

Abstract
The allosteric coupling of the highly conserved nucleotide- and substrate-binding domains of Hsp70 has been studied intensively. In contrast, the role of the disordered, highly variable C-terminal region of Hsp70 remains unclear. In many eukaryotic Hsp70s, the extreme C-terminal EEVD motif binds to the tetratricopeptide-repeat domains of Hsp70 co-chaperones. Here, we discovered that the TVEEVD sequence of Saccharomyces cerevisiae cytoplasmic Hsp70 (Ssa1) functions as a SUMO-interacting motif. A second C-terminal motif of ∼15 amino acids between the α-helical lid and the extreme C terminus, previously identified in bacterial and eukaryotic organellar Hsp70s, is known to enhance chaperone function by transiently interacting with folding clients. Using structural analysis, interaction studies, fibril formation assays, and in vivo functional assays, we investigated the individual contributions of the α-helical bundle and the C-terminal disordered region of Ssa1 in the inhibition of fibril formation of the prion protein Ure2. Our results revealed that although the α-helical bundle of the Ssa1 substrate-binding domain (SBDα) does not directly bind to Ure2, the SBDα enhances the ability of Hsp70 to inhibit fibril formation. We found that a 20-residue C-terminal motif in Ssa1, containing GGAP and GGAP-like tetrapeptide repeats, can directly bind to Ure2, the Hsp40 co-chaperone Ydj1, and α-synuclein, but not to the SUMO-like protein SMT3 or BSA. Deletion or substitution of the Ssa1 GGAP motif impaired yeast cell tolerance to temperature and cell-wall damage stress. This study highlights that the C-terminal GGAP motif of Hsp70 is important for substrate recognition and mediation of the heat shock response.
Keywords: amyloid, chaperone, heat shock protein (HSP), nuclear magnetic resonance (NMR), protein structure, Saccharomyces cerevisiae, stress response, SUMO-interacting motif (SIM), prion, proline-rich motif
Introduction
Heat shock protein 70 (Hsp70) 6 is an indispensable part of the protein quality control system in the cell and facilitates folding of nascent polypeptides, inhibits protein aggregation, and assists refolding of misfolded and aggregated proteins, transport of proteins across membranes, and regulation of assembly and disassembly of macromolecular complexes (1, 2). The Hsp70 family shares a highly conserved architecture, consisting of two individual domains, an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD), connected by a flexible linker (see Fig. 1A) (3). Intensive research has revealed that the physiological functions of Hsp70 proteins are achieved by allosteric cooperation between the NBD and SBD driven by an ATP-hydrolysis functional cycle assisted by the co-chaperone heat shock protein 40 (Hsp40) and nucleotide exchange factors (NEFs) (4, 5). The SBD can be further divided into a substrate-binding β-sheet domain (SBDβ), an α-helical lid domain (SBDα), and a C-terminal intrinsically disorder region (C-IDR) (see Fig. 1A) (6). There is highly regulated and coordinated interdomain communication between the SBD and NBD, mediated through an amino acid network and domain interfaces (7, 8). The SBDα of Hsp70 family members generally consists of five α-helixes followed by a C-IDR at the extreme C-terminal end. The first α-helix rests on the SBDβ, and the last four α-helixes form a helix-bundle structure that acts as a lid to cover the SBDβ. The SBDα and C-IDR make up the C-terminal domain (CTD) of Hsp70. The Hsp70 NBD and SBDβ are highly conserved in all species, whereas the SBDα is less conserved, and the C-IDR is highly variable.
Figure 1.

Structure of Ssa1 CTD. A, schematic of Ssa1 domain structure. B, ribbon representation of the Ssa1(523–622) structure. Red, intensity ratio < mean − S.D. caused by addition of Ure2 with ratio 1:1; yellow, proline or severely overlapped residues. C, superimposition of the SBDα of yeast Ssa1 (gray), E. coli DnaK (green), and human HspA1A (yellow). D, the GGAP-like motif and EEVD motif in eukaryotic cytosolic members of the Hsp70 family. The C-terminal GGAP-like repetitive sequence is indicated by a jagged line. The numbers indicate residues of yeast Ssa1.
The structure of the SBDα has been found to fluctuate during the functional cycle of Hsp70. The SBDα influences the affinity and kinetic interaction parameters of Escherichia coli DnaK with its substrate and is proposed to possess one or several sites for transient interaction with protein substrates, helping Hsp70 to refold proteins (9). The SBDα of Hsp70 is also crucial for binding to the J domain of the co-chaperone HSJ1a by providing a suitable conformation for binding, rather than directly binding to the J domain (10). Up to now, the function of the C-IDR remains much less well-understood, in contrast to the increasingly detailed picture of the allosteric mechanism controlling substrate binding of Hsp70 (11). The C-IDR generally consists of 30–40 amino acids. Some eukaryotic cytosolic Hsp70s end with a conserved tetratricopeptide repeat domain interaction motif (-EEVD), which mediates binding with co-chaperones, such as Hsp40, CHIP, HIP, and HOP, that facilitate the assembly of a variety of Hsp70 complexes (11–13). Smock et al. (11) have identified a ∼15-residue motif in the C terminus of DnaK that can directly bind misfolded protein substrates, providing a weak, auxiliary binding site for denatured substrate proteins and enhancing the protein refolding efficiency of DnaK. This motif generally occurs in bacterial Hsp70s and is also found in eukaryotic organellar Hsp70s and archaea, whereas other sequence variations have been hypothesized to substitute for this motif in eukaryotic cytoplasmic Hsp70 C-IDRs (11).
In studies of the effects of Hsp70 on amyloid fibril formation, it has been reported that the isolated Hsp70 SBD can inhibit the amyloid fibril formation of α-synuclein and Ure2, whereas the presence of the NBD modulates this fibril inhibition activity (14–16). Ure2 is a nitrogen metabolism regulator in Saccharomyces cerevisiae, which can give rise to the prion state [URE3] (17). Ure2 contains two distinct domains, a relatively flexible N-terminal prion domain and a C-terminal globular functional domain (18). The yeast Hsp70, Ssa1, is one of nine cytosolic Hsp70s (Ssa1-4, Ssb1-2, Sse1-2, and Ssz1) in S. cerevisiae (19). Ssa proteins are involved in a variety of cellular processes, such as translation, translocation, and general protein folding (20–22). SSA1 and SSA2 are constitutively expressed, and their gene products are 96% identical, whereas SSA3 and SSA4 are heat shock—inducible, and their gene products are both 80% identical to Ssa1 and Ssa2. Ssa1 can inhibit the fibril formation of the amyloid protein Ure2 without ATP (23). It also blocks Sup35 amyloid fibril formation in vitro with the assistance of the Hsp40 Ydj1 (24).
Previous work in our lab demonstrated that in the absence of the NBD, the Ssa1 SBD can delay fibril formation of Ure2, whereas deletion of the C-IDR significantly decreases the inhibition activity, and further truncation of the α-helical bundle leads to further reduction in inhibition activity (16). This is similar to the case of human Hsp70, where the SBD alone can inhibit the fibril formation of α-synuclein, and the SBDα enhances the inhibition effect (15). However, the role of the Ssa1 C-IDR and SBDα in Ure2 fibril inhibition is not clear. Here, we determined the NMR structures of the Ssa1 SBDα and SBDβ domains, which are the first structures for yeast canonical Hsp70s reported to date. Combining structural and biochemical results, we demonstrate that the Ssa1 C-IDR directly interacts with Ure2 through a 20-residue GGAP motif in the C-IDR, whereas the intact SBDα enhances the Ssa1 inhibition ability without directly binding to Ure2. Further, we detected a role of the GGAP motif in mediating response to stress in vivo.
Results
The C-terminal GGAP motif of Ssa1 directly binds to Ure2
The domain structure of Ssa1 is shown in Fig. 1A. Previous results from our lab indicate that mutants of the Ssa1 SBD with different degrees of truncation of the SBDα have varying degrees of fibril inhibition ability, with Ssa1(382–554) ≈ Ssa1(382–582) < Ssa1(382–602) ≈ Ssa1(382–612) < Ssa1(382–622) ≈ Ssa1(382–642) (16). This indicates the importance of the SBDα and C-IDR in the inhibition of Ure2 fibril formation. To obtain structural insight into the role of the SBDα and C-IDR in fibril inhibition, we first resolved the NMR structure of Ssa1(523–622) (Table 1), which represents the Ssa1 CTD lacking 20 residues of disordered sequence at the C-terminal end (Fig. 1A). This is the first reported structure of an SBDα domain for the Ssa family of yeast Hsp70s. The overall structure of Ssa1(523–622) compromises four α-helices (αB: 523–551, αC: 558–577, αD: 583–598, and αE: 599–605) and a flexible C-terminal tail (positions 606–622) (Fig. 1, B and C). This structure is similar to the same region in DnaK (PDB code 1DKZ) and HspA1A (PDB code 4PO2) (6, 25). The helices αB–αE form an α-helical bundle, showing a 3.7 Å root-mean-square deviation compared with the SBDα of E. coli DnaK and a 1.5 Å root-mean-square deviation compared with the SBDα of human HspA1A (hHsp70) (Fig. 1C), indicating that the α-helical bundle of the yeast Ssa1 SBDα is more similar to that of hHsp70 than DnaK. This reflects the higher sequence identity (∼45%) between the SBDα of Ssa1 and hHsp70 versus identity of ∼25% between Ssa1 and DnaK. Interestingly, the αE in the α-helical bundle of Ssa1 seems to be slightly more compact than in hHsp70 but less compact than in DnaK (Fig. 1C).
Table 1.
The experimental restraints and structural statistics for the 20 lowest energy structures of Ssa1 SBDα(523–622) and SBDβ(382–554)
| SBDα(523–622) | SBDβ(382–554) | |
|---|---|---|
| Distance restraints | ||
| Intraresidue | 715 | 1169 |
| Sequential | 451 | 693 |
| Medium | 321 | 310 |
| Long-range | 144 | 839 |
| Ambiguous | 800 | 1887 |
| Total | 2431 | 4898 |
| Hydrogen-bond restraints | 108 | 100 |
| Dihedral-angle restraints | ||
| φ | 83 | 115 |
| ψ | 83 | 122 |
| χ1 | 66 | |
| Total | 166 | 303 |
| Violations | ||
| NOE violations (>0.3 Å) | 0 | 0 |
| Torsion angle violations (>5°) | 0 | 0 |
| PROCHECK statistics (%)a | ||
| Most favored regions | 96.5 | 89.1 |
| Additional allowed regions | 3.0 | 9.1 |
| Generously allowed regions | 0.2 | 0.8 |
| Disallowed regions | 0.3 | 1.0 |
| Root-mean-square deviation from mean structure (Å) | ||
| Backbone heavy atoms | ||
| All residueb | 0.64 ± 0.10 | 0.82 ± 0.10 |
| Regular secondary structurec | 0.56 ± 0.12 | 0.56 ± 0.09 |
| All heavy atoms | ||
| All residue | 1.09 ± 0.08 | 1.28 ± 0.09 |
| Regular secondary structure | 1.03 ± 0.09 | 0.94 ± 0.09 |
a Residues used to calculate PROCHECK statistics of SBDα(523–622) and SBDβ(382–554) include 523–622 and 392–542, respectively.
b Residues used to calculate root-mean-square deviation values of all residues include 523–608 and 392–542, respectively.
c Regular secondary structure regions used here for SBDα(523–622) include residues 523–551, 558–577, and 583–605 and for SBDβ(382–554) include residues 397–402, 406–411, 419–427, 434–441, 451–459, 471–477, 483–489, 495–500, 509–521, and 523–531.
We then titrated Ssa1(523–622) with Ure2, by monitoring the 2D 1H-15N HSQC spectra of 15N-labeled Ssa1(523–622). The NH peak intensity ratios for titration with Ure2 were mapped onto the structure of Ssa1(523–622) (Fig. 1B). Nine NH peaks, mainly corresponding to the region containing residues Gly609–Gly621 at the C terminus, show a significant decrease in intensity, indicating that this sequence can directly bind to Ure2. To further understand the role of the entire C-IDR of Ssa1 in the interaction with Ure2, we assigned the backbone resonances of Ssa1(523–642) (Fig. S1A), which contains the intact C terminus. Ssa1(523–642) was then titrated with Ure2, resulting in significant signal decrease of the NH peaks (Fig. 2, A and B). Chemical shift index analysis (Fig. S1B) and the negative steady-state HN-NOE value of Phe623–Asp642 (Fig. 2C) indicate that this segment is disordered; therefore a modeled structure of Ssa1(523–642) was derived based on the NMR-derived structure of Ssa1(523–622), onto which the titration result was mapped (Fig. 2D). We find that the NH peaks with decreased intensity during Ure2 titration mainly belong to a GGAP motif (sequence: 609GGAP GGAA GGAP GGFP GGAP628) in the flexible C-terminal region (Fig. 1D). Interestingly, the NH signal intensities for the segment 630APEAEGP636 did not decrease during Ure2 titration, although this is also a Gly/Ala/Pro-rich sequence. The dissociation constant (KD) for Ssa1(523–642) and Ure2 was measured as 14.7 ± 3.6 μm by FRET (Fig. 2E), demonstrating a direct interaction between Ssa1(523–642) and Ure2. This binding affinity is significantly weaker than that reported for Ure2 and full-length Ssa1 (KD ∼40 nm) (23), indicating that the primary substrate-binding site in the SBDβ is required for high affinity binding, and so the GGAP motif represents a secondary binding site that may regulate specificity of binding. The GGAP motif may also contribute to the kinetics of the chaperone mechanism in similar ways as the C-IDR of DnaK (11). To confirm the interaction of the GGAP motif with Ure2, we further titrated the 15N-labeled peptide of the Ssa1 C-terminal GGAP motif, 609GGAP GGAA GGAP GGFP GGAP628, with Ure2. Significant intensity decrease of NH signals clearly confirmed that the isolated peptide can also bind to Ure2 (Fig. 2G).
Figure 2.

Titration of Ssa1(523–642) and GGAP motif with Ure2. A, 1H-15N HSQC spectra of Ssa1(523–642) titrated with Ure2 at a 1:2 ratio. B, bar diagram of intensity ratio versus residue number. C, steady-state 1H-15N NOE value per residue of Ssa1(523–642). D, mapping of intensity ratio and CSP onto the Ssa1(523–642) structure. Red, intensity ratio < mean − S.D. in B; cyan, proline and unassigned residues; green, CSP > 0.003 ppm in F. E, KD fitting (14.7 ± 3.6 μm) of binding of Ssa1(523–642) to Ure2 measured by FRET. F, bar diagram of CSP versus residue number. G, 1H-15N HSQC spectra of the 20-residue GGAP motif in Ssa1 C-IDR titrated with Ure2 at a 1:2 ratio.
In addition to residues of the GGAP motif, Asp555 and Gln559 in loop L12 also give decreased intensities of NH signals because of addition of Ure2, but the intensity decreases do not occur for other L12 residues. Because the L12 loop is structurally close to the C-IDR (Fig. 2D), Asp555 and Gln559 may interact with Ure2. The N-terminal residues 524EEDEKE529 show slight chemical shift perturbations (CSPs) upon Ure2 addition (Fig. 2, D and F). These residues are all charged, with a net negative charge of −4. This means that these residues with CSP upon Ure2 addition may also have a nonspecific electrostatic interaction with Ure2. Because residues 524-EEDEKE-529 are located at the N terminus of the αB helix of the Ssa1 SBD and are located far from the substrate-binding site and the C-IDR in the structure, it seems unlikely that they contribute to substrate binding in the full-length Hsp70, except where the substrate has a long positively charged tail and can bind this negatively charged sequence.
To investigate the interaction specificity of the GGAP motif with Ure2, we also titrated Ssa1(523–642) with yeast Ydj1 and SMT3, human α-synuclein, and BSA. NH-signal intensities of the C-terminal EEVD motif are significantly decreased because of the addition of Ydj1 at a ratio of 1:3, indicating a direct interaction between the EEVD motif and Ydj1 (Fig. 3, A and B). In addition, the NH-signal intensities of the GGAP motif residues decreased by almost 50% after addition of Ydj1 (Fig. 3B), indicating a second binding site in the Ssa1 C-IDR for Ydj1. Titration of Ssa1(523–622) with Ydj1 at a ratio of 1:7 also decreased the NH-signal intensities of the GGAP motif residues by 40% (Fig. 3C), suggesting that the presence of the EEVD motif can enhance the binding of the GGAP motif to Ydj1 and that these two motifs bind Ydj1 combinatorially.
Figure 3.

Titration of Ssa1 SBD(523–642) with Ydj1 or BSA. A, 1H-15N HSQC spectra of Ssa1(523–642) titrated with Ydj1 at a ratio of 1:3. B, bar diagram of intensity ratio versus residue number of Ssa1(523–642) in A. C, bar diagram of intensity ratio of Ssa1(523–622) caused by addition of Ydj1 with ratio 1:7. D and E, bar diagram of intensity ratio of Ssa1(523–642) caused by addition of α-synuclein (D) and SMT3 (E) both with ratio 1:4. F, bar diagram of CSP versus residue number of Ssa1(523–642) caused by addition of SMT3 as in E. G, bar diagram of intensity ratio of Ssa1(523–642) caused by addition of BSA at a ratio of 1:3.
Interestingly, the NH-signal intensities of the GGAP motif residues also decreased after addition of α-synuclein, without decrease of the NH-signal intensities of the EEVD residues (Fig. 3D). Unexpectedly, the NH-signal intensities of the C-terminal six residues (-TVEEVD642) decreased after addition of the SUMO-like protein SMT3, without decrease of the NH-signal intensities of the GGAP motif residues (Fig. 3E). The NH-signals of the C-terminal six residues (-TVEEVD) have a significant CSP caused by the addition of SMT3 (Fig. 3F). We then titrated 15N-labeled SMT3 with the C-terminal peptide of Ssa1 (-PEAEGPTVEEVD) and observed significant CSPs (Fig. S2, A and B). A dissociation constant (KD) of 240 μm was obtained by fitting the NMR titration data (Fig. S2C). The CSP results were mapped onto the SMT3 structure (PDB code 1L2N) (26) to identify the SMT3 binding site for the TVEEVD peptide (Fig. S2D). Residues with large CSPs form a hydrophobic cavity (Leu26, Ile35, Phe37, Ile39, and Ala51) surrounded by positively charged residues (Lys38, Lys40, Lys41, Arg46, Arg47, and Arg55). This site is same as the binding site in SMT3 for other proteins containing the SUMO-interacting motif (SIM) (27), indicating that the TVEEVD sequence of Ssa1 is a newly identified SIM.
In a control experiment, the NH signals of Ssa1(523–642) did not show any significant CSPs or intensity decrease upon addition of BSA (Fig. 3G). Thus, the Ssa1 C-terminal GGAP motif has a degree of specificity to recognize certain interacting proteins, providing an additional interacting site within Ssa1 in addition to the SBDβ.
Blocking of the Ssa1 substrate site upon truncation of the α-helical bundle
After characterization of the role of the Ssa1 C-IDR, especially the GGAP motif (Gly609–Pro628) in Ure2 binding and fibril inhibition (16), we next wanted to figure out the role of the α-helical bundle in the fibril inhibition of Ssa1. As mentioned above, the inhibition ability of Ssa1 SBD truncations has been found to decrease with the degree of truncation (16), which seems to indicate that Ssa1(523–642) enhances function in a length-dependent manner. However, the structure of C-terminal truncation mutants of DnaK (PDB code 1BPR) and bovine Hsp70 (PDB code 1YUW) indicate that if only αB is present, it will collapse into the hydrophobic binding site of SBDβ (Fig. 4A).
Figure 4.
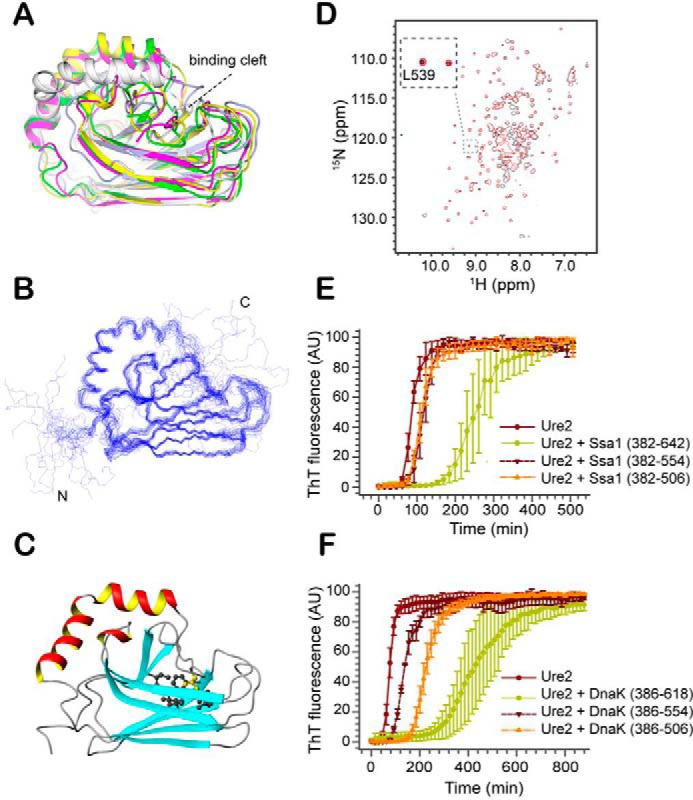
Effect of SBD truncation on the structure and fibril inhibition ability of Ssa1. A, structural superimposition of truncation mutants of SBDβ of different Hsp70 homologues, including S. cerevisiae Ssa1(382–554) (magenta), E. coli DnaK (white, PDB code 1BPR), G. kaustophilus DnaK (green, PDB code 2V7Y), rat Hsc70 (light blue, PDB code 1CKR), and bovine Hsc70 (yellow, PDB code 1YUW). B, the backbone ensemble of 20 structures of Ssa1(382–554). C, ribbon representation of the Ssa1(382–554) structure. Side chain of Leu539 (yellow) and surrounding hydrophobic side chains are shown by ball-and-stick model. The structural figures (A–C) were generated using MOLMOL (67) and PyMOL (68). D, overlay of the 2D 1H-15N HSQC spectra of Ssa1(382–554) and Ssa1(382–582). E, fibril formation of 30 μm Ure2 in the absence or presence of 50 μm Ssa1(382–642), Ssa1(382–554), and Ssa1(382–506), as indicated. F, fibril formation of 30 μm Ure2 in the absence or presence of 50 μm DnaK(384–638), DnaK(384–554), and DnaK(384–506), as indicated. E and F, the buffer conditions were 50 mm Tris-HCl, pH 8.4, 200 mm NaCl. The error bars indicate the standard error from at least three parallel experiments.
To confirm this in the case of the Ssa1 SBD, based on the chemical shift assignments of Ssa1(382–554) (28), we determined the solution structure of Ssa1(382–554) (Fig. 4, B and C, and Table 1). This is also the first reported structure of an SBDβ domain for the Ssa family of yeast Hsp70s. Similar to the DnaK and bovine Hsp70 truncation mutants, the secondary structure elements β1-β8, αA, and part of αB (residues 524–535) of Ssa1(382–554) are well-preserved, whereas the C-terminal residues 536–554 of αB are now in a disordered conformation. The side-chain methyl group of residue Leu539 binds in the hydrophobic cavity of the SBDβ and blocks the substrate-binding site (Fig. 4C). Such a conformation has also been found in the truncation SBD mutants of DnaK (PDB code 1BPR) and rat Hsp70 (PDB code 1CKR), which share a similar conformation, with the unfolded C terminus collapsed into the substrate-binding site (Fig. 4A). However, the positions of αB differ in these three structures (Fig. S3A). Interestingly, in similar truncation mutants of Geobacillus kaustophilus DnaK (PDB code 2V7Y) and bovine Hsp70 (PDB code 1YUW), the corresponding Leu residue does not block the cavity (Fig. S3A). It seems that in the G. kaustophilus DnaK structure, Leu504 (shown in red in Fig. S3B) is slightly too distant to bind to the SBDβ binding site, and such binding might require transition of αB to a coil conformation, which is not energetically favored. In the structure of bovine Hsc70 (PDB code 1YUW), Ser541, instead of Leu542, is positioned onto the binding site (Fig. S3A).
Because the αB collapses into the substrate-binding cavity, it will inhibit substrate binding and decrease the fibril inhibition ability. The 2D 1H-15N HSQC spectra of Ssa1(382–582) shows that it has similar spectra to that of Ssa1(382–554) with good NH signals (Fig. 4D). Especially the NH signals of Leu539 in the two mutants are well-overlapped, indicating that Leu539 is blocking the substrate-binding site in the same way in both structures. The NMR results explain the similarly weak ability of mutants Ssa1(382–554) and Ssa1(382–582) to inhibit Ure2 fibril formation (16).
The α-helical bundle of Hsp70 enhances inhibition of fibril formation
Because the αB collapses into the substrate-binding cavity, we expected that further deletion of αB would enhance the fibril inhibition activity of Ssa1 SBDβ. We compared the inhibition activities of Ssa1(382–506), Ssa1(382–554), and Ssa1(382–622), which were in the following sequence: Ssa1(382–506) ≈ Ssa1(382–554) < Ssa1(382–622) (Fig. 4E). We observed by size-exclusion chromatography that Ssa1(382–506) tends to form oligomers, as indicated by an elution volume in a Superdex 200 column (10/300 GL) of 13.9 ml versus 15.6 ml for Ssa1(382–554) (Fig. S4A), although the molecular mass of Ssa1(382–506) (13.3 kDa) is smaller than that of Ssa1(382–554) (18.8 kDa). The oligomeric state of Ssa1(382–506) was also confirmed by its 1H-15N HSQC spectrum, in which the signal intensities are significantly more heterogeneous than for Ssa1(382–554) (Fig. S4B). The lower-than-expected ability of Ssa1(382–506) to inhibit fibril formation can be explained by its tendency to form oligomers, which may also block its substrate cavity.
Because the SBDβ of DnaK has been demonstrated to be monomeric (29), we explored the role of the α-helical bundle in inhibition of Ure2 fibril formation using truncation mutants of DnaK, including DnaK(386–506), DnaK(386–554), and DnaK(386–618), which correspond to Ssa1(382–506), Ssa1(382–554), and Ssa1(382–622), respectively. The results clearly show that the DnaK SBDβ (residues 386–506) has stronger inhibition ability than DnaK(386–554) but weaker ability than DnaK(386–618) (Fig. 4F). This confirms that the Hsp70 C-terminal α-helical bundle enhances the inhibition ability of Hsp70 SBDβ. The reasons for this enhancement may be multiple. First, the SBDα plays a pivotal role in regulating the kinetics of substrate binding and release (4). Second, it has been reported that the SBDα of DnaK contacts the protein substrate, because the SBDα can approach the bound protein substrate according to cross-linking results (30, 31). Our results indicate that the isolated SBDα of Hsp70 does not bind to the protein substrate, but it is likely to interact nonspecifically with the substrate after the substrate binds to the β-domain of Hsp70 and thus facilitate binding of substrate to Hsp70. Third, the SBDα may also promote substrate binding by stabilizing the conformation of the SBDβ in a state that facilitates substrate binding, consistent with the report that the structure of the isolated DnaK SBDβ is highly flexible, with a KD of ∼600 μm for peptide substrate, whereas addition of the SBDα enhances the affinity to give a KD of ∼11 μm (32).
The GGAP motif contributes to the stress response of yeast
To study the in vivo function of the C-IDR GGAP motif of Ssa1, we constructed a GGAP deletion mutant, ΔGGAP, in which the 20 residues of the GGAP motif are deleted. We also constructed another Ssa1 mutant, GGAG, in which the four Pro residues of the GGAP motif are all mutated to Gly. We then compared the phenotypic effects of these mutants to the WT protein in terms of prion propagation and response to stress, when they represented the only source of cytosolic Hsp70-Ssa in the cell.
Yeast is a well-established model to investigate prion propagation, which can be monitored by utilizing colony color and growth assays on selective media. [URE3] propagation was assessed based on the color of colonies when WT Ssa1 or the ΔGGAP or GGAG mutant was expressed as the sole source of Ssa in the cell (33, 34). Prion status of cells was confirmed through mating assays and curing on guanidine HCl. Cells containing Ssa2 as the sole source of Ssa can grow without adenine and produce white colonies on YPD, indicating a [URE3] prion state, whereas cells containing WT Ssa1 as the sole source of Ssa require adenine to grow and produce red colonies on YPD, indicating a nonprion state (Fig. 5A) (35). Like WT Ssa1, the ΔGGAP and GGAG mutants were not able to maintain propagation of [URE3] (Fig. 5A), suggesting that if the ability of Ssa1 to actively cure or inability to propagate [URE3] is due to gain of function compared with Ssa2, this is maintained in both mutants. Additionally, both Hsp70 mutants behave similarly to WT in terms of propagation of another yeast prion, [PSI+] (data not shown).
Figure 5.

Effects of Ssa1 ΔGGAP and GGAG mutations in S. cerevisiae [URE3] strain under different conditions. A, assessment of [URE3] propagation. Single colonies containing Ssa2, WT Ssa1, ΔGGAP, or GGAG mutant Ssa1 as the sole source of Ssa were streaked on YPD and −ADE plates, which were then incubated at 30 °C for 2 days. Red colonies on YPD and nonviability to grow on −ADE plates indicate a [ure3-0] state; white colonies on YPD and viability on −ADE plates indicate a [URE3] state. B, growth assay at elevated temperatures. Fresh cultures were spotted onto YPD after a one-fifth serial dilution. The plates were incubated at 30, 37, or 39 °C for 2 days. C, acquired thermotolerance assay of ΔGGAP and GGAG mutations. Yeast cells were cultured at 30 °C to reach an A600 of 0.4 before pretreating at 39 °C for 1 h. Then 39 °C pretreated cells were heat-shocked at 47 °C for a time course. −1, cultures without 39 °C incubation; 0–4, cultures were first incubated 39 °C for 1 h and then moved to 47 °C incubator for 0, 10, 20, 30, or 40 min. D, expression levels of Ssa1 and Hsp104. The experiment was repeated three times, consistently showing that the mutant Ssa1 proteins are expressed at a similar level to WT and that slightly more Hsp104 is expressed in cells with GGAP deletion or substitution. E, assessment of ΔGGAP and GGAG mutations under cell-wall damage reagent SDS and oxidative damage reagent H2O2.
In terms of response to heat stress, ΔGGAP and GGAG mutants both show reduced viability at 39 °C on YPD, especially the ΔGGAP mutant (Fig. 5B), which demonstrates the importance of the GGAP motif in maintaining the function of Hsp70 in the heat shock response. The decrease in heat-shock tolerance of ΔGGAP and GGAG mutants can also be observed from an acquired thermotolerance assay, in which the yeast cells are pretreated at 39 °C followed by exposure to 47 °C and then grown on YPD or YPD with 3 mm guanidine HCl (Fig. 5C). Pretreatment at 39 °C will induce overexpression of Hsp70 and primarily Hsp104, whereas the function of Hsp104 can be inhibited by guanidine HCl. Yeast cells expressing GGAG or ΔGGAP mutants of Ssa1 were found to be less thermotolerant with 3 mm guanidine HCl (Fig. 5C), suggesting that deficiency in Hsp104 activity, through working with the Hsp70 chaperone machinery, may contribute to the temperature sensitivity of the GGAP mutants. Hsp104 expression is slightly enhanced in ΔGGAP and GGAG mutants, particularly in ΔGGAP (Fig. 5D). The slight expression enhancement may partially compensate for the functional impairment of the mutants, and/or it suggests that the C-terminal GGAP motif of Ssa1 might interact or influence Hsp104 abundance in some way.
We also assessed the growth of GGAP-deficient cells under other stress conditions, including a cell wall–damaging agent (SDS) and oxidative stress (H2O2) (Fig. 5E). Again, the most significant phenotypic changes were seen with the ΔGGAP mutant, which was clearly more sensitive to SDS compared with WT (Fig. 5E). However, neither mutant seemed to show an obvious effect on yeast response to oxidative stress (Fig. 5E). Together, these results indicate that the removal or modification of the GGAP motif does not alter the abundance of Hsp70, but there is clearly a role for this motif in vivo for maintaining Hsp70 function during heat shock and certain other stress responses.
Discussion
Compared with the NBD, SBDβ, and SBDα of Hsp70, studies of the Hsp70 C-terminal disordered region, which is least conserved among the domains of Hsp70, are more limited. Many eukaryotic cytosolic Hsp70s, including yeast Ssa1, have a C-terminal EEVD motif, which facilitates intermolecular interactions and chaperone assembly (13, 36, 37), as well as a C-IDR sequence between the α-helical bundle and the C-terminal EEVD motif. A GGAP-like motif (such as GGMP) frequently occurs in the Hsp70 C-IDR sequence and is observed in species including Brugia, Trypanosoma, Plasmodium, Rattus norvegicus, and Homosapiens, in both Hsp and Hsc groups (Fig. 1D) (36–41). The GGMPGGM motif was reported to be related to parasite virulence of Toxoplasma gondii (42). However, the general functions of the C-IDR sequence have remained unclear.
Our results address the function of this region, demonstrating by NMR and FRET that the Ssa1 GGAP motif in the C-IDR can directly interact with substrates such as the prion protein Ure2. Deletion or mutation of the Ssa1 GGAP motif was found to decrease the ability of yeast to respond to heat and cell-wall damage stress, demonstrating a cellular role of the GGAP motif in the stress response. Consistent with this, Smock et al. (11) have identified a conserved, disordered C-terminal motif in prokaryotic Hsp70s, including DnaK, that can nonspecifically interact with substrates and that enhances the DnaK chaperone activity and cellular survival upon stress. Although the motif observed in DnaK is different from the one observed in eukaryotic Hsp70s, it seems nevertheless that the function of the C-IDR of Hsp70 is universal from prokaryotic cells to eukaryotic cells.
It was recently discovered that the Ssa1-Hsp104 chaperone system is involved in the recovery of yeast after heat shock by disassembling the stress granules formed during the heat shock response (43). Preconditioned cells can be protected from promiscuous interactions with misfolded proteins by up-regulation of the chaperones, preventing the mislocalization of P-body (liquid-like) components to stress granules (solid-like) (44). Interestingly, the Gly/Pro-rich P domain of Pab1, a defining marker of stress granules in yeast, can tune biophysical properties of Pab1 and induce phase separation in vitro and in vivo, although the P domain is not required for demixing (45). Consistent with these studies, our results imply that a significant function of the Ssa1 GGAP motif in the stress response may be related to its promiscuous substrate-binding ability, enhancing the function of Ssa1 during disassembly of stress granules and cell recovery from stress.
The GGAP-like motif also exists widely in non-Hsp70 proteins. In the UniProt data bank, we found 81 protein clusters containing GGAP-like motifs when using three repeats of GGAP (GGAP × 3) to search the database with the expectation value threshold setting to 1.0 (Table 2). An E-threshold of 1.0 when used in the search will result in sequences with no more than two residue differences from the 12-residue query sequence. More than 20 of the protein clusters have known function, including the sodium/hydrogen exchanger (UniRef50_U6MSL8), para-aminobenzoate synthase (UniRef50_U6MNR9), RNA helicase, and Hsp70 family members. More than 500 protein clusters were obtained when using GGPP × 3, 162 clusters using GGMP × 3, 51 clusters using GGFP × 3, and 34 clusters using GGVP × 3 (Table 2). Thus, the GGPP motif occurs most frequently in proteins among these GGAP-like motifs. GGMP and GGAP motifs also exist more widely than other GGAP-like motifs. This study provides experimental evidence that these motifs may play important roles by nonspecifically interacting with their partners. However, the mechanism of such interactions still needs to be elucidated. It also remains unclear whether or not the GGAP-like motif can interact with other biomolecules such as DNA, RNA, and lipids.
Table 2.
Protein sequence clusters containing GGAP-like motif in UniProt databank
The following parameters were used to search sequences in the UniProt databank: target database: UniRef50; E-threshold: 1; matrix: auto; filtering: none; gapped: yes; hits: 500.
| Query sequence (× 3) | GGAP | GGVP | GGMP | GGFP | GGPP | GGAS | EGAP | GEAP |
|---|---|---|---|---|---|---|---|---|
| Total number of protein clusters | 81 | 34 | 162 | 51 | 500 | 12 | 62 | 7 |
| Clusters with known function | 23 | 20 | 82 | 26 | 137 | 3 | 13 | 5 |
It is well-established that the EEVD motif of yeast Ssa1 can bind the class B Hsp40 Sis1 by electrostatic interaction, and this interaction is functionally important for its chaperone activity (13, 46). Similarly, the human B-type J proteins and the Hsp70 EEVD motif have a functional interaction (12, 47). Interaction between the EEVD domain and class A Hsp40s has been reported for mammalian Hsp70 and also in vivo for Ssa1 (48, 49). Our NMR titration experiments detected a direct interaction between the EEVD motif of yeast Ssa1 and Ydj1. Consistent with our results, deletion of the EEVD motif causes a slight decrease in stimulation of the refolding activity of Ssa1 by Ydj1 (12). At the same time, our results suggest that the GGAP motif of Ssa1 also participates in the interaction of Ssa1 and Ydj1, but with more limited contribution compared with the EEVD motif. Hsp40 mainly interacts with the Hsp70 NBD via the J domain (5), and the α-helical bundle of SBD is also involved in interaction with the J domain in the case of human Hsp70 and HSJ1a (10). The newly characterized interaction of the C-terminal GGAP and EEVD motifs of Ssa1 with Ydj1 suggests that the C-IDR of Ssa1 may also play a role in the positioning of Ydj1.
Ssa1 and its functional partner Sse1 have been identified to be targets of SUMO (50, 51). Similarly, the human Hsp70 homologues HspA1A, HspA1B, and HspA8 were also identified to be targets of SUMO (52). In this study, we unexpectedly discovered that the C-terminal TVEEVD peptide of Ssa1 can specifically bind to the same site of the SMT3 protein as for other SIMs (Fig. S2), which is required for efficient activity of SUMO modifiers (53). Thus, the C-terminal peptide TVEEVD of Ssa1 may play a role in the SUMOylation of Ssa1, as well as the SUMOylation-related processes regulated by Hsp70.
Experimental procedures
Plasmids and yeast strains
The SSA1 gene and its GGAP mutations were constructed in the pC210 plasmid (35). The plasmids were transferred into a [URE3] (MATα, kar1-1, SUQ5, PDAL5::ADE2, his3Δ202, leu2Δ1, trp1Δ63, ura3-52 ssa1::KanMX, ssa2::HIS3, ssa3::TRP1, ssa4::ura3–2f/pJ401 (strain 1161, a gift from Daniel Masison) (54). Then colonies were streaked on plates containing 5-fluoroorotic acid to eliminate the resident URA3 plasmid containing WT Ssa1.
Protein expression and purification
The constructs containing genes for dnaK, ssa1, and ure2 were described previously (16, 55, 56). For all Hsp70 variants, Ure2 and α-synuclein used in this paper, the gene of each construct was cloned into a modified pET28a plasmid, which was fused to a His-tagged SMT3 protein followed by a ULP1 cleavage site at the N terminus of the protein of interest. The plasmids were used to transform E. coli BL21-CodonPlus (DE3)-RIL cells. The cells were grown at 37 °C in 2YT medium containing 50 μg/ml kanamycin and 33 μg/ml chloramphenicol until the absorbance at 600 nm of the medium reached 0.6–0.8, and then protein expression was induced overnight at 16 °C with 0.5 mm isopropyl-β-d-thiogalactopyranoside. The cells were harvested by centrifugation at 4800 × g at 4 °C for 30 min, resuspended in buffer A (50 mm Tris-HCl, pH 8.4, 200 mm NaCl), and lysed using a JNBIO JN-3000 PLUS high-pressure cell press. The debris was removed by centrifugation (35,000 × g, 4 °C, 30 min), and the supernatant was applied onto a Ni2+-affinity column (chelating Sepharose fast-flow resin; Pharmacia/GE) and washed with buffer A containing 30 mm imidazole. Protein was eluted using buffer A containing 300 mm imidazole and dialyzed against buffer A to reduce the imidazole concentration to lower than 10 mm, followed by SUMO protease ULP1 treatment (2 h at 4 °C) to cleave the His-tagged SMT3 protein. The protease cleavage leaves an extra serine as the N-terminal residue of the protein of interest. An additional Ni2+-affinity chromatography step was applied to remove uncleaved protein, the SMT3 protein, and ULP1. For Hsp70 chaperone variants and cleaved SMT3, the proteins were further purified by gel filtration chromatography using a SuperdexTM 75 column (GE Healthcare). Protein concentrations were determined by UV absorption at 280 nm. The oligomerization states of Hsp70 chaperone variants were analyzed by a Superdex 200 column (10/300 GL; GE Healthcare).
Expression and purification of the Ssa1 C-terminal GGAP motif was similar to Ssa1 variants, except that the product of the first Ni2+-affinity column was concentrated using an ultrafiltration tube (Amicon Ultra 15 ml 3K; Millipore) and dialyzed against buffer B (20 mm NaH2PO4–Na2HPO4, 50 mm NaCl, pH 6.5) twice at 4 °C. Then the concentrated protein was cleaved using ULP1, the solution was concentrated again, and the flow through containing the Ssa1 C-terminal GGAP motif was collected. Ydj1 was expressed and purified under the same conditions as described previously (57), except that the cells were lysed using a high-pressure cell press instead of sonication.
15N- and 15N/13C-labeled proteins and peptides were prepared using the same procedures except that cells were grown in M9 minimal medium containing 15NH4Cl and [13C]glucose as the sole nitrogen and carbon sources, respectively.
NMR spectroscopy
NMR experiments were performed at 298 K on a Varian INOVA 600 MHz spectrometer equipped with a triple resonance cryo probe. NMR samples consisted of 0.2–1.0 mm 15N- or 15N/13C-labeled protein in 20 mm NaH2PO4–Na2HPO4, 50 mm NaCl, 1 mm DTT, 1 mm EDTA, and 10% (v/v) D2O at pH 6.5. Two-dimensional 1H-15N and 1H-13C HSQC and three-dimensional HNCO, HN(CA)CO, CBCA(CO)NH, and HNCACB experiments were performed for backbone assignment. C(CCO)NH, HBHA(CO)NH, HCCH-TOCSY, and CCH-TOCSY experiments were performed for side-chain assignments. These through-bond experiments correlate nuclei (such as HN and CO) connected by a limited number of chemical bonds. Three-dimensional 1H-15N and 1H-13C NOESY–HSQC spectra were collected to generate distance restraints. All data were processed with NMRPipe (58) and analyzed with NMRViewJ (59). The steady-state heteronuclear HN-NOE values of Ssa1 SBD were measured using standard pulse programs (60).
Structure calculations
The NMR structures of Ssa1(523–622) and Ssa1(382–554) were initially calculated with the program CYANA (61) and then refined using CNS (62) with manual assignments as well as semiautomated NOE assignments by SANE (63). Backbone dihedral angle restraints obtained using TALOS-N (64), as well as hydrogen bond restraints according to the regular secondary structure patterns, were also incorporated into the structural calculation. From 100 CNS-calculated structures, the 50 lowest energy conformers of Ssa1(523–622) and Ssa1(382–554) were selected for further water refinement using CNS and RECOORDScript (65). The resulting 20 energy-minimized conformers were used to represent the solution structure of Ssa1(523–622) and Ssa1(382–554). The quality of the determined structures (Table 1) was analyzed using PROCHECK-NMR (66) and MOLMOL (67). Structural figures were created with MOLMOL (67) and PyMOL (68).
NMR titration
For NMR titration, a protein mixture containing 15N-labeled Ssa1 CTD (523–622 or 523–642) or Ssa1 GGAP peptide (609–628) was monitored using 2D 1H-15N HSQC spectra. For 15N-labeled Ssa1 CTDs or the Ssa1 C-terminal GGAP motif titrated with Ure2, the buffer was 20 mm NaH2PO4–Na2HPO4, 50 mm NaCl, and 10% (v/v) D2O at pH 7.0. For 15N-labeled Ssa1 CTDs titrated with Ydj1, α-synuclein, SMT3, or BSA or 15N-labeled SMT3 titrated with PEAEGPTVEEVD, the buffer was 20 mm NaH2PO4–Na2HPO4, 50 mm NaCl, and 10% (v/v) D2O at pH 6.5. For all the titration and control NMR experiments, the 15N-labeled control protein sample and the 15N-labeled protein sample mixed with titrating proteins were simultaneously exchanged with the same buffer before measurements to ensure that the buffer conditions were identical in the two samples. For the titration of GGAP peptide, the Ure2 protein solution was exchanged with the same buffer that was used for SMT3 fusion GGAP peptide before ULP1 digestion, to ensure that the buffer conditions were identical.
In vitro amyloid fibril formation
The kinetics of amyloid fibril formation of Ure2 were monitored using thioflavin T (Sigma–Aldrich) as previously described (16). A 150-μl protein sample in buffer A containing 20 μm thioflavin T was added to a 96-well plate with an optical flat bottom (Corning, Inc., Corning, NY) and covered with a PlateMax film (Corning, Inc.). The fluorescence was measured using a microplate reader (SpectraMax M3; Molecular Devices) at 30 °C, with continuous shaking between readings. The samples were excited at 450 nm, and the emission was measured at 485 nm, with a reading interval of 15 min. For each sample at least four replicates were measured.
FRET
In this paper, we used two kinds of fluorescent dye: Cy3 Mono NHS-ester and Cy5 Mono NHS-ester (GE Healthcare). Cy3 was used as a donor to label Ure2, whereas Cy5 was used as a receptor to label Ssa1 variants. For labeling, 300 μl of protein solution in buffer A was mixed with the corresponding dye dissolved in DMSO at 3-fold molar excess and incubated at 4 °C overnight (shaken gently in the dark). The supernatant was collected after centrifugation at 14,000 × g at 4 °C for 30 min. The labeled protein was separated from unbound dye using a PD-10 desalting column (GE Healthcare). Protein concentration and labeling efficiency were determined by measuring the absorbance of the labeled protein at 280 nm (A280) and at the λmax for the dye (Amax), according to the manufacturer's instructions for the dye. The labeling efficiency of each protein was close to 1.0, meaning approximately one dye molecule per protein molecule. For FRET data collection, a series of 150-μl protein samples in buffer A containing 2 μm Cy3-labeled Ure2 with constant concentration and Cy5-labeled Ssa1 variant (both labeled by corresponding dyes) with different concentration ratios to Ure2 were added to a 96-well plate with an optical flat bottom (Corning, Inc.), covered with a PlateMax film (Corning, Inc.) and incubated at 25 °C for 30 min. The same samples containing Cy5-labeled Ssa1 variant at the same series of concentrations but without Ure2 were used as a control group. The fluorescence was measured using a microplate reader (SpectraMax M3; Molecular Devices). The parameters were as follows: excitation = 532 nm; emission: start = 540 nm, stop = 750 nm, step = 1 nm, cutoff = 550 nm. The fluorescence data of control groups were subtracted to obtain the fluorescence from FRET. The FRET efficiency under different concentrations of Cy5-labeled Ssa1 was calculated using the equation,
| (Eq. 1) |
where E represents the FRET efficiency, and F(D) and F(DA) represent the donor fluorescence in the absence or the presence of Cy5–Ssa1. The donor fluorescence intensity was calculated as an average from 565 to 575 nm. The FRET efficiencies were plotted against Cy5–Ssa1 concentrations and fitted to the following formula to obtain the dissociation constant KD,
| (Eq. 2) |
where E is the FRET efficiency, x is the Cy5–Ssa1 concentration, and a and b are constants.
Yeast growth assay
Yeast strains were cultured in 5 ml of YPD or SC medium with appropriate selection at 30 °C overnight. The following morning, the yeast cultures were diluted into 6 ml of fresh medium at an A600 of 0.2 and cultured again until the A600 reached 0.5. A one-fifth serial dilution was performed in a 96-well plate and then replicated onto YPD or appropriate medium. The plates were incubated at 30 °C or elevated temperatures (37 or 39 °C) for 2 days. For growth assays with oxidant or cell wall–damaging agents, the cells (A600 of 0.5) were spotted onto SC-H2O2 (0, 1, and 2 mm) or onto YPD containing SDS (0, 0.005, and 0.01%, w/v).
Acquired thermotolerance assay
Single colonies were inoculated and cultured in 5 ml of YPD or SC selective medium at 30 °C, 200 rpm overnight. The next day, yeast cultures were diluted into 7 ml of the same media to reach an A600 of 0.2. The cells were cultured until reaching an A600 of 0.4. A 1-ml aliquot was immediately transferred to a 1.5-ml Eppendorf tube and placed on ice. The rest of the culture was heat-shocked at 39 °C with shaking for 1 h to induce Hsp104. Subsequently, 0.5-ml volumes of cell aliquots were maintained at 47 °C in an incubator with shaking at 200 rpm for 0, 10, 20, 30, or 40 min. As described above, a one-fifth serial dilution of the aliquots of each time point were carried out. Finally, YPD and YPD-3 mm guanidine HCl plates with spotted cells were incubated in a 30 °C incubator for 2 days.
Monitoring the presence of the prion phenotype in S. cerevisiae
Monitoring of yeast prion status was carried out as described by Jones and Masison (34). The presence or absence of the [PSI+] and [URE3] prion states can be monitored in yeast strains by a simple color assay. Briefly, single colonies streaked on YPD and −ADE plates, which were then incubated at 30 °C for 2 days or at room temperature for 5–7 days. [psi−] and [ure3-0] cells show red colonies on YPD and are lethal on −ADE plates; [PSI+] and [URE3] cells show white colonies on YPD and are viable on −ADE plates.
Immunoblotting
Yeast strains were cultured in 25 ml of YPD medium from an A600 of 0.2 to an A600 of 0.6–0.8. The cells were collected by centrifugation and were resuspended in 750 μl of Y cell lysis reagent (Sigma; C4482), which was supplemented with protease inhibitor mixture (Sigma; P8215) and 10 mm DTT. Then 0.5-mm soda lime glass beads were added into tubes to assist lysis. Supernatants were centrifuged at 4 °C at 16,200 × g for 10 min to obtain clarified lysates. Finally, 10 μg of lysates were loaded onto 4–20% gradient gels and blotted by anti-Hsp70 (Stressgen; SPA-882) and anti-Hsp104 (a gift from Prof. John Glover, University of Toronto). This experiment was repeated three times, and the results were consistent.
Author contributions
W. G., L. X., G. W. J., and S. P. conceptualization; W. G., W. H., and H. W. formal analysis; W. G., S. W., H. Z., G. W. J., and S. P. supervision; W. G., S. W., H. Z., G. W. J., and S. P. funding acquisition; W. G., W. H., L. X., H. W., S. W., H. Z., J.W., G. W. J., and S. P. validation; W. G., W. H., L. X., and H. W. investigation; W. G. and L. X. visualization; W. G., L. X., and S. P. writing-original draft; W. G., W. H., L. X., H. W., S. W., H. Z., J.W., G. W. J., and S. P. writing-review and editing; S. P. project administration.
Supplementary Material
Acknowledgments
We thank Prof. Yingang Feng and Dr. Tao Cai for helpful discussion regarding the NMR structure calculation and bioinformatics analysis of the GGAP motif.
This work was supported by National Key R&D Program of China Grant 2017YFA0504000; National Natural Science Foundation of China Grants 31570780, 31200578, 31470747, 31770829, 31300631, and 21673278; Beijing Natural Science Foundation Grant 5172026; funds from the National Laboratory of Biomacromolecules and the Chinese Academy of Sciences Centre of Excellence in Biomacromolecules; a John and Pat Hume Ph.D. scholarship from Maynooth University (to L. X.); and Science Foundation Ireland Grant SFI/13/ISCA/2845 (to the G. W. J. laboratory). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
The atomic coordinates and structure factors (codes 5Z8Q and 5Z8I) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shifts (accession number 36162) have been deposited in the BioMagResBank database.
- Hsp70
- 70-kDa heat shock protein
- Hsp40
- heat shock protein 40
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- IDR
- intrinsically disordered region
- C-IDR
- C-terminal IDR
- CTD
- C-terminal domain
- PDB
- Protein Data Bank
- SUMO
- small ubiquitin-like modifier
- SIM
- SUMO-interacting motif
- HSQC
- heteronuclear single quantum coherence
- CSP
- chemical shift perturbation
- TOCSY
- total correlation spectroscopy.
References
- 1. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., and Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- 2. Clerico E. M., Tilitsky J. M., Meng W., and Gierasch L. M. (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 427, 1575–1588 10.1016/j.jmb.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertelsen E. B., Chang L., Gestwicki J. E., and Zuiderweg E. R. (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. U.S.A. 106, 8471–8476 10.1073/pnas.0903503106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zuiderweg E. R., Bertelsen E. B., Rousaki A., Mayer M. P., Gestwicki J. E., and Ahmad A. (2013) Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 328, 99–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alderson T. R., Kim J. H., and Markley J. L. (2016) Dynamical structures of Hsp70 and Hsp70–Hsp40 complexes. Structure 24, 1014–1030 10.1016/j.str.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang P., Leu J. I., Murphy M. E., George D. L., and Marmorstein R. (2014) Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One 9, e103518 10.1371/journal.pone.0103518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mayer M. P., and Kityk R. (2015) Insights into the molecular mechanism of allostery in Hsp70s. Front. Mol. Biosci. 2, 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu L., Gong W., Cusack S. A., Wu H., Loovers H. M., Zhang H., Perrett S., and Jones G. W. (2018) The β6/β7 region of the Hsp70 substrate-binding domain mediates heat-shock response and prion propagation. Cell Mol. Life Sci. 75, 1445–1459 10.1007/s00018-017-2698-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mayer M. P. (2013) Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 38, 507–514 10.1016/j.tibs.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 10. Gao X. C., Zhou C. J., Zhou Z. R., Wu M., Cao C. Y., and Hu H. Y. (2012) The C-terminal helices of heat shock protein 70 are essential for J-domain binding and ATPase activation. J. Biol. Chem. 287, 6044–6052 10.1074/jbc.M111.294728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smock R. G., Blackburn M. E., and Gierasch L. M. (2011) Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J. Biol. Chem. 286, 31821–31829 10.1074/jbc.M111.265835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu H. Y., Ziegelhoffer T., Osipiuk J., Ciesielski S. J., Baranowski M., Zhou M., Joachimiak A., and Craig E. A. (2015) Roles of intramolecular and intermolecular interactions in functional regulation of the Hsp70 J-protein co-chaperone Sis1. J. Mol. Biol. 427, 1632–1643 10.1016/j.jmb.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li J., Wu Y., Qian X., and Sha B. (2006) Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. Biochem. J. 398, 353–360 10.1042/BJ20060618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pemberton S., Madiona K., Pieri L., Kabani M., Bousset L., and Melki R. (2011) Hsc70 protein interaction with soluble and fibrillar α-synuclein. J. Biol. Chem. 286, 34690–34699 10.1074/jbc.M111.261321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang C., Cheng H., Hao S., Zhou H., Zhang X., Gao J., Sun Q. H., Hu H., and Wang C. C. (2006) Heat shock protein 70 inhibits α-synuclein fibril formation via interactions with diverse intermediates. J. Mol. Biol. 364, 323–336 10.1016/j.jmb.2006.08.062 [DOI] [PubMed] [Google Scholar]
- 16. Xu L. Q., Wu S., Buell A. K., Cohen S. I., Chen L. J., Hu W. H., Cusack S. A., Itzhaki L. S., Zhang H., Knowles T. P., Dobson C. M., Welland M. E., Jones G. W., and Perrett S. (2013) Influence of specific HSP70 domains on fibril formation of the yeast prion protein Ure2. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110410 10.1098/rstb.2011.0410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wickner R. B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- 18. Lian H. Y., Jiang Y., Zhang H., Jones G. W., and Perrett S. (2006) The yeast prion protein Ure2: structure, function and folding. Biochim. Biophys. Acta 1764, 535–545 10.1016/j.bbapap.2005.11.016 [DOI] [PubMed] [Google Scholar]
- 19. Peisker K., Chiabudini M., and Rospert S. (2010) The ribosome-bound Hsp70 homolog Ssb of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1803, 662–672 10.1016/j.bbamcr.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 20. Frydman J. (2001) Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu. Rev. Biochem. 70, 603–647 10.1146/annurev.biochem.70.1.603 [DOI] [PubMed] [Google Scholar]
- 21. Bush G. L., and Meyer D. I. (1996) The refolding activity of the yeast heat shock proteins Ssa1 and Ssa2 defines their role in protein translocation. J. Cell Biol. 135, 1229–1237 10.1083/jcb.135.5.1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beckmann R. P., Mizzen L. E., and Welch W. J. (1990) Interaction of Hsp 70 with newly synthesized proteins: implications for protein folding and assembly. Science 248, 850–854 10.1126/science.2188360 [DOI] [PubMed] [Google Scholar]
- 23. Savistchenko J., Krzewska J., Fay N., and Melki R. (2008) Molecular chaperones and the assembly of the prion Ure2p in vitro. J. Biol. Chem. 283, 15732–15739 10.1074/jbc.M800728200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krzewska J., and Melki R. (2006) Molecular chaperones and the assembly of the prion Sup35p, an in vitro study. EMBO J. 25, 822–833 10.1038/sj.emboj.7600985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., and Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheng W., and Liao X. (2002) Solution structure of a yeast ubiquitin-like protein Smt3: the role of structurally less defined sequences in protein-protein recognitions. Protein Sci. 11, 1482–1491 10.1110/ps.0201602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., and Chen Y. (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 14373–14378 10.1073/pnas.0403498101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu W., Wu H., Zhang H., Gong W., and Perrett S. (2015) Resonance assignments for the substrate binding domain of Hsp70 chaperone Ssa1 from Saccharomyces cerevisiae. Biomol. NMR Assign. 9, 329–332 10.1007/s12104-015-9603-5 [DOI] [PubMed] [Google Scholar]
- 29. Mayer M. P., Schröder H., Rüdiger S., Paal K., Laufen T., and Bukau B. (2000) Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7, 586–593 10.1038/76819 [DOI] [PubMed] [Google Scholar]
- 30. Schlecht R., Erbse A. H., Bukau B., and Mayer M. P. (2011) Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 18, 345–351 10.1038/nsmb.2006 [DOI] [PubMed] [Google Scholar]
- 31. Marcinowski M., Höller M., Feige M. J., Baerend D., Lamb D. C., and Buchner J. (2011) Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 18, 150–158 10.1038/nsmb.1970 [DOI] [PubMed] [Google Scholar]
- 32. Pellecchia M., Montgomery D. L., Stevens S. Y., Vander Kooi C. W., Feng H. P., Gierasch L. M., and Zuiderweg E. R. (2000) Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Biol. 7, 298–303 10.1038/74062 [DOI] [PubMed] [Google Scholar]
- 33. Schlumpberger M., Prusiner S. B., and Herskowitz I. (2001) Induction of distinct [URE3] yeast prion strains. Mol. Cell Biol. 21, 7035–7046 10.1128/MCB.21.20.7035-7046.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones G. W., and Masison D. C. (2003) Saccharomyces cerevisiae Hsp70 mutations affect [PSI+] prion propagation and cell growth differently and implicate Hsp40 and tetratricopeptide repeat cochaperones in impairment of [PSI+]. Genetics 163, 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwimmer C., and Masison D. C. (2002) Antagonistic interactions between yeast [PSI(+)] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol. Cell Biol. 22, 3590–3598 10.1128/MCB.22.11.3590-3598.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu S. J., Liu F. H., Hu S. M., and Wang C. (2001) Different combinations of the heat-shock cognate protein 70 (hsc70) C-terminal functional groups are utilized to interact with distinct tetratricopeptide repeat-containing proteins. Biochem. J. 359, 419–426 10.1042/bj3590419, 10.1042/0264-6021:3590419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Demand J., Lüders J., and Höhfeld J. (1998) The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell Biol. 18, 2023–2028 10.1128/MCB.18.4.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piano A., Franzellitti S., Tinti F., and Fabbri E. (2005) Sequencing and expression pattern of inducible heat shock gene products in the European flat oyster, Ostrea edulis. Gene 361, 119–126 10.1016/j.gene.2005.06.034 [DOI] [PubMed] [Google Scholar]
- 39. Cui Z., Liu Y., Luan W., Li Q., Wu D., and Wang S. (2010) Molecular cloning and characterization of a heat shock protein 70 gene in swimming crab (Portunus trituberculatus). Fish Shellfish Immunol. 28, 56–64 10.1016/j.fsi.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 40. Cho E. S., and Jeong H. D. (2012) Effect of environmental impact to molecular expression of heat-shock protein (HSP70) in oyster Crassostrea gigas from Gamak bay, Korea. J. Environ. Biol. 33, 609–615 [PubMed] [Google Scholar]
- 41. Fu W., Zhang F., Liao M., Liu M., Zheng B., Yang H., and Zhong M. (2013) Molecular cloning and expression analysis of a cytosolic heat shock protein 70 gene from mud crab Scylla serrata. Fish Shellfish Immunol. 34, 1306–1314 10.1016/j.fsi.2013.02.027 [DOI] [PubMed] [Google Scholar]
- 42. Lyons R. E., and Johnson A. M. (1998) Gene sequence and transcription differences in 70 kDa heat shock protein correlate with murine virulence of Toxoplasma gondii. Int. J. Parasitol. 28, 1041–1051 10.1016/S0020-7519(98)00074-5 [DOI] [PubMed] [Google Scholar]
- 43. Cherkasov V., Hofmann S., Druffel-Augustin S., Mogk A., Tyedmers J., Stoecklin G., and Bukau B. (2013) Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 23, 2452–2462 10.1016/j.cub.2013.09.058 [DOI] [PubMed] [Google Scholar]
- 44. Kroschwald S., Maharana S., Mateju D., Malinovska L., Nüske E., Poser I., Richter D., and Alberti S. (2015) Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 4, e06807 10.7554/eLife.06807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riback J. A., Katanski C. D., Kear-Scott J. L., Pilipenko E. V., Rojek A. E., Sosnick T. R., and Drummond D. A. (2017) Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168, 1028–1040.e19 10.1016/j.cell.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aron R., Lopez N., Walter W., Craig E. A., and Johnson J. (2005) In vivo bipartite interaction between the Hsp40 Sis1 and Hsp70 in Saccharomyces cerevisiae. Genetics 169, 1873–1882 10.1534/genetics.104.037242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu H. Y., Ziegelhoffer T., and Craig E. A. (2015) Functionality of class A and class B J-protein co-chaperones with Hsp70. FEBS Lett. 589, 2825–2830 10.1016/j.febslet.2015.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jones G., Song Y., Chung S., and Masison D. C. (2004) Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol. Cell Biol. 24, 3928–3937 10.1128/MCB.24.9.3928-3937.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Freeman B. C., Myers M. P., Schumacher R., and Morimoto R. I. (1995) Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 14, 2281–2292 10.1002/j.1460-2075.1995.tb07222.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hannich J. T., Lewis A., Kroetz M. B., Li S. J., Heide H., Emili A., and Hochstrasser M. (2005) Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J. Biol. Chem. 280, 4102–4110 10.1074/jbc.M413209200 [DOI] [PubMed] [Google Scholar]
- 51. Zhou W., Ryan J. J., and Zhou H. (2004) Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J. Biol. Chem. 279, 32262–32268 10.1074/jbc.M404173200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wen D., Xu Z., Xia L., Liu X., Tu Y., Lei H., Wang W., Wang T., Song L., Ma C., Xu H., Zhu W., Chen G., and Wu Y. (2014) Important role of SUMOylation of Spliceosome factors in prostate cancer cells. J. Proteome Res. 13, 3571–3582 10.1021/pr4012848 [DOI] [PubMed] [Google Scholar]
- 53. Gareau J. R., and Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 10.1038/nrm3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sharma D., and Masison D. C. (2008) Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179, 1301–1311 10.1534/genetics.108.089458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang H., Yang J., Wu S., Gong W., Chen C., and Perrett S. (2016) Glutathionylation of the bacterial Hsp70 chaperone DnaK provides a link between oxidative stress and the heat shock response. J. Biol. Chem. 291, 6967–6981 10.1074/jbc.M115.673608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Perrett S., Freeman S. J., Butler P. J., and Fersht A. R. (1999) Equilibrium folding properties of the yeast prion protein determinant Ure2. J. Mol. Biol. 290, 331–345 10.1006/jmbi.1999.2872 [DOI] [PubMed] [Google Scholar]
- 57. Lian H. Y., Zhang H., Zhang Z. R., Loovers H. M., Jones G. W., Rowling P. J., Itzhaki L. S., Zhou J. M., and Perrett S. (2007) Hsp40 interacts directly with the native state of the yeast prion protein Ure2 and inhibits formation of amyloid-like fibrils. J. Biol. Chem. 282, 11931–11940 10.1074/jbc.M606856200 [DOI] [PubMed] [Google Scholar]
- 58. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 59. Johnson B. A., and Blevins R. A. (1994) NMRView: a computer program for visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 60. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., and Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 10.1021/bi00185a040 [DOI] [PubMed] [Google Scholar]
- 61. Herrmann T., Güntert P., and Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 10.1016/S0022-2836(02)00241-3 [DOI] [PubMed] [Google Scholar]
- 62. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 10.1107/S0907444998003254, 10.1107/S0108767398011465 [DOI] [PubMed] [Google Scholar]
- 63. Duggan B. M., Legge G. B., Dyson H. J., and Wright P. E. (2001) SANE (structure assisted NOE evaluation): an automated model-based approach for NOE assignment. J. Biomol. NMR 19, 321–329 10.1023/A:1011227824104 [DOI] [PubMed] [Google Scholar]
- 64. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 10.1007/s10858-013-9741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., and Bonvin A. M. (2005) RECOORD: a recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672 10.1002/prot.20408 [DOI] [PubMed] [Google Scholar]
- 66. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 67. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 68. DeLano W. L. (2002) The PyMOL User's Manual, Delano Scientific, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.