

Abstract
Background
This study was performed to test the therapeutic potential of obestatin, an autocrine anabolic factor regulating skeletal muscle repair, to ameliorate the Duchenne muscular dystrophy (DMD) phenotype.
Methods and results
Using a multidisciplinary approach, we characterized the ageing‐related preproghrelin/GPR39 expression patterns in tibialis anterior (TA) muscles of 4‐, 8‐, and 18‐week‐old mdx mice (n = 3/group) and established the effects of obestatin administration at this level in 8‐week‐old mdx mice (n = 5/group). The findings were extended to in vitro effects on human immortalized DMD myotubes. An analysis of TAs revealed an age‐related loss of preproghrelin expression, as precursor of obestatin, in mdx mice. Administration of obestatin resulted in a significant increase in tetanic specific force (33.0% ± 1.5%, P < 0.05), compared with control mdx mice. Obestatin‐treated TAs were characterized by reduction of fibres with centrally located nuclei (10.0% ± 1.2%, P < 0.05) together with an increase in the number of type I fibres (25.2% ± 1.7%, P < 0.05) associated to histone deacetylases/myocyte enhancer factor‐2 and peroxisome proliferator‐activated receptor‐gamma coactivator 1α axis, and down‐regulation of ubiquitin E3‐ligases by inactivation of FoxO1/4, indexes of muscle atrophy. Obestatin reduced the level of contractile damage and tissue fibrosis. These observations correlated with decline in serum creatine kinase (58.8 ± 15.2, P < 0.05). Obestatin led to stabilization of the sarcolemma by up‐regulation of utrophin, α‐syntrophin, β‐dystroglycan, and α7β1‐integrin proteins. These pathways were also operative in human DMD myotubes.
Conclusions
These results highlight the potential of obestatin as a peptide therapeutic for preserving muscle integrity in DMD, thus allowing a better efficiency of gene or cell therapy in a combined therapeutic approach.
Keywords: Duchenne muscular dystrophy, Skeletal muscle cell atrophy, Pharmacological modifier, Obestatin signalling, Skeletal muscle
Introduction
Duchenne muscular dystrophy (DMD) is a degenerative disorder caused by mutations in the X‐linked dystrophin gene, affecting 1 in 3500 newborn males. DMD patients suffer from progressive muscle wasting with clinical symptoms first detected between 2 and 5 years of age. In skeletal muscle, the dystrophin‐glycoprotein complex is localized within the membrane along muscle fibres and at myotendinous and neuromuscular junctions (NMJs). This complex includes dystroglycans, sarcoglycans, and syntrophins that link the muscle cell cytoskeleton to the extracellular matrix and maintains sarcolemma integrity.1 Dystrophin deficiency results in the disruption of the dystrophin glycoprotein complex‐extracellular matrix linkage that leads to the loss of sarcolemma integrity and tearing of the sarcolemma. This causes an influx of Ca2+ into muscle cells, activation of Ca2+‐dependent proteases, and muscle degeneration during contraction.2 Muscle degeneration is accompanied by regeneration through the activation of muscle satellite cells; however, regenerative capacity in DMD patients becomes progressively impaired, associated in humans at least partly to an exhaustion of satellite cell proliferative potential. The muscle itself cannot counteract this structural damage, ultimately leading to loss of muscle fibres, infiltration of adipose tissue, and increased fibrosis, all resulting in loss of muscle mass and function.3 As the disease progresses, patients lose ambulation in their early teens, along with scoliosis, contractures, respiratory, and cardiac impairment, and their life expectancy is reduced to 30–40 years.4, 5, 6
Different potential therapeutic approaches are presently undergoing clinical evaluation.5, 7, 8 However, current strategies for the treatment of dystrophic skeletal muscle, whether these are gene or cell‐based, face the bottleneck that targeted fibres often degenerate before they are fully rescued, thus causing the loss of the therapeutic agent.9, 10, 11 A potential approach would be the use of autocrine/paracrine signals involved in muscle regeneration to enhance endogenous satellite cell function and temporarily protect the fibres from degenerating while gene or cell therapy is applied. In this sense, obestatin, a 23‐amino acid peptide derived from a polypeptide called preproghrelin, exerts an autocrine function in skeletal muscle to control the myogenic programme through the G protein‐coupled receptor GPR39.12 In addition, obestatin injection or its overexpression in damaged muscle significantly improves muscle regeneration in healthy mice.13 Mechanistically, in injured muscles from healthy mice, obestatin enhances muscle regeneration by regulating multiple steps of myogenesis: myoblast proliferation, cell cycle exit, differentiation, and recruitment‐to fuse and form multinucleated hypertrophic myotubes. This action is co‐ordinated by the interplay between G protein‐dependent and β‐arrestin‐dependent mechanisms.14 The obestatin‐associated mitogenic action is determined by G protein‐dependent activation defining the intricate pathways related to the ERK1/2 and JunD axis, and the transactivation of epidermal growth factor receptor through the β‐arrestin signal complex determines the cell cycle exit and the development and progression of differentiation through a kinase hierarchy determined by the Akt, Ca2+/calmodulin‐dependent protein kinase II (CaMKII), c‐Jun, and p38 axes. In addition to its well‐established role in the control of muscle regeneration, obestatin participates in the specification of muscle fibre identity by inducing skeletal muscle remodelling toward an oxidative phenotype more resistant to damage and enhanced in muscle strength.15 Obestatin acts through both class II histone deacetylases (HDAC)/myocyte enhancer factor‐2 (Mef2) and peroxisome proliferator‐activated receptor‐gamma coactivator 1α (PGC1α) mechanisms, thereby controlling the establishment of oxidative muscle fibres. The obestatin/GPR39 system is able to counteract deregulations in proteostasis, e.g. those associated to glucocorticoid‐induced myotube atrophy, and to restore efficient basal homoeostasis.16 In cell transplantation therapy, e.g. myoblast‐based therapy by xenotransplanting primary human myoblasts into immunodeficient mice, obestatin not only enhances the efficiency of engraftment but also facilitates an even distribution of myoblasts within the host muscle, probably by enhancing migration.17
In this study, we assessed the therapeutic potential of the factor obestatin for focal treatment of dystrophic DMD muscles using in the dystrophin deficient mdx mouse. We found that, in addition to the established role in the control of muscle regeneration, obestatin‐treated mdx mice show skeletal muscle remodelling toward an oxidative phenotype, with improved muscle strength and reduced skeletal muscle pathology. We also show that after obestatin treatment, there is an increased expression of two critical sarcolemma complexes in dystrophin‐deficient skeletal muscle that have been shown to modify disease progression: the utrophin glycoprotein complex and α7β1 integrin. These pathways were also operative in a human context, because some features of the DMD phenotype were reversed by obestatin in human immortalized DMD myoblasts/myotubes. Together, our findings indicate that obestatin is a promising candidate to optimize current treatments for DMD.
Material and methods
Materials
Rat/mouse obestatin and human obestatin was obtained from BCN Peptides (Barcelona, Spain). Antibodies used are listed in Table S1. All other chemical reagents were from Sigma Chemical Co. (St. Louis, MO, USA).
Animals
This study used 4‐, 8‐, and 18‐week‐old male C57BL/10ScSn‐Dmdmdx/J mice (mdx mice) obtained from The Jackson Laboratory (Bar Harbor, ME, USA). For protein expression assays, we used 4‐, 8‐, and 18‐week‐old male mdx mice. To examine the effects of exogenous obestatin in dystrophic muscles, 8‐week‐old male mdx mice were used. Experiments were performed in accordance with the University of Santiago de Compostela guidelines for animal handling and animal care as determined by the University of Santiago de Compostela Animal Care Committee according to the guidelines of the Spanish Royal Decree 53/2013, Directive 2010/63/EU, and FELASA Guidelines.
Obestatin dosing
The experimental protocol used in the present work was previously described.15 Animals, 8‐week‐old male mdx mice, were assigned to one of the following experimental groups (n = 5 per group): (i) control group {[vehicle administration: phosphate‐buffered saline (PBS)]; n = 5} and (ii) obestatin‐treated group (n = 5). Obestatin treatment was performed via injection of 20 μL of obestatin solution in PBS (pH 6.3) (500 nmol/kg body weight) into the target muscles every 72 h during 30 days. Control group was treated with 20 μL of PBS under the same conditions. Injections were performed inserting the needle of a 0.3 mL/29 gauge syringe (Terumo Myjector V‐100 insulin syringe) at 1 mm in the distal part of the muscle, in a region that is not taken into account for histological analysis, as previously described.15 After 30 days, mice were anaesthetized by intraperitoneal injection of 2.2 mg ketamine/0.4 mg xylazine/0.22 mg acepromazine per 10 g of animal body weight.13 Muscle force was measured, and blood samples were collected from tail bleeds in all animals. Animals were sacrificed by cervical dislocation under anaesthesia, and tibialis anterior (TA) muscles from different groups were excised, weighed, and processed for subsequent analyses.
Measurements of muscle force in vivo
Muscle force was measured in a living animal as previously described.15 Briefly, TA muscle contractile performance was measured in vivo in anaesthetized mice using 1305A Whole Animal System (Aurora Scientific, Inc., ON, Canada). TA contractions were evoked by electrical stimulation of the common peroneal nerve using one electrode placed on the popliteal fossa. The force‐frequency curves were determined by stepwise increasing stimulation frequency, pausing 120 s between stimuli to avoid effects due to fatigue. Developed force was expressed as specific force.
Cell culture and differentiation of human immortalized Duchenne muscular dystrophy myoblasts
Muscle biopsy was obtained from the MyoBank‐AFM bank of tissues for research, a partner in the EU network EuroBioBank, in accordance with European recommendations and French legislation. Deltoid muscle biopsy was obtained from a 6‐year‐old male patient suffering from DMD (duplication of exon 2). Primary cells were isolated from the deltoid muscle as described previously.18, 19 Myoblasts are cultivated in growth medium containing Medium 199 : Dulbecco's modified Eagle's medium (1:4, v:v; Lonza, Pontevedra, Spain) supplemented with 20% foetal bovine serum (v/v), 25 μg/μL fetuin, 5 ng/mL hEGF, 0.5 ng/mL bFGF, 0.2 μg/mL dexamethasone (Sigma‐Aldrich, MO, USA), and 50 μg/mL gentamycin (Invitrogen, Thermo Fisher Scientific, MA, USA). Immortalization and selection processes were performed as previously described.20 Briefly, TERT and Cdk4 cDNAs were cloned into different lentiviral vectors containing puromycin and neomycin selection markers, respectively. Myoblasts were transduced with both TERT and Cdk4 lentiviral vectors, with a MOI of 3–5 in the presence of 4 μg/mL of polybrene (Sigma‐Aldrich). Transduced cell cultures were selected with puromycin (0.2 μg/mL) and/or neomycin (0.3 mg/mL) for 8 days. Transduced cells were seeded at clonal density, and selected individual clones were isolated from each population. To confirm that immortalized cell line maintained their myogenic signature, the expression of several myogenic markers (desmin, neural cell adhesion molecule, and MyoD) was assessed in proliferating primary and immortalized cell line. Immortalized cells retain the characteristics of the unmodified parental population.21 In addition, the ability to differentiate into myotubes, using immunostaining with MF20 antibody, which recognizes all skeletal‐muscle myosin heavy chains (MHC), was investigated. Differentiation into myotubes was initiated at 90% confluence by switching to differentiation medium [DM; Dulbecco's modified Eagle's medium supplemented 50 μg/mL gentamycin (Invitrogen)] for 3 days.
Histology and immunofluorescence analysis
Muscle samples were mounted in tissue freezing medium (tragacanth paste) and snap frozen in nitrogen‐cooled isopentane. The sections, 10 μm thick, were mounted on Histobond Adhesion Microslides (Marienfeld, Lauda‐Königshofen, Germany). For the haematoxylin and eosin (HE), succinate dehydrogenase (SDH), and Masson's trichrome stains, serial cryostat sections were stained following standard protocols. Muscle sections for immunofluorescence analysis were permeabilized and blocked with PBS with Triton X‐100 and Tween 20 (PBT) (1% Triton X‐100, 1% Tween‐20, 5% heat inactivated goat serum, 0.2% bovine serum albumin in PBS) for 30 min and incubated with primary antibodies diluted in PBT overnight at 4°C, washed with PBS, and then incubated with a mixture of appropriate secondary antibodies for 1 h at room temperature. Primary and secondary antibodies used are listed in Table S1. 4′,6‐Diamidino‐2‐phenylindole was used to counterstain the cell nuclei (Life Technologies, Thermo Fisher Scientific, MA, USA). Compiled images were used to reconstruct a view of the entire TA muscle. This compilation was used for quantification of centrally located nuclei (CLN), number of myofibres, cross‐sectional area (CSA), and fibre type analysis using ImageJ64 analysis software. Formalin‐fixed kidneys and livers were paraffin‐embedded, and 4 μm longitudinal and sagittal sections were cut and stained with HE following a standard protocol. For analysis of cultured cells, the DMD cells were cultured on coverslips and differentiated into myotubes. The intact cells were fixed with ethanol, washed, permeabilized, and blocked with PBT, and then stained with primary antibody diluted in PBT overnight at 4°C. The cells were then washed and incubated with secondary antibody for 1 h at room temperature. 4′,6‐Diamidino‐2‐phenylindole was used to counterstain the cell nuclei (Life Technologies). Five fields from three independent experiments were randomly selected for each treatment. The differentiation grade was evaluated based on the number of MHC positive cells above total nuclei. The number of nuclei within individual myotubes (≥3 nuclei) was counted for 20–50 myotubes. The digital images of the cell cultures were acquired with a Leica TCS‐SP8 spectral confocal microscope (Leica Microsystems, Heidelberg, Germany).
Immunoblot analysis
Protein was isolated from cells or tissue using ice‐cold radioimmunoprecipitation assay buffer [50 mmol/L Tris–HCl (pH 7.2), 150 mmol/L NaCl, 1 mmol/L EDTA, 1% (v/v) NP‐40, 0.25% (w/v) Na‐deoxycholate, protease inhibitor cocktail (Sigma Chemical), phosphatase inhibitor cocktail (Sigma Chemical)]. The lysates were clarified by centrifugation (14 000× g for 15 min at 4°C), and the protein concentration was quantified using the QuantiProTM BCA assay kit (Sigma Chemical). For immunoblotting, equal amounts of protein were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Primary and secondary antibodies used are listed in Table S1. Immunoreactive bands were detected by enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Thermo Fisher Scientific, Rockford, IL, USA).
RNA extraction, cDNA synthesis, and quantitative real‐time PCR
Total RNA was extracted from TA muscle using TRIzol reagent according to the manufacturer's instructions (MRC, Cincinnati, OH, USA). After DNase I treatment (Thermo Fisher Scientific, Waltham, MA, USA), 0.5 μg of total RNA was reverse transcribed using High‐Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). Real‐time PCR was carried out in Applied Biosystems StepOnePlusTM Real‐Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) using Luminaris HiGreen qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The 2−ΔΔCt method was used to analyse the relative changes in each gene's expression normalized against the housekeeping 40S ribosomal protein S20 rRNA expression.22 Sequences of the primers used in this study were as follows: AchRα, Fw 5′‐CCACAGACTCAGGGGAGAAG‐3′ and Rv 5′‐AACGGTGGTGTGTGTTGATG‐3′; AchRε, Fw 5′‐CAATGCCAATCCAGACACTG‐3′ and Rv 5′‐CCCTGCTTCTCCTGACACTC‐3′; AchRδ, Fw 5′‐CATCGAGTGGATCATCATTGAC‐3′ and Rv 5′‐CGGCGGATGATAAGGTAGAA‐3′; muscle‐specific receptor tyrosine kinase (MuSK), Fw 5′‐TGAGAACTGCCCCTTGGAACT‐3′and Rv 5′‐GGGTCTATCAGCAGGCAGCTT‐3′; ErbB1, Fw 5′‐GACCTTCACATCCTGCCAGT‐3 and Rv 5′GCATGGAGGTCAGTCCAGTT‐3′; and ribosomal protein S20, Fw 5′‐CCATGGCATTTAAAGATACCG‐3′ and Rv 5′‐AACCTTCTCCAGCGACTTCAC‐3′.
Blood measurements
Blood samples were collected from tail bleeds under general anaesthesia at the end of the treatment. The blood was collected in heparinized tubes, serum was isolated, and creatine kinase (CK), alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, bilirubin, and urea levels were performed by the Central Laboratory at Hospital Clínico Universitario de Santiago de Compostela (Santiago de Compostela, Spain).
Statistical analysis
All values are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism (version 5.0b; GraphPad Software, San Diego, CA, USA). Statistical significance was assessed by one‐way analysis of variance with the Bonferroni post‐test or Student's unpaired t‐test, with P < 0.05 being considered significant (*,#).
Results
Obestatin treatment improves muscle strength in mdx mice
As a first step, we investigated the protein expression of preproghrelin, as precursor of obestatin, and GPR39 in the TA muscles of mdx mice at different ages (4‐, 8‐, and 18‐week‐old mdx mice) by immunoblot analysis. As shown in Figure 1A, we found an inverse relationship between muscle preproghrelin and GPR39 protein expression with ageing. The age‐related changes prompted us to examine the effects of exogenous obestatin in 8‐week‐old mdx mice. Obestatin treatment was performed via intramuscular injection into the TA muscles every 72 h during 30 days [500 nmol/kg body weight (n = 5); referred as obestatin group; Figure 1B] with sampling at 12 weeks the age, as previously described.15 Results from this group were compared with control vehicle‐treated mdx mice (PBS; n = 5; now referred as control group) under the same conditions. As functional strength is one the measurable outcomes frequently used for DMD patients, we investigated force production in the TA muscles. No significant differences in twitch force were found between the obestatin‐treated and control groups (20.0 ± 1.5 mN/mm2 and 20.5 ± 1.7 mN/mm2, respectively; Figure 1C). In contrast, mean tetanic specific force was higher in obestatin‐treated TA regarding control TA muscles (51.8 ± 0.4 mN/mm2 and 39.0 ± 3.2 mN/mm2, respectively; Figure 1D), representing a 33.0% ± 1.5% increase in relation to control muscles. The force‐frequency curves for TA muscle displayed improved force generation beginning at 140 Hz and continuing through 200 Hz relative to PBS (Figure 1E).
Figure 1.
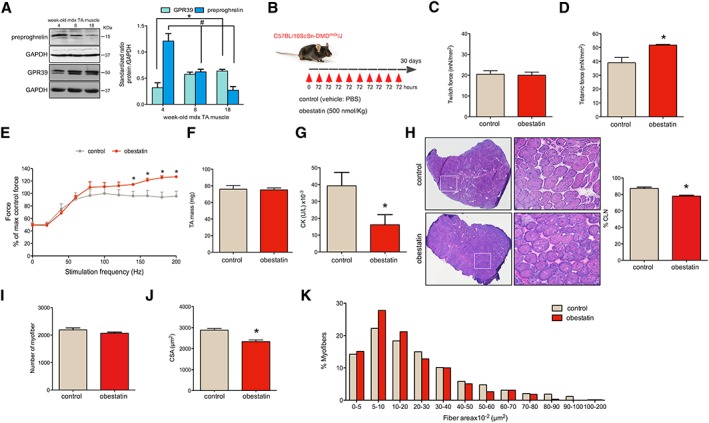
Obestatin treatment improves Duchenne muscular dystrophy (DMD) phenotype in mdx mice. (A) Protein levels of preproghrelin, precursor of obestatin, and GPR39 in the tibialis anterior (TA) muscles of mdx mice of different ages (4‐, 8‐, and 18‐week‐old mdx mice; n = 3 per age group). Protein levels were analysed by immunoblot, and data were expressed as arbitrary units (mean ± SEM; *,# P < 0.05 vs. control values) obtained from intensity scans. (B) A schematic diagram of intramuscular administration of obestatin (500 nmol/kg body weight each 72 h; n = 5) or vehicle (control; n = 5) in TA from mdx mice. (C) Effect of intramuscular injection of obestatin or vehicle on twitch force at 30 days. (D) Effect of intramuscular injection of obestatin or vehicle on tetanic force at 30 days. (E) Force‐frequency curve of TA muscles in obestatin‐treated or vehicle‐treated groups. (F) TA muscle weight after 30 days of treatment with vehicle or obestatin. (G) Serum creatine kinase (CK) levels after 30 days of treatment with vehicle or obestatin. (H) Left panel, representative haematoxylin and eosin staining from vehicle‐treated and obestatin‐treated muscles TA. Right panel, quantitation of myofibres with CNL from TA muscles. Data are shown as % of myofibres with centrally located nuclei (CLN). (I) Quantification of number of myofibres in TA muscles after 30 days of treatment with vehicle or obestatin. (J) Cross‐sectional area of muscle fibres from TA muscles after intramuscular injection of obestatin or vehicle at 30 days (mean ± SEM; n = 5 animals per group; * P < 0.05 vs. control values). (K) Distribution of fibre diameter from vehicle‐treated and obestatin‐treated mice. Data are expressed as % of myofibres. From (C) to (K), data are represented as mean ± SEM and * P < 0.05 vs. control values. CSA, cross‐sectional area; PBS, phosphate‐buffered saline.
The wet‐weights of TA muscles were measured. Interestingly, we did not observe significant differences in the average muscle mass of the TA treated with obestatin relative to control (Figure 1F). Furthermore, we analysed the serum levels of CK, a skeletal muscle enzyme released during fibre degeneration (Figure 1G). We observed a significant decrease in the average CK levels of mdx mice treated with obestatin relative to control mice (16 180.0 ± 5968.0 U/L and 39 290.0 ± 7979 U/L, respectively), suggesting a decrease in the level and severity of muscle damage. The TA muscles from obestatin‐treated or control mdx mice were stained with HE and analysed for the presence of CLN (Figure 1H), myofibre number (Figure 1I), and CSA (Figure 1J and 1K). A 10% ± 1.2% decrease in CLN percentage was observed in the obestatin‐treated mdx TA muscle compared with control (Figure 1H). We found that the average number of myofibres in the obestatin‐treated mdx mice (2067.0 ± 40.5) was only slightly lower compared with control animals (2191.0 ± 68.0) (Figure 1I), but this difference was not statistically significant. CSA was 23.5% ± 3.2% larger in control mice compared with obestatin‐treated mice (Figure 1J): mice treated with obestatin presented an increase in the percentage of small myofibres between 5 and 20 × 102 μm2 (Figure 1K). Together, these data show that mdx mice treated with obestatin present an improvement in functional outcome measures (muscle strength) and a small increase in the number of small diameter fibres.
Obestatin regulates oxidative fibre expression
On histological examination of TA (fast/glycolytic muscle) from obestatin‐treated animals, we observed a marked increase in the relative density of SDH positive fibres (SDH+, oxidative fibres), compared with control mdx mice, with a corresponding decrease in SDH negative fibres (SDH−, Figure 2A). Quantitation of the total fibre number revealed an increase of 23.0% ± 1.9% in SDH+ fibres, oxidative fibre type, in obestatin‐treated group relative to control group. On the other hand, the number of SDH− fibres was decreased by 23.0% ± 6.5% in obestatin‐treated TA muscles relative to control muscles. This oxidative fibre‐type switch was associated with significant increase in the expression of proteins involved in the mitochondrial biogenesis pathway, such as Cytochrome C (40.3% ± 7.0%), mitochondrial uncoupling protein 3 (136.5% ± 7.1%), mitochondrial transcription factor A (81.1% ± 17.0%), PGC1α (58.5% ± 14.0%), and carnitine palmitoyltransferase‐1 (50.36% ± 13.1%), in obestatin‐treated mdx mice as compared with control animals (Figure 2B and 2C).
Figure 2.
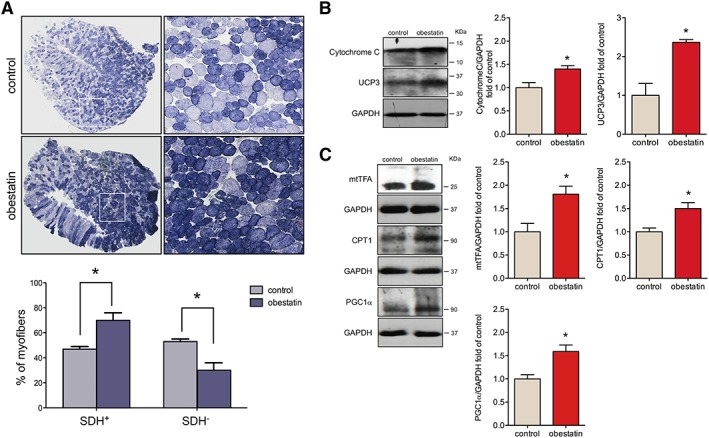
Obestatin treatment increases oxidative fibre density. (A) Upper panel, representative succinate dehydrogenase (SDH) staining from tibialis anterior (TA) from control muscles and obestatin‐treated muscles at 30 days. Bottom panel, quantitation of SDH+ and SDH− muscle fibres from TA muscles. Data were expressed as mean ± SEM (* P < 0.05 vs. control values). (B) Immunoblot analysis of Cytochrome C and uncoupling protein 3 (UCP3) in TA muscles after intramuscular injection of obestatin or control (phosphate‐buffered saline) at 30 days. (C) Immunoblot analysis of mitochondrial transcription factor A (mtTFA), carnitine palmitoyltransferase‐1 (CPT1), and proliferator‐activated receptor‐gamma coactivator 1α (PGC1α) in TA muscles after intramuscular injection of obestatin or control (phosphate‐buffered saline) at 30 days. In (B) and (C), protein level was expressed as fold of control TA muscles, and immunoblots are representative of the mean value. Data were expressed as mean ± SEM obtained from intensity scans (* P < 0.05 vs. control values).
The slow type I and the fast type IIa, IIb, and IIx constitute the four basic fibre types, classically identified based on their MHC isoform content.23, 24 Muscle fibre‐type assessment by immunofluorescence (Figure 3A) revealed that obestatin increased by 25.2% ± 1.7% the number of type I fibres relative to controls (Figure 3A). These TA muscles had a reduction in type IIx fibres compared with control, while no significant differences were observed in type IIa and IIb fibres (Figure 3A). The CSA of type I fibres was significantly increased in obestatin‐treated TA muscles compared with control mice (four‐fold increase; Figure 3A). Interestingly, the type IIb fibres had a significantly larger CSA in obestatin‐treated mice than control mice, whereas no significant differences were observed in type IIa and IIx (Figure 3A).
Figure 3.
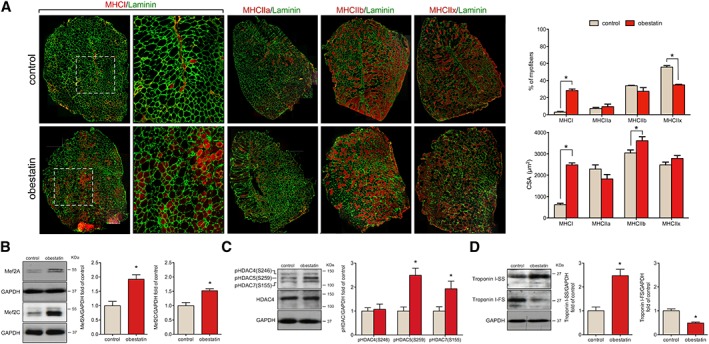
Obestatin stimulates fast to slow twitch fibre type shifting in mdx mice. (A) Left panel, representative images of control‐treated and obestatin‐treated tibialis anterior (TA) muscles showing myosin heavy chains (MHC) expression. Mice muscle serial cross section incubated with a primary antibody against MHCI, MHCIIa, MHCIIb, or MHCIIx, followed by incubation with appropriate fluorescent‐conjugated secondary antibody. Right panel, quantitation of fibre types (upper panel) and cross‐sectional area (CSA) (bottom panel). Data are shown as mean ± SEM of five animals per group (* P < 0.05 vs. control values). (B) Immunoblot analysis of Mef2A and Mef2C in TA muscles after intramuscular injection of obestatin or vehicle (phosphate‐buffered saline, control) at 30 days. (C) Immunoblot analysis of pHDAC4(S246), pHDAC5(S259), pHDAC7(S155), and HDAC4 in TA muscles after intramuscular injection of obestatin or control (phosphate‐buffered saline) at 30 days. (D) Expression of the slow‐fibre‐specific troponin I‐SS and fast‐fibre‐specific troponin I‐FS in TA muscles after intramuscular injection of obestatin or vehicle at 30 days. In (B) to (D), protein level was expressed as fold of control TA muscles, and immunoblots are representative of the mean value. Data were expressed as mean ± SEM (n = 5 per group) obtained from intensity scans (* P < 0.05 vs. control values). HDAC, histone deacetylases; Mef2, myocyte enhancer factor‐2.
Slow and oxidative myofibre identity is regulated by the balance between positive and negative signalling by Mef2 and the class II HDAC proteins, respectively.25 Indeed, western blot analysis of obestatin‐treated TA muscles demonstrated the up‐regulation of Mef2A and Mef2C protein expression (92.0% ± 16.0% and 52.0% ± 7.0%, respectively; Figure 3B). However, the activity of Mef2 is tightly regulated through association with HDACs, which act as signal‐dependent repressors of gene expression. HDAC phosphorylation in response to differentiation signals or motor innervation provides docking sites for the 14‐3‐3 chaperone protein that leads to nuclear export of HDACs enabling Mef2 to activate the slow myofibre gene programme.26, 27, 28 Consistent with this, obestatin treatment resulted in markedly elevated levels of HDAC5 [pHDAC5(S259); 149.2% ± 29.5%] and HDAC7 [pHDAC7(S155); 93% ± 32.0%] phosphorylation, but no significant differences were observed in HDAC4 phosphorylation (Figure 3C). Consistent with the role of Mef2 as a transcriptional regulator of the slow‐fibre phenotype, the expression of slow troponin I (troponin I‐SS) was much higher in obestatin‐treated muscles (147.0% ± 27%) than in control muscles, whereas fast troponin I (troponin I‐FS) expression was decreased (51.0% ± 4.0%; Figure 3D). Therefore, the phosphorylation of HDAC5/7 potentially regulated by obestatin signalling provides a mechanism for the modulation of Mef2 target genes to promote slow and oxidative myofibres.
Obestatin modulates molecular markers of protein synthesis and degradation
In terms of protein degradation, obestatin treatment led to a 43.6% ± 7.6% and 42.8% ± 14.0% decrease in the expression of the ubiquitin E3‐ligases MuRF1 and MAFbx, respectively (Figure 4A). One mechanism by which obestatin reduces the expression of the ubiquitin E3 ligases is the phosphorylation and subsequent nuclear exclusion of FoxO family members.16 As shown in Figure 4B, obestatin‐treated mice showed a striking increase in FoxO1 phosphorylation at T24 [pFoxO1 (T24); 130.4% ± 18.0%] and FoxO4 phosphorylation at T28 [pFoxO4 (T28); 378.6% ± 59.5%] related to control mice but did not change basal FoxO3a phosphorylation at T32 [pFoxO3 (T32)]. This suggests that post‐translational modifications of FoxO4 and FoxO1 could play a role in directing FoxO activity in response to obestatin and, thus, to control the E3 ubiquitin ligases MuRF1 and MAFbx.
Figure 4.
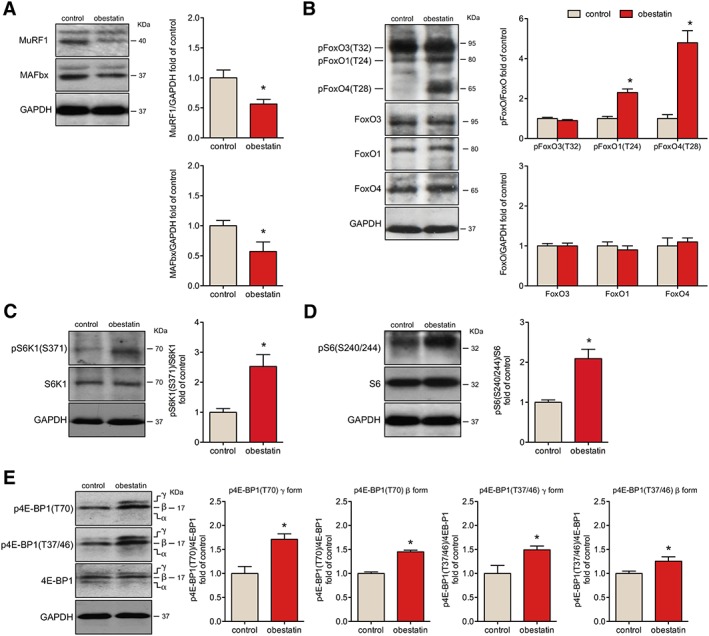
Obestatin treatment protects from atrophy with induction of protein synthesis signalling in mdx mice. (A) Immunoblot analysis of MuRF1 and MAFbx in tibialis anterior (TA) muscles after intramuscular injection of obestatin or control. (B) Immunoblot analysis of the phosphorylation partner of FoxO3, FoxO1, and FoxO4 in TA muscles after intramuscular injection of obestatin or vehicle (phosphate‐buffered saline, control) at 30 days. (C) Analysis of pS6K1(S6371) and S6K1 in TA muscles after intramuscular injection of obestatin or vehicle at 30 days. (D) Immunoblot analysis of pS6(S240/244) and S6 in TA muscles after intramuscular injection of obestatin or vehicle. (E) Analysis of p4E‐BP1(T70), p4E‐BP1(T37/46), and 4E‐BP1 in TA muscles after intramuscular injection of obestatin or vehicle (phosphate‐buffered saline, control) by immunoblot. In (A) to (E), protein level was expressed as fold of control TA muscles. Immunoblots are representative of the mean value (n = 5 per group). Data were expressed as mean ± SEM obtained from intensity scans (* P < 0.05 vs. control values).
The down‐regulation of MuRF1 and MAFbx expression was concomitant with the activation of positive regulators of protein synthesis. Obestatin increased S6K1 phosphorylation at S371 [pS6K1(S371)] and its downstream target, the ribosomal protein S6 phosphorylation at S240/244 [pS6(S240/244)] by 152.9% ± 39.0% and 109.0% ± 23.4%, respectively, compared with untreated mice (Figure 4C and 4D). Furthermore, obestatin markedly promoted 4E‐BP1 hyperphosphorylation at T70 [p4E‐BP1(T70)], especially concerning the γ form (71.2% ± 11.4%), altering the basal phosphorylation of β and γ forms at T37/T46 [p4E‐BP1(T37/46); 49.4% ± 7.8% and 25.5% ± 9.2%, respectively] (Figure 4E). Consequently, obestatin signalling regulates critical components of the anabolic machinery for protein synthesis, S6K1 and 4E‐BP1,29 in dystrophic conditions.
Obestatin‐treated mdx mice have increased sarcolemma adhesion protein components
The improvements in mdx mice under obestatin treatment could be associated with restoration or increased abundance of sarcolemma adhesion protein components. Analysis by immunoblot of some of these proteins showed that obestatin treatment increased the expression of utrophin by 74.0% ± 20.3% compared with untreated mice (Figure 5A). This up‐regulation was concomitant with increased expression of α‐syntrophin and β‐dystroglycan compared with untreated mice (38.8% ± 6.9% and 96.1% ± 26.0%, respectively; Figure 5A) but did not change basal neuronal nitric oxide synthase (NOS1) expression. In addition, obestatin treatment led to increased levels of α7‐integrin and β1D‐integrin (35.2% ± 4.8% and 37.9% ± 5.8% compared with control mice, respectively; Figure 5A). Confocal imaging analysis of histological sections from obestatin‐treated TAs showed a noticeable increase in the expression of utrophin in the extrasynaptic sarcolemma (Figure 5B). In contrast, utrophin was expressed at low levels on the extrasynaptic sarcolemma, being mainly restricted to NMJ in untreated muscles (Figure 5B). Importantly, we found an increase in α7‐integrin and β1D‐integrin proteins at the sarcolemma in obestatin‐treated mice when compared with untreated (Figure 5C and 5D, respectively). These data, together with the significant decrease in CK, suggest that obestatin treatment protects the fibres by reducing the degradation of the sarcolemma adhesion components in skeletal muscle of mdx mice, thereby increasing membrane stability.
Figure 5.
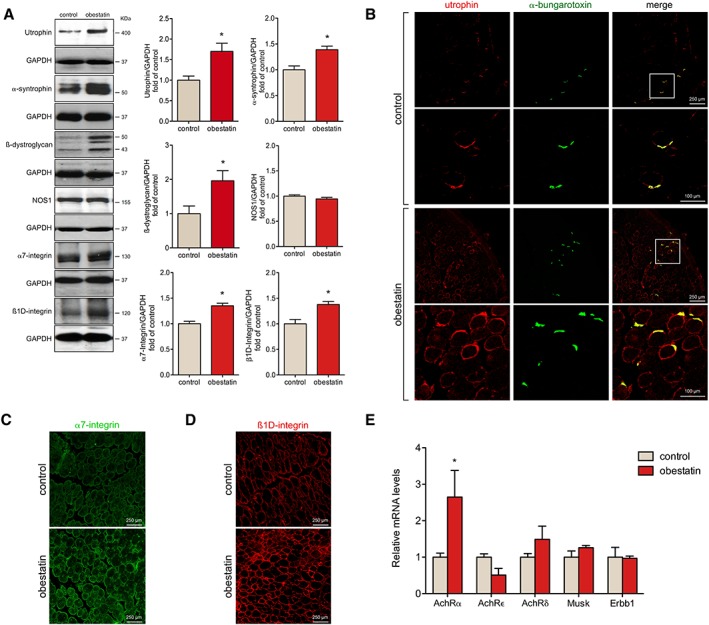
Obestatin regulates the complex of proteins that acts as link between the muscle fibres and the extracellular matrix. (A) Expression analysis of utrophin, α‐syntrophin, ß‐dystroglycan, nitric oxide synthase (NOS1), α7‐integrin, and ß1D‐integrin in tibialis anterior (TA) muscles after intramuscular injection of obestatin or control at 30 days. Protein level was expressed as fold of control. Immunoblots are representative of the mean value. Data were expressed as mean ± SEM obtained from intensity scans (* P < 0.05 vs. control values). (B) Representative immunofluorescence images of obestatin‐treated or control TA muscles [phosphate‐buffered saline (PBS)] at 30 days showing utrophin and neuromuscular junctions (α‐bungarotoxin) expression. Mouse muscle serial cross sections were incubated with primary antibodies against utrophin and α‐bungarotoxin, followed by incubation with appropriate fluorescent‐conjugated secondary. (C) Representative immunofluorescence images from obestatin‐treated or control TA muscles showing α7‐integrin expression at 30 days. (D) Representative immunofluorescence images of obestatin‐treated or control (PBS) TA muscles showing ß1D‐integrin expression at 30 days. (E) Relative mRNA levels of acetylcholine receptor (AchR) subunits α, δ, ε, and Musk, Erbb1 in obestatin‐treated (500 nmol/kg/72 h) or control TA muscles (PBS, control) at 30 days (n = 5 per group). Data were expressed as mean ± SEM (* P < 0.05 vs. control values).
Obestatin enhances acetylcholine receptor α gene
We further tested whether obestatin regulates the NMJ gene expression, based on the regulatory role of PGC1α in the NMJ program.30 To test this hypothesis, we measured the expression levels of acetylcholine receptor (AchR) α subunit (AchRα), ε subunit (AchRε), δ subunit (AchRδ), muscle‐specific receptor tyrosine kinase (MuSK), and receptor tyrosine‐protein kinase erbb‐1 (ErbB1) by quantitative real‐time PCR. We detected increased levels of AchRα (165.0% ± 73%) but not AchRε, AchRδ, MuSK, or ErbB1 in obestatin‐treated mice relative to controls (Figure 5E).
Obestatin alleviates the phenotype of muscular dystrophy
We determined the rate of interstitial fibrotic regions by Masson's trichrome staining. While fibrosis typically only becomes prominent in older mdx mice, even the mild degree of tissue fibrosis developed at 12 weeks of age was reduced in TAs of obestatin‐treated mice (Figure 6A). Consistent with this, the expression of fibrosis‐associated molecules, connective tissue growth factor (CTGF), α‐smooth muscle actin (α‐SMA), and collagen type I (Coll I) proteins was decreased by 45.0% ± 5.0%, 33% ± 6.0%, and 26.5% ± 8.4% in obestatin‐treated muscles, compared with control mice, respectively (Figure 6B). To further assess the extent of damaged myofibres, we quantified the number of IgG‐positive damaged fibres by immunofluorescence31 and found a 16‐fold decrease in the number of damaged fibres in obestatin‐treated muscles (Figure 6C).
Figure 6.
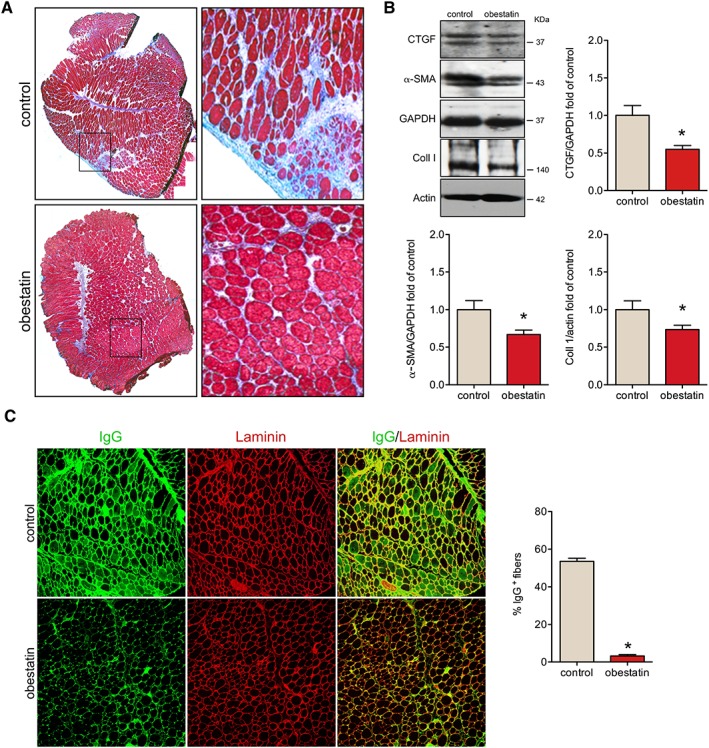
Obestatin regulates muscle tissue fibrosis and necrosis in mdx mice. (A) Representative images of the histological analysis from sections stained with Masson's trichrome staining are shown from obestatin‐treated or control tibialis anterior (TA) muscles (control) at 30 days. (B) Expression analysis of connective tissue growth factor (CTGF), α‐smooth muscle actin (α‐SMA), and collagen type I (Coll I) in TA muscles after intramuscular injection of obestatin or control. Protein level was expressed as fold of control TA muscles, and immunoblots are representative of the mean value. Data were expressed as mean ± SEM (n = 5 per group) obtained from intensity scans (* P < 0.05 vs. control values). (C) Left panel, representative images of IgG staining and laminin of obestatin‐treated TA muscles or control. Right panel, quantification of IgG+ fibres in obestatin‐treated or control (phosphate‐buffered saline) TA muscles at 30 days (n = 5 per group). Data were expressed as mean ± SEM (* P < 0.05 vs. control values).
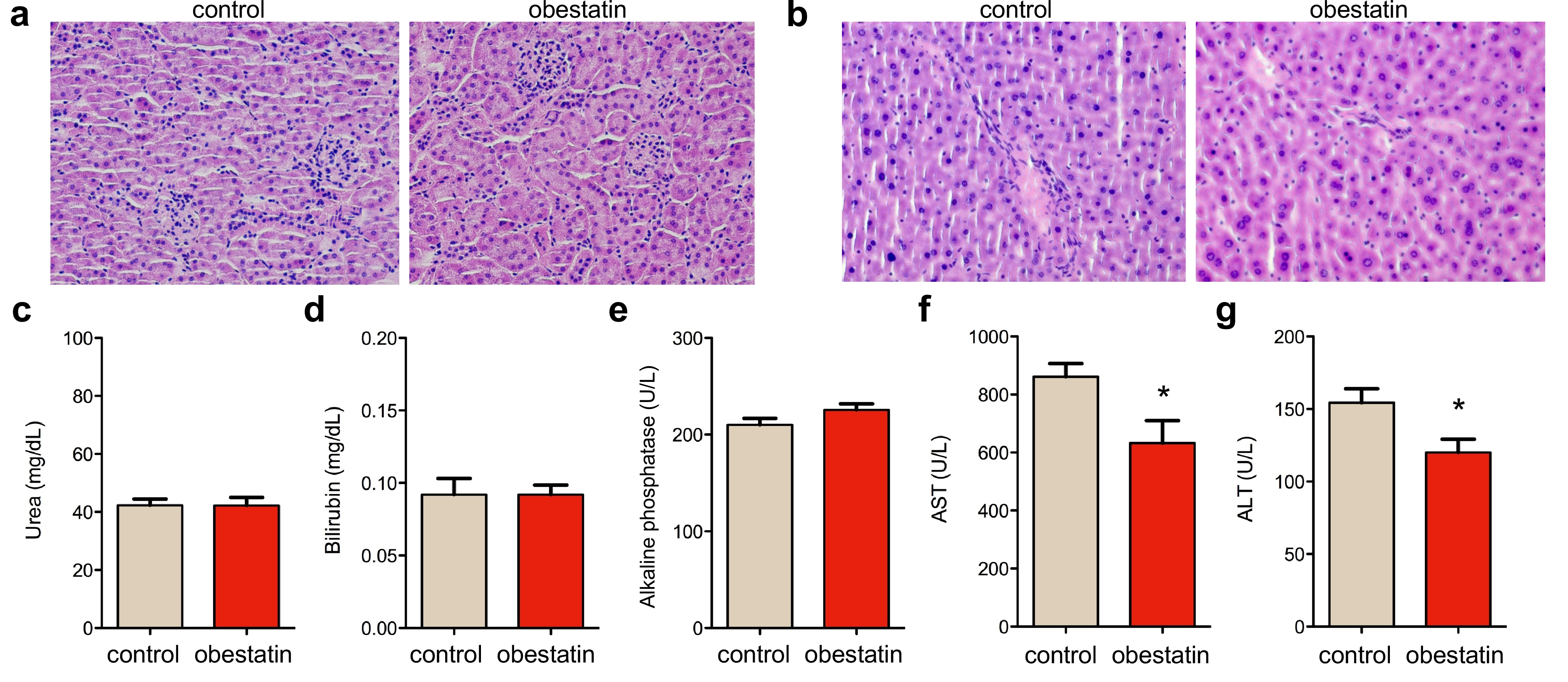
In order to assess potential toxicity of obestatin treatment, plasma was collected from mice at the end of treatment, and organs were harvested for toxicological analysis. Compared with untreated mdx mice, histological HE staining of kidney and liver tissue sections of obestatin‐treated mice showed no detectable effect on cells in those tissues nor any increase in the amount of infiltrating immune cells, suggesting the absence of any toxic effect (Figure S1a and S1b, respectively). Serum levels of urea, bilirubin, and alkaline phosphatase showed no significant difference compared with mdx untreated mice (Figure S1c to S1e, respectively). Interestingly, AST and ALT liver enzymes were significantly reduced in obestatin‐treated mice compared with untreated mdx controls (Figure S1f and S1g, respectively). Overall, these results indicate that during the time course of the experiment, obestatin did not induce any overt hepatic or renal toxicity in mdx mice.
Obestatin regulates sarcolemma adhesion protein components in human immortalized Duchenne muscular dystrophy myoblasts
To validate whether obestatin signalling and effect are conserved between human and mouse, we used an in vitro cell culture model of human DMD skeletal muscle cells, which is the only model available to assess signalling in a human context (for details, see Material and methods section). To assess the effect of human obestatin on differentiation, DMD myoblasts were switched to DM supplemented with obestatin at a range of concentrations (0.01–100 nM) for 3 days. As shown in Figure 7A, the protein levels of myogenin, as detected by immunoblot, were up‐regulated at a maximal level in the presence of 10 nM obestatin. Insulin (1.72 μM) was used as the positive control based on its role in the control of skeletal muscle growth and in the regulation of muscle mass.14 To investigate whether obestatin stimulated hypertrophic growth, the DMD cells were treated with obestatin in DM for 3 days (Figure 7B). The myotube area was larger in the obestatin‐treated cells (DM + obestatin; 992.32 ± 40.31 μm2) as compared with untreated conditions (DM; 687.42 ± 31.02 μm2) or insulin‐treated cells (DM + insulin; 879.76 ± 44.40 μm2) at 72 h after differentiation (Figure 7B). Furthermore, the number of myonuclei per MHC‐positive myotube (MHC+) was 20 ± 1.79 in obestatin‐treated cells vs. 5 ± 0.19 in control cells or 8 ± 0.67 in insulin‐treated cells (Figure 7C). Immunoblot analysis of total‐MHC, slow‐MHC, and fast‐MHC demonstrated a robust increase in the protein levels of total‐MHC and slow‐MHC in the presence of 10 nM obestatin (75.0% ± 2.2% and 52% ± 6.0% compared with untreated cells; Figure 7D) with a significant decrease in the levels of fast‐MHC (Figure 7D). Immunoblot analyses of utrophin, α‐syntrophin, and ß‐dystroglycan revealed elevated protein expression in obestatin‐treated cells as compared with untreated cells at 72 h of differentiation (36.7% ± 3.0%, 42.1% ± 3.0%, and 26.9% ± 6.5%, respectively; Figure 7E). The protein levels of NOS1, α7‐integrin, and ß1D‐integrin were also up‐regulated at 72 h after differentiation (16.7% ± 2.0%, 30.5% ± 3.0%, and 32.7% ± 2.0%; Figure 7E). Thus, human DMD myotubes underwent a functional switch towards an oxidative fibre type orchestrated by obestatin signalling during in vitro myogenesis. In addition, the up‐regulation of the expression of sarcolemma adhesion components is also confirmed in this system, thereby increasing membrane stability.
Figure 7.
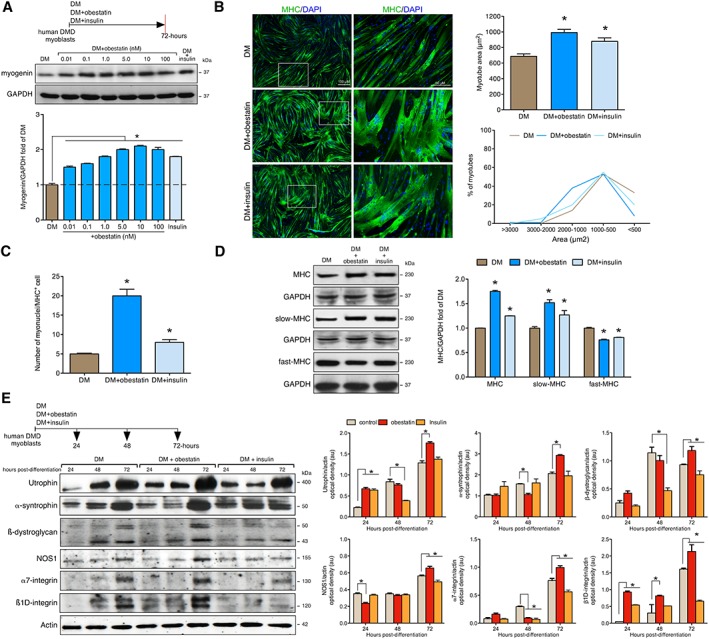
Obestatin signalling regulates differentiation of human Duchenne muscular dystrophy (DMD) cells. (A) Dose–response effect of obestatin (0.01–100 nM) or insulin (1.72 μM) on differentiating human DMD cells. Levels of myogenin were represented as a fold of respective expression in differentiation medium (DM) (72 h post‐differentiation). (B) Left panel, immunofluorescence detection of myosin heavy chains (MHC) and 4′,6‐diamidino‐2‐phenylindole (DAPI) in human DMD myotube cells under DM (control), DM + obestatin (10 nM), or DMD + insulin (1.72 μM) at the 72 h point after stimulation. Right panel, the differentiation grade was evaluated based on the myotube area and distribution. Data were expressed as mean ± SEM (* P < 0.05 vs. control values). (C) Quantification of the number of myonuclei in MHC+ cells in human DMD myotube cells under DM (control), DM + obestatin (10 nM), or DMD + insulin (1.72 μM) at the 72 h point after stimulation. Data were expressed as mean ± SEM (* P < 0.05 vs. control values). (D) Immunoblot analysis of MHC, slow‐MHC and fast‐MHC, in human DMD myotubes at the 72 h point under DM (control), DM + obestatin (10 nM), or DMD + insulin (1.72 μM). (E) Effect of obestatin (10 nM) on utrophin, α‐syntrophin, ß‐dystroglycan, nitric oxide synthase (NOS1), α7‐integrin, and ß1D‐integrin expression on differentiating human DMD cells. In (A), (D), and (E), protein level was expressed as optical density arbitrary units obtained from intensity scans. Immunoblots are representative of the mean value. Data were expressed as mean ± SEM (* P < 0.05 vs. control values).
Discussion
In this report, we have shown that obestatin ameliorates the DMD phenotype. Obestatin treatment results in a fast‐to‐slow fibre type switch that translates into an increase in oxidative fibres. Obestatin acts through both HDAC/Mef2 and PGC1α mechanisms, thereby controlling the establishment of oxidative muscle fibres. Moreover, obestatin signalling regulates muscle atrophy through regulation of ubiquitin E3‐ligases expression, MAFbx, and MuRF1, through inactivation of FoxO4 and FoxO1. Notably, obestatin leads to a stabilization of the sarcolemma of mdx skeletal muscle through the expression of utrophin, α‐syntrophin, β‐dystroglycan, and α7β1‐integrin proteins. This observation correlates with the decrease in the levels of serum CK, AST, and ALT, signs that indicate a partial rescue of muscle tissue necrosis. Furthermore, treatment with obestatin reduces muscle fibrosis. All these effects provoke a substantial improvement in skeletal muscle physiological function, in terms of specific force production. These actions acquire more relevance considering the age‐related loss of preproghrelin expression, and thus obestatin, in dystrophin deficient mdx mice.
Obestatin attenuates histopathological and biochemical features of muscular dystrophy in mdx mice. Rates of myofibre central nucleation, tissue fibrosis, and tissue necrosis (evaluated as IgG+ myofibres) were reduced in mdx mice treated with obestatin. In addition, CK, ALT, and AST levels were significantly reduced compared with untreated mice, which also strongly suggest a decrease in muscle fibre damage. This improvement correlates with improved muscle function because obestatin‐treated mice show a significant increase in muscle strength, as determined by generation of specific force. Because obestatin controls the fibre type determination,15 some of this gain‐of‐function may be linked to a conversion of fast‐twitch to slow‐twitch fibres, which are more resistant to degeneration. The slow and oxidative myofibre identity is governed by the balance between positive and negative signalling by Mef2 and class II HDACs, respectively.25 Obestatin increases class II HDAC phosphorylation, thus leading to nuclear export of HDACs and allowing sustained activation of Mef2, which promotes the establishment of slow and oxidative myofibres, i.e. MHC type I and troponin I‐SS. The increase in oxidative fibre density triggered by obestatin signalling is accompanied by significant changes in known regulators of mitochondrial function, such as Cytochrome C, uncoupling protein 3, and carnitine palmitoyltransferase‐1. This improved mitochondrial capacity is concomitant to an increased expression of PGC1α, a master regulator of mitochondrial gene expression involved in activation of mitochondrial biogenesis and oxidative metabolism.32 Mechanistically, PGC1α regulates fibre type switching through the coactivation of Mef2.33 Furthermore, Mef2 regulates PGC1α expression through an autoregulatory loop.28 Therefore, Mef2 operates as a nodal point for the control of multiple downstream transcriptional regulators of the slow‐fibre phenotype and PGC1α expression. Our data show that obestatin treatment triggers an up‐regulation of AchRα, a component of the NMJ gene program. This observation correlates with previous observations made with PGC1α transgenic mice in which PGC1α was shown to regulate the NMJ genes.30 In addition, obestatin has an anti‐atrophic effect by regulating the expression levels of the two E3 ubiquitin ligases, hallmarks of skeletal muscle atrophy, MAFbx and MuRF1. Although the exact mechanism of this effect is unknown in DMD, it is clear that obestatin inhibits the function of the pro‐atrophic transcription factors FoxO1 and FoxO4 and blunts the induction of the atrogenes MAFbx and MuRF1. Interestingly, the obestatin action is not due to an effect on the FoxO3, which appears to be phosphorylated or inactivated under basal conditions. Recent data have shown that Akt/mTOR activation combined with PKD/CAMKII/AMPK inactivation by the obestatin signalling is required to inhibit FoxO‐dependent atrogenes in catabolic processes provoked by glucocorticoids.16 This is consistent with the activation of S6K1 and by eIF4E availability, through phosphorylation of 4E‐BP1, key regulators of protein synthesis, which shift the balance from protein degradation to protein synthesis in obestatin‐treated mice. Despite these results, which suggest a role for obestatin in the regulation of muscle hypertrophy, the mean value of CSA was larger in control than in obestatin‐treated mdx mice. It is tempting to assume that the obestatin/GPR39 system operates at first to induce the adaptive response of dystrophic myofibres by regulating fibre‐type switch or transition in myosin isoform expression. In this scenario, the activation of positive regulators of protein synthesis reflects a reprogramming of gene transcription that leads to a remodelling of the fibre contractile properties (fast‐slow transitions) and the metabolic profile (glycolytic‐oxidative transitions). Indeed, here, we show that in obestatin‐treated muscles, the number of MHCI+ myofibres increased with the concomitant reduction of the MHCII+ myofibres, MHCIIb, and MHCIIx, while the average number of myofibres remains unchanged. The fact that obestatin signalling was insufficient to increase muscle growth in this situation, despite being able to act on S6K1 and 4E‐BP1, implies that there are other Akt/mTOR‐dependent signalling events that are required for muscle growth.34
The absence of dystrophin leads to a decrease and a delocalization of dystrophin‐associated proteins with the consequent sarcolemma instability and damage as muscle fibres detach from the laminin‐rich basal lamina during contraction.35 An alternative approach to restoring dystrophin deficiency is to up‐regulate the expression of utrophin based on the evidence for functional redundancy between these two proteins.36 Indeed, utrophin shares a high degree of sequence identity with dystrophin and also associates with members of the dystrophin‐associated protein complex.37 Utrophin is restricted to neuromuscular and myotendinous junctions in adult muscle, but during embryonic development and in patients with DMD, it is localized at the sarcolemma when dystrophin is absent or present only at low levels. Studies in animal models have provided compelling evidence that utrophin functions in these scenarios directly to protect the muscle against dystrophic degeneration.36 Our results indicate that obestatin signalling regulates utrophin expression. Specifically, mdx mice treated for 30 days with obestatin showed an increase in utrophin levels compared with control mdx mice, which are localized throughout the entire length of the sarcolemma. These results are consistent with the promotion of the slow oxidative myogenic programme by obestatin signalling. Indeed, metabolic changes to the muscle can also influence utrophin expression.38 Given the demonstrated role of PGC1α in regulating utrophin transcription,39, 40, 41 we speculate that inhibition of HDAC activity through obestatin signalling results in increased Mef2 levels and stimulation of PGC1α expression leading to utrophin up‐regulation. In addition, the up‐regulation of utrophin expression and its localization at the sarcolemma may stimulate the expression and correct localization of the dystrophin glycoprotein complex, i.e. α‐syntrophin and ß‐dystroglycan, except for NOS1, which does not show significant differences with control mice. It has been previously established that dystrophin localizes NOS1 to the sarcolemma, whereas utrophin does not.42 The implication of this is uncertain, given that there may be compensatory pathways for the lack of NOS1.36, 43 In addition to the enhancement of structural proteins like utrophin, obestatin also regulates α7β1 integrin expression, a laminin‐binding protein that contributes to the overall integrity of the sarcolemma.44 The α7β1 integrin has structural and signalling functions that contribute to muscle development and physiology.45 Loss of the α7 integrin in dystrophin‐deficient mdx mice leads to a severe dystrophic phenotype,44 while transgenic overexpression of the α7β1 integrin ameliorates disease pathology and extends the longevity of severe dystrophic models.46 Taken together, our data suggest that the up‐regulation of utrophin and α7β1 macromolecular complexes in obestatin‐treated mdx mice protect myofibres from contraction‐induced muscle injury. This action stabilizes the link between the sarcolemma and the extracellular matrix in mdx mice.
The convenience and cost effectiveness of the mdx mouse leaves it as the model of choice or research into DMD.47 This convenience is partly counterbalanced by the fact that the mdx myopathy is plainly not an exact reproduction of DMD.48 To remain as close as possible to the human pathology, human immortalized DMD myoblasts were used to validate obestatin‐signalling pathway. We report here that obestatin favours the recruitment and fusion of DMD myoblasts into myotubes, as indicated by the increased number of myonuclei in MHC+ cells, with increased expression of slow‐MHC and hypertrophic effect. Furthermore, we show that obestatin acts to increase levels of both utrophin and α7β1 integrin complexes during the myogenic differentiation. These findings support and emphasize the role of the obestatin signalling into the myogenic programme in DMD.
Although there are several promising therapeutic approaches under investigation for DMD, this pathology will require a multidisciplinary approach to address its many features. Our results illustrate how obestatin can restore to some extent muscle integrity and function in DMD. These data position obestatin as a potential DMD therapeutic candidate not only as an ameliorative treatment to slow the muscle damage but also as part of combinatorial treatment strategies. In this sense, obestatin‐mediated fibre restoration would allow the maintenance of vector or cell content in the decisive period between injection and transgene expression in the dystrophic muscles. For instance, the stability of the pre‐conditioned muscle fibre could help mitigate the loss of therapeutic vectors in gene therapy and guarantee a higher therapeutic benefit in restoration of dystrophin in the muscles of DMD patients.9, 10, 11 The results presented so far provide compelling evidence for a potential way of treating DMD.
Conflict of interest
The authors declare no competing interests.
Funding
This work was supported by grants from Instituto de Salud Carlos III, European Regional Development Fund (ISCIII and Fondos FEDER; MINECO, Spain; PI15/01537), Duchenne Parent Project Spain, and Association Française contre les Myopathies (AFM‐Téléthon). Xunta de Galicia funds J. Gonzalez through a pre‐doctorate research scholarship.
Supporting information
Figure S1. Histological HE staining of kidney (a) and liver (b) tissue sections of mdx mice after intramuscular injection in TA of obestatin or control. Serum urea (c), bilirubin (d), alkaline phosphatase (e), AST (f) and ALT (g) levels were measured at the end of the treatment in mdx controls, and mdx mice treated with obestatin. Data were expressed as mean ± SEM (* P< 0.05 versus control values; n = 5 per group).
Table S1. Supporting Information
{kind=link}
Acknowledgements
Marta Picado Barreiro from IDIS (Santiago de Compostela, Spain) is greatly acknowledged, as well as the platform for immortalization of human cells from the Myology Institute (Paris, France). The Central Laboratory at Hospital Clínico Universitario de Santiago de Compostela (Santiago de Compostela, Spain) is greatly acknowledged for assistance with the clinical biochemistry assays. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle.49
González‐Sánchez, J. , Sánchez‐Temprano, A. , Cid‐Díaz, T. , Pabst‐Fernández, R. , Mosteiro, C. S. , Gallego, R. , Nogueiras, R. , Casabiell, X. , Butler‐Browne, G. S. , Mouly, V. , Relova, J. L. , Pazos, Y. , and Camiña, J. P. (2018) Improvement of Duchenne muscular dystrophy phenotype following obestatin treatment. Journal of Cachexia, Sarcopenia and Muscle, 9: 1063–1078. 10.1002/jcsm.12338.
References
- 1. Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E. Molecular organization at the glycoprotein‐complex‐binding site of dystrophin. Three dystrophin‐associated proteins bind directly to the carboxy‐terminal portion of dystrophin. Eur J Biochem 1994;220:283–292. [DOI] [PubMed] [Google Scholar]
- 2. Gillis JM. Membrane abnormalities and Ca homeostasis in muscles of the mdx mouse, an animal model of the Duchenne muscular dystrophy: a review. Acta Physiol Scand 1996;156:397–406. [DOI] [PubMed] [Google Scholar]
- 3. O'Brien KF, Kunkel LM. Dystrophin and muscular dystrophy: past, present, and future. Mol Genet Metab 2001;74:75–88. [DOI] [PubMed] [Google Scholar]
- 4. Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet 2001;66:17–40. [DOI] [PubMed] [Google Scholar]
- 5. Walter MC, Reilich P. Recent developments in Duchenne muscular dystrophy: facts and numbers. J Cachexia Sarcopenia Muscle 2017;8:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emery AE. The muscular dystrophies. Lancet 2002;359:687–695. [DOI] [PubMed] [Google Scholar]
- 7. Pichavant C, Aartsma‐Rus A, Clemens PR, Davies KE, Dickson G, Takeda S, Wilton SD, Wolff JA, Wooddell CI, Xiao X, Tremblay JP. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther 2011;19:830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet 2013;14:373–378. [DOI] [PubMed] [Google Scholar]
- 9. Le Hir M, Goyenvalle A, Peccate C, Précigout G, Davies KE, Voit T, Garcia L, Lorain S. AAV genome loss from dystrophic mouse muscles during AAV‐U7 snRNA‐mediated exon‐skipping therapy. Mol Ther 2013;21:1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peccate C, Mollard A, Le Hir M, Julien L, McClorey G, Jarmin S, Le Heron A, Dickson G, Benkhelifa‐Ziyyat S, Piétri‐Rouxel F, Wood MJ. Antisense pre‐treatment increases gene therapy efficacy in dystrophic muscles. Hum Mol Genet 2016;25:3555–3563. [DOI] [PubMed] [Google Scholar]
- 11. Godfrey C, Desviat LR, Smedsrød B, Piétri‐Rouxel F, Denti MA, Disterer P, Lorain S, Nogales‐Gadea G, Sardone V, Anwar R, Andaloussi SE. Delivery is key: lessons learnt from developing splice‐switching antisense therapies. EMBO Mol Med 2017;9:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gurriarán‐Rodríguez U, Santos‐Zas I, Al‐Massadi O, Mosteiro CS, Beiroa D, Nogueiras R, Nogueiras R, Crujeiras AB, Seoane LM, Señarís J, García‐Caballero T, Gallego R. The obestatin/GPR39 system is up‐regulated by muscle injury and functions as an autocrine regenerative system. J Biol Chem 2012;287:38379–38389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gurriarán‐Rodríguez U, Santos‐Zas I, González‐Sánchez J, Beiroa D, Moresi V, Mosteiro CS, Lin W, Viñuela JE, Señarís J, García‐Caballero T, Casanueva FF. Action of obestatin in skeletal muscle repair: stem cell expansion, muscle growth, and microenvironment remodeling. Mol Ther 2015;23:1003–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santos‐Zas I, Gurriarán‐Rodríguez U, Cid‐Díaz T, Figueroa G, González‐Sánchez J, Bouzo‐Lorenzo M, Mosteiro CS, Señarís J, Casanueva FF, Casabiell X, Gallego R. β‐Arrestin scaffolds and signaling elements essential for the obestatin/GPR39 system that determine the myogenic program in human myoblast cells. Cell Mol Life Sci 2016;73:617–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santos‐Zas I, Cid‐Díaz T, González‐Sánchez J, Gurriarán‐Rodriguez U, Seoane‐Mosteiro C, Porteiro B, Nogueiras R, Casabiell X, Relova JL, Gallego R, Mouly V. Obestatin controls skeletal muscle‐type determination. Sci Rep 2017;7:2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cid‐Díaz T, Santos‐Zas I, González‐Sánchez J, Gurriarán‐Rodríguez U, Mosteiro CS, Casabiell X, García‐Caballero T, Mouly V, Pazos Y, Camiña JP. Obestatin controls the ubiquitin‐proteasome and autophagy‐lysosome systems in glucocorticoid‐induced muscle cell atrophy. J Cachexia Sarcopenia Muscle 2017;8:974–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Santos‐Zas I, Negroni E, Mamchaoui K, Mosteiro CS, Gallego R, Butler‐Browne GS, Pazos Y, Mouly V, Camiña JP. Obestatin increases the regenerative capacity of human myoblasts transplanted intramuscularly in an immunodeficient mouse model. Mol Ther 2017;25:2345–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edom F, Mouly V, Barbet JP, Fiszman MY, Butler‐Browne GS. Clones of human satellite cells can express in vitro both fast and slow myosin heavy chains. Dev Biol 1994;164:219–229. [DOI] [PubMed] [Google Scholar]
- 19. Chaouch S, Mouly V, Goyenvalle A, Vulin A, Mamchaoui K, Negroni E, Di Santo J, Butler‐Browne G, Torrente Y, Garcia L, Furling D. Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: validation of an exon‐skipping approach to restore dystrophin in Duchenne muscular dystrophy cells. Hum Gene Ther 2009;20:784–790. [DOI] [PubMed] [Google Scholar]
- 20. Mamchaoui K, Trollet C, Bigot A, Negroni E, Chaouch S, Wolff A, Kandalla PK, Marie S, Di Santo J, St Guily JL, Muntoni F. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle 2011;1:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorley M, Duguez S, Mazza EMC, Valsoni S, Bigot A, Mamchaoui K, Harmon B, Voit T, Mouly V, Duddy W. Skeletal muscle characteristics are preserved in hTERT/cdk4 human myogenic cell lines. Skelet Muscle 2016;6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 23. Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 2013;45:2191–2199. [DOI] [PubMed] [Google Scholar]
- 24. Bassel‐Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem 2006;75:19–37. [DOI] [PubMed] [Google Scholar]
- 25. Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel‐Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow‐twitch myofibers. J Clin Invest 2007;117:2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKinsey TA, Zhang CL, Lu J, Olson EN. Signal‐dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000;408:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKinsey TA, Zhang CL, Lu J, Olson EN. Signaling chromatin to make muscle. Curr Opin Cell Biol 2002;14:763–772. [DOI] [PubMed] [Google Scholar]
- 28. Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development 2007;134:4131–4140. [DOI] [PubMed] [Google Scholar]
- 29. Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care 2010;13:225–229. [DOI] [PubMed] [Google Scholar]
- 30. Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC‐1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 2007;21:770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bentzinger CF, von Maltzahn J, Dumont NA, Stark DA, Wang YX, Nhan K, Frenette J, Cornelison DD, Rudnicki MA. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J Cell Biol 2014;205:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Correia JC, Ferreira DM, Ruas JL. Intercellular: local and systemic actions of skeletal muscle PGC‐1s. Trends Endocrinol Metab 2015;26:305–314. [DOI] [PubMed] [Google Scholar]
- 33. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB. Transcriptional co‐activator PGC‐1 alpha drives the formation of slow‐twitch muscle fibers. Nature 2002;418:797–801. [DOI] [PubMed] [Google Scholar]
- 34. Marabita M, Baraldo M, Solagna F, Ceelen JJM, Sartori R, Nolte H, Nemazanyy I, Pyronnet S, Kruger M, Pende M, Blaauw B. S6K1 is required for increasing skeletal muscle force during hypertrophy. Cell Rep 2016;17:501–513. [DOI] [PubMed] [Google Scholar]
- 35. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 1993;90:3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fairclough MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet 2013;14:373–378. [DOI] [PubMed] [Google Scholar]
- 37. Miura P, Jasmin JB. Utrophin up‐regulation for treating Duchenne or Becker muscular dystrophy: how close are we? Trends Mol Med 2006;12:122–129. [DOI] [PubMed] [Google Scholar]
- 38. Miura P, Andrews M, Holcik M, Jasmin BJ. IRES‐mediated translationof utrophin A is enhanced by glucocorticoid treatment in skeletal muscle cells. PLoS One 2008;3:e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Angus LM, Chakkalakal JV, Méjat A, Eibl JK, Bélanger G, Megeney LA, Chin ER, Schaeffer L, Michel RN, Jasmin BJ. Calcineurin‐NFAT signaling, together with GABP and peroxisome PGC‐1{alpha}, drives utrophin gene expression at the neuromuscular junction. Am J Physiol Cell Physiol 2005;289:C908–C917. [DOI] [PubMed] [Google Scholar]
- 40. Hollinger K, Gardan‐Salmon D, Santana C, Rice D, Snella E, Selsby JT. Rescue of dystrophic skeletal muscle by PGC‐1alpha involves restored expression of dystrophin‐associated protein complex components and satellite cell signaling. Am J Physiol Regul Integr Comp Physiol 2013;305:R13–R23. [DOI] [PubMed] [Google Scholar]
- 41. Selsby JT, Morine KJ, Pendrak K, Barton ER, Sweeney HL. Rescue of dystrophic skeletal muscle by PGC‐1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS One 2012;7:e30063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li D, Bareja A, Judge L, Yue Y, Lai Y, Fairclough R, Davies KE, Chamberlain JS, Duan D. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci 2010;123:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van den Bergen JC. Studying the role of dystrophin‐associated proteins in influencing Becker muscular dystrophy disease severity. Neuromuscul Disord 2015;25:231–237. [DOI] [PubMed] [Google Scholar]
- 44. Rooney JE, Welser JV, Dechert MA, Flintoff‐Dye NL, Kaufman SJ, Burkin DJ. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci 2006;119:2185–2195. [DOI] [PubMed] [Google Scholar]
- 45. Song WK, Wang W, Sato H, Bielser DA, Kaufman SJ. Expression of alpha 7 integrin cytoplasmic domains during skeletal muscle development: alternate forms, conformational change, and homologies with serine/threonine kinases and tyrosine phosphatases. J Cell Sci 1993;106:1139–1152. [DOI] [PubMed] [Google Scholar]
- 46. Burkin DJ, Wallace GQ, Nicol KJ, Kaufman DJ, Kaufman SJ. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol 2001;152:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J 2013;280:4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 2015;8:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Histological HE staining of kidney (a) and liver (b) tissue sections of mdx mice after intramuscular injection in TA of obestatin or control. Serum urea (c), bilirubin (d), alkaline phosphatase (e), AST (f) and ALT (g) levels were measured at the end of the treatment in mdx controls, and mdx mice treated with obestatin. Data were expressed as mean ± SEM (* P< 0.05 versus control values; n = 5 per group).
Table S1. Supporting Information