

Abstract
The Frizzled (FZD) proteins belong to class F of G protein–coupled receptors (GPCRs) and are essential for various pathways involving the secreted lipoglycoproteins of the wingless/int-1 (WNT) family. A WNT-binding cysteine-rich domain (CRD) in FZDs is N-terminally located and connected to the seven transmembrane domain–spanning receptor core by a linker domain that has a variable length in different FZD homologs. However, the function and importance of this linker domain are poorly understood. Here we used systematic mutagenesis of FZD6 to define the minimal N-terminal domain sufficient for receptor surface expression and recruitment of the intracellular scaffold protein Dishevelled (DVL). Further, we identified a triad of evolutionarily conserved cysteines in the FZD linker domain that is crucial for receptor membrane expression and recruitment of DVL. Our results are in agreement with the concept that the conserved cysteines in the linker domain of FZDs assist with the formation of a common secondary structure in this region. We propose that this structure could be involved in agonist binding and receptor activation mechanisms that are similar to the binding and activation mechanisms known for other GPCRs.
Keywords: G protein–coupled receptor (GPCR), Wnt signaling, membrane protein, protein structure, mutagenesis, β-catenin, DVL, FZD6, G protein signaling, linker domain
Introduction
Class F of G protein–coupled receptors (GPCRs) 2 consists of 10 Frizzled homologs (FZD1–10) and Smoothened (SMO) (1). FZDs, the focus of this study, bind WNT proteins with their extracellular cysteine-rich domain (CRD) to initiate a complex network of WNT signaling pathways (2). The CRD is seen as the orthosteric binding site for WNTs and is functionally implicated in receptor dimerization and signal initiation (3–6). Structurally, the CRD is connected to the seven transmembrane (7TM) core of the receptor through a linker domain of variable length and low degree of conservation among the representatives of class F (5). A central intracellular interaction partner of FZDs is the scaffold protein Dishevelled (DVL), of which three isoforms exist in mammals (7, 8). DVL, which does not interact with SMO, is involved in several branches of WNT signaling, including β-catenin–dependent and –independent pathways. The mechanisms of interaction between FZDs and DVL are complex and multimodal. Originally, it was shown that DVL binds an unconventional PDZ ligand domain in helix 8 of FZDs with the consensus motif KTXXXW (9). In addition, three amino acids C-terminal of the KTXXXW sequence, the flanking regions of intracellular loop 3, and electrochemical interaction with the phospholipids of the membrane were shown to be important for DVL membrane recruitment (10–12). The current model, however, includes rather prominent DVL–DEP (Dishevelled, Egl-10, pleckstrin) domain interactions with FZDs, and it was recently shown that the DVL–PDZ and the DVL–DIX (derived from dishevelled and axin) domains are dispensable for FZD–DVL recruitment (13–15). Mutagenesis has further revealed sites in other regions of FZDs to regulate FZD–DVL contact (16, 17). Even though some of these results might not necessarily pinpoint direct interaction sites, such as Tyr-2502.39 at the lower end of TM 2 of FZD4, which is most likely involved in defining an overall conformation enabling FZD to interact with DVL (13, 16).
So far, the dynamics and the regulation of the FZD–DVL interaction remain unclear. Recently, it has been proposed that FZD–DVL interactions appear to be WNT-induced when feeding into the WNT/β-catenin pathway, whereas FZD–DVL binding could be more static when signaling is pushed toward β-catenin–independent, planar cell polarity–like signaling pathways (15). WNT-induced DEP domain swapping in DVL leads to reduced DVL–FZD interaction, which appears to be an underlying mechanism of low-density lipoprotein receptor–related protein 5/6 (LRP5/6)–based signalosome formation (13, 18).
In this study, we focus on FZD6, a receptor that is generally not signaling in collaboration with LRP5/6 to activate WNT/β-catenin signaling (19). Instead, FZD6 is known to activate β-catenin–independent signaling; for example, through planar cell polarity–like signaling or activation of heterotrimeric G proteins of the Gi/o and Gq/11 families (20–25). To define the role of the extracellular CRD and the linker region for receptor surface expression and functionality toward DVL, we use systematic mutagenesis. Furthermore, we apply pharmacological inhibition of porcupine, which is required for WNT acylation and thereby inhibits secretion of WNTs from cells (26). Mutagenesis clearly defines the linker domain and especially a triad of conserved cysteines as crucial for receptor surface expression. Neither removal of the CRD nor pretreatment with a porcupine inhibitor affect the ability of FZD6 to recruit DVL to the membrane and to induce a phosphorylation-dependent shift in the electrophoretic mobility of DVL. On the other hand, Cys-to-Ala mutation of any of the three linker cysteines completely disrupts FZD6 cell surface expression and the receptor's functionality toward DVL. Thus, our findings emphasize the functional importance of the linker domain for surface expression of FZDs and receptor function.
Results
Design of N-terminal deletion mutants of FZD6
The N terminus of FZD6 consists of a short signaling sequence, the CRD, and a linker region that connects the CRD to the 7TM domain (1). The receptor model (Fig. 1A) presents an overlay of the crystal structure of full-length SMO (27) and the XWNT-8/mFZD8-CRD structure (3), visualizing the areas of putative WNT-CRD binding sites in relation to the full-length class F receptor and pinpointing the position of the linker, including an antiparallel β-sheet in the linker domain. The human FZD6 constructs used in this study contain an N-terminal signal sequence, a FLAG epitope tag, and a C-terminal His tag to allow careful validation of full-length receptor expression. To assess the role of the N-terminal part of the receptor that contains the WNT binding site, we created deletion mutants lacking the CRD or the CRD and the linker region, shown schematically in Fig. 1, B and C. For the construct lacking the CRD, we aimed to delete the whole CRD. To preserve the disulfide bond in the linker region, the deletion was made after Arg-155. For removal of the linker domain, the intention was to maintain the last cysteine of the linker (Cys-185), which, according to the SMO model, creates a disulfide bond with a cysteine in extracellular loop (ECL) 1 (Cys-260). Therefore, the receptor was truncated after Cys-181 (Fig. 1C).
Figure 1.
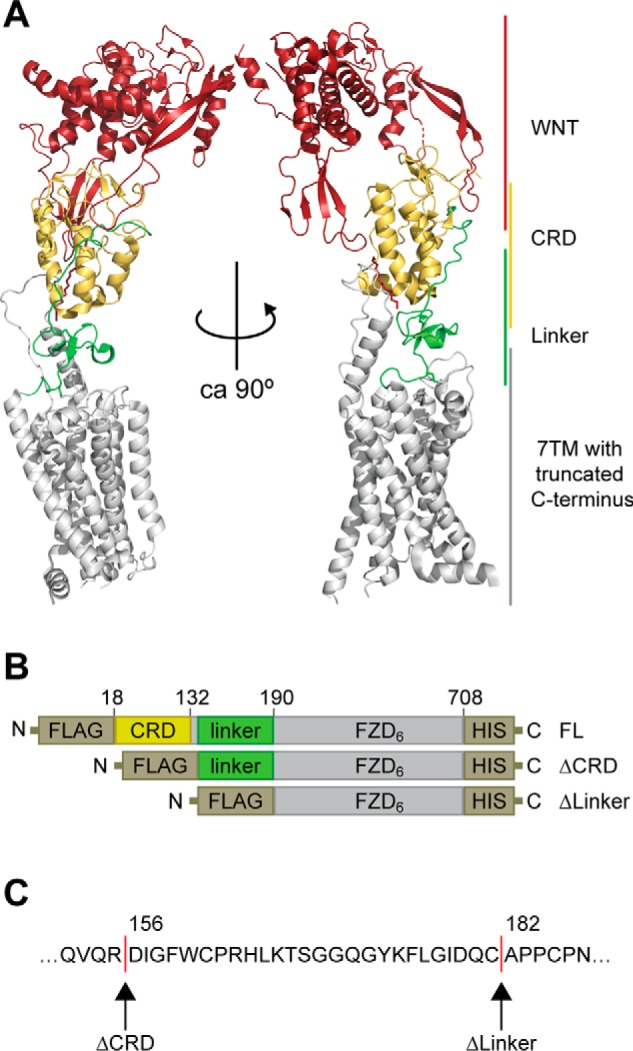
Design of N-terminal deletion mutants of FZD6. A, an overlay of the full-length SMO structure (PDB code 5V57) and the XWNT8-mFZD8-CRD structure (PDB code 4F0A) to visualize domain arrangement in FZDs. XWNT8, red/firebrick; CRD, yellow; linker region, green; 7TM core, gray. B, schematic of constructs for full-length human FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker). All three constructs carry an N-terminal FLAG and a C-terminal His tag. Numbers refer to amino acid numbering in the human FZD6 (UniProtKB accession number O60353 (FZD6_HUMAN)). C, the amino acid sequence defines the N-terminal amino acid of ΔCRD-FZD6 (Asp-156) and ΔLinker-FZD6 (Ala-182). Structures were rendered using PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC).
Expression of N-terminal deletion mutants of FZD6
The full-length (FL) receptor (1–708 aa), FZD6 lacking the CRD (ΔCRD-FZD6, 156–708 aa), and the construct lacking both the CRD and linker region (ΔLinker-FZD6, 182–708 aa) were expressed to similar levels in transiently transfected HEK293T cells, as shown by SDS-PAGE/immunoblotting using an anti-FZD6 antibody with a C-terminal epitope or an anti-FLAG or anti-His antibody (Fig. 2A). Immunoblot detection of the FZD6 constructs by an anti-FZD6 antibody indicates that FZD6 and ΔCRD-FZD6 are more extensively posttranslationally modified compared with ΔLinker-FZD6 (Fig. 2A), potentially pointing to reduced maturation and glycosylation in the latter construct. Indeed, glycosylation site prediction using the NetOGlyc 4.0 and NetNGlyc 1.0 applications suggests that the linker domain of human FZD6 could be O-glycosylated at Ser-168 (28, 29). Indirect immunocytochemistry using conditions with and without membrane permeabilization prior antibody staining revealed that all of the FZD6 constructs are expressed (Fig. 2B), although only full-length and ΔCRD-FZD6, but not ΔLinker-FZD6, were localized at the cell surface (Fig. 2, C and D), corroborating what was surmised from the electrophoretic mobility of the constructs. Thus, removal of both the CRD and the linker domain corrupted proper membrane embedding.
Figure 2.
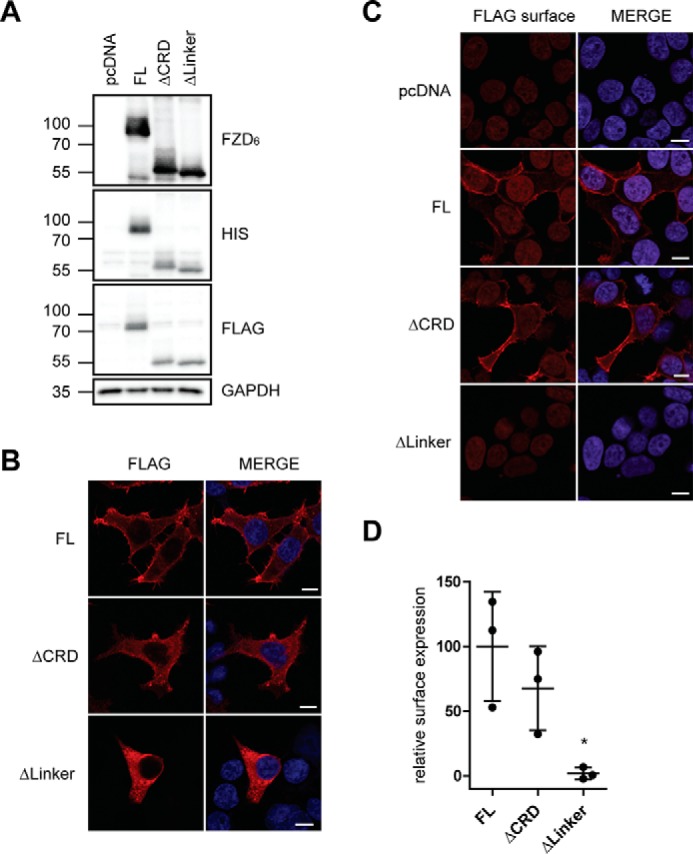
Expression of N-terminal deletion mutants of FZD6. A, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker). Cell lysates were analyzed by immunoblotting using anti-FZD6, anti-FLAG, and anti-His antibodies for detection. Anti-GAPDH served as a loading control. B and C, HEK293T cells were transfected with empty vector (pcDNA), FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker). Confocal photomicrographs present staining of indirect immunofluorescence using an anti-FLAG antibody in permeabilized (B) and nonpermeabilized (C, FLAG surface) HEK293T cells. Scale bars = 10 μm. D, surface expression of transfected FZD6 constructs was quantified using a cell ELISA based on detection of anti-FLAG under nonpermeabilized conditions. The scatter dot plot represents mean with S.D. of three independent experiments performed in triplicates. Statistical analysis was done by one-way ANOVA. *, p < 0.05.
DVL2 signaling and recruitment induced by FZD6 deletion mutants
To functionally validate the receptor constructs, we analyzed the DVL shift induced by full-length and ΔCRD-FZD6. The electrophoretic mobility shift is a validated consequence of DVL activation (30, 31). HEK293T cells, similar to many other cell types, constitutively secrete WNTs, resulting in an autocrine signaling loop. To address the influence of autocrine WNT signaling through the overexpressed FZD6 constructs on FZD–DVL signaling, we pretreated the cells with the porcupine inhibitor C59 (26). The effectiveness of the C59 treatment was verified by increasing doses of C59, resulting in reduced formation of phosphorylated and shifted, endogenously expressed DVL2 (Fig. S1A, open triangle) in a dose-dependent manner. Upon overexpression in HEK293T cells, full-length and ΔCRD-FZD6 induced an electrophoretic mobility shift of DVL, visible as a minor (Fig. 3A, open triangle) and major retardation (Fig. 3A, filled triangle) on the PAGE, similar to what was previously shown for FZD5 (31). We refer to this as shifted and hypershifted DVL, respectively. ΔLinker-FZD6 was not capable of altering the electrophoretic mobility of DVL. Thus, we decided to analyze the ability of full-length and ΔCRD-FZD6 to shift DVL in more detail (Fig. 3, B and C). There was no difference in FZD-induced DVL shift/hypershift between full-length and ΔCRD-FZD6 (Fig. 3, B and C). We performed the experiments in parallel under conditions where endogenous secretion of WNTs was blocked by C59 (Fig. 3, B and C, and Fig. S1B). As expected, the basal shift of DVL was reduced, resulting in a stronger band of the unshifted form of DVL. Despite the presence of C59, full-length and ΔCRD-FZD6 resulted in a weak induction of shifted DVL. The hypershift (Fig. 3B, filled triangle), which was observed in the absence of C59, was hardly affected by C59 pretreatment (Fig. 3, B and C). Thus, both full-length and ΔCRD-FZD6 exhibit the ability to shift and hypershift DVL in the presence and absence of C59. These findings indicate that the induction of DVL shift and hypershift by FZD6 is CRD-independent and can be achieved in a ligand-independent manner. Furthermore, we analyzed the ability of full-length, ΔCRD-FZD6, and ΔLinker-FZD6 to recruit DVL to the membrane, similar to previous studies (10, 13, 22, 32). In agreement with previous results, overexpression of an HA-tagged DVL2 (DVL2-HA) alone resulted in the typical cytosolic aggregates (33, 34) (Fig. 4A). Cotransfection of the different FZD6 constructs together with DVL2-HA showed that receptor constructs that are able to reach the plasma membrane also recruit DVL2-HA, irrespective of the presence of the CRD (Fig. 4A). ΔLinker-FZD6 was neither recruiting DVL2-HA to the membrane nor colocalizing with DVL2-HA intracellularly. C59 treatment did not affect FZD–DVL recruitment mediated by full-length or ΔCRD-FZD6 (Fig. 4B). Furthermore, the findings with recruitment of full-length DVL2-HA can be recapitulated with the isolated GFP-tagged DEP domain derived from DVL2. Although DVL2-DEP-GFP is evenly distributed in the cell and the nucleus in the absence of overexpressed FZD, coexpression of FZD6 as well as ΔCRD-FZD6 resulted in distinct membrane recruitment of DVL2-DEP-GFP (Fig. 4C). ΔLinker-FZD6, however, did not affect DEP-GFP localization. In agreement with the findings using full-length DVL2, the DEP-GFP construct was also recruited to co-expressed full-length or ΔCRD-FZD6 in the presence of C59 (Fig. 4D). Thus, FZD6 can recruit the DEP-GFP construct irrespective of the presence or absence of the CRD in a ligand-independent manner.
Figure 3.

DVL2 signaling induced by FZD6 deletion mutants. A, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker). Cell lysates were analyzed for the FZD-induced electrophoretic mobility shift from unshifted (fastest-migrating, bottom band; red triangle), shifted (open triangle), and hypershifted (filled triangle) DVL2 using an anti-DVL2 antibody to detect endogenously expressed DVL2. Expression of FZD constructs was detected by an anti-FZD6 antibody; anti-α-tubulin antibody was used as a loading control. B, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), and ΔCRD-FZD6 (ΔCRD) and kept overnight in the absence or presence of C59. Cell lysates were analyzed for the FZD-induced electrophoretic mobility shift from unshifted (fastest-migrating band; red triangle), shifted (open triangle), and hypershifted (filled triangle) DVL2 using an anti-DVL2 antibody to detect endogenously expressed DVL2. C, the three prominent DVL2 bands were quantified by densitometry. Bottom band, unshifted (red triangle); middle band, shifted (open triangle); top band, hypershifted (filled triangle). The scatter dot plot represents normalized values from three independent experiments for the mean ratio of middle/bottom or top/bottom bands with S.D. Tubulin detection was not included in the calculations. For details, see “Experimental procedures.” Statistical analysis was done by one-way ANOVA with Tukey's multiple comparisons post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Figure 4.

DVL recruitment induced by FZD6 deletion mutants. A, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker) in combination with DVL2-HA. Proteins were visualized using indirect immunocytochemistry using an anti-FLAG (red, FZD6) or an anti-HA (green, DVL2) antibody. Cells were counterstained with the nuclear stain DAPI (blue). B, cells were treated with C59 (1 μm, overnight). C, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), ΔCRD-FZD6 (ΔCRD), and ΔLinker-FZD6 (ΔLinker) together with the GFP-tagged DEP domain of DVL2 (DVL2-DEP-GFP). FZD6 constructs were stained with an anti-FLAG antibody. Cells were counterstained with the nuclear stain DAPI (blue). Proteins were visualized using indirect immunocytochemistry with an anti-FLAG antibody (FZD6) or GFP fluorescence (DVL2-DEP). D, cells were treated with C59 (1 μm, overnight). Scale bars = 10 μm. C and D, the fluorescence (fl) intensity profile (DVL2-DEP-GFP, green; FZD6 constructs, red) along the dashed line was quantified in ImageJ with the plot profile tool. The maximal value of the fluorescent intensity was normalized to 100%.
Identification of a well-conserved triad of cysteines in the linker domain
Based on our findings regarding linker domain deletion, the following question arose: what linker domain–intrinsic features are important for the structural integrity of the receptor and its ability to be embedded in the plasma membrane? The crystal structure of the full-length SMO (PDB code 5V57 (27)) indicates that the linker domain adopts an antiparallel β-sheet that is stabilized by a disulfide bond (Fig. 5A, red). Although Cys-193 and Cys-213 in SMO (corresponding to FZD6 Cys-161 and Cys-181) form a linker-internal disulfide bond, Cys-217 (Cys-185 in FZD6) is linking to ECL1 by forming a disulfide bond with Cys-295 (Cys-260 in FZD6). Detailed inspection of all 11 published SMO crystal structures and the only Frizzled (FZD4) structure of both full-length and ΔCRD constructs indicated that all protein structures contain an antiparallel β-sheet in the linker domain (Fig. 5B) (27, 35–39). Furthermore, family-wide sequence alignment of the extracellular domains and ECL1 of all class F receptors showed that this triad of cysteines (Cys-161, Cys-181, and Cys-185 in human FZD6; Fig. 5C) and the cysteine in ECL1 (Cys-260 in FZD6, Fig. 5D) are fully conserved.
Figure 5.

Identification of a well-conserved triad of cysteines in the linker domain. A, close-up of the SMO structure shown in Fig. 1A. CRD, yellow; linker, green; 7TM core, gray. In addition, the linker domain cysteines corresponding to human FZD6 Cys-161, Cys-181, and Cys-185 and Cys-260 in ECL1 are shown as red sticks. B, the table summarizes information about the presence of the antiparallel β-sheet in the linker domain from all published SMO crystal structures and one FZD4 structure (PDB code 6BD4). FL, CRD present; ΔCRD, CRD absent. C and D, alignment of the extracellular linker domains and ECL1 of all human class F receptor homologs shows a high degree of conservation among the cysteines in the linker domain (C) and the cysteine in ECL1 (D). The bar graphs show the degree of conservation between the compared sequences. Numbers identify Cys-161, Cys-181, and Cys-185 in human FZD6. Increasing intensity of blue indicates a higher degree of conservation. Alignment was done using MAFFT with default settings. Structures were rendered using PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC).
Cys-161 defines the minimal length of a functional FZD6 construct
We have introduced more subtle truncations varying in the N terminus of the linker region by either including Cys-161 together with Trp-160 (FZD6 (Δ1–159)), exposing Cys-161 as a terminal amino acid (FZD6 (Δ1–160)), or removing Cys-161 (FZD6 (Δ1–161)) (Fig. 6A). All three constructs are expressed at similar levels as the full-length receptor when overexpressed in HEK293 cells. However, in accordance to what we observed (Fig. 2A) for the ΔCRD and the ΔLinker constructs of FZD6, only the full-length FZD6, FZD6 (Δ1–159), and FZD6 (Δ1–160) showed a double band indicative of posttranslational modifications, whereas FZD6 (Δ1–161) migrated as a single band (Fig. 6B). Quantification of receptor surface expression using a cell ELISA assay in nonpermeabilized cells showed that FZD6 (Δ1–159) and FZD6 (Δ1–160), but not FZD6 (Δ1–161), are expressed at the cell surface (Fig. 6, C and D). Additionally, only constructs containing Cys-161 both display the FZD-induced electrophoretic mobility shift (Fig. 6B) and recruit DVL (Fig. 6E). Thus, these mutants indicated that the highly conserved N-terminal Cys-161 in the linker region is required to maintain proper membrane insertion of the receptor.
Figure 6.

Cys-161 defines the minimal length of a functional FZD6 construct. A, schematic presentation of FZD6 ΔCRD constructs with differences in the N-terminal amino acid. The cysteines in the linker domain are highlighted in red. B, HEK293T cells were transfected with empty vector (pcDNA), full-length FZD6 (FL), and N-terminal deletion mutants. The electrophoretic mobility shift of endogenous DVL2 was assessed by immunoblotting using an anti-DVL2 antibody. The filled triangle marks the band of hypershifted DVL2 induced by FZD overexpression. Protein expression was verified with an anti-FZD6 antibody; anti-GAPDH was used as a loading control. C, HEK293T cells were transfected with various N-terminal deletion mutants. Confocal photomicrographs of HEK293T cells transiently expressing truncated FZD6 with indirect immunofluorescence using fluorophore-labeled secondary antibody against the anti-FLAG antibody on nonpermeabilized cells (FLAG surface). D, surface expression of the transfected FZD6 constructs was quantified using a cell ELISA based on detection of anti-FLAG under nonpermeabilized conditions. The scatter dot plot represents means with S.D. of three independent experiments performed in triplicates. Statistical analysis was done by one-way ANOVA. Significance levels denoting differences between FL FZD6 and mutant receptor surface expression are given as follows: **, p < 0,01. E, confocal photomicrographs of DVL2 recruitment in HEK293T cells transfected with FZD6 N-terminal deletion mutants and DVL2-HA and stained with anti-FLAG (red, FZD6) and anti-HA (green, DVL2) antibodies. Scale bars = 10 μm.
A well-conserved triad of cysteines in the linker domain is crucial for receptor function
To address the relevance of the triad of cysteines in the linker domain in the full-length and ΔCRD-FZD6, we introduced Cys-to-Ala mutations for each of the cysteines individually (Fig. 7A). Constructs with single point mutations of any of the three cysteines in the linker domain failed to reach the plasma membrane, as assessed by indirect immunocytochemistry in nonpermeabilized cells and by cell ELISA for quantitative assessment (Fig. 7, B and C). Furthermore, the electrophoretic mobility shift (Fig. 7D) that is inherently connected with FZD-induced DVL plasma membrane recruitment was not observed in cells expressing any of the cysteine mutations in the full-length or ΔCRD-FZD6 constructs (Fig. 7E).
Figure 7.

A well-conserved triad of cysteines in the linker domain is crucial for receptor function. A, schematic of the Cys-to-Ala mutations in FL FZD6 (top) and ΔCRD-FZD6 (bottom). Cys-161, Cys-181, and Cys-185 were individually mutated in both receptor constructs. B, confocal images of surface expression of FZD6 Cys-to-Ala constructs, which were transiently transfected into HEK293T cells and detected with indirect immunofluorescence using a fluorophore-conjugated secondary antibody against the anti-FLAG antibody on nonpermeabilized cells (FLAG surface). FL FZD6 was used as a positive control. C, surface expression was quantified using a cell ELISA based on detection of anti-FLAG under nonpermeabilized conditions. The scatter dot plot represents mean with S.D. of three independent experiments performed in triplicates. ***, p < 0.001. D, cell lysates were analyzed for the electrophoretic mobility shift of DVL2 by immunoblotting using anti-DVL2, anti-FZD6, and anti-α-tubulin (loading control) antibodies. E, photomicrographs of the DVL2 recruitment assay performed on HEK293T cells transfected with Cys-to-Ala mutants and DVL2-HA and detected with anti-FLAG (red, FZD6) and anti-HA (green, DVL2) antibody. Scale bars = 10 μm.
Discussion
FZDs need to be exposed at the cell surface of the cell receiving the WNT signal to be accessible for WNT stimulation either in an autocrine or a paracrine manner. Intracellularly, DVL is a central mediator of WNT/FZD signaling, acting at the crossroads of β-catenin–dependent and –independent signaling pathways, and the mechanisms of DVL recruitment to FZDs define a field of intense research (5, 10, 11, 13, 14, 16, 18, 22, 40, 50). The activation of DVL can be assessed as a phosphorylation-dependent electrophoretic mobility shift of DVL, which is most prominently seen upon overexpression of casein kinase 1 (15, 30, 31, 42, 43). In addition, overexpression of FZDs also results in phosphorylation and shift of DVL, which, however, appears to be independent of CK1 and is qualitatively different from the CK1-induced shift (31). Thus, the current model of FZD–DVL interaction combines direct interaction of the DEP domain with FZD and subsequent DVL phosphorylation (13, 15); both events appear intrinsically interconnected and are, as of now, not experimentally dissociable. However, it remains unclear to which degree agonist stimulation of FZDs is required for FZD–DVL interaction and which exact mechanisms lead from receptor activation to DVL phosphorylation. In fact, the ligand dependence of the FZD–DVL complex remains a mystery even though circumstantial evidence indicates that WNT stimulation could lead to dissociation of DVL from the receptor (13). On the other hand, recent results employing a DVL recruitment bioluminescence resonance energy transfer (BRET) assay in cells overexpressing FZD4, LRP6, and TSPAN12 and stimulated with recombinant Norrin (a FZD4-selective, WNT-unrelated agonist) suggest that FZD stimulation actually promotes FZD–DVL recruitment (44). From this study, we conclude that no agonist input is required for the basal interaction between overexpressed FZD and DVL because the ΔCRD-FZD6, a construct devoid of the orthosteric binding site for WNTs, is capable of recruiting DVL, even in the absence of WNTs. We interpret these findings in two ways: agonist input is not required to stabilize an FZD conformation that provides a high-affinity binding site for DVL, and the CRD is not required for the formation of a functional FZD–DVL complex. Furthermore, we speculate that DVL could prefer the ligand-free state of FZD over WNT-bound FZD, which would be supported by the report of WNT-induced DVL dissociation (13). However, in that case, we would have expected to observe an increase in DVL membrane recruitment or the FZD-induced hypershift of DVL in the presence of C59, which we were not able to detect.
Results with CRD deletion in SMO are contradictory. Several reports indicated that ΔCRD-SMO constructs are not able to activate the glioma-associated oncogene homolog (GLI)-luciferase reporter assay (45, 46). This stands in contrast to the data where deletion of the CRD did not affect SMO activity (47). In Drosophila melanogaster, deletion of the CRD in SMO resulted in loss of normal receptor activity (48). Interestingly, it was also reported that deletion of the CRD in SMO conveys constitutive activation of downstream GLI signaling (36). On the other hand, removal of the CRD from D. melanogaster FZDs did not result in enhanced basal activity, whereas these constructs still mediated modest transcriptional activity of the reporter assay when cotransfected with wingless/WNT (49).
Here we present a detailed characterization of the function of the linker domain connecting the N-terminal CRD with the 7TM core of the receptor. In our hands and with the assays employed, we could not detect more enhanced constitutive activity of FZD6 upon removal of the CRD. In the process of defining the minimal extracellular domain of a functional FZD, we also identified a triad of conserved cysteine residues, which turned out to be crucial for receptor surface expression. Our mutagenesis experiments support the idea that the cysteine-based formation of disulfide bonds and subsequent stabilization of the linker β-sheets observed in SMO and FZD4 could be a common, well-conserved feature of class F receptors. Overall, the formation of a secondary structure in the linker domain could render this region less flexible than surmised previously (6). Interestingly, several proline residues, which introduce turns in the peptide chain, are frequent in the linker domain and partially conserved, such as Pro-162, Pro-184, and Pro-186 in FZD6 (Fig. 5C). These prolines could contribute to stabilization of a secondary structure in this region.
As pointed out above, linker domain glycosylation could play a role in receptor surface expression. So far, we have not defined whether either mutation of the cysteines in the linker domain and subsequent destruction of the linker domain's secondary structure causes loss of surface expression or whether the mutation-induced loss of glycosylation and maturation is the main cause of reduced membrane exposure (Fig. 7, B and D). However, the O-glycosylation site Ser-168 in the linker domain is not conserved among class F receptors, not even in the closely related FZD3. This suggests that the structural impact of destroying disulfide bonds rather than loss of glycosylation causes the trafficking phenotype in the FZD6 cysteine mutants.
As presented in Fig. 5B, all reported SMO crystal structures, irrespective of the presence or absence of the CRD, contain the antiparallel β-sheet in the linker domain (27, 35–38), suggesting that this secondary structure is not a packing or crystallization artifact and that it is maintained under different experimental conditions not requiring the CRD for stabilization. In the process of revision of this manuscript, the first crystal structure of a class F receptor, FZD4, was published (39). The presented data further corroborate our conclusions regarding the importance of a structured linker domain. As shown in Fig. 5B and Fig. S2, the FZD4 structure contains a short antiparallel β-sheet in the linker domain based on the conserved Cys triad and the Cys in ECL1. This information is of importance to understand how the linker domain could contribute to agonist binding to FZDs and the agonist-induced dynamics of FZDs leading to receptor activation. The question of whether the CRD is indeed required for WNT-induced FZD activation has been raised before (49, 50) and remains a matter of debate, especially because the current state of knowledge places the orthosteric WNT binding site on the CRD (3). The debate around the importance of the CRD for WNT-dependent signaling originated from experiments where the mutant phenotype of D. melanogaster embryos caused by depletion of FZDs was at least partially rescued by expression of ΔCRD-FZD constructs (50–52). In addition, ΔCRD-FZD constructs mediated WNT/β-catenin signaling, as assessed by the transcriptional TOPflash reporter assay, albeit to a lesser degree than the full-length receptor (49). In the case of FZD6, it was suggested previously that removal of the CRD abolished the receptor's ability to counteract WNT/β-catenin (19). However, in the latter experiments, no controls for protein expression, surface embedding, or structural integrity of the deletion mutants were provided, making it difficult to interpret the loss of activity in the ΔCRD-FZD6 versus loss of surface expression.
Even though much more work is required to shed light on the function and dynamics of the CRD/linker in relation to the receptor core, our findings emphasize a central function of the linker domain for cell surface expression of the receptors. In the context of our data, however, it is experimentally challenging to distinguish whether receptor dysfunction is due to reduced surface expression of the mutant or due to a linker-intrinsic mechanism affecting ligand–receptor interaction. It is attractive to speculate that the linker domain does not only maintain receptor surface expression but that it also has a role in the agonist-induced dynamics and a potential two-step agonist binding mode, as described for class B receptors (53, 54). Very recently it was shown for FZD4 that the linker domain indeed has a role in defining functional high-affinity binding of the WNT-unrelated, FZD4-selective ligand Norrin (44). The study supports our speculation of a CRD- and linker-mediated two-step binding mode also in the case of FZD-WNT interaction. Unfortunately, it remains technically challenging to assess WNT-FZD binding using the full-length receptors in a lipid environment because of the lipophilic nature and instability of WNT proteins, preventing us from directly testing this concept (55, 56).
The importance of the linker cysteines for receptor function is further underlined by previous reports identifying FZD4 familial exudative vitreoretinopathy (FEVR) mutations in Cys-181 and Cys-204 of human FZD4 (57–59). These C181R, C204R, C204Y mutations (corresponding to Cys-161 and Cys-185 in human FZD6, respectively) manifested with a trafficking phenotype and poor receptor surface expression (57, 58). Also, it was shown that the C204R FZD4 mutant failed to mediate Norrin-stimulated β-catenin signaling (58). In agreement with FEVR as a loss-of-function phenotype of the FZD4 signaling system, Cys-181 and Cys-204 are causally associated with the disease (57–59). On the other hand, mutations in the three cysteines in the linker domain and the cysteine in ECL1, all predicted to result in loss of signaling, are only rarely associated with cancers. In fact, among the almost 64,000 samples included in the cBioPortal for Cancer Genomics to date (60), only two FZD5 mutations were found to be associated with cutaneous melanoma (C192G, TCGA-EB-A42Y-01) and with papillary renal cell carcinoma (C222R, TCGA-GL-6846-01) and two SMO mutations in lung adenocarcinoma (C213F, P-0002215-T01-IM3) and in malignant peripheral nerve sheath tumors (C295S, TCGA-QQ-A8VB-01). None of the other class F members presented with similar cysteine mutations in the cancer material.
In summary, our findings highlight the importance of the hitherto poorly defined function of the linker domain connecting the CRD with the 7TM core of class F receptors. Although we show, with the example of FZD6, that this linker region, and especially several highly conserved cysteines, are essential for receptor surface expression and recruitment of DVL, we speculate that maintenance of the secondary structure of this region has importance for receptor function. The physiological and family-wide relevance of our findings is underlined by the disease-causing mutations in the cysteines of the linker domain of FZD4 associated with FEVR.
Experimental procedures
Cloning of FZD constructs and used plasmids
The full-length version of FZD6 with an N-terminal cleavable hemagglutinin signal sequence (MKTIIALSYIFCLVF), a FLAG epitope (DYKDDDD), and a C-terminal decahistidine His tag was subcloned using a β2-adrenergic receptor construct in pcDNA3.1(+) with primers 5′-AAA AGC TTG CCA CCA TGA AGA CGA TCA TCG CCC TGA-3′ (forward) and 5′-TTT GAA TTC GTC ATC ATC GTC CTT GTA GTC GGC GAA CAC C-3′ (reverse) and HindIII and EcoRI restriction sites (the original signal sequence consisting of 18 amino acids was removed). Additionally, the FZD6 sequence was cloned into this construct with primers 5′-AAA GAA TTC CAC AGT CTC TTC ACC TGT GAA C-3′ (forward) and 5′-TTT CTA GAT CAA GTA TCT GAA TGA CAA CCA CCT CC-3′ (reverse) with EcoRI and XbaI restriction sites from the FZD6-GFP plasmid (23). The His6 tag was added to the C terminus via site-directed mutagenesis using primers 5′-GGG AGG TGG TTG TCA TTC AGA TAC TCA TCA CCA TCA CCA TCA CTG ATC TAG AGG GCC CGT TTA AAC CC-3′ (forward) and 5′-GGG TTT AAA CGG GCC CTC TAG ATC AGT GAT GGT GAT GGT GAT GAG TAT CTG AAT GAC AAC CAC CTC CC-3′ (reverse). The truncated versions were cloned from the full-length construct with restriction sites EcoRI and XbaI using primers 5′-AAA GAA TTC GAC ATT GGA TTT TGG TGT CCA AGG C-3′ (forward) for ΔCRD and 5′-AAA GAA TTC GCG CCT CCA TGC CC-3′ (forward) for the ΔLinker together with 5′-TTT TCT AGA TCA GTG ATG GTG ATG GTG ATG-3′ (reverse) and inserted into the construct with the signal sequence and FLAG tag. The truncation mutants FZD6 Δ1–159, FZD6 Δ1–160, and FZD6 Δ1–161 were made in a similar way with primers 5′-AAA GAA TTC TGG TGT CCA AGG CAT CTT AAG AC-3′, 5′-AAA GAA TTC TGT CCA AGG CAT CTT AAG ACT TCT G-3′, and 5′-AAA GAA TTC CCA AGG CAT CTT AAG ACT TCT GG-3′, respectively. The Cys-to-Ala mutant C161A was made from the full length using primers 5′-CAA AGA GAC ATT GGA TTT TGG GCT CCA AGG CAT CTT AAG ACT TC-3′ (forward) and 5′-GAA GTC TTA AGA TGC CTT GGA GCC CAA AAT CCA ATG TCT CTT TG-3′ (reverse) and from ΔCRD with primers 5′-GAA TTC GAC ATT GGA TTT TGG GCT CCA AGG CAT CTT AAG ACT TC-3′ (forward) and 5′-GAA GTC TTA AGA TGC CTT GGA GCC CAA AAT CCA ATG TCG AAT TC-3′ (reverse). C181A was made from the full-length and ΔCRD FZD6 with primers 5′-CTG GGA ATT GAC CAG GCT GCG CCT CCA TGC CC-3′ (forward) and 5′-GGG CAT GGA GGC GCA GCC TGG TCA ATT CCC AG-3′ (reverse) and C185A with primers 5′-GAC CAG TGT GCG CCT CCA GCC CCC AAC ATG TAT TT-3′ (forward) and 5′-GAC CAG TGT GCG CCT CCA GCC CCC AAC ATG TAT TT-3′ (reverse). DVL2-HA and DVL2-DEP-GFP were kind gifts from Mariann Bienz (13).
Cell culture, treatments, and transfection
HEK293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium with 1% penicillin/streptomycin, 1% l-glutamine, and 10% fetal bovine serum (all from Invitrogen) in a humidified 5% CO2 incubator at 37 °C. Untransfected cells were treated with 2-[4-(2-methylpyridin-4-yl)phenyl]-N-[4-(pyridin-3-yl)phenyl]acetamide (C59, Abcam, ab142216) 1 day after seeding. The medium was changed to serum-free medium containing 1 μm C59 for approximately 24 h. DMSO (Merck, 8.02912.1000) was used as a control. In transfected cells, the medium change to C59-containing serum free-medium was done 24 h after transfection. Treatment with C59 was maintained for approximately 24 h. Cells were typically transfected 24 h after seeding with Lipofectamine 2000 (Invitrogen, 11668019).
Immunoblotting
HEK293T cells were seeded in 24-well plates and transfected with 0.3 μg of FZD and 0.05 μg of DVL DNA per well; control conditions were balanced with pcDNA. The DNA amount for DVL2-DEP-GFP was decreased to 0.03 μg. Cells were lysed 48 h after transfection in Laemmli buffer. Lysates were sonicated and separated by SDS-PAGE/immunoblotting using 8% or 10% gels. Transfer to a polyvinylidene difluoride membrane was done with the Trans-Blot® Turbo Transfer System (Bio-Rad). Membranes were incubated in 5% low-fat milk in TBST and subsequently in primary antibodies overnight at 4 °C. The next day, the membranes were washed three times in TBST (25 mm Tris, 150 mm NaCl, 0.05% Tween 20, pH 7.6), incubated with the suitable secondary antibody conjugated to horseradish peroxidase (Pierce), washed, and developed using ClarityTM Western ECL Substrate (Bio-Rad) according to the manufacturer's instructions. Primary antibodies were as follows: FZD6 (Novus Biologicals, NBP1-89702, rabbit, dilution 1:1000), His (Abcam, ab18184, mouse, 1:1000), FLAG (Sigma-Aldrich, F7425, rabbit, 1:1000), GAPDH (Cell Signaling Technology, 2118, rabbit, 1:4000), α-tubulin (Sigma-Aldrich, T6199, mouse, 1:2000), and DVL2 (Cell Signaling Technology, 3216, rabbit, 1:1000). The FZD-induced electrophoretic mobility shift of DVL2 was quantified by densitometry of the three predominant DVL2 bands in ImageLab (Bio-Rad). After background subtraction, the ratios of the values of top/bottom and middle/bottom bands were calculated. Those ratios were normalized by the average of the ratio values from each corresponding dataset (with/without treatment, separately for each experiment).
Indirect immunofluorescence and confocal microscopy
One day prior to transfection, 100,000 cells/well were seeded on coverslips coated with poly-d-lysine (Sigma-Aldrich, P0899) in 24-well plates. Cells were fixed with 4% paraformaldehyde in PBS, washed three times with PBS, and blocked with PBTA (3% BSA, 0.25% Triton X-100, and 0.01% NaN3 in PBS) for 1 h. Then they were incubated with the primary antibodies anti-HA (Covance, MMS-101P, mouse, 1:1000) and anti-FLAG (Sigma-Aldrich, F7425, rabbit, 1:1000) in PBTA overnight at 4 °C. The next day, the cells were washed three times with PBS, blocked in PBTA for 30 min, incubated with fluorophore-conjugated secondary antibody (Fitzgerald, 1:500) diluted in PBTA for 2 h at room temperature in the dark, washed three times with PBS, incubated with DAPI for 5 min, washed with PBS, and mounted using glycerol/gelatin (Sigma). For the surface staining, the primary anti-FLAG antibody was added to the medium at a concentration of 1:500 and incubated for 7 min at room temperature. Cells were washed in PBS, fixed with 4% PFA, washed three times with PBS, blocked for 30 min with PBTA, and detected as described above. Staining was visualized using a Zeiss LSM710 confocal microscope.
Cell ELISA
For quantification of cell surface receptor expression, 5 × 104 HEK293T cells were plated in 96-well plates coated with 0.1 mg/ml poly-d-lysine. Cells were transfected with 0.1 μg of the indicated constructs and maintained for an additional 24 h. Cells were then incubated with an anti-FLAG antibody (Sigma-Aldrich, F7425, rabbit, 1:1000) in 1% BSA/PBS for 1 h at 4 °C. Following incubation, cells were washed five times with 0.5% BSA in PBS and probed with an horseradish peroxidase–conjugated goat anti-rabbit antibody at a 1:4000 dilution in 1% BSA/PBS for 1 h at 4 °C. The cells were washed five times with 0.5% BSA/PBS, and 100 μl of the peroxidase substrate 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich, T8665) was added (30 min at room temperature). After acidification with 100 μl of 2 m HCl, the yellow color was read at 450 nm using a POLARstar Omega plate reader (BMG Labtech). The basal fluorescence detected in pcDNA-transfected HEK293T cells was subtracted from all data, and mean values were normalized to FL FZD6.
MAFFT alignment
The multiple alignment of the extracellular domains and the ECL1 of the human class F receptors was done in MAFFT version 7, which is available online (https://mafft.cbrc.jp/alignment/server/). 3 Default settings were used.
Statistical analysis
For graphical and statistical analysis, GraphPad Prism 5 and 6 software was used. For cell ELISA, values from technical triplicates were averaged, and pcDNA (background) was subtracted from the experimental conditions. Values were normalized to surface expression of full-length FZD6, which was set to 100%. Statistical significance was determined by one-way ANOVA with Dunnett′s multiple comparison test (Figs. 2D, 6D, and 7C). The data are presented as mean ± S.D. of three independent experiments, performed in triplicates. Significance levels are given as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. For the DVL2 shift (Fig. 3, B and C, and Fig. S1B), statistical significance was determined from three independent experiments by one-way ANOVA with Tukey's multiple comparisons test. Significance levels are given as follows: *p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
J. V., R. K. S., and G. S. conceptualization; J. V. data curation; J. V., M. K.-J., and G. S. formal analysis; J. V. and M. K.-J. validation; J. V., M. K.-J., and G. S. investigation; J. V., M. K.-J., and G. S. visualization; J. V., M. K.-J., and R. K. S. methodology; J. V. and G. S. writing-original draft; J. V., M. K.-J., R. K. S., and G. S. writing-review and editing; R. K. S. resources; R. K. S. and G. S. supervision; G. S. funding acquisition; G. S. project administration.
Supplementary Material
Acknowledgments
We thank Mariann Bienz for providing plasmids and Greg Slodkowicz and Madan M. Babu for consultation regarding sequence alignment and population genetics. We also thank Dr. Prahnesh Akshayalingam Venkataraman for helpful discussions.
This work was supported by grants from Karolinska Institutet (2-70/2014-76), the Swedish Research Council (2013-5708 and 2015-02899), the Swedish Cancer Society (CAN2014/659 and 2017/561), the Knut and Alice Wallenberg Foundation (KAW2008.0149), Stiftelsen Olle Engkvist Byggmästare (2016/193), SSMF (Travel Grant 2016/193 to J. V.), the Marie Curie ITN WntsApp (Grant 608180), the Deutsche Forschungsgemeinschaft (German Research Foundation, grant KO 5463/1-1), and NIGMS, National Institutes of Health Grant RO1-GM083118 (to R. K. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- GPCR
- G protein–coupled receptor
- SMO
- Smoothened
- FZD
- Frizzled
- CRD
- cysteine-rich domain
- 7TM domain
- seven transmembrane–spanning domain
- DVL
- Dishevelled
- DEP
- Dishevelled, Egl-10 and pleckstrin domain
- ECL
- extracellular loop
- aa
- amino acids
- FL
- full-length
- HEK
- human embryonic kidney
- HA
- hemagglutinin
- FEVR
- familial exudative vitreoretinopathy
- C59
- 2-[4-(2-methylpyridin-4-yl)phenyl]-N-[4-(pyridin-3-yl)phenyl]acetamide
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole
- ANOVA
- analysis of variance.
References
- 1. Schulte G. (2010) International Union of Basic and Clinical Pharmacology: LXXX: the class Frizzled receptors. Pharmacol. Rev. 62, 632–667 10.1124/pr.110.002931 [DOI] [PubMed] [Google Scholar]
- 2. Grainger S., and Willert K. (2018) Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 10, e1422 10.1002/wsbm.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janda C. Y., Waghray D., Levin A. M., Thomas C., and Garcia K. C. (2012) Structural basis of Wnt recognition by Frizzled. Science 337, 59–64 10.1126/science.1222879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nile A. H., Mukund S., Stanger K., Wang W., and Hannoush R. N. (2017) Unsaturated fatty acyl recognition by Frizzled receptors mediates dimerization upon Wnt ligand binding. Proc. Natl. Acad. Sci. U.S.A. 114, 4147–4152 10.1073/pnas.1618293114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeBruine Z. J., Eric Xu H. E., and Melcher K. (2017) Assembly and Architecture of the Wnt/β-catenin signalosome at the membrane. Br. J. Pharmacol. 174, 4564–4574 10.1111/bph.14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeBruine Z. J., Ke J., Harikumar K. G., Gu X., Borowsky P., Williams B. O., Xu W., Miller L. J., Xu H. E., and Melcher K. (2017) Wnt5a promotes Frizzled-4 signalosome assembly by stabilizing cysteine-rich domain dimerization. Genes Dev. 31, 916–926 10.1101/gad.298331.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mlodzik M. (2016) The Dishevelled protein family: still rather a mystery after over 20 years of molecular studies. Curr. Top Dev. Biol. 117, 75–91 10.1016/bs.ctdb.2015.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao C., and Chen Y. G. (2010) Dishevelled: The hub of Wnt signaling. Cell Signal. 22, 717–727 10.1016/j.cellsig.2009.11.021 [DOI] [PubMed] [Google Scholar]
- 9. Umbhauer M., Djiane A., Goisset C., Penzo-Méndez A., Riou J. F., Boucaut J. C., and Shi D. L. (2000) The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/β-catenin signalling. EMBO J. 19, 4944–4954 10.1093/emboj/19.18.4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tauriello D. V., Jordens I., Kirchner K., Slootstra J. W., Kruitwagen T., Bouwman B. A., Noutsou M., Rüdiger S. G., Schwamborn K., Schambony A., and Maurice M. M. (2012) Wnt/β-catenin signaling requires interaction of the Dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc. Natl. Acad. Sci. U.S.A. 109, E812–E820 10.1073/pnas.1114802109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simons M., Gault W. J., Gotthardt D., Rohatgi R., Klein T. J., Shao Y., Lee H. J., Wu A. L., Fang Y., Satlin L. M., Dow J. T., Chen J., Zheng J., Boutros M., and Mlodzik M. (2009) Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat. Cell Biol. 11, 286–294 10.1038/ncb1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertalovitz A. C., Pau M. S., Gao S., Malbon C. C., and Wang H. Y. (2016) Frizzled-4 C-terminus distal to KTXXXW motif is essential for normal Dishevelled recruitment and Norrin-stimulated activation of Lef/Tcf-dependent transcriptional activation. J. Mol. Signal. 11, 1 10.5334/1750-2187-11-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gammons M. V., Renko M., Johnson C. M., Rutherford T. J., and Bienz M. (2016) Wnt signalosome assembly by DEP domain swapping of Dishevelled. Mol. Cell 64, 92–104 10.1016/j.molcel.2016.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gammons M. V., Rutherford T. J., Steinhart Z., Angers S., and Bienz M. (2016) Essential role of the Dishevelled DEP domain in a Wnt-dependent human-cell-based complementation assay. J. Cell Sci. 129, 3892–3902 10.1242/jcs.195685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paclíková P., Bernatik O., Radaszkiewicz T. W., and Bryja V. (2017) N-terminal part of Dishevelled DEP domain is required for Wnt/β-catenin signaling in mammalian cells. Mol. Cell Biol. 37, MCB.00145-17 10.1128/MCB.00145-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strakova K., Matricon P., Yokota C., Arthofer E., Bernatik O., Rodriguez D., Arenas E., Carlsson J., Bryja V., and Schulte G. (2017) The tyrosine Y250(2.39) in Frizzled 4 defines a conserved motif important for structural integrity of the receptor and recruitment of Disheveled. Cell Signal. 38, 85–96 10.1016/j.cellsig.2017.06.018 [DOI] [PubMed] [Google Scholar]
- 17. Pau M. S., Gao S., Malbon C. C., Wang H. Y., and Bertalovitz A. C. (2015) The intracellular loop 2 F328S Frizzled-4 mutation implicated in familial exudative vitreoretinopathy impairs Dishevelled recruitment. J. Mol. Signal. 10, 5 10.5334/1750-2187-10-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bilic J., Huang Y. L., Davidson G., Zimmermann T., Cruciat C. M., Bienz M., and Niehrs C. (2007) Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622 10.1126/science.1137065 [DOI] [PubMed] [Google Scholar]
- 19. Golan T., Yaniv A., Bafico A., Liu G., and Gazit A. (2004) The human Frizzled 6 (HFz6) acts as a negative regulator of the canonical Wnt β-catenin signaling cascade. J. Biol. Chem. 279, 14879–14888 10.1074/jbc.M306421200 [DOI] [PubMed] [Google Scholar]
- 20. Sheldahl L. C., Park M., Malbon C. C., and Moon R. T. (1999) Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 9, 695–698 10.1016/S0960-9822(99)80310-8 [DOI] [PubMed] [Google Scholar]
- 21. Kilander M. B., Dahlström J., and Schulte G. (2014) Assessment of Frizzled 6 membrane mobility by FRAP supports G protein coupling and reveals WNT-Frizzled selectivity. Cell Signal. 26, 1943–1949 10.1016/j.cellsig.2014.05.012 [DOI] [PubMed] [Google Scholar]
- 22. Kilander M. B., Petersen J., Andressen K. W., Ganji R. S., Levy F. O., Schuster J., Dahl N., Bryja V., and Schulte G. (2014) Disheveled regulates precoupling of heterotrimeric G proteins to Frizzled 6. FASEB J. 28, 2293–2305 10.1096/fj.13-246363 [DOI] [PubMed] [Google Scholar]
- 23. Petersen J., Wright S. C., Rodríguez D., Matricon P., Lahav N., Vromen A., Friedler A., Strömqvist J., Wennmalm S., Carlsson J., and Schulte G. (2017) Agonist-induced dimer dissociation as a macromolecular step in G protein-coupled receptor signaling. Nat. Commun. 8, 226 10.1038/s41467-017-00253-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie Z., Khair M., Shaukat I., Netter P., Mainard D., Barré L., and Ouzzine M. (2018) Non-canonical Wnt induces chondrocyte de-differentiation through Frizzled 6 and DVL-2/B-raf/CaMKIIα/syndecan 4 axis. Cell Death Differ. 25, 1442–1456 10.1038/s41418-017-0050-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y., Guo N., and Nathans J. (2006) The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 26, 2147–2156 10.1523/JNEUROSCI.4698-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Proffitt K. D., Madan B., Ke Z., Pendharkar V., Ding L., Lee M. A., Hannoush R. N., and Virshup D. M. (2013) Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 73, 502–507 10.1158/0008-5472.CAN-12-2258 [DOI] [PubMed] [Google Scholar]
- 27. Zhang X., Zhao F., Wu Y., Yang J., Han G. W., Zhao S., Ishchenko A., Ye L., Lin X., Ding K., Dharmarajan V., Griffin P. R., Gati C., Nelson G., Hunter M. S., et al. (2017) Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 8, 15383 10.1038/ncomms15383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., and Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 10.1002/pmic.200300771 [DOI] [PubMed] [Google Scholar]
- 29. Steentoft C., Vakhrushev S. Y., Joshi H. J., Kong Y., Vester-Christensen M. B., Schjoldager K. T., Lavrsen K., Dabelsteen S., Pedersen N. B., Marcos-Silva L., Gupta R., Bennett E. P., Mandel U., Brunak S., Wandall H. H., et al. (2013) Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488 10.1038/emboj.2013.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernatik O., Ganji R. S., Dijksterhuis J. P., Konik P., Cervenka I., Polonio T., Krejci P., Schulte G., and Bryja V. (2011) Sequential activation and inactivation of Dishevelled in the Wnt/β-catenin pathway by casein kinases. J. Biol. Chem. 286, 10396–10410 10.1074/jbc.M110.169870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernatík O., Šedová K., Schille C., Ganji R. S., ervenka I., Trantírek L., Schambony A., Zdráhal Z., and Bryja V. (2014) Functional analysis of dishevelled-3 phosphorylation identifies distinct mechanisms driven by casein kinase 1 and frizzled5. J. Biol. Chem. 289, 23520–23533 10.1074/jbc.M114.590638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cong F., Schweizer L., and Varmus H. (2004) Wnt signals across the plasma membrane to activate the β-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development 131, 5103–5115 10.1242/dev.01318 [DOI] [PubMed] [Google Scholar]
- 33. Schwarz-Romond T., Fiedler M., Shibata N., Butler P. J., Kikuchi A., Higuchi Y., and Bienz M. (2007) The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 14, 484–492 10.1038/nsmb1247 [DOI] [PubMed] [Google Scholar]
- 34. Bryja V., Schulte G., Rawal N., Grahn A., and Arenas E. (2007) Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cell Sci. 120, 586–595 10.1242/jcs.03368 [DOI] [PubMed] [Google Scholar]
- 35. Huang P., Zheng S., Wierbowski B. M., Kim Y., Nedelcu D., Aravena L., Liu J., Kruse A. C., and Salic A. (2018) Structural basis of Smoothened activation in Hedgehog signaling. Cell 174, 312–324.e16 10.1016/j.cell.2018.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Byrne E. F. X., Sircar R., Miller P. S., Hedger G., Luchetti G., Nachtergaele S., Tully M. D., Mydock-McGrane L., Covey D. F., Rambo R. P., Sansom M. S. P., Newstead S., Rohatgi R., and Siebold C. (2016) Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 10.1038/nature18934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang C., Wu H., Evron T., Vardy E., Han G. W., Huang X. P., Hufeisen S. J., Mangano T. J., Urban D. J., Katritch V., Cherezov V., Caron M. G., Roth B. L., and Stevens R. C. (2014) Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355 10.1038/ncomms5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang C., Wu H., Katritch V., Han G. W., Huang X. P., Liu W., Siu F. Y., Roth B. L., Cherezov V., and Stevens R. C. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 10.1038/nature12167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang S., Wu Y., Xu T. H., de Waal P. W., He Y., Pu M., Chen Y., DeBruine Z. J., Zhang B., Zaidi S. A., Popov P., Guo Y., Han G. W., Lu Y., Suino-Powell K., et al. (2018) Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 560, 666–670 10.1038/s41586-018-0447-x [DOI] [PubMed] [Google Scholar]
- 40. Arthofer E., Hot B., Petersen J., Strakova K., Jäger S., Grundmann M., Kostenis E., Gutkind J. S., and Schulte G. (2016) WNT stimulation dissociates a Frizzled 4 inactive-state complex with Gα12/13. Mol. Pharmacol. 90, 447–459 10.1124/mol.116.104919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wong H. C., Bourdelas A., Krauss A., Lee H. J., Shao Y., Wu D., Mlodzik M., Shi D. L., and Zheng J. (2003) Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol. Cell 12, 1251–1260 10.1016/S1097-2765(03)00427-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strutt H., Price M. A., and Strutt D. (2006) Planar polarity is positively regulated by casein kinase Iϵ in Drosophila. Curr. Biol. 16, 1329–1336 10.1016/j.cub.2006.04.041 [DOI] [PubMed] [Google Scholar]
- 43. Cong F., Schweizer L., and Varmus H. (2004) Casein kinase Iϵ modulates the signaling specificities of dishevelled. Mol. Cell Biol. 24, 2000–2011 10.1128/MCB.24.5.2000-2011.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bang I., Kim H. R., Beaven A. H., Kim J., Ko S. B., Lee G. R., Lee H., Im W., Seok C., Chung K. Y., and Choi H. J. (2018) Biophysical and functional characterization of Norrin signaling through Frizzled4. Proc. Natl. Acad. Sci. U.S.A. 115, 8787–8792 10.1073/pnas.1805901115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rana R., Carroll C. E., Lee H. J., Bao J., Marada S., Grace C. R., Guibao C. D., Ogden S. K., and Zheng J. J. (2013) Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat. Commun. 4, 2965 10.1038/ncomms3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aanstad P., Santos N., Corbit K. C., Scherz P. J., Trinh le A., Salvenmoser W., Huisken J., Reiter J. F., and Stainier D. Y. (2009) The extracellular domain of Smoothened regulates ciliary localization and is required for high-level Hh signaling. Curr. Biol. 19, 1034–1039 10.1016/j.cub.2009.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taipale J., Cooper M. K., Maiti T., and Beachy P. A. (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897 10.1038/nature00989 [DOI] [PubMed] [Google Scholar]
- 48. Nakano Y., Nystedt S., Shivdasani A. A., Strutt H., Thomas C., and Ingham P. W. (2004) Functional domains and sub-cellular distribution of the Hedgehog transducing protein Smoothened in Drosophila. Mech. Dev. 121, 507–518 10.1016/j.mod.2004.04.015 [DOI] [PubMed] [Google Scholar]
- 49. Povelones M., and Nusse R. (2005) The role of the cysteine-rich domain of Frizzled in Wingless-Armadillo signaling. EMBO J. 24, 3493–3503 10.1038/sj.emboj.7600817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen C. M., Strapps W., Tomlinson A., and Struhl G. (2004) Evidence that the cysteine-rich domain of Drosophila frizzled family receptors is dispensable for transducing wingless. Proc. Natl. Acad. Sci. U.S.A. 101, 15961–15966 10.1073/pnas.0407103101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen W. S., Antic D., Matis M., Logan C. Y., Povelones M., Anderson G. A., Nusse R., and Axelrod J. D. (2008) Asymmetric homotypic interactions of the atypical cadherin flamingo mediate intercellular polarity signaling. Cell 133, 1093–1105 10.1016/j.cell.2008.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu J., and Mlodzik M. (2008) The frizzled extracellular domain is a ligand for Van Gogh/Stbm during nonautonomous planar cell polarity signaling. Dev. Cell 15, 462–469 10.1016/j.devcel.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y., Sun B., Feng D., Hu H., Chu M., Qu Q., Tarrasch J. T., Li S., Sun Kobilka T., Kobilka B. K., and Skiniotis G. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 10.1038/nature22394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Castro M., Nikolaev V. O., Palm D., Lohse M. J., and Vilardaga J. P. (2005) Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 16084–16089 10.1073/pnas.0503942102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Willert K., Brown J. D., Danenberg E., Duncan A. W., Weissman I. L., Reya T., Yates J. R. 3rd, and Nusse R. (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452 10.1038/nature01611 [DOI] [PubMed] [Google Scholar]
- 56. Dijksterhuis J. P., Baljinnyam B., Stanger K., Sercan H. O., Ji Y., Andres O., Rubin J. S., Hannoush R. N., and Schulte G. (2015) Systematic mapping of WNT-Frizzled interactions reveals functional selectivity by distinct WNT-Frizzled pairs. J. Biol. Chem. 290, 6789–6798 10.1074/jbc.M114.612648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Milhem R. M., Ben-Salem S., Al-Gazali L., and Ali B. R. (2014) Identification of the cellular mechanisms that modulate trafficking of frizzled family receptor 4 (FZD4) missense mutants associated with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 55, 3423–3431 10.1167/iovs.14-13885 [DOI] [PubMed] [Google Scholar]
- 58. Zhang K., Harada Y., Wei X., Shukla D., Rajendran A., Tawansy K., Bedell M., Lim S., Shaw P. X., He X., and Yang Z. (2011) An essential role of the cysteine-rich domain of FZD4 in Norrin/Wnt signaling and familial exudative vitreoretinopathy. J. Biol. Chem. 286, 10210–10215 10.1074/jbc.M110.194399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Omoto S., Hayashi T., Kitahara K., Takeuchi T., and Ueoka Y. (2004) Autosomal dominant familial exudative vitreoretinopathy in two Japanese families with FZD4 mutations (H69Y and C181R). Ophthalmic Genet. 25, 81–90 10.1080/13816810490514270 [DOI] [PubMed] [Google Scholar]
- 60. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.