

Abstract
Nicotinic acetylcholine receptors (nAChRs) containing α6 and β4 subunits are expressed by dorsal root ganglion neurons and have been implicated in neuropathic pain. Rodent models are often used to evaluate the efficacy of analgesic compounds, but species differences may affect the activity of some nAChR ligands. A previous candidate α-conotoxin-based therapeutic yielded promising results in rodent models, but failed in human clinical trials, emphasizing the importance of understanding species differences in ligand activity. Here, we show that human and rat α6/α3β4 nAChRs expressed in Xenopus laevis oocytes exhibit differential sensitivity to α-conotoxins. Sequence homology comparisons of human and rat α6β4 nAChR subunits indicated that α6 residues forming the ligand-binding pocket are highly conserved between the two species, but several residues of β4 differed, including a Leu–Gln difference at position 119. X-ray crystallography of α-conotoxin PeIA complexed with the Aplysia californica acetylcholine-binding protein (AChBP) revealed that binding of PeIA orients Pro13 in close proximity to residue 119 of the AChBP complementary subunit. Site-directed mutagenesis studies revealed that Leu119 of human β4 contributes to higher sensitivity of human α6/α3β4 nAChRs to α-conotoxins, and structure–activity studies indicated that PeIA Pro13 is critical for high potency. Human and rat α6/α3β4 nAChRs displayed differential sensitivities to perturbations of the interaction between PeIA Pro13 and residue 119 of the β4 subunit. These results highlight the potential significance of species differences in α6β4 nAChR pharmacology that should be taken into consideration when evaluating the activity of candidate human therapeutics in rodent models.
Keywords: nicotinic acetylcholine receptors (nAChR), neuroinflammation, neuroscience, neurotransmitter receptor, neurotoxin, α1;-conotoxin, nicotinic subunit α6, nicotinic subunit β4, nuclear magnetic resonance spectroscopy, X-ray crystallography, small peptide, analgesic, neuropathic pain
Introduction
Nicotinic acetylcholine receptors (nAChRs) 5 are ligand-gated ion channels formed by the pentameric assembly of individual subunits. There are 16 genes in mammals that encode these subunits and are represented by the Greek symbols α1–α7, α9, α10, β1–β4, δ, ϵ, and γ (1). nAChRs are expressed by neurons in both the central and peripheral nervous systems and are involved in diverse physiological processes (2), including fast synaptic transmission (3), the modulation of neurotransmitter release (4–10), and numerous immunological processes (11, 12).
Native nAChRs containing the α6 subunit are broadly classified into two subtype categories: those that contain the β2 subunit and those that contain the β4 subunit. The α6β2* subtype (the asterisk denotes the potential presence of additional subunits in native receptors) has a limited distribution profile in the nervous system but is abundantly expressed in certain regions of the brain and spinal cord (13–19). The α6β4* subtype probably has an even more restricted expression profile. Functional evidence for α6β4* nAChR expression in sensory neurons of rat and mouse dorsal root ganglia (DRG) has been demonstrated (20, 21), although their functional role in these cells is mostly unknown.
DRG contain neurons that perform a wide range of sensory functions, including proprioception and the detection of harmful or painful stimuli. Recent evidence suggests that α6β4 nAChRs represent a new molecular target for the treatment of neuropathic pain. Development of neuropathic pain-like symptoms has been shown to be inversely correlated with CHRNA6 expression levels in the DRG of mice (21). Strains with high levels of CHRNA6 expression show lower levels of mechanical allodynia in several neuropathic and inflammatory pain models, and those with low levels of CHRNA6 expression are more susceptible to developing neuropathic pain. Intriguingly, CHRNA6 null mice show no analgesic responses to nicotine, whereas mice with a gain-of-function mutation show increased analgesic responses. In addition, a history of chronic pain syndromes in humans correlates with levels of CHRNA6 expression (21).
Rodent models of neuropathic pain are often used to study mechanisms of nociception as well as to evaluate potential therapeutics that can modulate the transmission and perception of pain. However, rodent models can be compromised by a number of species-related factors that complicate translation of results obtained in rodent studies to human clinical trials. One of these factors is the difference in ligand sensitivity between human and rodent receptors and ion channels.
Conotoxins are small peptides found in the venom of carnivorous marine snails of the Conus genus and are used by these mollusks to capture prey. α-Conotoxins (α-Ctxs) belong to a subclass of conotoxins and are antagonists of nAChRs. Some α-Ctxs are capable of distinguishing among the various nAChR subtypes (22). α-Ctx Vc1.1 (23, 24) is an example of a ligand developed as a treatment for neuropathic pain that showed promising results in rodent models but failed to produce similar levels of analgesia in human clinical trials. It was later demonstrated that decreased sensitivity of human versus rat nAChRs to Vc1.1 might have contributed to this outcome. It is therefore important to determine how species differences influence receptor sensitivity to ligands. To this end, we have examined the interaction of human and rat α6β4 nAChRs with α-Ctxs to elucidate molecular determinants of ligand potency for this receptor subtype. Through structure–activity studies of PeIA, site-directed mutagenesis of α6 and β4 subunits, X-ray crystallography, and NMR studies of PeIA, we have identified important molecular determinants of α-Ctx potency for human and rat α6β4 nAChRs. The information obtained from these studies offers important insights into the pharmacology of α6β4 nAChRs and might facilitate the development of selective ligands that interact with this potential neuropathic pain target.
Results
α-Conotoxins PeIA, PnIA, and TxIB inhibit human α6/α3β4 nAChRs more potently than rat α6/α3β4 nAChRs
We chose three α-Ctxs with different sequences to assess their ability to distinguish between human and rat α6/α3β4 nAChRs, namely PeIA (26), PnIA (27), and TxIB (28) (Fig. 1). When tested on human and rat α6/α3β4 nAChRs heterologously expressed in Xenopus oocytes, we found that all three α-Ctxs showed higher potency on the human homolog. The IC50 values for inhibition of human α6/α3β4 nAChRs were ∼15-fold lower for PeIA, ∼50-fold lower for PnIA, and >30-fold lower for TxIB than those obtained for rat α6/α3β4 nAChRs (Fig. 2 (A–C) and Table 1). These results suggest that intrinsic differences between human and rat α6/α3β4 nAChRs confer differential sensitivities to α-Ctxs.
Figure 1.

Sequence comparison of select 4/7 framework α-Ctxs. Conotoxins are classified into various subclasses based on the number of cysteine residues present in the sequence and the number of amino acid residues between them (63). Peptides of the α-Ctx subclass contain two disulfide-connected pairs of cysteines. Thus, for example, α-Ctxs with four residues between Cys2 and Cys3 and seven between Cys8 and Cys16 belong to the 4/7 framework subclass. Residues in black are variable, and those in red are conserved among this α-Ctx set. The disulfide bonds between Cys residues are depicted with lines. *, C-terminal amidation.
Figure 2.

α-Ctxs distinguish between human and rat α6/α3β4 nAChRs. X. laevis oocytes expressing human or rat α6/α3β4 nAChRs were subjected to TEVC electrophysiology, and the IC50 values were determined for inhibition of ACh-evoked currents by PeIA (A), PnIA (B), and TxIB (C). Values are provided in Table 1. Error bars, S.D. from at least four oocytes for each IC50 determination.
Table 1.
IC50 values for inhibition of human and rat α6/α3β4 nAChRs by PeIA, PnIA, and TxIB
Values in parentheses indicate the 95% CI of the data obtained from at least four oocytes per IC50 determination.
| α-Ctx | IC50 |
|
|---|---|---|
| Human α6/α3β4 | Rat α6/α3β4 | |
| nm | ||
| PeIA | 9.9 (8.2–11.9) | 154 (143–166) |
| PnIA | 149 (135–164) | 8,456 (5,419–13,290) |
| TxIB | 402 (338–477) | >10,000 |
Sequences of human and rat α6 subunits are highly conserved, but several residues of β4 that form the ligand-binding pocket differ
A sequence alignment of human and rat α6 subunits indicates that the extracellular domains are similar, and in fact residues that form the canonical ligand-binding pocket are strictly conserved (Fig. S1). The closest residue to the ligand-binding pocket that differs in the α6 sequences is located at position 177. In humans, this residue is Ile, but in rat, a Val is present. Analysis of the β4 subunits revealed more significant sequence differences occurring at positions 110, 118, and 119. In human β4, these residues are Leu, Val, and Leu, and in rat, they are Val, Ile, and Gln. These three nonconserved residues are all located in the ligand-binding pocket and might contribute to the species differences in α-Ctxs potencies for inhibition of human and rat α6/α3β4 nAChRs.
The β4 subunit is a major determinant of the species differences in PeIA, PnIA, and TxIB potencies for human versus rat α6/α3β4 nAChRs
A series of experiments was performed to determine whether differences in the amino acid sequences of human and rat β4 subunits are important for α-Ctx potency for α6/α3β4 nAChRs. In the first experiment, one group of oocytes was injected with cRNAs encoding human α6/α3 and rat β4 subunits, and another group was injected with cRNAs encoding rat α6/α3 and human β4 subunits. The potencies for PeIA, PnIA, and TxIB were then reassessed on these hybrid combinations with the hypothesis that the IC50 values for receptors composed of human (H) α6/α3 and rat (R) β4 subunits would be similar to Rα6/α3Rβ4 nAChRs. Likewise, the IC50 values for inhibition of Rα6/α3Hβ4 and Hα6/α3Hβ4 nAChRs would also be similar. Consistent with these predictions, the IC50 curves for inhibition of Hα6/α3Rβ4 by PeIA, PnIA, and TxIB are right-shifted toward those for Rα6/α3Rβ4 nAChRs (Fig. 3 (A–C) and Table 2). Conversely, the IC50 curves for inhibition of Rα6/α3Hβ4 are left-shifted toward Hα6/α3Hβ4 nAChRs (Fig. 3 (A–C) and Table 2). These results indicate that the β4 subunit is an important determinant of the species differences in α-Ctx potency for inhibition of α6/α3β4 nAChRs.
Figure 3.
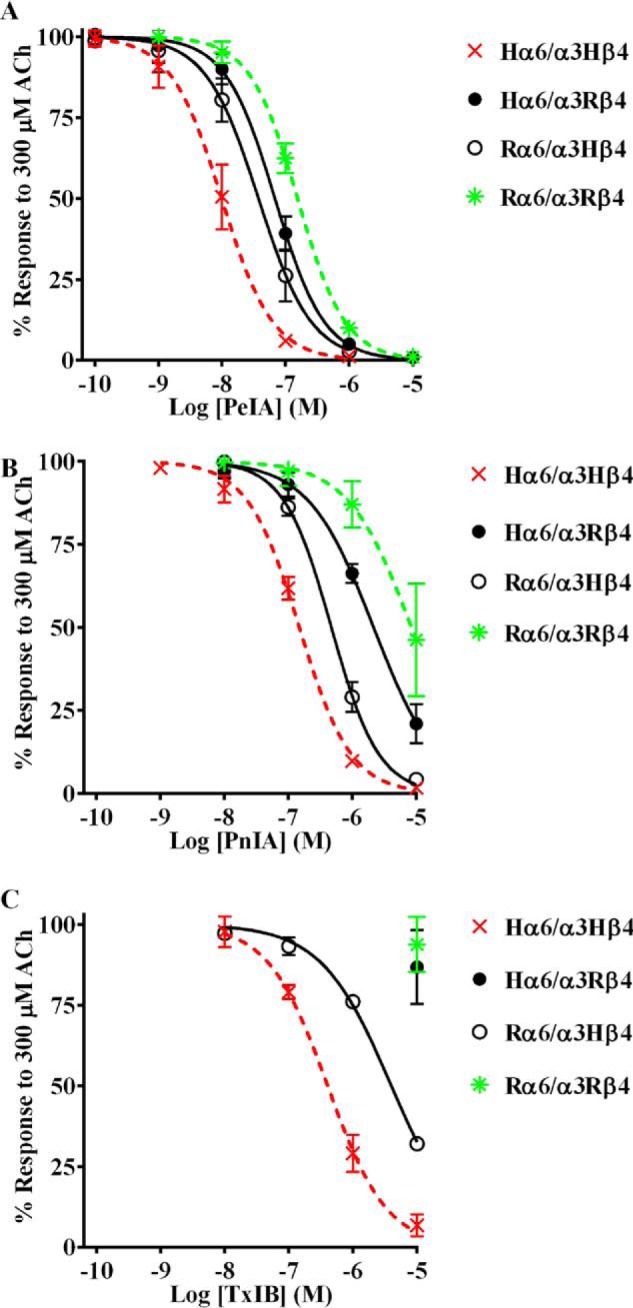
Potency of α-Ctx PeIA on α6/α3β4 nAChRs formed by switching species subunits. Different combinations of H and R α6/α3 and β4 subunits were expressed in X. laevis oocytes and subjected to TEVC electrophysiology as described under “Experimental procedures.” A–C, inhibition curves for ACh-evoked currents by PeIA, PnIA, and TxIB were obtained from oocytes expressing human α6/α3 with rat β4 subunits (closed circles) or rat α6/α3 with human β4 subunits (open circles). Data for inhibition of human (dashed red) and rat (dashed green) α6/α3β4 nAChRs by PeIA, PnIA, and TxIB were previously presented and shown for ease of comparison. Values are provided in Table 2. Error bars, S.D. from at least four oocytes for each IC50 determination.
Table 2.
PeIA IC50 values for inhibition of α6/α3β4 nAChRs formed by mixing human and rat α6/α3 and β4 subunits
Values in parentheses indicate the 95% CI of the data obtained from four oocytes per IC50 determination. H, human; R, rat.
| nAChR | IC50 |
|---|---|
| nm | |
| Hα6/α3Hβ4 | 9.9 (8.2–11.9)a |
| Hα6/α3Rβ4 | 68.1 (59.8–77.6) |
| Rα6/α3Rβ4 | 154 (143–166)a |
| Rα6/α3Hβ4 | 37.5 (30.1–46.2) |
a Data from Table 1 shown for ease of comparison.
As additional evidence in support of the importance of the β4 subunit for conferring differential α-Ctx potencies, we tested the same three α-Ctxs on human and rat α6/α3β2β3 nAChRs. In the α6β2β3 nAChR, the canonical α-Ctx ligand-binding site is located at the interface between the α6 and β2 subunits. We aligned the sequences of human and rat β2 subunits and found that residues known to interact with α-Ctxs are strictly conserved (29, 30), and in fact only three conservative differences were noted in the entire extracellular ligand-binding domain sequence (Fig. S2). When we tested PeIA on human and rat α6/α3β2β3 nAChRs, we found that the IC50 values for the two species were nearly identical (Fig. 4A and Table 3). Similar results were obtained for PnIA and TxIB (Fig. 4 (B and C) and Table 3). In all comparisons, a <2-fold difference in the IC50 values was found.
Figure 4.

α-Ctxs PeIA, PnIA, and TxIB do not distinguish between human and rat α6/α3β2β3 nAChRs. X. laevis oocytes expressing human or rat α6/α3β2β3 nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values were determined for inhibition of ACh-evoked currents by PeIA (A), PnIA (B), and TxIB (C). Values are provided in Table 3. Error bars, S.D. from at least four oocytes for each IC50 determination.
Table 3.
IC50 values for inhibition of human and rat α6/α3β2β3 nAChRs by PeIA, PnIA, and TxIB
Values in parentheses indicate the 95% CI of the data obtained from four oocytes per IC50 determination.
| α-Ctx | IC50 |
|
|---|---|---|
| Human | Rat | |
| nm | ||
| PeIA | 16.0 (14.2–18.1) | 16.3 (13.2–20.2) |
| PnIA | 2.8 (2.0–3.8) | 3.3 (3.0–3.6) |
| TxIB | 291 (255–332) | 207 (175–244) |
Site-directed mutagenesis of human β4 implicates residue 119 as a critical determinant of high-potency PeIA binding to human α6/α3β4 nAChRs
The high sequence similarity of human and rat α6 and β4 subunits allowed us to focus on select residues that might contribute to the species differences in α-Ctx potency for human and rat α6/α3β4 nAChRs. We used site-directed mutagenesis to generate receptor mutants where nonconserved residues of α6 and β4 subunits were switched to the residues found in the homologous positions of the other species and then assessed the effects of these mutations on α6/α3β4 nAChR sensitivity to PeIA. In the α6 subunit, there is an Ile-Val difference between human and rat sequences, respectively, at position 177. We found that when Ile177 of human α6 was switched to Val, the IC50 value of PeIA was similar to the value obtained for human α6/α3β4 nAChRs (Fig. 5A; Table 4), indicating that this residue contributes very little to the species difference in PeIA potency. Next, residues Leu110, Val118, and Leu119 of human β4 were individually switched to Val, Ile, and Gln, respectively, and expressed with human α6/α3 subunits. Similar to the α6I177V mutation, L110V and V118I mutations in the β4 subunit had very little impact on the IC50 value of PeIA (Fig. 5A and Table 4). However, when the α6/α3 subunit was expressed with the β4L119Q mutant subunit, the IC50 value increased by ∼10-fold, and the curve shifted to the right toward that of rat α6/α3β4 nAChRs (Fig. 5A and Table 4). To determine whether residues 118 and 119 might play a combined role in PeIA binding, we made a double V118I,L119Q mutant human β4 subunit and reassessed the potency of PeIA. The sensitivity of the α6/α3β4V118I,L119Q mutant to PeIA was similar to that of the α6/α3β4L119Q single mutant, indicating no added effect with the combined mutations (Fig. 5A and Table 4). To further assess the influence of position 119 on PeIA potency, we made an additional human β4 mutant where Leu119 was changed to Phe, the residue found in the homologous positions of human and rat β2 subunits (Fig. S2). The IC50 value for inhibition of α6/α3β4L119F mutant nAChRs by PeIA was similar (<2-fold difference) to the value for receptors with native β4 subunits (Fig. 5A and Table 4). Mutation experiments were also performed for rat α6/α3β4 nAChRs and, similar to the results found for human receptor α6V177I, β4V110L, β4I118V, and β4Q119F mutations, had very little effect on the sensitivity of rat α6/α3β4 nAChRs to PeIA relative to nonmutated receptors (Fig. 5B and Table 4). However, in contrast to the results obtained with human α6/α3β4L119Q and α6/α3β4V118I,L119Q nAChRs, mutation of the homologous residues of rat β4 had no effect on the sensitivity of rat α6/α3β4 nAChRs to PeIA (Fig. 5B and Table 4). The results of these receptor mutation experiments indicate that position 119 of human β4 plays an important role in the interaction between PeIA and human α6/α3β4 nAChRs, but other factors appear to be involved in determining the lower potency of PeIA for rat α6/α3β4 nAChRs.
Figure 5.

Residue Leu119 of human β4 is an important determinant of PeIA potency for human α6/α3β4 nAChRs. X. laevis oocytes expressing mutant α6/α3β4 nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values for inhibition of ACh-evoked currents by PeIA were determined. A, concentration–response curves for inhibition of human α6I177V/α3β4, α6/α3β4L110V, α6/α3β4V118I, α6/α3β4L119F α6/α3β4L119Q (blue), and α6/α3β4V118I,L119Q nAChRs by PeIA. B, concentration–response curves for inhibition of rat α6V177I/α3β4, α6/α3β4V110L, α6/α3β4I118V, α6/α3β4Q119L (blue), α6/α3β4Q119F, and α6/α3β4I118V,Q119L nAChRs by PeIA. Values are provided in Table 4. Data for inhibition of human (dashed red) and rat (dashed green) α6/α3β4 nAChRs by PeIA were previously presented and shown for ease of visual comparison. Error bars, S.D. from at least four oocytes for each IC50 determination.
Table 4.
Effect of α6/α3 and β4 subunit mutation on the IC50 values for inhibition of human and rat α6/α3β4 nAChRs by PeIA
Values in parentheses indicate the 95% CI of the data obtained from four oocytes per IC50 determination. Note that only the human β4L119Q mutation substantially (>3-fold) affects PeIA potency.
| nAChR | IC50 |
|---|---|
| nm | |
| Human | |
| α6/α3β4 | 9.9 (8.2–11.9)a |
| α6I177V/α3β4 | 14.6 (12.3–17.3) |
| α6/α3β4L110V | 15.7 (13.9–17.8) |
| α6/α3β4V118I | 10.6 (8.6–13.0) |
| α6/α3β4L119Q | 94.3 (87.8–101) |
| α6/α3β4L119F | 13.0 (10.7–15.7) |
| α6/α3β4V118I,L119Q | 70.7 (66.1–75.6) |
| Rat | |
| α6/α3β4 | 154 (143–166)a |
| α6V177I/α3β4 | 88.7 (76.3–103) |
| α6/α3β4V110L | 96.3 (85.9–108) |
| α6/α3β4I118V | 119 (105–134) |
| α6/α3β4Q119L | 200 (166–250) |
| α6/α3β4Q119F | 241 (212–274) |
| α6/α3β4I118V,Q119L | 116 (101–133) |
a Data from Table 1 shown for ease of comparison.
X-ray crystallography of PeIA complexed with Aplysia californica AChBP reveals close spatial proximity of residue Pro13 of PeIA to Met119 of the AChBP complementary subunit
The marine mollusk Aplysia californica expresses a water-soluble protein called AChBP that functions to modulate ACh-mediated synaptic transmission in this organism (31). X-ray crystallography studies of the AChBP have been used to elucidate binding interactions between ligands and nAChRs (32–35). Several high-resolution crystal structures have been reported for the AChBP complexed with α-Ctxs (36–39) but not with PeIA. To gain further insights into the interactions between PeIA and human α6β4 nAChRs, we performed X-ray crystallography of the AChBP complexed with PeIA. The resulting 2.34 Å resolution structure of the AChBP–PeIA complex is shown in Fig. 6 (A–C). An examination of the complex reveals that PeIA residue Pro13 is oriented toward the complementary subunit and in close proximity to Met119 of the AChBP (Fig. 6C). In the α6β4 receptor complex, the complementary subunit corresponds to the β4 subunit. A sequence alignment of the human β4 ligand-binding domain with the AChBP indicates that Met119 of the AChBP is homologous with human β4Leu-119 (Fig. S3). The proximity of Met119 of the AChBP to Pro13 of PeIA suggests that an interaction might occur between these two residues, and functional analysis of human α6/α3β4L119M mutant nAChRs expressed in oocytes indicates that a Met in position 119 influences PeIA activity (Fig. S3).
Figure 6.

X-ray crystallography of PeIA complexed with the A. californica AChBP. A and B, cartoon rendition of the AChBP–PeIA structure shown from the side (A) and from the top or extracellular view (B). The AChBP is shown in green, and PeIA is shown in red. Residues 1–10 of PeIA are recognized as forming two α-helices split between residues His5 and Pro6. An electron density for a fifth PeIA molecule was observed but omitted because the clarity of the electron density was such that only the peptide backbone structure could be determined accurately, not the side-chain positions. C, cartoon rendition of two subunits showing the location of residues Ala110, Val118, and Met119 (yellow) in the complementary (−) subunit (cyan) and Leu177 (yellow) of the principal subunit (green). Note that the side chain of Met119 is oriented toward PeIA, whereas the side chains of Ala110 and Val118 are oriented away from PeIA. Pro13 of PeIA is depicted as a stick model. The distances between the γ-carbon of PeIA Pro13 and the δ-sulfur and ϵ-carbon of Met119 were 3.6 and 5.4 Å, respectively. D, cartoon rendition of the AChBP–PeIA structure with positions 177 of the principal (+) subunit and 110, 118, and 119 of the complementary (−) subunit mutated to the residues found in the homologous positions of human α6 and β4 subunits, respectively. The distance between the γ-carbon of PeIA Pro13 and the γ-carbon of Leu119 of the complementary subunit was 4.0 Å. Note that position 177 is located outside the ligand-binding pocket and therefore unlikely to directly interact with PeIA.
Structure–activity studies identify Pro13 as important for PeIA potency on human and rat α6/α3β4 nAChRs
The crystal structure of PeIA complexed with the AChBP suggests that PeIA residue Pro13 probably interacts with the β4 subunit. Therefore, we used analogs of PeIA where Pro13 was substituted with different amino acids to probe the interaction between this residue and position 119 of the β4 subunit. Four analogs, including [P13A]PeIA and [P13O]PeIA, described previously (40), as well as two newly synthesized analogs, [P13Q]PeIA and [P13R]PeIA, were tested on human α6/α3β4 nAChRs expressed in oocytes and evaluated for changes in potencies relative to native PeIA. Substitution of Pro13 with 4-hydroxyproline (Hyp) or Gln resulted in small decreases (∼3-fold) in the potency of PeIA for human α6/α3β4 nAChRs (Fig. 7A and Table 5). However, substitution of Pro with Ala or Arg resulted in more substantial decreases in potency by ∼7- and ∼20-fold, respectively (Fig. 7A and Table 5). The rank order potency of these analogs is [P13O]PeIA = [P13Q]PeIA > [P13A]PeIA > [P13R]PeIA. The IC50 values for these analogs were then determined for rat α6/α3β4 nAChRs. Although all analogs showed reduced potency, relative to native PeIA, there were differences in the magnitude of the changes for rat compared with human α6/α3β4 nAChRs. The potency of [P13A]PeIA for rat α6/α3β4 nAChRs was only 3-fold lower (Fig. 7B and Table 5) compared with the 10-fold lower potency observed for the human homolog. Analogs [P13Q]PeIA and [P13R]PeIA showed ∼53- and ∼335-fold lower potencies for rat α6/α3β4 compared with ∼3- and ∼20-fold lower potencies (Fig. 7B and Table 5), respectively, for human α6/α3β4 nAChRs. Substitution of Pro13 with Hyp resulted in ∼4-fold lower potency on rat α6/α3β4 nAChRs (Fig. 7B and Table 5) similar to the ∼3-fold decrease observed for human α6/α3β4 nAChRs. Thus, the rank order potency for these analogs on rat α6/α3β4 nAChRs is [P13A]PeIA > [P13O]PeIA > [P13Q]PeIA > [P13R]PeIA.
Figure 7.

PeIA residue Pro13 is an important determinant of PeIA potency for α6/α3β4 nAChRs. X. laevis oocytes expressing human or rat α6/α3β4 nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values were determined for inhibition of ACh-evoked currents by analogs of PeIA. A, concentration–response curves for inhibition of human α6/α3β4 nAChRs by analogs of PeIA. B, concentration–response curves for inhibition of rat α6/α3β4 nAChRs by analogs of PeIA. Values are provided in Table 5. Error bars, S.D. from at least four individual oocytes for each IC50 determination. Data for inhibition of human α6/α3β4 (dashed red) and rat α6/α3β4 nAChRs (dashed green) by PeIA were presented previously and shown for ease of visual comparison.
Table 5.
IC50 values for inhibition of human and rat α6/α3β4 nAChRs by analogs of PeIA
Values in parentheses indicate the 95% CI of the data obtained from four oocytes per IC50 determination. Log(IC50 ratio) = log(analog/PeIA).
| α-Ctx | Human |
Rat |
||
|---|---|---|---|---|
| IC50 | Log (IC50 ratio) | IC50 | Log (IC50 ratio) | |
| nm | nm | |||
| PeIA | 9.9 (8.2–11.9)a | 154 (143–166)a | ||
| [P13O]PeIA | 32.6 (28.2–37.7) | 0.5 | 688 (540–876) | 0.7 |
| [P13A]PeIA | 73.3 (66.8–80.5) | 0.9 | 381 (317–456) | 0.4 |
| [P13Q]PeIA | 34.0 (26.1–44.4) | 0.5 | 8,231 (5,784–11,710) | 1.7 |
| [P13R]PeIA | 188 (159–222) | 1.3 | 51,780 (29,710–90,270) | 2.5 |
a Data from Table 1 shown for ease of comparison.
NMR analysis of PeIA demonstrates that the backbone structure and side-chain positions are unchanged with Ala substitution of Pro13
One possible effect of substituting PeIA Pro13 with other amino acids is that changes in the tertiary structure of the peptide might occur. To determine whether large changes in the backbone structure or repositioning of the side chains could account for the loss of potency observed with the P13A substitution, we performed NMR analysis on [P13A]PeIA and compared its structure with that of native PeIA. A pattern of negative secondary shifts across residues Ala7–Val10 indicated the presence of a short helix (Fig. 8A) in agreement with the NMR solution structure of PeIA determined previously (41). Analysis of the secondary Hα shifts, as calculated by subtracting random coil Hα shifts from the peptide Hα shifts, suggested a slight difference in [P13A]PeIA across the C-terminal section of the peptide from residue Asn11 to Cys16. Therefore, we calculated the three-dimensional solution NMR structure of [P13A]PeIA to determine whether there were changes in the overall structure of the peptide. A total of 98 distance restraints were determined from NOESY data collected in aqueous solution at 290 K, along with 14 dihedral angle restraints. Restraints for two hydrogen bonds (Val10 HN–Pro6 CO and His12 HN–Cys8 CO) were also added based upon preliminary structures and amide chemical shift/temperature coefficients (42). The ensemble of the 20 lowest-energy structures overlay well (root mean square deviation 0.60 ± 0.16 Å for the backbone atoms) (Fig. 8B). A comparison with native PeIA revealed no apparent effect of the P13A substitution on either peptide backbone (root mean square deviation 1.02 Å, backbone atoms) or side-chain orientation (Fig. 8C). These structural data suggest that the losses of potency observed with the P13A-substituted analog are not due to large changes in tertiary structure of the peptide.
Figure 8.

NMR indicates that Ala substitution of Pro13 has minimal impact on peptide structure. A, secondary αH shifts of native PeIA and the [P13A]PeIA analog in aqueous solution at 290 K. The horizontal axis represents the sequence of PeIA. B, backbone superposition of the 20 lowest-energy structures of [P13A]PeIA. C, ribbon diagram of [P13A]PeIA (blue) overlaid with PeIA (gray, PDB obtained from ConoServer (25)). An α-helix is present from residue 7 to 10, and the side chains of residue 13 are shown as sticks.
A direct interaction between PeIA Pro13 and position 119 of β4 is involved in the binding of PeIA to human α6/α3β4 nAChRs
The close proximity of PeIA Pro-13 and Met-119 of the AChBP complementary subunit and the lower potencies observed with the PeIA analogs suggest that Pro-13 and position 119 of the β4 subunit might directly interact. Double mutant paradigms where ligand mutants are paired with receptor mutants can be used to examine pairwise interactions between ligand and receptor residues. Therefore, to determine whether an interaction occurs between Pro-13 and position 119, we tested the PeIA analogs on the β4 subunit mutants and found that the IC50 value of [P13A]PeIA for inhibition of human α6/α3β4L119Q mutant receptors was higher than the values obtained for the α6/α3β4:[P13A]PeIA and α6/α3β4L119Q:PeIA combinations (Fig. 9A and Table 6). Similar results were found when [P13Q]PeIA and [P13R]PeIA were tested on the α6/α3β4L119Q mutant (Fig. 9 (B and C) and Table 6). In each case, the -fold increase in the IC50 value for each double peptide–receptor mutant combination was larger than the sum of the each individual mutant IC50. These data are consistent with a direct interaction between residue 13 of PeIA and residue 119 of human β4.
Figure 9.

Structure–activity studies identify a critical interaction between PeIA Pro13 and β4Leu-119 for high potency on human α6/α3β4 nAChRs. X. laevis oocytes expressing human nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values were determined for inhibition of ACh-evoked currents by α-Ctxs. A–C, concentration–response curves for inhibition of α6/α3β4L119Q mutant nAChRs by [P13A]PeIA, [P13R]PeIA, and [P13Q]PeIA. The IC50 values are provided in Table 6. Error bars, S.D. from at least four individual oocytes for each IC50 determination. Dashed lines, data previously presented and shown for ease of visual comparison.
Table 6.
IC50 values for inhibition of human α6/α3β4 mutants and α6/α3β2 nAChRs by α-Ctxs
Values in parentheses indicate the 95% CI of the data obtained from at least four oocytes per IC50 determination. The log(IC50 ratio) was calculated by dividing the IC50 value for each nAChR:α-Ctx combination (nAChR:PeIA analog, nAChR mutant:α-Ctx, or nAChR mutant:PeIA analog) by the IC50 for the WT:WT combination; negative values indicate higher potency, and positive values indicate lower potency. The -fold change in IC50 = nAChR:PeIA analog/WT:WT, nAChR mutant:α-Ctx/WT:WT, or nAChR mutant:PeIA analog/WT:WT. Note that for the latter comparison, the IC50 values are greater than the sum of each single nAChR mutant or PeIA analog combination (nAChR:PeIA analog/WT:WT or nAChR mutant:PeIA/WT:WT). Differences between IC50 values are considered substantial if ≥0.5 log units or ≥3-fold. WT, WT receptor or WT α-Ctx.
| nAChR:α-Ctx | IC50 | Log (IC50 ratio) | Change in IC50 | |
|---|---|---|---|---|
| nm | -fold | |||
| α6/α3β4:PeIA | 9.9 (8.2–11.9)a | |||
| α6/α3β4L119Q:PeIA | 94.3 (87.8–101)a | 1.0 | 10 | |
| α6/α3β4:[P13A]PeIA | 73.3 (66.8–80.5)a | 0.9 | 7 | |
| α6/α3β4L119Q:[P13A]PeIA | 244 (223–267) | 1.4 | 25 | |
| α6/α3β4:[P13R]PeIA | 188 (159–222)a | 1.3 | 19 | |
| α6/α3β4L119Q:[P13R]PeIA | 365 (331–403) | 1.6 | 37 | |
| α6/α3β4:[P13Q]PeIA | 34.0 (26.1–44.4)a | 0.5 | 3 | |
| α6/α3β4L119Q:[P13Q]PeIA | 363 (287–460) | 1.6 | 37 | |
| α6/α3β4L119F:PeIA | 13.0 (10.7–15.7)a | 0.1 | 1 | |
| α6/α3β4L119F:[P13A]PeIA | 56.7 (52.8–61.3) | 0.8 | 6 | |
| α6/α3β4L119F:[P13R]PeIA | 208 (170–256) | 1.3 | 21 | |
| α6/α3β2β3:PeIA | 16.0 (14.2–18.1)a | |||
| α6/α3β2β3:[P13A]PeIA | 30.6 (25.5–36.7) | 0.3 | 2 | |
| α6/α3β2β3:[P13R]PeIA | 8.9 (8.2–9.6) | −0.3 | 0.6 | |
| α6/α3β4:PnIA | 149 (135–164)a | |||
| α6/α3β4L119F:PnIA | 94.6 (87.9–102) | −0.2 | 0.6 | |
a Data previously shown for ease of comparison.
Position 119 of human and rat β4 and human β2 subunits interacts differentially with residue 13 of α-Ctxs to influence binding
The results shown in Fig. 5 indicate that mutating residue 119 of β4 to Phe has no impact on the potency of PeIA for human or rat α6/α3β4 nAChRs. However, we note that there are some differences in the potencies of the Pro13-substituted PeIA analogs on human α6/α3β4 and those previously found for rat α6/α3β2β3 nAChRs. PeIA shows a ∼7-fold loss of potency on human α6/α3β4 nAChRs with the P13A substitution (Fig. 7A and Table 5), yet rat α6/α3β2β3 nAChRs are equally sensitive to PeIA and the P13A analog (40). Furthermore, PnIA, which also has a Pro in position 13, is significantly more potent on human α6/α3β2β3 than α6/α3β4 nAChRs, in contrast to PeIA, which is equally potent on both subtypes (Fig. 3 (A and B) and Table 3). These differences in potency suggest that the binding interactions between Pro13 of α-Ctxs and position 119 of the β2 and β4 subunits may not be equivalent. Therefore, we tested [P13A]PeIA for its ability to inhibit the α6/α3β4L119F mutant and compared the IC50 value with those for human α6/α3β4 and α6/α3β2β3 nAChRs. The Leu to Phe mutation in position 119 of β4 had no effect on the loss of PeIA potency for α6/α3β4 nAChRs caused by the P13A substitution (Fig. 10A and Table 6). Similar results were observed with the [P13R]PeIA analog (Fig. 10A and Table 6). In each case, the higher IC50 values for inhibition of the α6/α3β4L119F mutant by [P13A]PeIA and [P13R]PeIA were similar to those for α6/α3β4 nAChRs. By contrast, the IC50 values of [P13A]PeIA and [P13R]PeIA on human α6/α3β2β3 nAChRs were unchanged relative to the values for native PeIA (Fig. 10B and Table 6). Additionally, there were no differences between the IC50 values of PnIA for the α6/α3β4L119F mutant and α6/α3β4 nAChRs (Fig. 10C and Table 6). These results are in contrast to those found when PnIA was tested on rat α6/α3β4Q119F mutant receptors; the IC50 value was ∼12-fold lower than the IC50 value for rat α6/α3β4 nAChRs (Fig. 11A and Table 7). Similarly, the IC50 value for PnIA was ∼9-fold lower on the rat α6/α3β4Q119L mutant (Fig. 11A and Table 7), also in contrast to the lack of effect observed for human α6/α3β4L119Q nAChRs (Fig. 10C and Table 6). Nevertheless, similar to the results obtained for the human α6/α3β4L119F mutant, rat α6/α3β4Q119F nAChRs were less sensitive to [P13R]PeIA and [P13Q]PeIA than to native PeIA (Fig. 11B and Table 7). These results indicate that with respect to ligand potency, the interactions between residue 13 of α-Ctxs and position 119 of β2 and β4 subunits are not equivalent. Furthermore, the interaction between PeIA Pro13 and position 119 of the β subunit is involved in the ability of PnIA to discriminate between rat, but not human, α6/α3β4 and α6/α3β2β3 nAChRs.
Figure 10.

α-Ctx potency for human α6/α3β4 nAChRs is not affected by mutation of Leu119 to Phe. X. laevis oocytes expressing human nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values were determined for inhibition of ACh-evoked currents by α-Ctxs. A, concentration–response curves for inhibition of α6/α3β4L119F mutant nAChRs by [P13A]PeIA and [P13R]PeIA. B, concentration–response curves for inhibition of α6/α3β2β3 nAChRs by [P13A]PeIA and [P13R]PeIA. Note that P13A and P13R substitutions of PeIA have very little effect on PeIA potency for inhibition of the α6/α3β2β3 subtype. C, concentration–response curves for inhibition of α6/α3β4L119F mutant and α6/α3β2β3 nAChRs by PnIA. Note that mutating Leu119 of the β4 subunit to Phe has no effect on PnIA potency for α6/α3β4 nAChRs. The IC50 values are provided in Table 6. Error bars, S.D. from at least four individual oocytes for each IC50 determination. The ACh concentrations used were 300 μm for α6/α3β4, α6/α3β4L119Q, and α6/α3β4L119F nAChRs and 100 μm for the α6/α3β2β3 subtype. Dashed lines, data previously presented and shown for ease of visual comparison.
Figure 11.

Structure–activity studies of PnIA and PeIA demonstrate interaction between position 119 of rat β4 and α-Ctxs. X. laevis oocytes expressing rat nAChRs were subjected to TEVC electrophysiology as described under “Experimental procedures,” and the IC50 values were determined for inhibition of ACh-evoked currents by α-Ctxs. A, concentration–response curves for inhibition of α6/α3β4Q119L and α6/α3β4Q119F mutant nAChRs by PnIA. The IC50 values are provided in Table 7. Error bars, S.D. from at least four individual oocytes for each IC50 determination. Dashed lines, data previously presented for PnIA on rat α6/α3β2β3 and α6/α3β4 nAChRs and shown for ease of visual comparison. B, representative current traces showing inhibition of α6/α3β4Q119F and α6/α3β4Q119L by 10 μm PnIA. Traces in black are control responses, and those in red are responses in the presence of PnIA. C, response analysis of [P13Q]PeIA and [P13R]PeIA inhibition of ACh-evoked currents mediated by mutant α6/α3β4Q119L and α6/α3β4Q119F nAChRs. The currents mediated by α6/α3β4Q119L and α6/α3β4Q119F nAChRs in the presence of 10 μm [P13Q]PeIA were 82 ± 5% (n = 4) and 69 ± 5% (n = 5), respectively, of control responses and were significantly larger than those obtained for α6/α3β4 nAChRs (47 ± 5% (n = 4)). The responses of α6/α3β4Q119L and α6/α3β4Q119F nAChRs after exposure to 10 μm [P13R]PeIA were also significantly larger than those obtained with α6/α3β4 nAChRs under the same conditions (98 ± 2% (n = 4) and 86 ± 4% (n = 4), respectively, of control responses compared with 58 ± 3% (n = 4) for α6/α3β4 nAChRs). Statistical significance was determined using an analysis of variance and Fisher's least significant difference test (***, p ≤ 0.001; ****, p ≤ 0.0001). Error bars, S.D. for the indicated number of individual replicates. D, representative current traces for inhibition of mutant α6/α3β4Q119L and α6/α3β4Q119F nAChRs by the indicated PeIA analogs. Traces in black are control responses, and those in red are responses in the presence of the α-Ctxs.
Table 7.
IC50 values for inhibition of rat α6/α3β4 mutants and α6/α3β2β3 nAChRs by α-Ctxs
Values in parentheses indicate the 95% CI of the data obtained from at least four oocytes per IC50 determination. The log(IC50 ratio) was calculated by dividing the IC50 value for each nAChR:α-Ctx combination (nAChR:PeIA analog, nAChR mutant:α-Ctx, or nAChR mutant:PeIA analog) by the IC50 for the WT:WT combination; negative values indicate higher potency, and positive values indicate lower potency. The -fold change in IC50 = nAChR:PeIA analog/WT:WT, nAChR mutant:α-Ctx/WT:WT, or nAChR mutant:PeIA analog/WT:WT. Differences between IC50 values are considered substantial if ≥0.5 log units or ≥3-fold. WT, WT receptor or WT α-Ctx.
| nAChR:α-Ctx | IC50 | Log (IC50 ratio) | Change in IC50 |
|---|---|---|---|
| nm | -fold | ||
| α6/α3β4:PeIA | 154 (143–166)a | ||
| α6/α3β4Q119L:PeIA | 203 (166–250)a | 0.1 | 1 |
| α6/α3β4:[P13Q]PeIA | 8,231 (5,784–11,710)a | 1.7 | 53 |
| α6/α3β4Q119L:[P13Q]PeIA | >10,000 | >1.8 | >65 |
| α6/α3β4:[P13R]PeIA | 51,780 (29,710–90,270)a | 2.5 | 336 |
| α6/α3β4Q119L:[P13R]PeIA | >10,000 | >1.8 | >65 |
| α6/α3β4Q119F:PeIA | 241 (212–274)a | 0.2 | 2 |
| α6/α3β4Q119F:[P13Q]PeIA | 35,170 (24,000–51,520) | 2.4 | 228 |
| α6/α3β4Q119F:[P13R]PeIA | >10,000 | >1.8 | >65 |
| α6/α3β2β3:PnIA | 3.3 (3.0–3.6)a | ||
| α6/α3β4:PnIA | 8,456 (5,419–13,290)a | ||
| α6/α3β4Q119L:PnIA | 942 (744–1,192) | −1.0 | 0.1 |
| α6/α3β4Q119F:PnIA | 684 (533–878) | −1.1 | 0.1 |
a Data previously shown for ease of comparison.
Discussion
Compounds expected to have analgesic properties are often tested in rodent models of pain with the expectation (and hope) that the results will be translatable to humans. However, several factors can influence whether a drug that is effective in rodents will also produce the same efficacy in humans. Some of these factors include bioavailability, metabolism, and, importantly, sensitivity of the molecular target to the compound of interest. Species differences in the amino acid sequences of target molecules can have substantial effects on ligand potency. For example, α-Ctx Vc1.1 was an effective analgesic in rodent models of neuropathic pain but failed to show similar efficacy in human clinical trials (23–25). A single amino acid difference between human and rat α9 nAChR subunits was later shown to confer higher potency for rat over human α9α10 nAChRs, a potential analgesic target of Vc1.1 (43).
We recently reported that human α3β4 nAChRs are essentially insensitive to the α3β4 antagonist α-Ctx AuIB but show increased sensitivity to other α-Ctxs compared with rat α3β4 nAChRs (44, 45). Understanding how species differences influence ligand potency for α6β4 nAChRs is potentially critical for the successful development of analgesic ligands, with minimal off-target effects, for treating neuropathic pain in humans. In this study, we report that human and rat α6/α3β4 nAChRs show differential sensitivity to α-Ctxs PeIA, PnIA, and TxIB (Fig. 2 and Table 1). To evaluate the contributions of the α6 and β4 subunits to the higher α-Ctx potency for human α6/α3β4 nAChRs, we expressed human α6/α3 subunits with rat β4 subunits in Xenopus oocytes and found that the IC50 curves for inhibition of this hybrid combination by all three tested α-Ctxs are substantially right-shifted toward those for Rα6/α3Rβ4 nAChRs (Fig. 3 and Table 2). Likewise, when rat α6/α3 is expressed with human β4, the IC50 curves are left-shifted toward those for Hα6/α3Hβ4 nAChRs (Fig. 3 and Table 2). These experiments suggest that intrinsic properties of human β4 are important determinants of higher α-Ctx potency for α6β4 nAChRs. Complementary experiments were also conducted on oocytes expressing human or rat α6/α3β2β3 nAChRs to determine whether the β2 subunit might also produce species differences in α-Ctx potency. However, the IC50 values for PeIA, PnIA, and TxIB differed by less than ∼2-fold, indicating that species differences between human and rat contribute to the differential sensitivities of α6/α3β4, but not α6/α3β2β3, nAChRs to these α-Ctxs (Fig. 4 and Table 3).
There are several factors that might contribute to the higher α-Ctx potency for human α6/α3β4 nAChRs. First, there are three nonconserved residues of human and rat β4 subunits that form the ligand-binding pocket and might directly interact with ligands potentially affecting ligand affinity. Nonconserved residues outside the ligand-binding pocket might also contribute, albeit indirectly, to ligand affinity by altering the tertiary structure of the subunit. This, in turn, might affect the way the subunits associate with each other to create the ligand-binding surfaces. Here, we have examined the effect of substituting nonconserved residues of human α6 and β4 subunits with residues found in the homologous positions of the respective rat subunits. These residues include Ile177 of human α6 and Leu110, Val118, and Leu119 of human β4 (Fig. S1). We observed that mutation of Ile177 to Val in the α6 sequence has no effect on PeIA potency, but substantial effects were found with mutations of human β4. When Leu119 was mutated to Gln, the potency of PeIA for α6/α3β4 nAChRs was reduced by ∼10-fold, but when Leu119 was mutated to Phe, the potency was unchanged (Fig. 5A and Table 4). PeIA potency was also reduced by ∼4-fold when Leu119 was mutated to Met (Fig. S3). Additional mutations of human β4 were made to determine whether other nonconserved residues contributed to species differences in PeIA potency. We found that the potency of PeIA was not affected by either L110V or V118I mutations. A double mutant combining the V118I and L119Q mutations was then made to determine whether an interaction between Ile and Gln could potentially influence PeIA potency, but no further decrease in PeIAs IC50 value, compared with that for the α6/α3β4L119Q single mutant, was observed. It should be noted that mutation of Leu to Val and Val to Ile are conservative changes that might not result in easily observable effects on PeIA potency. Nevertheless, these experiments demonstrate that of the three nonconserved ligand-binding pocket residues between human and rat β4, only the Leu–Gln difference at position 119 contributes to the higher affinity of PeIA for human α6/α3β4 nAChRs. Similar experiments were conducted for rat α6/α3β4 nAChRs, but strikingly, we found that none of the mutations in rat subunits substantially changed the potency of PeIA, including the β4Q119L mutation (Fig. 5B and Table 4). Therefore, we took a different approach to further examine the interaction of PeIA with α6/α3β4 nAChRs.
X-ray crystallography studies of PeIA complexed with the AChBP showed that PeIA Pro-13 is in close proximity to Met-119 of the complementary subunit. Based on this observation, we synthesized analogs of PeIA where Pro13 was substituted with Hyp, Gln, Ala, or Arg to determine whether Pro13 and position 119 of the β4 subunit interacted (Fig. 7 (A and B) and Table 5). Substitution of Pro13 with Hyp or Gln reduced the potency of PeIA for human α6/α3β4 nAChRs but by only ∼3-fold. More substantial decreases in potency resulted when Pro13 was substituted with Ala (∼7-fold) or Arg (∼20-fold). Although all four PeIA analogs showed reduced potencies for rat α6/α3β4 nAChRs, they did not show the same magnitude of change found for human α6/α3β4 nAChRs. The most substantial differences were found with the P13Q and P13R substitutions (Fig. 7 (A and B) and Table 5). [P13Q]PeIA showed a ∼53-fold reduction in potency for rat α6/α3β4 nAChRs, much larger than the ∼3-fold reduction observed for human α6/α3β4. The P13R substitution rendered rat α6/α3β4 nAChRs essentially insensitive to PeIA and resulted in a ∼335-fold reduction in potency relative to native PeIA. The [P13A]PeIA analog showed the largest reduction in potency for human (∼7-fold) compared with rat (∼3-fold) α6/α3β4 nAChRs (Fig. 7 (A and B) and Table 5). The results obtained with these PeIA analogs reveal additional species differences in ligand binding and suggest that the ligand-binding surfaces of human and rat α6/α3β4 nAChRs are intrinsically different despite having highly conserved ligand-binding domain sequences.
Additional support for an interaction between PeIA Pro13 and position 119 of the β4 subunit was obtained by determining the potencies of the PeIA analogs on the human α6/α3β4L119Q (Fig. 8 (A–C) and Table 6) and rat α6/α3β4Q119L mutants (Fig. 11B and Table 7). These double ligand–receptor mutant combinations resulted in additional reductions in PeIA potency for both species, lending support for a direct interaction between PeIA Pro13 and position 119 of the β4 subunit.
To ensure that the reduced potencies observed with the PeIA analogs were not the result of changes in the overall structure of the peptide due to substitution of rigid Pro, we examined the structure of [P13A]PeIA and compared it with that of the native peptide using NMR. Although some differences in the αH shifts of residues 10–15 were observed, the overall backbone structure appeared unchanged relative to the native peptide (Fig. 8). Furthermore, the orientations of the Pro13 and Ala13 side chains were similar in the native peptide and the analog, respectively, suggesting that the losses in potencies observed with the PeIA analogs are not due to repositioning of the side chains. Last, the PeIA potency differences observed for human α6/α3β4 and α6/α3β4L119Q nAChRs are not due to changes in the potency or efficacy of the agonist acetylcholine (Fig. S4).
Zhangsun et al. showed that mutation of Phe119 to Gln in the rat β2 subunit results in increased potency of LvIA for α3β2 nAChRs (46). By contrast, we found that for human α6/α3β4 nAChRs, mutation of β4Leu-119 to Gln decreased the potency of PeIA by ∼10-fold, and mutation to Phe had no effect (Fig. 5A and Table 4). These results initially suggested that Phe can substitute for Leu in position 119 with respect to PeIA binding and might offer a “protective” effect against the reduction in potencies observed with the [P13A]PeIA and [P13R]PeIA analogs because the IC50 values for [P13A]PeIA and [P13R]PeIA on human α6/α3β2β3 nAChRs are unchanged relative to native PeIA (Fig. 10B and Table 6). However, when [P13A]PeIA and [P13R]PeIA were tested on the human α6/α3β4L119F mutant, the loss of PeIA potency was similar to that found for nonmutated receptors (Fig. 10A and Table 6). Additionally, the rat α6/α3β4Q119F mutant as well as the α6/α3β4Q119L mutant also showed lower sensitivity to [P13R]PeIA and [P13Q]PeIA (Fig. 11 (B and C) and Table 7). These results suggest that position 119 of human β2 and β4 and rat β4 are not equivalent with respect to ligand binding even when the same amino acid is present (by mutation) in the receptor. Additional evidence for the nonequivalency of position 119 for ligand binding was obtained with PnIA. There was no difference in PnIA potency between human α6/α3β4 and the α6/α3β4L119F mutant and, consequently, no change in the ability of PnIA to discriminate between human α6/α3β2β3 and α6/α3β4 nAChRs (Fig. 10C and Table 6). However, PnIA showed substantially increased potency on rat α6/α3β4Q119F and α6/α3β4Q119L mutants (Fig. 11A and Table 7). In summary, the results of these receptor and ligand mutation studies argue for species- and subtype-specific ligand–receptor interactions that are context-dependent. Therefore, we urge caution directly comparing ligand–receptor interactions among species as well as generalizing results obtained for different nAChR subtypes of the same species. Furthermore, the fact that both PeIA and PnIA have a Pro residue in position 13 but only PnIA shows differential potencies for mutant rat, but not human, α6/α3β4 nAChRs also argues for caution when generalizing interactions between receptor residues and residues of different α-Ctxs.
The α6β4 subtype is an emerging, novel target for the treatment of neuropathic pain, but very little information is available concerning the interaction of ligands with this subtype at the molecular level. Ligands selective for α6β4 nAChRs might be critical to avoid off-target effects that can occur due to interactions with closely related subtypes, particularly α3β4. In this study, we have identified β4Leu-119 as an important residue of the human β4 subunit that interacts with α-Ctxs. To our knowledge, this is the first report to functionally identify this key interaction. The information contained in this study might ultimately guide the design of ligands that target α6β4 nAChRs for the treatment of neuropathic pain.
Experimental procedures
Oocyte two-electrode voltage-clamp electrophysiology
Xenopus laevis frogs were obtained from Xenopus Express (RRID:SCR_016373). Protocols for the isolation of oocytes from the frogs were approved by the University of Utah institutional animal care and use committee. Methods describing the preparation of cRNA encoding human and rat nAChR subunits for expression of nAChRs in Xenopus oocytes have been described previously (44). The human and rat α6/α3 constructs were generated by replacing the extracellular ligand-binding domain of the α3 subunit with that of the α6 subunit as described previously (47, 48). These constructs were used because injection of oocytes with cRNAs encoding human α6 and β4 subunits resulted in no functionally expressed receptors across multiple donors (data not shown). The rat α6/α3 construct was used for comparison and has been previously shown to display similar sensitivities to α-Ctxs compared with nonmutated α6 (49). Preliminary experiments varying the ratio of rat α6/α3 to β4 subunit cRNAs by 10:1 or 1:10 to favor the formation of receptors with different stoichiometries had no effect on PeIA potency (data not shown). In all subsequent experiments, the oocytes were injected with equal ratios of cRNA for all subunit combinations.
Data analysis
Concentration–response data were obtained from a minimum of four oocytes. α-Ctx concentrations of ≤1 μm were applied to the oocyte by continuous perfusion, and the ACh responses in the presence of the α-Ctxs were normalized to the average of at least three control responses. α-Ctxs were applied only after the ACh response-to-response variation was <10%. The variance of the responses is provided as the ±S.D. and shown with error bars. α-Ctxs were routinely retested on oocytes from different donors to ensure data reproducibility. To estimate the IC50 value for inhibition of the ACh responses by a given α-Ctx, the normalized data were analyzed by nonlinear regression and fit using a four-parameter logistic equation in Prism (RRID:SCR_002798) (GraphPad Software Inc., La Jolla, CA). The IC50 values are presented with corresponding 95% confidence intervals to evaluate the precision of the IC50 estimate. Although in many cases, the confidence intervals are nonoverlapping, for the purposes of this study, the difference between two IC50 values is considered significant if ≥3-fold. For α-Ctx–receptor combinations where inhibition by the maximal α-Ctx concentration tested was less than ∼40%, the response after a 5-min static bath exposure to 10 μm α-Ctx was compared with control response, and the means were compared using a one-way analysis of variance and Fisher's least significant difference to determine significance. Significance was determined at the 95% level (p < .05). Concentration–response curves for activation of nAChRs were obtained according to the following procedures. ACh was applied in ascending concentrations, and the current amplitudes for each individual oocyte were analyzed by nonlinear regression and fit using a four-parameter logistic equation in Prism to obtain the calculated plateau value for activation. All ACh-evoked responses were then normalized to the calculated plateau value to obtain a percentage response and then analyzed with the same four-parameter logistic equation. Data for the ACh curves were collected using oocytes from three different donors to ensure reproducibility. Acetylcholine chloride (catalog no. A6625), potassium chloride (catalog no. P3911), and BSA (catalog no. A2153) were purchased from Sigma-Aldrich. Sodium chloride (catalog no. S271), calcium chloride dihydrate (catalog no. C79), magnesium chloride hexahydrate (catalog no. M33), sodium hydroxide (catalog no. S313), and HEPES (catalog no. BP310) were purchased from Fisher Scientific.
Site-directed mutagenesis of α6/α3 and β4 subunits
cDNAs for human α6/α3 and human and rat β4 subunits in the pGEMHE vector were used as starting templates. cDNAs for rat α6/α3 were in the pT7TS vector. Oligonucleotide primers were designed to individually span positions 177 of the α6/α3 subunit and 110, 118, and 119 of the β4 subunit and included nucleotide substitutions to mutate each respective amino acid residue. The double mutant constructs were made using a single primer pair that mutated positions 118 and 119 in the same PCR. All oligonucleotides were synthesized by the DNA/Peptide Facility, part of the Health Sciences Center Cores at the University of Utah. Pfu Turbo (catalog no. 600250) DNA polymerase (Agilent Technologies, Santa Clara, CA) was used to extend the primers. The PCR conditions were as follows: 95 °C for 120 s for denaturation followed by 30 cycles of 95 °C for 30 s, 65–78 °C (depending on the primers used) for 7 min, 72 °C for 60 s, and a final extension step for 10 min at 72 °C. The reaction was then digested with DpnI (catalog no. R0176S) to remove template cDNA (New England Biolabs, Ipswich, MA). Chemically competent DH5α (New England Biolabs, catalog no. C2987I) or 10-β (New England Biolabs, catalog no. C3019I) cells were used for transformation. The cells were grown at 37 °C for 1 h after transformation and then plated on agar plates containing ampicillin and maintained at 37 °C overnight. Several colonies were selected from each plate and individually grown overnight at 37 °C in Luria–Bertani medium containing ampicillin (Fisher Scientific, catalog no. BP1760). cDNA was isolated from the cells using a Qiaprep Spin Miniprep Kit (catalog no. 27104) (Qiagen, Valencia, CA) followed by sequencing at the University of Utah DNA Sequencing Core Facility to verify incorporation of the mutations. cDNAs for human α6/α3 and human and rat β4 were linearized overnight at 37 °C using the restriction enzyme NheI (New England Biolabs, catalog no. R0131S), and rat α6/α3 cDNA was linearized using SalI (New England Biolabs, catalog no. R0138T) using the same protocol. Following linearization, the cDNAs were purified and eluted with water using the Qiaquick PCR purification kit (Qiagen, catalog no. 28104). cRNA was then prepared from the linearized cDNA using Ambion's mMESSAGE mMACHINE T7 Transcription Kit (Fisher Scientific, catalog no. AM1344) and purified using a Qiagen RNeasy Mini Kit (Qiagen, catalog no. 74104).
Peptide synthesis
The synthesis of α-Ctxs TxIB, [P13Q]PeIA, and [P13R]PeIA was performed according to methods described previously (40). Synthesis of α-Ctxs PnIA, PeIA, [P13A]PeIA, and [P13O]PeIA was performed as described by Cartier et al. (50). Correct synthesis of [P13Q]PeIA and [P13R]PeIA was verified by matrix-assisted laser desorption TOF MS. The calculated monoisotopic masses for [P13Q]PeIA and [P13R]PeIA are 1682.63 and 1710.67 Da, and the observed masses were 1682.74 and 1710.81 Da, respectively.
X-ray crystallography
The AChBP from A. californica was expressed and purified as described previously (51, 52). Briefly, AChBP was expressed with an N-terminal FLAG epitope tag and secreted from stably transfected human embryonic kidney 293S cells lacking the N-acetyglucosaminyltransferase I (GnTI−) gene (53). The protein was purified with FLAG antibody resin and eluted with FLAG peptide (Sigma-Aldrich, catalog no. F3290). Affinity-purified protein was further characterized by size-exclusion chromatography in a Superdex 200 16/60 gel filtration column (GE Healthcare) in 25 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 0.02% NaN3 (w/v). From this process, the pentameric association could be ascertained, and monomeric subunits and trace contaminants could be removed. Purified AChBP pentamers were then concentrated using a Millipore YM50 Centricon ultrafiltration unit (Fisher Scientific) to a final concentration of ∼5 mg/ml.
Complex formation and crystallization
The PeIA–AChBP complex was formed by dissolving 10 μmol of lyophilized PeIA with 50 μl of purified and concentrated protein at a concentration of 5 mg/ml. The PeIA–AChBP complex co-crystals were obtained by the vapor-diffusion hanging-drop method. Concentrated protein complex was mixed in a 1 μl:1 μl solution consisting of 0.1 m Tris-HCl (pH 8.0), 0.25 m MgCl2, 20% (w/v) PEG 4000, incubated at 22 °C, and suspended over 500 μl of the same solution. Crystals of 0.3 × 0.3 × 0.2 mm final size appeared after a few weeks.
X-ray diffraction data collection
PeIA–AChBP complex co-crystals were transferred to a cryoprotectant solution consisting of 0.1 m Tris-HCl (pH 8.0), 0.25 m MgCl2, 12% (w/v) PEG 4000, and 10% (v/v) glycerol and flash-cooled in liquid nitrogen. A full set of X-ray diffraction data were collected at 278 °C at beamline 8.2.1 (Advanced Light Source, Berkeley, CA). Diffraction data were processed and scaled using HKL2000 (54). Final data statistics are given in Table S1.
Structure refinement
The PeIA–AChBP complex structure was solved by the molecular replacement method using PHASER (55) using the AChBP/α-Ctx BuIA structure (PDB entry 4EZ1) as a search model. The electron density maps were manually fitted in COOT (56) with iterative structure refinement done using phenix.refine (RRID:SCR_014224) (57), resulting in a final model with Rwork and Rfree of 19.6 and 22.8%, respectively. Refinement statistics are listed in Table S1. Atomic coordinates and structure factors have been deposited in the PDB (entry 5JME). Cartoon representations of the structures were generated using PyMOL (RRID:SCR_000305) (58). It should be noted that there was density for a fifth PeIA in the data obtained. However, although the density clearly showed the position of the peptide backbone, it was not sufficient for accurate positioning of the side chains. Out of an abundance of caution, it was decided to omit the final toxin chain to avoid any possible misinterpretation.
NMR spectroscopy and structure calculations
Peptide samples (1.0 mg) were dissolved in 550 μl of 10% D2O, 90% H2O (pH ∼3), and spectra were recorded on a Bruker Advance III 600-MHz spectrometer equipped with a cryoprobe. Data were collected at 290 K using TOCSY, NOESY H-N HSQC, and H-C HSQC experiments. Spectra were acquired with mixing times of 80 ms (TOCSY) or 200 ms (NOESY) and 4,096 data points in F2 and 512 in F1. Chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate at 0 ppm. Spectra were processed with Topspin version 3.5 (Bruker Biospin) and assigned using the program CcpNmr Analysis (59). Structure calculations of [P13A]PeIA were based upon distance restraints derived from NOESY spectra and on backbone dihedral angle restraints generated using TALOS+ (60). A family of 20 lowest-energy structures consistent with the experimental restraints was calculated using CYANA (61) and assessed using Molprobity (RRID:SCR_014226) (62). Experimental restraints and stereochemical quality assessment outcomes are provided in Table S2.
Author contributions
A. J. H., T. T. T., and J. M. M. conceptualization; A. J. H., T. T. T., J. B., J. B. G., and P. J. H. data curation; A. J. H., T. T. T., J. B., J. B. G., S. B. C., P. J. H., and D. J. C. formal analysis; A. J. H., T. T. T., J. B., J. B. G., S. B. C., P. J. H., and D. J. C. validation; A. J. H., T. T. T., J. B., C. H. M., F. H., J. B. G., S. B. C., and P. J. H. investigation; A. J. H. and T. T. T. methodology; A. J. H., T. T. T., D. J. C., and J. M. M. writing-original draft; A. J. H., T. T. T., D. J. C., and J. M. M. writing-review and editing; T. T. T., S. B. C., D. J. C., and J. M. M. supervision; T. T. T., D. J. C., and J. M. M. funding acquisition; T. T. T., D. J. C., and J. M. M. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Quentin Kaas (University of Queensland, Brisbane, Australia) for critical reading of the manuscript. We acknowledge the contribution to the manuscript by the scientists and support staff of the Berkeley Center for Structural Biology (Berkeley Hills, CA) as well as those at the University of Utah DNA Sequencing and DNA/Peptide Synthesis Core Facilities (Salt Lake City, UT). The Berkeley Center for Structural Biology is supported in part by grants from NIGMS, NIH, and the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under contract DE-AC02-05CH11231.
This work was supported by National Institutes of Health (NIH) Grants R01 GM103801 and P01 GM48677 (to J. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- α-Ctx
- α-conotoxin
- AChBP
- acetylcholine binding protein
- DRG
- dorsal root ganglion
- CI
- confidence interval
- H
- human
- M
- mouse
- Hyp
- 4-hydroxyproline
- PDB
- Protein Data Bank
- TEVC
- two-electrode voltage clamp.
References
- 1. Albuquerque E. X., Pereira E. F., Alkondon M., and Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120 10.1152/physrev.00015.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dani J. A., and Bertrand D. (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 47, 699–729 10.1146/annurev.pharmtox.47.120505.105214 [DOI] [PubMed] [Google Scholar]
- 3. Bradaïa A., and Trouslard J. (2002) Fast synaptic transmission mediated by α-bungarotoxin-sensitive nicotinic acetylcholine receptors in lamina X neurones of neonatal rat spinal cord. J. Physiol. 544, 727–739 10.1113/jphysiol.2002.028894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Exley R., McIntosh J. M., Marks M. J., Maskos U., and Cragg S. J. (2012) Striatal α5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J. Neurosci. 32, 2352–2356 10.1523/JNEUROSCI.4985-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salminen O., Murphy K. L., McIntosh J. M., Drago J., Marks M. J., Collins A. C., and Grady S. R. (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol. Pharmacol. 65, 1526–1535 10.1124/mol.65.6.1526 [DOI] [PubMed] [Google Scholar]
- 6. Clarke P. B., and Reuben M. (1996) Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br. J. Pharmacol. 117, 595–606 10.1111/j.1476-5381.1996.tb15232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaiser S., and Wonnacott S. (2000) α-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol. Pharmacol. 58, 312–318 10.1124/mol.58.2.312 [DOI] [PubMed] [Google Scholar]
- 8. Sharma G., and Vijayaraghavan S. (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38, 929–939 10.1016/S0896-6273(03)00322-2 [DOI] [PubMed] [Google Scholar]
- 9. Lu Y., Grady S., Marks M. J., Picciotto M., Changeux J. P., and Collins A. C. (1998) Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. J. Pharmacol. Exp. Ther. 287, 648–657 [PubMed] [Google Scholar]
- 10. Grady S. R., Meinerz N. M., Cao J., Reynolds A. M., Picciotto M. R., Changeux J. P., McIntosh J. M., Marks M. J., and Collins A. C. (2001) Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: a function mediated by a different nAChR than dopamine release from striatum. J. Neurochem. 76, 258–268 [DOI] [PubMed] [Google Scholar]
- 11. Hone A. J., and McIntosh J. M. (2018) Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 592, 1045–1062 10.1002/1873-3468.12884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bagdas D., Gurun M. S., Flood P., Papke R. L., and Damaj M. I. (2018) New insights on neuronal nicotinic acetylcholine receptors as targets for pain and inflammation: a focus on α7 nAChRs. Curr. Neuropharmacol. 16, 415–425 10.2174/1570159X15666170818102108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cox B. C., Marritt A. M., Perry D. C., and Kellar K. J. (2008) Transport of multiple nicotinic acetylcholine receptors in the rat optic nerve: high densities of receptors containing α6 and β3 subunits. J. Neurochem. 105, 1924–1938 10.1111/j.1471-4159.2008.05282.x [DOI] [PubMed] [Google Scholar]
- 14. Marritt A. M., Cox B. C., Yasuda R. P., McIntosh J. M., Xiao Y., Wolfe B. B., and Kellar K. J. (2005) Nicotinic cholinergic receptors in the rat retina: simple and mixed heteromeric subtypes. Mol. Pharmacol. 68, 1656–1668 [DOI] [PubMed] [Google Scholar]
- 15. Gotti C., Guiducci S., Tedesco V., Corbioli S., Zanetti L., Moretti M., Zanardi A., Rimondini R., Mugnaini M., Clementi F., Chiamulera C., and Zoli M. (2010) Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J. Neurosci. 30, 5311–5325 10.1523/JNEUROSCI.5095-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang K., Buhlman L., Khan G. M., Nichols R. A., Jin G., McIntosh J. M., Whiteaker P., Lukas R. J., and Wu J. (2011) Functional nicotinic acetylcholine receptors containing α6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J. Neurosci. 31, 2537–2548 10.1523/JNEUROSCI.3003-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Champtiaux N., Han Z. Y., Bessis A., Rossi F. M., Zoli M., Marubio L., McIntosh J. M., and Changeux J. P. (2002) Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J. Neurosci. 22, 1208–1217 10.1523/JNEUROSCI.22-04-01208.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whiteaker P., McIntosh J. M., Luo S., Collins A. C., and Marks M. J. (2000) 125I-α-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol. Pharmacol. 57, 913–925 [PubMed] [Google Scholar]
- 19. Cordero-Erausquin M., Pons S., Faure P., and Changeux J. P. (2004) Nicotine differentially activates inhibitory and excitatory neurons in the dorsal spinal cord. Pain 109, 308–318 10.1016/j.pain.2004.01.034 [DOI] [PubMed] [Google Scholar]
- 20. Hone A. J., Meyer E. L., McIntyre M., and McIntosh J. M. (2012) Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J. 26, 917–926 10.1096/fj.11-195883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wieskopf J. S., Mathur J., Limapichat W., Post M. R., Al-Qazzaz M., Sorge R. E., Martin L. J., Zaykin D. V., Smith S. B., Freitas K., Austin J. S., Dai F., Zhang J., Marcovitz J., Tuttle A. H., et al. (2015) The nicotinic alpha6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Sci. Transl. Med. 7, 287ra72 10.1126/scitranslmed.3009986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giribaldi J., and Dutertre S. (2018) α-Conotoxins to explore the molecular, physiological and pathophysiological functions of neuronal nicotinic acetylcholine receptors. Neurosci. Lett. 679, 24–34 10.1016/j.neulet.2017.11.063 [DOI] [PubMed] [Google Scholar]
- 23. Sandall D. W., Satkunanathan N., Keays D. A., Polidano M. A., Liping X., Pham V., Down J. G., Khalil Z., Livett B. G., and Gayler K. R. (2003) A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 42, 6904–6911 10.1021/bi034043e [DOI] [PubMed] [Google Scholar]
- 24. Satkunanathan N., Livett B., Gayler K., Sandall D., Down J., and Khalil Z. (2005) α-Conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 1059, 149–158 10.1016/j.brainres.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 25. Kaas Q., Yu R., Jin A. H., Dutertre S., and Craik D. J. (2012) ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330 10.1093/nar/gkr886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McIntosh J. M., Plazas P. V., Watkins M., Gomez-Casati M. E., Olivera B. M., and Elgoyhen A. B. (2005) A novel α-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J. Biol. Chem. 280, 30107–30112 10.1074/jbc.M504102200 [DOI] [PubMed] [Google Scholar]
- 27. Fainzilber M., Hasson A., Oren R., Burlingame A. L., Gordon D., Spira M. E., and Zlotkin E. (1994) New mollusc-specific α-conotoxins block Aplysia neuronal acetylcholine receptors. Biochemistry 33, 9523–9529 10.1021/bi00198a018 [DOI] [PubMed] [Google Scholar]
- 28. Luo S., Zhangsun D., Wu Y., Zhu X., Hu Y., McIntyre M., Christensen S., Akcan M., Craik D. J., and McIntosh J. M. (2013) Characterization of a novel α-conotoxin from conus textile that selectively targets α6/α3β2β3 nicotinic acetylcholine receptors. J. Biol. Chem. 288, 894–902 10.1074/jbc.M112.427898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cuny H., Yu R., Tae H. S., Kompella S. N., and Adams D. J. (2018) α-Conotoxins active at α3-containing nicotinic acetylcholine receptors and their molecular determinants for selective inhibition. Br. J. Pharmacol. 175, 1855–1868 10.1111/bph.13852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dutertre S., Nicke A., and Lewis R. J. (2005) β2 subunit contribution to 4/7 α-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 280, 30460–30468 10.1074/jbc.M504229200 [DOI] [PubMed] [Google Scholar]
- 31. Smit A. B., Syed N. I., Schaap D., van Minnen J., Klumperman J., Kits K. S., Lodder H., van der Schors R. C., van Elk R., Sorgedrager B., Brejc K., Sixma T. K., and Geraerts W. P. (2001) A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 411, 261–268 10.1038/35077000 [DOI] [PubMed] [Google Scholar]
- 32. Celie P. H., van Rossum-Fikkert S. E., van Dijk W. J., Brejc K., Smit A. B., and Sixma T. K. (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907–914 10.1016/S0896-6273(04)00115-1 [DOI] [PubMed] [Google Scholar]
- 33. Thompson A. J., Metzger S., Lochner M., and Ruepp M. D. (2017) The binding orientation of epibatidine at α7 nACh receptors. Neuropharmacology 116, 421–428 10.1016/j.neuropharm.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Bourne Y., and Taylor P. (2006) Structural characterization of agonist and antagonist-bound acetylcholine-binding protein from Aplysia californica. J. Mol. Neurosci. 30, 101–102 10.1385/JMN:30:1:101 [DOI] [PubMed] [Google Scholar]
- 35. Lin B., Xiang S., and Li M. (2016) Residues responsible for the selectivity of α-conotoxins for Ac-AChBP or nAChRs. Mar. Drugs 14, E173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Talley T. T., Olivera B. M., Han K. H., Christensen S. B., Dowell C., Tsigelny I., Ho K. Y., Taylor P., and McIntosh J. M. (2006) α-Conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as α3β2 and α7 nicotinic acetylcholine receptors. J. Biol. Chem. 281, 24678–24686 10.1074/jbc.M602969200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dutertre S., Ulens C., Büttner R., Fish A., van Elk R., Kendel Y., Hopping G., Alewood P. F., Schroeder C., Nicke A., Smit A. B., Sixma T. K., and Lewis R. J. (2007) AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 26, 3858–3867 10.1038/sj.emboj.7601785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Celie P. H., Kasheverov I. E., Mordvintsev D. Y., Hogg R. C., van Nierop P., van Elk R., van Rossum-Fikkert S. E., Zhmak M. N., Bertrand D., Tsetlin V., Sixma T. K., and Smit A. B. (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 12, 582–588 10.1038/nsmb951 [DOI] [PubMed] [Google Scholar]
- 39. Xu M., Zhu X., Yu J., Yu J., Luo S., and Wang X. (2017) The crystal structure of Ac-AChBP in complex with α-conotoxin LvIA reveals the mechanism of its selectivity towards different nAChR subtypes. Protein Cell 8, 675–685 10.1007/s13238-017-0426-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hone A. J., Ruiz M., Scadden M., Christensen S., Gajewiak J., Azam L., and McIntosh J. M. (2013) Positional scanning mutagenesis of alpha-conotoxin PeIA identifies critical residues that confer potency and selectivity for α6/α3β2β3 and α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 288, 25428–25439 10.1074/jbc.M113.482059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daly N. L., Callaghan B., Clark R. J., Nevin S. T., Adams D. J., and Craik D. J. (2011) Structure and activity of α-conotoxin and PeIA at nicotinic acetylcholine receptor subtypes and GABAB receptor-coupled N-type calcium channels. J. Biol. Chem. 286, 10233–10237 10.1074/jbc.M110.196170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cierpicki T., and Otlewski J. (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21, 249–261 10.1023/A:1012911329730 [DOI] [PubMed] [Google Scholar]
- 43. Yu R., Kompella S. N., Adams D. J., Craik D. J., and Kaas Q. (2013) Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 56, 3557–3567 10.1021/jm400041h [DOI] [PubMed] [Google Scholar]
- 44. Hone A. J., McIntosh J. M., Azam L., Lindstrom J., Lucero L., Whiteaker P., Passas J., Blázquez J., and Albillos A. (2015) α-Conotoxins identify the α3β4* subtype as the predominant nicotinic acetylcholine receptor expressed in human adrenal chromaffin cells. Mol. Pharmacol. 88, 881–893 10.1124/mol.115.100982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luo S., Kulak J. M., Cartier G. E., Jacobsen R. B., Yoshikami D., Olivera B. M., and McIntosh J. M. (1998) α-Conotoxin AuIB selectively blocks α3 β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 18, 8571–8579 10.1523/JNEUROSCI.18-21-08571.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhangsun D., Zhu X., Wu Y., Hu Y., Kaas Q., Craik D. J., McIntosh J. M., and Luo S. (2015) Key residues in the nicotinic acetylcholine receptor β2 subunit contribute to α-conotoxin LvIA binding. J. Biol. Chem. 290, 9855–9862 10.1074/jbc.M114.632646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuryatov A., Olale F., Cooper J., Choi C., and Lindstrom J. (2000) Human α6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology 39, 2570–2590 10.1016/S0028-3908(00)00144-1 [DOI] [PubMed] [Google Scholar]
- 48. McIntosh J. M., Azam L., Staheli S., Dowell C., Lindstrom J. M., Kuryatov A., Garrett J. E., Marks M. J., and Whiteaker P. (2004) Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 65, 944–952 10.1124/mol.65.4.944 [DOI] [PubMed] [Google Scholar]
- 49. Hone A. J., Scadden M., Gajewiak J., Christensen S., Lindstrom J., and McIntosh J. M. (2012) α-Conotoxin PeIA[S9H,V10A,E14N] potently and selectively blocks α6β2β3 versus α6β4 nicotinic acetylcholine receptors. Mol. Pharmacol. 82, 972–982 10.1124/mol.112.080853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cartier G. E., Yoshikami D., Gray W. R., Luo S., Olivera B. M., and McIntosh J. M. (1996) A new α-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J. Biol. Chem. 271, 7522–7528 10.1074/jbc.271.13.7522 [DOI] [PubMed] [Google Scholar]
- 51. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., and Bourne Y. (2005) Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 24, 3635–3646 10.1038/sj.emboj.7600828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Talley T. T., Yalda S., Ho K. Y., Tor Y., Soti F. S., Kem W. R., and Taylor P. (2006) Spectroscopic analysis of benzylidene anabaseine complexes with acetylcholine binding proteins as models for ligand-nicotinic receptor interactions. Biochemistry 45, 8894–8902 10.1021/bi060534y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reeves P. J., Callewaert N., Contreras R., and Khorana H. G. (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424 10.1073/pnas.212519299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 55. Storoni L. C., McCoy A. J., and Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 10.1107/S0907444903028956 [DOI] [PubMed] [Google Scholar]
- 56. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 57. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeLano W. L. (2017) The PyMOL Molecular Graphics System, Version 2.0, Schroedinger, LLC [Google Scholar]
- 59. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 60. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ikeya T., Terauchi T., Güntert P., and Kainosho M. (2006) Evaluation of stereo-array isotope labeling (SAIL) patterns for automated structural analysis of proteins with CYANA. Magn. Reson. Chem. 44, S152–S157 10.1002/mrc.1815 [DOI] [PubMed] [Google Scholar]
- 62. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McIntosh J. M., Santos A. D., and Olivera B. M. (1999) Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu. Rev. Biochem. 68, 59–88 10.1146/annurev.biochem.68.1.59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.