 (4.9) |
| MIP‐1α | 22.2 (8.1) | 28.4 (2.7) | 23.9 (3.2) |
| MIP‐2 | 155.0 (18.4) | 145 (13.7) | 193 (24.4) |
| IL‐12(p70) | 5.7 (0.52) | 7.9 (1.5) | 5.6 (0.43) |
| IL‐12(p40) | n.d. | n.d. | n.d. |
| IL‐13 | n.d. | n.d. | n.d. |
All values are pg/mL.
Different from control value (P < 0.05).
Different from C26 CM value (P < 0.05); n.d., not detected.
Discussion
Cancer cachexia is initiated by tumour‐derived factors.10 There is general consensus that many of these factors are proteins.5, 8, 9, 11 We previously showed that LIF is not only sufficient to induce myotube atrophy, but it is entirely responsible for atrophy due to treatment with C26 tumour cell CM.23 We also found a direct parallel between blood LIF levels and cachexia in C26 tumour‐bearing mice. In the present study, we used a C26 Lif null tumour cell line to test whether tumour‐derived LIF is required for cachexia in vivo. Importantly, we found that LIF is responsible for the majority of cancer‐induced wasting and morbidity in the C26 mouse, and further, we identified cytokines in the circulation of these mice that are regulated by LIF. This is the first demonstration by CRISPR/Cas9 knockout of a tumour‐derived factor that is required for the majority of cachexia in vivo.
In cell culture, we found that in contrast to CM made from C26 tumour cells, CM made from C26Lif−/− tumour cells did not cause myotube atrophy. This confirmed our previous finding that a mLIF blocking antibody completely prevented myotube atrophy due to C26 CM treatment. In mice bearing C26Lif−/− tumours, cachexia, as measured by tumour‐free body mass and the mass of organs such as epididymal fat, spleen, and different muscle types, shows amelioration of wasting by more than half compared with mice with C26 tumours. Because wasting was not completely abolished in mice with C26Lif−/− tumours, this suggests that interactions of the tumour and host produce factors that contribute to wasting in vivo that are independent from tumour‐derived LIF.
In vivo, it was evident that the C26Lif−/− tumours did not grow as fast as C26 tumours. This is consistent with the slower DT of C26Lif−/− tumour cells (20 h) compared with C26 cells (15 h) in culture (Figure S1 ). Therefore, LIF may act either directly or indirectly, as an autocrine growth factor for the C26 tumour in vivo. In order to study cachexia when the two tumour types were the same size, mice with C26Lif−/− tumours were sacrificed between 1 and 2 weeks after the mice with C26 tumours, and this meant that C26Lif−/− mice had tumours for a longer period of time. Nevertheless, at the time of sacrifice, mice with C26Lif−/− tumours had a healthy appearance in contrast to severely cachectic C26 mice (Figure 3B) that consistently show hunched posture, piloerection, reduced activity, and grooming behaviour (e.g. other literatures29, 30, 31).
The healthier presentation of the C26Lif−/− mice at the time of sacrifice is also consistent with normalization of most cytokines that were elevated in the C26 mice, notably LIF, IL‐6, G‐CSF, IL‐10, TNFα, and IL‐1β. These data also suggest that there is a dependence on LIF for the elevation of these cytokines in C26 mice. Further, any atrophy in mice with Lif knockout tumours is not due to these cytokines because they were not elevated. The highest levels of these putative LIF‐dependent cytokines in C26 mice were IL‐6 and G‐CSF. LIF can elicit cytokine expression in multiple cell types. We showed that it can induce IL‐6 secretion in cultured myotubes.23 It is also known to induce IL‐6 secretion in chondrocytes and blood monocytes, and it induces IL‐6 in synoviocytes, neuronal, and epithelial cells.32 The effect of LIF on G‐CSF is not previously known. The combined effect of Lif knockout on the low levels of IL‐6 and G‐CSF, inducers of white blood cell activation, may provide a molecular explanation for the finding that mice with the C26Lif−/− tumours do not show outward signs of sickness compared with C26 mice. In addition, the normalization of G‐CSF levels in mice with the C26Lif−/− tumours may explain the significant amelioration of splenomegaly.
Our finding that IP‐10, MCP‐1, and KC (Cxcl‐1) levels were significantly increased in the blood of C26Lif−/− compared with C26 or control mice indicates two important points: first, that these proteins are regulated by other factors besides LIF in C26Lif−/− mice, and second, that these chemokines may contribute to the mild cachexia in C26Lif−/− mice compared with no‐tumour controls. Increased circulating MCP‐1 was recently found to associate with cachexia in pancreatic cancer patients.33 Interestingly, the increase in blood levels of MCP‐1, as well as IP‐10, in mice with C26Lif−/− tumours is in contrast to the 10‐fold reduction in these secreted factors in C26Lif−/− CM compared with C26 CM (Table 1) suggesting that the increased blood levels may derive from the host rather than the tumour. KC was unique in that it was unchanged in C26 mice but was five‐fold higher in C26Lif−/− mice compared with controls. It is possible that the additional time required to achieve the same size Lif knockout C26 tumour as the wild‐type C26 tumour involves longer exposure to the inflammatory action of MCP‐1, IP‐10, and KC in C26Lif−/− tumour mice, thereby causing some myopathy and mild cachexia. In this regard, we have shown that continuous exposure to low doses of cachectic factors can be more effective at muscle atrophy than larger doses applied at one time.7 However, it is also possible that the mild cachectic features of the C26Lif−/− mice are due to factors not measured.
In contrast to skeletal muscle and adipose tissue, the mouse myocardium appears to be unaffected by LIF levels because heart mass was reduced by ~25% in both C26 and C26Lif−/− mice. This LIF‐independent heart atrophy is consistent with the study by Schafer et al. who found that Ataxin‐10 in C26 tumour secretions is involved in cardiac myocyte atrophy.5
We began to investigate LIF as a causative agent in C26 cancer cachexia because our transcription factor reporter screening showed that only the Stat reporter was functionally activated in muscle cells treated with C26 CM, LIF was increased in C26 CM, and an LIF blocking antibody prevented myotube atrophy.23 Now, by abolishing tumour‐derived LIF in C26 tumour‐bearing mice, we provide direct support for the hypothesis that LIF is a major cachectic factor in the C26 mouse, and these results support that idea suggested in several early studies that LIF may be a major cachectic factor in nude mice bearing human LIF secreting tumour cells. Work published as early as 1989 suggested that LIF could be a cachectic factor because it was secreted from multiple human cancer cell lines, it was a strong inhibitor of lipoprotein lipase activity, it was sufficient to cause cachexia, and there was a strong correlation between LIF levels and whole body wasting in nude mice carrying LIF‐secreting human tumours.34, 35, 36, 37 These papers suggested that further studies were needed to confirm LIF as a causative cancer cachexia factor,36, 37 which we provide here, at least in the C26 tumour‐bearing mouse.
Not all preclinical models of cachexia involve LIF. The Lewis Lung Carcinoma (LLC) is another well‐studied mouse model of cancer cachexia, and several tumour‐secreted proteins, other than LIF, have been implicated in causing wasting from LLC.8, 11 Our own evaluation of CM from LLC shows that they secrete very low levels of LIF, not enough to contribute to wasting (see Figure S3 ). In addition, recent work suggests that active components from tumours that produce cachexia may arise via secretion in exosomal vesicles, but these factors have not been verified in vivo.38, 39 Whether the active contents are protein or inhibitory RNA, the proof of their tumour origin for the initiation of cachexia will require ablation of the genes responsible in the tumour itself.
In conclusion, by knocking out Lif in the C26 tumour, the present study demonstrates that tumour‐secreted LIF is required for the majority of cancer cachexia in vivo, and it implicates a role of this factor in regulating other cytokines, most notably IL‐6 and G‐CSF, in C26 tumour‐bearing mice. This approach of gene editing tumour cells will be of great value in leading the study of cachexia from other tumour models and in cancer patients.
Conflict of interest
Susan Kandarian, Rachel Nosacka, Andrea Delitto, Andrew Judge, Sarah Judge, John Ganey, Jesse Moreira, and Robert Jackman declare that they have no conflict of interest.
Supporting information
Figure S1. The number of viable C26 and C26Lif−/− cells counted daily for 7 days. The doubling time (DT) for C26 cells was 15 hours and for C26Lif−/− cells was 20 hours.
Figure S2. Diameter of differentiated myotubes treated with C26Lif−/− CM for 3 days or C26Lif−/− CM + mouse LIF (1500 pg/mL). Myotubes at time 0 were 4‐days differentiated. LIF added to C26Lif−/− CM rescued atrophy at 24, 48, and 72 hours. *significantly different compared to C26Lif−/− CM (P < 0.05).
Figure S3. Data for mouse LIF in Lewis Lung Carcinoma (LLC) cells. (A) Low levels of mouse LIF in differentiation medium (DM) and in LLC medium made in DM. For comparison, mLIF in C26 medium was 1500 pg/ml (Figure 2(A)). (B) Diameter of myotubes treated with DM (Control) or 33% LLC conditioned medium (CM) plus IgG or LLC CM plus mouse LIF antibody (mLIF Ab). No effect on diameter of myotubes treated with LLC plus mLIF antibody but complete prevention of atrophy when myotubes treated with C26 CM plus mLIF antibody23. *significantly different compared to control (P < 0.05).
Acknowledgements
This work was supported by NIAMS R01 AR060217 to S. C. K. and R. W. J. and NIAMS R01 R01AR060209 to A. R. J., and by the Dudley Allen Sargent Research Fund. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.40
Kandarian, S. C. , Nosacka, R. L. , Delitto, A. E. , Judge, A. R. , Judge, S. M. , Ganey, J. D. , Moreira, J. D. , and Jackman, R. W. (2018) Tumour‐derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour‐bearing mice. Journal of Cachexia, Sarcopenia and Muscle, 9: 1109–1120. 10.1002/jcsm.12346.
References
- 1. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 2. Johns N, Hatakeyama S, Stephens NA, Degen M, Degen S, Frieauff W, et al. Clinical classification of cancer cachexia: phenotypic correlates in human skeletal muscle. PLoS One 2014;9:e83618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol 2013;31:1539–1547. [DOI] [PubMed] [Google Scholar]
- 4. Delitto D, Judge SM, George TJ Jr, Sarosi GA, Thomas RM, Behrns KE, et al. A clinically applicable muscular index predicts long‐term survival in resectable pancreatic cancer. Surgery 2017;161:930–938. [DOI] [PubMed] [Google Scholar]
- 5. Schafer M, Oeing CU, Rohm M, Baysal‐Temel E, Lehmann LH, Bauer R, et al. Ataxin‐10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol Metab 2016;5:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 7. Jackman RW, Floro J, Yoshimine R, Zitin B, Eiampikul M, El‐Jack K, et al. Continuous release of tumor‐derived factors improves the modeling of cachexia in muscle cell culture. Front Physiol 2017;8:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petruzzelli M, Wagner EF. Mechanisms of metabolic dysfunction in cancer‐associated cachexia. Genes Dev 2016;30:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rubin H. Cancer cachexia: its correlations and causes. Proc Natl Acad Sci U S A 2003;100:5384–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 2017;8:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, et al. MAP 3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 2016;7:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bilir C, Engin H, Can M, Temi YB, Demirtas D. The prognostic role of inflammation and hormones in patients with metastatic cancer with cachexia. Med Oncol 2015;32:56. [DOI] [PubMed] [Google Scholar]
- 14. Loumaye A, Thissen JP. Biomarkers of cancer cachexia. Clin Biochem 2017;50:1281–1288. [DOI] [PubMed] [Google Scholar]
- 15. Dingemans AM, de Vos‐Geelen J, Langen R, Schols AM. Phase II drugs that are currently in development for the treatment of cachexia. Expert Opin Investig Drugs 2014;23:1655–1669. [DOI] [PubMed] [Google Scholar]
- 16. Onesti JK, Guttridge DC. Inflammation based regulation of cancer cachexia. Biomed Res Int 2014;2014:168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 2017;8:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tseng YC, Kulp SK, Lai IL, Hsu EC, He WA, Frankhouser DE, et al. Preclinical investigation of the novel histone deacetylase inhibitor AR‐42 in the treatment of cancer‐induced cachexia. J Natl Cancer Inst 2015;107:djv274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukawa T, Yan‐Jiang BC, Min‐Wen JC, Jun‐Hao ET, Huang D, Qian CN, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 2016;22:666–671. [DOI] [PubMed] [Google Scholar]
- 20. Ben‐David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient‐derived xenografts undergo mouse‐specific tumor evolution. Nat Genet 2017;49:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med 2010;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Penna F, Busquets S, Argiles JM. Experimental cancer cachexia: evolving strategies for getting closer to the human scenario. Semin Cell Dev Biol 2016;54:20–27. [DOI] [PubMed] [Google Scholar]
- 23. Seto DN, Kandarian SC, Jackman RW. A key role for leukemia inhibitory factor in C26 cancer cachexia. J Biol Chem 2015;290:19976–19986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 2011;6:e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Graf U, Casanova EA, Cinelli P. The role of the leukemia inhibitory factor (LIF)—pathway in derivation and maintenance of murine pluripotent stem cells. Genes (Basel) 2011;2:280–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alter J, Rozentzweig D, Bengal E. Inhibition of myoblast differentiation by tumor necrosis factor alpha is mediated by c‐Jun N‐terminal kinase 1 and leukemia inhibitory factor. J Biol Chem 2008;283:23224–23234. [DOI] [PubMed] [Google Scholar]
- 28. Jo C, Kim H, Jo I, Choi I, Jung SC, Kim J, et al. Leukemia inhibitory factor blocks early differentiation of skeletal muscle cells by activating ERK. Biochim Biophys Acta 2005;1743:187–197. [DOI] [PubMed] [Google Scholar]
- 29. Aulino P, Berardi E, Cardillo VM, Rizzuto E, Perniconi B, Ramina C, et al. Molecular, cellular and physiological characterization of the cancer cachexia‐inducing C26 colon carcinoma in mouse. BMC Cancer 2010;10:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cornwell EW, Mirbod A, Wu CL, Kandarian SC, Jackman RW. C26 cancer‐induced muscle wasting is IKKbeta‐dependent and NF‐kappaB‐independent. PLoS One 2014;9:e87776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Judge SM, Wu CL, Beharry AW, Roberts BM, Ferreira LF, Kandarian SC, et al. Genome‐wide identification of FoxO‐dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014;14:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villiger PM, Geng Y, Lotz M. Induction of cytokine expression by leukemia inhibitory factor. J Clin Invest 1993;91:1575–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Talbert EE, Lewis HL, Farren MR, Ramsey ML, Chakedis JM, Rajasekera P, et al. Circulating monocyte chemoattractant protein‐1 (MCP‐1) is associated with cachexia in treatment‐naive pancreatic cancer patients. J Cachexia Sarcopenia Muscle 2018;9:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gascan H, Anegon I, Praloran V, Naulet J, Godard A, Soulillou JP, et al. Constitutive production of human interleukin for DA cells/leukemia inhibitory factor by human tumor cell lines derived from various tissues. J Immunol 1990;144:2592–2598. [PubMed] [Google Scholar]
- 35. Mori M, Yamaguchi K, Abe K. Purification of a lipoprotein lipase‐inhibiting protein produced by a melanoma cell line associated with cancer cachexia. Biochem Biophys Res Commun 1989;160:1085–1092. [DOI] [PubMed] [Google Scholar]
- 36. Mori M, Yamaguchi K, Honda S, Nagasaki K, Ueda M, Abe O, et al. Cancer cachexia syndrome developed in nude mice bearing melanoma cells producing leukemia‐inhibitory factor. Cancer Res 1991;51:6656–6659. [PubMed] [Google Scholar]
- 37. Iseki H, Kajimura N, Ohue C, Tanaka R, Akiyama Y, Yamaguchi K. Cytokine production in five tumor cell lines with activity to induce cancer cachexia syndrome in nude mice. Jpn J Cancer Res 1995;86:562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci U S A 2014;111:4525–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS, et al. Pathogenesis of pancreatic cancer exosome‐induced lipolysis in adipose tissue. Gut 2016;65:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The number of viable C26 and C26Lif−/− cells counted daily for 7 days. The doubling time (DT) for C26 cells was 15 hours and for C26Lif−/− cells was 20 hours.
Figure S2. Diameter of differentiated myotubes treated with C26Lif−/− CM for 3 days or C26Lif−/− CM + mouse LIF (1500 pg/mL). Myotubes at time 0 were 4‐days differentiated. LIF added to C26Lif−/− CM rescued atrophy at 24, 48, and 72 hours. *significantly different compared to C26Lif−/− CM (P < 0.05).
Figure S3. Data for mouse LIF in Lewis Lung Carcinoma (LLC) cells. (A) Low levels of mouse LIF in differentiation medium (DM) and in LLC medium made in DM. For comparison, mLIF in C26 medium was 1500 pg/ml (Figure 2(A)). (B) Diameter of myotubes treated with DM (Control) or 33% LLC conditioned medium (CM) plus IgG or LLC CM plus mouse LIF antibody (mLIF Ab). No effect on diameter of myotubes treated with LLC plus mLIF antibody but complete prevention of atrophy when myotubes treated with C26 CM plus mLIF antibody23. *significantly different compared to control (P < 0.05).