

Abstract
Cyanobacteriochromes (CBCRs) are photochromic proteins in cyanobacteria that act as photosensors. CBCRs bind bilins as chromophores and sense nearly the entire visible spectrum of light, but the regulation of the chromophorylation of CBCRs is unknown. Slr1393 from Synechocystis sp. PCC 6803 is a CBCR containing three consecutive GAF (cGMP phosphodiesterase, adenylyl cyclase, and FhlA protein) domains, of which only the third one (Slr1393g3) can be phycocyanobilin-chromophorylated. The protein Slr2111 from Synechocystis sp. PCC 6803 includes a cystathionine β-synthase (CBS) domain pair of an as yet unknown function at its N terminus. CBS domains are often characterized as sensors of cellular energy status by binding nucleotides. In this work, we demonstrate that Slr2111 strongly interacts with Slr1393 in vivo and in vitro, which generates a complex in a 1:1 molar ratio. This tight interaction inhibits the chromophorylation of Slr1393g3, even if the chromophore is present. Instead, the complex stability and thereby the chromophorylation of Slr1393 are regulated by the binding of nucleotides (ATP, ADP, AMP) to the CBS domains of Slr2111 with varying affinities. It is demonstrated that residues Asp-53 and Arg-97 of Slr2111 are involved in nucleotide binding. While ATP binds to Slr2111, the association between the two proteins gets weaker and chromophorylation of Slr1393 are enabled. In contrast, AMP binding to Slr2111 leads to a stronger association, thereby inhibiting the chromophorylation. It is concluded that Slr2111 acts as a sensor of the cellular energy status that regulates the chromophorylation of Slr1393 and thereby its function as a light-driven histidine kinase.
Keywords: cyanobacteria, protein-protein interaction, photosynthesis, photosynthetic pigment, photoreceptor, phototransduction, adenosine-binding protein, co-immunoprecipitation, cystathionine beta-synthase domain, phycocyanobilin, regulation
Introduction
Sensing of light is important for many organisms, especially phototrophic organisms. One class of typical sensor proteins are the cyanobacteriochromes (CBCRs) 2 (1), proteins related to the red/far-red phytochromes that utilize an open-chain tetrapyrrole (bilin) as chromophore. The absorption maxima of the different CBCRs cover nearly the entire visible spectrum (1, 2); the red/green-type CBCR GAF domains that photoconverts between red (parental) and green (photoproduct) forms are widely spread and are found in many cyanobacteria (3–7). The structural and functional complexity of CBCRs has been intensively studied (1, 2, 8, 9). However, as the chromophorylation of CBCRs and similar phytochromes is an autocatalytic reaction, attention has not yet been paid to the chromophorylation regulation, and, accordingly, nothing is known about the regulation of chromophorylation. The gene slr1393 from Synechocystis sp. PCC6803 (Synechocystis) encodes a protein (974 aa) of the CBCR photoreceptor family. The gene product is composed of three GAF (cGMP phosphodiesterase, adenylyl cyclase, and FhlA protein) domains, a PAS (Per, ARNT, and SIM protein domain) domain, and a histidine kinase (HK) domain. GAF3 (Slr1393g3, amino acids 423–599) is the sole domain that can bind PCB autocatalytically. It yields a photochromic protein with absorption maxima at 649 nm (parental form) and 536 nm (photoproduct form) (10), which thereby light-regulates the activity of the C terminally located HK domain that binds ATP and autophosphorylates Slr1393. Its autophosphorylation activity is up-regulated when the GAF3 domain is in its photoproduct state (5).
The cystathionine β-synthase (CBS) domain folds with a three-stranded β-sheet and two α-helices packed according to a β1-α1-β2-β3-α2 topology, resulting in a quite compact structure. In most resolved structures, the CBS domains occur in tandem pairs as functional units (11), forming a so-called CBS pair or Bateman module (12). The CBS domain is an evolutionarily conserved protein domain that is present in the proteome of archaebacteria, prokaryotes, and eukaryotes (13). Some proteins consist of two or four tandem CBS domains, and others carry only one CBS domain; in plant-derived proteins, CBS domains occur more frequently in two CBS domains than in a single arrangement (14).
CBS domains are found in cytosolic and membrane proteins and perform various functions. Normally, CBS domains are fused to other functional domain(s), which may indicate their probable functions that involve binding of ligands, such as metal ions or nucleotides, so causing structural and functional changes and regulating a large number of cellular functions (13, 15, 16). In addition, CBS domains activate or inhibit protein functions by binding adenosine nucleotides (15, 17–19), acting as autoinhibitory regulatory elements in some proteins. The adenosine-binding capacity with different affinities is considered as being related to an energy-sensing process. Regulation of kinases is a well understood process for CBS domains. AMP kinase (AMPK) is activated by AMP and prevented by ATP (20). As shown for the CBS domain of AtCLCa (member of the chloride channel family from Arabidopsis thaliana), when ATP is bound in this protein, AtCLCa activity is diminished by 60%; however, AMP, if present, competes with ATP and prevents inhibition (21). A similar effect was identified in inosine 5-monophosphate dehydrogenase (22), this regulation of the catalytic function of inosine 5-monophosphate dehydrogenase may occur through allosteric stimulation by ATP (15). Also, the activity of CBS-PPase is inhibited by ADP and AMP, and is activated up to 1.6-fold by ATP (17). In all cases, CBS domains are an internal part of a large protein with specific functions, however, to date, no report reveals how CBS domains influence the functions of separate interacting proteins.
Slr2111 (157 aa) from the unicellular cyanobacterium Synechocystis sp. PCC 6803 contains two domains: a tandem arrangement of two conservative CBS domains (spanning positions 1–115), and a segment assigned to the aspartate aminotransferase (AAT) superfamily (fold type I) of the pyridoxal phosphate-dependent enzymes (42 aa, positions 116–157). Data from a large-scale protein-protein interaction screening for Synechocystis sp. PCC 6803 indicate that Slr1393 might interact with Slr2111 (23), which in that screen was annotated as a hypothetical protein with unknown function.
Here, we report complex formation between the two independent proteins Slr1393 and Slr2111 in a 1:1 molar ratio, which is impaired by ATP but not by the Slr1393 chromophore PCB. This strong interaction reduces the PCB-chromophorylation of Slr1393g3. Also, co-immunoprecipitation indicates that Slr2111 interacts with the apo-form of Slr1393 in vivo. Slr2111, like other CBS domains, binds nucleotides such as ATP, ADP, and AMP. AMP-, ADP-, and ATP-binding have various strong allosteric effects on Slr2111. The mutation of amino acids in the adenosyl-binding pocket, D53A and R97A of Slr2111, significantly reduced the affinity for binding nucleotides. Both a protein-docking approach and the kinetics of PCB-binding Slr1393g3 reveal that Slr2111 binds at the entrance of the PCB-binding pocket of Slr1393g3. Although ATP-binding decreases the association between Slr2111 and Slr1393g3 and increases the chromophorylation, AMP-binding increases the association and decreases the chromophorylation. The differing binding strengths and direction of the effect depend on nucleotides entities and makes Slr1393 together with this CBS domain pair relevant to sensing the cellular energy status.
Results and discussion
Slr2111 interacts strongly with Slr1393g3 but not PCB-Slr1393g3
A large-scale survey of protein-protein interactions in Synechocystis sp. PCC 6803 has proposed an interaction between Slr2111 and Slr1393 (23). To prove the interaction, a bacterial two-hybrid, co-immunoprecipitation and a pulldown assay were performed.
Employment of the BacterioMatch II two-hybrid system confirmed the interaction between Slr2111 and Slr1393 (Fig. 1A). The gene slr2111 was cloned into pTRG and combined with the pBTs containing full-length slr1393 (coding for amino acids 1–974) or slr1393g3 (encoding only the GAF3 domain with amino acids 441–597) (Table S1). The results clearly identify for both proteins interactions that were of strong and comparable intensity (Fig. 1A). Therefore, Slr1393g3 is the domain interacting with Slr2111.
Figure 1.

Protein interaction of Slr2111 and Slr1393g3. A, protein interaction demonstrated by the BacterioMatch II Two-hybrid System. The reporter strain containing pBT-Slr1393g3 + pTRG-Slr2111 (A, IV) grew well in the presence of 3-AT and streptomycin. B, the wheel shows the details of the plate of A. The reporter strains II, VI, III, and V acted as negative controls, and I acted as the positive control. C, SDS-PAGE control of pulldown assay results; lane 1, elution fractions of GST-Slr2111 (43 kDa); lane 2, PCB-His6tag-Slr1393g3 (24 kDa); lane 3, elution fraction of PCB-His6tag-Slr1393g3 and GST-Slr2111 on GST affinity chromatography; lane 4, elution fraction of PCB-His6tag-Slr1393g3 and GST-Slr2111 on His6-tag affinity column. The bottom panel shows the Zn2+-induced fluorescence of the chromoprotein band on SDS-PAGE. D, co-immunoprecipitations of Slr1393 and Slr2111. Input: lysate from Synechocystis. Slr2111 antibodies were bound to protein A beads and used for immunoprecipitation from total Synechocystis sp. PCC 6803 soluble proteins. Immunoprecipitates with anti-Slr2111 antibodies or control immunoglobulin G (IgG) was assayed with SDS-PAGE without reducing agent and then Western blotted with anti-Slr1393. The bottom panel shows the Zn2+-induced fluorescence of the chromoprotein band on SDS-PAGE. E, gel filtration chromatography profiles of His6tag-Slr2111 (blue line), PCB-His6tag-Slr1393g3 (orange line), His6tag-Slr1393g3 (violet line), using a Superdex 200 column; elution buffer: KPB (20 mm, pH 7.4) containing NaCl (200 mm) (see “Materials and methods”). Blue line, peak corresponds to Slr2111 as dimer (57 kDa, calculated 45 kDa). Violet line, peak corresponds to Slr1393g3 as monomer (30 kDa, calculated 24 kDa). Orange line, two peaks correspond to PCB-Slr1393g3 as monomer (30 kDa, calculated 24 kDa) and dimer (53 kDa, calculated 48 kDa), respectively. Molecular markers (peaks from left to right of gray dashed line) were 66, 45, 29, and 12.4 kDa; F, the complex of Slr2111 and Slr1393g3 in a 1:1 ratio elutes with an apparent mass of 72 kDa (black line), and the mixture of Slr2111 and PCB-Slr1393g3 in a 1:1 ratio elutes with the respective apparent mass of 72 and 30 kDa (green line) under the same experimental conditions as in E. Gel chromatography identifies the complex of Slr2111 and Slr1393g3 as a mixture of heterodimer (72 kDa, calculated 47 kDa) and heterotetramer (72 kDa, calculated 94 kDa). G, absorption spectrum (black line) of the early peak of the green line in F purified by gel chromatography, absorption after irradiation with 530 nm (Z state, blue line), and 650-nm light (E state, red line) of the later peak of the green line in F were measured. Inset G, SDS-PAGE of the early peak (lane 1) and later peak (lane 2) of the green line in F; left panel 1′ and 2′ show the Zn2+-induced fluorescence of the chromoprotein band on SDS-PAGE.
For the pulldown assay, Slr1393g3 was expressed with an N-terminal His6-tag, assembled with PCB (i.e. PCB-His6tag-Slr1393g3) and mixed with Slr2111, tagged with an N-terminal GST (GST-Slr2111), and applied to a Ni2+ affinity column. Even in the presence of excessive GST-Slr2111, the molar ratio in the eluted portion of PCB-His6tag-Slr1393g3 and GST-Slr2111 is 2:1 (Fig. 1C, lane 4). In the alternative set-up, applying the GST-affinity pulldown of GST-Slr2111 and PCB-His6tag-Slr1393g3, the molar ratio of the elution portion of GST-Slr2111 and Slr1393g3-His6tag was nearly 0.7:1, even in the presence of excessive PCB-His6tag-Slr1393g3 (Fig. 1C, lane 3). However, the eluted portion of GST-Slr2111 and PCB-His6tag-Slr1393g3 showed no Zn2+-induced fluorescence on SDS-PAGE (as a proof for a bilin chromophore presence, Fig. 1C), its absorption spectrum did not exhibit the characteristic spectrum of PCB-Slr1393g3 (Fig. S1A), and simultaneously PCB-Slr1393g3 was detected in the flow-through portion of the pulldown assay (Fig. S1A). These findings indicate that Slr2111 only interacts with the apo-form, Slr1393g3, but not with the chromophore-assembled PCB-Slr1393g3. The results are understandable in view of the fact that under most conditions, the chromophorylation of Slr1393g3 is not complete, thereby a fraction of the protein remains in its apo-form (10, 24). As control, all possible combinations of proteins under study were probed in several combinations of the pulldown assays. All these combinations did not show any interaction (Fig. S1C).
To examine the interaction between these two proteinsin vivo, we performed co-immunoprecipitation experiments using Synechocystis sp. PCC 6803 extracts against anti-Slr2111 antibodies. The resulting immunoprecipitated materials were blotted and then probed with anti-Slr1393 antibodies. The results clearly demonstrate that Slr2111 interacts with Slr1393, and in particular only interacts with the apo-form of Slr1393 (Fig. 1D).
To determine the oligomeric state of Slr2111, Slr1393g3, PCB-Slr1393g3, and their complexes, mixtures of His6tag-Slr2111 and His6tag-Slr1393g3 as well as His6tag-Slr2111 and PCB-His6tag-Slr1393g3 were analyzed using gel filtration in a molar ratio of 1:1. Fig. 1, panels E and F, show the elution profiles of the individual components: Slr2111 is dimeric, whereas the apo-form, His6tag-Slr1393, is monomeric. Elution of the PCB-loaded form, PCB-His6tag-Slr1393, however, yields two peaks, the larger one corresponding to the molecular mass of the monomer, and the second one being eluted as dimer, indicating that only part of His6tag-Slr1393 is bound with PCB (Fig. 1E). If samples of Slr2111 and Slr1393 were mixed together and then loaded onto the gel filtration column, the peak assigned to the monomer of Slr1393g3 disappeared and a new peak with apparent molecular mass of 72 kDa appeared, indicating that His6tag-Slr2111 and His6tag-Slr1393g3 have formed a 1:1 molar ratio complex as heterodimer as well as heterotetramer. The peak of PCB-Slr1393g3 had decreased in intensity, but did not fully disappear with the oligomerization of His6tag-Slr2111 and PCB-His6tag-Slr1393g3, again indicating that Slr2111 cannot form a complex with PCB-Slr1393g3. Comparison of the peak intensities of PCB-His6tag-Slr1393g3 and PCB-His6tag-Slr1393g3 mixed with His6tag-Slr2111 reveals that the PCB-loaded fraction is only 59%. The absorption of the eluted sample of Slr2111 and PCB-Slr1393g3 (Fig. 1F, green line) indicates that the early eluted peak showed no PCB-Slr1393g3 absorbance, whereas the later peak showed a characteristic feature for the E state and Z state of PCB-Slr1393g3 after irradiation as well as a strong Zn2+-induced fluorescence (Fig. 1G, inset). The complex of His6tag-Slr1393g3 and His6tag-Slr2111 is 1:1 according to SDS-PAGE analysis (Fig. 1G).
The CBS domain protein Slr2111 binds adenosine derivatives
Slr2111 follows the canonical arrangement of many CBS-containing proteins, as it carries two CBS domains in tandem, CBS1 (11–60 aa) and CBS2 (76–116 aa); these units exhibit the common β1-α1-β2-β3-α2 arrangement (Fig. 2). Based on a GeneDoc search, we found that the sequence conservation of Slr2111 is high for the CBS1 and CBS2 domains (except α2 helix of CBS2). The alignment reveals that the third or fourth amino acid in the β2 sheet is highly conserved as Leu/Ile/Val of CBS1 and CBS2; the third amino acid in α2 of CBS1 is Asp, and fifth amino acid in α2 of CBS2 is Leu/Ile/Val. However, the sequence following β3 of CBS2 (being annotated as the AAT part by NCBI (Conserved Domains database accession number CL18945)) shows low identity to other CBS domains, so we conclude that the CBS2 of Slr2111 lacks α2. Canonical binding sites for adenosines of the CBS domain have been predicted from published structural data (18, 25). The crystal structures of CBS domains that bind adenosines show that the binding site of the nucleotide is highly apparent between α1 and β2 as well as α2 and β3 of CBS1, β1 and β2 as well as α2 and β3 of CBS2, and also the loop preceding β1 and β2 has been proposed as a binding site. In CBS1 of Slr2111 Leu-48, Thr-50 in β3, and Asp-53 in α2 of CBS1 are highly similar to other binding sites of adenosine, and Ile-77, Arg-97, and Ile-98 in CBS2 are conserved compared with other binding sites of adenosine (Fig. 2A, the putative binding site is indicated by violet and brown frames). Accordingly, Slr2111 was considered a promising target that might bind nucleotides.
Figure 2.

Sequence alignment of CBS domains and model building of Slr2111. A, sequence alignments were performed by GeneDoc. Nucleotide-binding sites are colored: green, amino acids binding ATP; blue, amino acids binding AMP; yellow, amino acids binding ADP; amino acids binding more than two kinds of nucleotides are shown in red. Possible binding sites of Slr2111 are highlighted in a violet frame. B–E, simulation of the Slr2111 structure (10–123 aa). The three-dimensional model was obtained from the SWISS-MODEL server, and a putative signal-transduction protein with CBS domains from B. ambifaria MC40–6 (PDB code 4fry) was used as reference structure (see “Materials and methods” for details). Side chains of aromatic residues are indicated for three phenylalanine residues (Phe-45, Phe-92, and Phe-114) (blue). Leu-48, Thr-50, Ile-77, and Ile-98 are shown in violet, Asp-53 and Arg-97 are shown in brown. D and E depicts the structure after rotation around 180 degrees of B and C, respectively. Two possible binding sites of adenine nucleotides are indicated. Image was generated in PyMOL.
Taking the crystal structure of Bammc 406_4587 (a putative signal-transduction protein with CBS domains from Burkholderia ambifaria MC40–6, PDB code 4fry) (26) as template, a three-dimensional model of Slr2111 was constructed (Fig. 2, B–E). Taking into account the found effects of adenosine nucleotide-binding on phenylalanine fluorescence (Table 1) and the identified, highly conserved binding pockets for AMP, ADP, or ATP in the CBS domains (Fig. 2). Based on sequence alignment two possible binding sites were proposed: one site being organized by Leu-48, Thr-50, Asp-53, Ile-77, Arg-97, and Ile-98. The surface of the model of Slr2111 similarly exhibits a fairly large and deep pocket with Ile-77 occupying the bottom (Fig. 2, B and C). The other possible binding site is near Thr-50, Arg-97, and Ile-98 of CBS2, and, again, the surface model presents a deep pocket with the potential for ligand binding (Fig. 2, D and E). The fluorescence titrations results (Table 1) suggest that phenylalanines (at least one of them) are located in or close by the binding sites. Phe-45 is close to site 1, and Phe-114 is close to site 2 (Fig. 2). Alternatively, one might assume that the site for binding ATP, ADP, or AMP is formed when the CBS domains oligomerize to a dimer or polymer (27, 28).
Table 1.
Dissociation constants Kd for Slr2111 and its variants binding AXPs
The proteins were kept at a fixed concentration (8 μm) and were titrated with increasing concentrations of AXPs (from 0.1 to 120 μm) to measure the fluorescence emission spectra between 280 and 400 nm. The data of the fluorescence quenching were analyzed using the modified Stern–Volmer plot (see “Materials and methods”). The fa is the fractional number of fluorophores accessible to the quencher. The Kd values and fa values are reported as the mean ± S.D. of three independent experiments.
| Protein | ATP |
ADP |
AMP |
|||
|---|---|---|---|---|---|---|
| Kd | fa | Kd | fa | Kd | fa | |
| μm | μm | μm | ||||
| Slr2111 | 4.9 ± 0.4 | 0.8 ± 0.0 | 2.8 ± 0.4 | 1.1 ± 0.0 | 1.8 ± 0.2 | 0.8 ± 0.0 |
| Slr2111(D53A) | 7.3 ± 0.3 | 0.9 ± 0.1 | 6.5 ± 0.2 | 0.6 ± 0.1 | 4.0 ± 0.4 | 0.6 ± 0.1 |
| Slr2111(R97A) | 5.3 ± 0.5 | 0.8 ± 0.0 | 3.1 ± 0.2 | 0.7 ± 0.1 | 3.2 ± 0.3 | 0.8 ± 0.0 |
| Slr2111(D53A/R97A) | 13.1 ± 2.4 | 0.3 ± 0.1 | 13.7 ± 1.5 | 0.4 ± 0.1 | 11.2 ± 2.0 | 0.4 ± 0.1 |
We quantified the affinity of Slr2111 for adenosine nucleotides by measuring its intrinsic fluorescence. This method identifies fluorescence quenching of aromatic amino acids upon binding adenosine nucleotides. The aromatic amino acids in Slr2111 include one tryptophan (Trp-143 in AAT), two tyrosines (Tyr-44 in CBS1 and Tyr-134 in AAT), and five phenylalanines (Phe-45 in CBS1, Phe-92 and Phe-114 in CBS2, Phe-150, and Phe-154 in AAT). As only phenylalanines are found in both CBS domains, we decided to follow changes in the intrinsic fluorescence at 290 nm (indicative for phenylalanines) upon titration with adenosyl nucleotides (λex = 260 nm for phenylalanines). Significant quenching of the fluorescence intensity of Slr2111 was recorded upon addition of ATP, ADP, and AMP (AXPs). The data of the fluorescence quenching were analyzed using the modified Stern-Volmer plot (see “Materials and methods”) yielding the dissociation constants, Kd, and the fractional number of fluorophores accessible to the quencher, fa, for Slr2111 binding AXPs (Table 1). The calculated Kd values are in the low micromolar range: 4.9 ± 0.4 μm for ATP, 2.8 ± 0.4 μm for ADP, and 1.8 ± 0.2 μm for AMP, respectively. Interestingly, the affinity of AMP is 2- to 3-fold stronger than that of ATP binding to Slr2111, and 1.5-fold stronger than that of ADP binding to Slr2111. The Kd constants for the adenosyl ligands were similar to those measured for adenosyl nucleotides in other CBS domains, e.g. AMPK (29), and alike for mtCBS-PPase (17), which binds ATP with lower affinity than AMP as some irons appeared.
To demonstrate whether these residues could influence the binding affinity of nucleotides, we selected two residues (Asp-53 and Arg-97) from the predicted important residues and mutated them into alanine. Similar mutations in ortholog proteins have been reported to induce severe functional changes: the mutation D727A of hCLC-5 abolishes ATP binding (30), also the effect of ATP binding is abolished by the D753A mutation of AtCLCa (21). The mutation of R279A of CBS domain of OpuC exhibits increased transport activity and D281A results in lower transport activity (31). Moreover, both Asp and Arg are charged amino acids, which might influence the surface charge of protein, we thus assumed that they may influence the binding of nucleotides. Hence, we chose Asp-53 and Arg-97 for mutation (Fig. 2) and studied their influence on nucleotide binding.
The fluorescence quenching results are shown in Table 1, Slr2111(D53A) influenced the binding of nucleotide, the binding affinity for ATP decreased 1.5-fold, for ADP decreased 2.3-fold, and for AMP decreased 2.2-fold. The variant R97A, instead, influences the binding affinity only slightly: for ATP and ADP, the Kd values are similar with Slr2111, just for AMP, the binding affinity decreased 1.8-fold. These results indicate that Arg-97 is probably near the binding site of AMP, whereas Asp-53, instead, is an important residue for nucleotide binding. The most interesting results were found for a combination of both mutations. As shown in Table 1, the binding affinity of ATP (compared with the WT protein) decreased 2.7-fold, for ADP it decreased 4.9-fold, and for AMP the effect was an even 6.2-fold decrease. Even though Arg-97 is assumed to be located only “near” the binding site, the combined mutagenesis identifies the importance of both amino acids for the function of Slr2111.
ATP can also bind to HK. The binding affinity of HKs for ATP is various: Kd of the HK domain of Thermotoga maritima CheA is 14 μm (32), Escherichia coli CheA is about 300 μm (33, 34), HK1 from Mycobacterium tuberculosis is 730 μm (35), and CheA from Salmonella typhimurium is 200–300 μm (36). Therefore, Kd of Slr2111 for ATP is lower than the HKs' Kd, which may indicates the binding affinity of Slr2111 is higher than HKs. Maybe Slr2111 binds more ATP and indirectly inhibit the activity of Slr1393's HKs.
Having confirmed the binding of adenosine derivatives by Slr2111, as similarly reported for other CBS domains, we assume a possible regulating function of Slr2111-AXPs. Given the available structures, it seems to be well-established that binding of adenosine derivatives to CBS domains usually requires the presence of two Bateman domains, which form homodimers; a control on Slr2111 by size-exclusion chromatography confirms this dimeric conformation (Fig. 1).
Binding of adenosyl derivatives leads to conformational changes of Slr2111
The far-UV CD (CD) spectra of Slr2111 upon addition of adenosine nucleotides revealed significant changes of protein secondary structures. The spectra of the complexed protein were compared with the individual components (protein and nucleotides) and with the plain sum of both components (Fig. 3). Isolated Slr2111 shows the features of a protein with high α-helical content (shoulder at 222 nm) and a small percentage of β-sheet (with an intense minimum at 208 nm). Upon addition of ATP, ADP, or AMP, remarkable changes in secondary structure become apparent, especially when “complex” spectra are compared with the sum of individual CD spectra of both Slr2111 and adenosine derivatives. The structural changes were largest in the presence of AMP (Fig. 3C), compared with effects induced by ATP (Fig. 3A) or ADP (Fig. 3B). The mutations of Slr2111 caused slight conformational changes on binding nucleotides, especially the double mutation of D53A/R97A (Fig. S2). For R97A alone, the conformational change upon binding ATP was larger than that upon WT protein-binding ATP, but the Kd value of Slr2111(R97A) for binding ATP is similar to that of Slr2111. Because Arg has a larger molecular size and a positively charged side chain, the mutation may cause a larger conformational change, although the effect may not yet generate sufficient influence on the binding affinity of ATP. Experiments with different Slr2111 protein concentrations (from 6 to 30 μm) showed similar changes upon the addition of adenosine ligands, suggesting that the changes observed are not protein-concentration dependent. Thus, although the microenvironment around Trp and Tyr residues scarcely undergo local structural changes upon binding of nucleotides (Fig. S3), the binding leads to the remarkable structural changes, e.g. in the secondary structure, of Slr2111.
Figure 3.
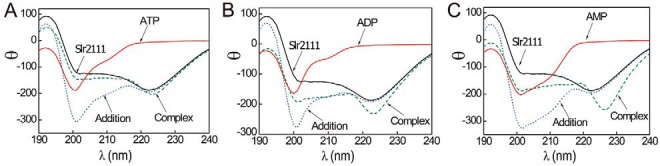
Far-UV CD of Slr2111 complexed with adenosine ligands. A, spectra of isolated Slr2111 (black line), the isolated ATP (red line), the addition of the spectra of both isolated species (blue dotted line), and the complex formed by equimolar (10 μm) amounts of both biomolecules (green dashed line). B, spectra of isolated Slr2111, the isolated ADP, the complex Slr2111/ADP, and the addition of both spectra of the isolated biomolecules (color coding as in A). C, spectra of isolated Slr2111, the isolated AMP, the complex Slr2111/AMP, and the addition of both spectra of the isolated biomolecules (color coding as in A). Experiments were carried out at 25 °C.
The inhibition of Slr2111 on chromophorylation of Slr1393 is adjusted by nucleotides
So far, the results indicate that Slr2111 forms a complex to Slr1393g3 only if the latter is in the nonchromophorylated form. It was thus of interest to test whether addition of PCB can break the complex of Slr2111 and Slr1393g3. The molar ratio of the complex of His6tag-Slr1393g3 and GST-Slr2111 with added PCB (stock solution in DMSO) was 0.72:1 (Fig. S4, lane 1), and the molar ratio of the complex of His6tag-Slr1393g3 and GST-Slr2111 with added DMSO (as control) containing no PCB was 0.74:1 (Fig. S4, lane 2). This indicates that the complex of Slr1393g3 and Slr2111 was hardly broken up and that only very little Slr1393g3 binding PCB (i.e. PCB-His6tag-Slr1393g3) was generated. This indicates that the complex of Slr2111 and Slr1393g3 is very stable and prevents covalent binding of PCB to Slr1393g3. When ATP (Fig. S4, lane 3), ADP (Fig. S4, lane 4), or AMP (Fig. S4, lane 5) were added to PCB-His6tag-Slr1393g3 and GST-Slr2111, the molar ratio of the complex of His6tag-Slr1393g3 and GST-Slr2111 changes; it becomes 0.5:1 in the presence of ATP, 1:1 in the presence of ADP, similarly, 1.1:1 in the presence of AMP. SDS-PAGE of the pulldown of the (nonchromophorylated) His6tag-Slr1393g3 and GST-Slr2111 in the presence of ATP (Fig. S4, lane 6), ADP (Fig. S4, lane 7), or AMP (Fig. S4, lane 8) shows the same trend. The molar ratio of the complex of His6tag-Slr1393g3 and GST-Slr2111 in the presence of ATP is 0.5:1, in the presence of ADP is 0.9:1, and in the presence of AMP is 1:1, respectively. Without adding AXPs, the molar ratio of the complex of His6tag-Slr1393g3 and GST-Slr2111 is 0.7:1 (Fig. S4, lane 9). This indicates that under pulldown conditions ATP disrupts a portion of the complex of His6tag-Slr1393g3 and GST-Slr2111, and GST-Slr2111 in the presence of AMP or ADP can bind more His6tag-Slr1393g3. In conclusion, the complex of Slr1393g3 and Slr2111 cannot be broken by PCB (see above), ADP, or AMP, but was unstable in the presence of ATP.
The high degree of sequence similarity and secondary structure prediction between CBS domains allows a docking simulation aimed at identification of the contact sites between Slr2111 and Slr1393g3 proteins. The docking of both proteins (by Hex 8.0, Fig. S5) reveals that the complex formed from both proteins blocks the entry site of the PCB chromophore into the Slr1393g3-binding pocket. We propose that Slr2111 regulates the chromophorylation of Slr1393 by blocking the entrance of the chromophore pocket.
Analysis of the chromophorylation kinetics of Slr1393g3 (10 μm) with PCB (0–2.3 μm) gave the following parameters: Km = 3.6 μm, Vmax = 22 μm/min, kcat = 2 min−1 (Fig. 4). To investigate the inhibition of Slr2111 on the PCB-chromophorylation of Slr1393g3, a series of Lineweaver-Burk plots were further analyzed, using concentrations of 0, 2, 4, 6, and 8 μm Slr2111 in the chromophorylation reaction. Four kinetic traces intersected the ordinate at the same point (Fig. 4A) indicating that Slr2111 acted as a competitive inhibitor on PCB-chromophorylation of Slr1393g3 with a Ki value of 7.3 μm (Fig. 4A, inset). When 8 μm Slr2111 was added, the line of Lineweaver-Burk plot did not intersect at the same point, possibly because other complicated effects such as allosteric effects at high concentrations of Slr2111 (15). Slr2111 with binding ATP or AMP were also tested by kinetic analysis. In this case, the slopes increased with increasing Slr2111/ATP concentration and the Lineweaver-Burk plots intersected on the ordinate, indicating a competitive inhibition with a Ki value of 3.8 μm (Fig. 4B, inset); this value is half of that for Slr2111.
Figure 4.

Lineweaver-Burk plots in the inhibition of Slr1393g3 by (A) Slr2111, (B) Slr2111/ATP, and (C) Slr2111/AMP. Numbers on the different lines indicate Slr2111 concentrations (0–8 μm). PCB was used at concentrations of 0, 0.5, 0.8, 1.1, 1.7, and 2.3 μm. The slopes of the solid lines by inhibitors (0–6 μm) was used to calculate for Ki with a secondary plot (insets A–C). Competitive inhibition was observed with respect to Slr2111, Slr2111/ATP, and Slr2111/AMP, the calculated Ki values were 7.3, 3.8, and 2.9 μm, respectively.
Addition of adenosine nucleotides, however, changed the interaction significantly. Having the ATP-bound form of Slr2111 present, the chromophorylation activity of Slr1393g3 was greatly inhibited already by lower concentrations of Slr2111/ATP (2–6 μm). Furthermore, the inhibitor Slr2111/ATP caused a biphasic inhibition versus concentration: at lower concentrations (2–6 μm) Slr1393g3 chromophorylation was inhibited, whereas at higher concentrations (8 μm) the inhibition became less effective. Similar kinetic patterns were observed for the addition of Slr2111/AMP: Lineweaver-Burk plots demonstrated that the inhibition by Slr2111/AMP was competitive with Slr1393g3 with a Ki value of 2.9 μm (Fig. 4C, inset). At concentrations of 2 and 4 μm Slr2111/AMP, the slope of the Lineweaver-Burk plots (Km/Vmax) slightly increased, and at higher concentrations of Slr2111/AMP (8 μm) the inhibition nearly disappeared. These data reveal that Slr2111 acts as a competitive inhibitor after binding nucleotides, and the inhibition of the adenosine nucleotide-loaded proteins, Slr2111/ATP and Slr2111/AMP, on PCB-chromophorylation of Slr1393g3, although different, were stronger than for the ligand-free Slr2111 protein (Fig. 4). The Ki values of Slr2111/ATP were bigger than Slr2111/AMP indicating that during PCB-chromophorylation of Slr1393, the inhibition of Slr2111/AMP is stronger than that of Slr2111/ATP.
Concluding remarks
Our findings demonstrate a complex formation between the CBS domain-containing protein Slr2111 and the CBCR Slr1393 in vitro and in vivo, which blocks the entrance of the chromophore into the binding site of 1393g3 and thereby inhibits the assembly of Slr1393g3 with its PCB chromophore. We, furthermore, could identify that adenosyl ligands (ATP/ADP/AMP) are bound by Slr2111 with different affinities and induce substantial structural rearrangements of the protein. Residues Asp-53 and Arg-97 were involved in nucleotide binding, thus identifying the nucleotide-binding site. Most interestingly, the strength of the complex is modulated by the binding of adenosyl ligands to Slr2111. Slr2111 has a competitive inhibition on Slr1393g3-binding PCB. Slr2111/AMP and Slr2111/ATP also show competitive inhibition on chromophorylation of Slr1393g3 at low concentrations. The fact that the Ki value of Slr2111/AMP is lower than that of Slr2111/ATP means that while AMP binds to Slr2111 the inhibition becomes stronger, and when ATP binds to Slr2111 the inhibition becomes weaker. Although AMP binds to Slr2111, due to the changes in conformation of Slr2111, Slr2111 binds more Slr1393g3. When, however, ATP binds to Slr2111, the complex stability of Slr1393g3 and Slr2111 decreased. Hence, whereas AMP makes the complex of Slr1393g3 and Slr2111 more stable and Slr2111/AMP shows stronger inhibition on the chromophorylation of Slr1393g3, ATP makes the complex weaker and gives Slr1393g3 a chance to bind PCB, which engenders Slr1393 becoming a photosensor and photoactivate the mosaicked HK, so transducing the photosensory signal transmission (Fig. 5). In conclusion, Slr2111 works through binding nucleotides and thereby regulates its Slr1393-binding affinity and in consequence, regulating also the chromophorylation of Slr1393. We have tried constructing the null mutant of the slr2111 gene, but failed, possibly indicating that apparently Slr2111 is an essential component for the lifestyle of Synechocystis.
Figure 5.
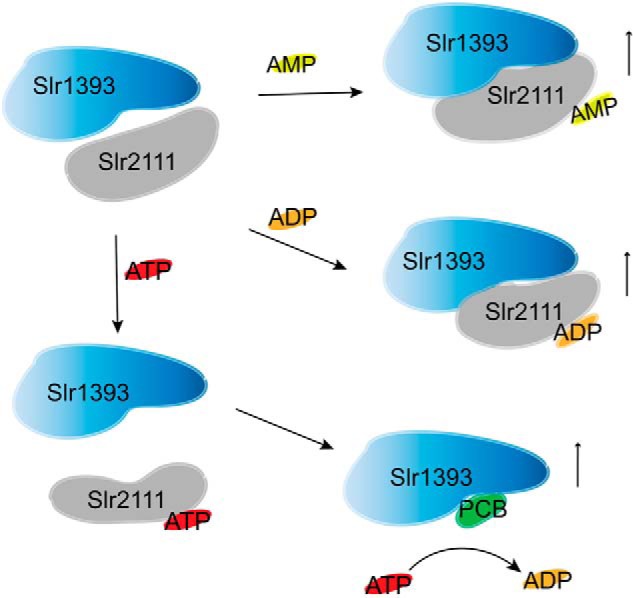
Model for interaction of Slr1393g3 with Slr2111 that regulates the chromophorylation of Slr1393. As levels of AMP and ADP increase in cells, Slr2111 binds free AMP and ADP, which leads to a conformation change of Slr2111 and is more convenient for binding Slr1393g3, so inhibiting PCB-chromophorylation of Slr1393. As the ATP level increased, the complex of Slr2111 and Slr1393 is more liable to dissociation, free Slr1393 is subject to PCB-chromophorylation, so cascading the photoactivation of HK and signal transmission. It should be noted that the HK converts ATP to ADP, thereby decreasing the level of ATP, which leads to the decrease of the chromophorylation of Slr1393g3. The vertical up arrows indicate that the yields of the products increase.
Many reports can be found in the literature suggesting that protein functions in one protein domain are regulated by a fused CBS domain (13, 15, 16). However, so far no example was identified for an interaction between two individual proteins, such that a CBS domain of one protein would regulate function of another, separate, but strongly interacting protein. CBCRs as related proteins to phytochromes show autocatalytic chromophorylation, no attention has been paid to the regulation of the chromophorylation. The results presented here are the first example that identify regulation of a light-sensing photoreceptor activity through control of assembly with its chromophore. The additional finding that this protein-protein complex formation and consequently the photoreceptor assembly is modulated by the presence of ATP, ADP, or AMP connects the regulatory functions of CBCRs to the energy level of the cyanobacterial cell.
Materials and methods
Cloning and expression
All genetic manipulations were performed according to standard protocols (37). Plasmid pACYC-ho1-pcyA for PCB has been reported before (38). To facilitate purification via Ni2+-affinity chromatography, slr1393g1 (coding for amino acids 51–202), slr1393g2 (248–398 aa), slr1393g3 (441–597 aa), and slr2111 from Synechocystis sp. PCC 6803 were cloned into pET30 (Novagen) vectors (Table S1). Site-directed mutations of slr2111 were generated via PCR: Slr2111(D53A), Slr2111(R97A), Slr2111(D53A/R97A) (Tables S1 and S2).
To facilitate purification by affinity chromatography on GSH-Sepharose, slr2111, slr2111cbs (1–115 aa), and slr2111aat (116–157 aa) were cloned into pGEX-6P-1 vector (Amersham Biosciences). In these constructs the GSH S-transferase (GST) tag is attached to the N terminus of the proteins.
For over-expression of the proteins, the respective plasmids (Table S2) were transformed into E. coli BL21 (DE3) (Novagen). For chromophorylation, pET-derived expression vectors for slr1393g3 were transformed into E. coli BL21 containing the PCB-generating plasmid (pACYC-ho1-pcyA). E. coli cells were cultured in Luria-Bertani (LB) medium at 37 °C and supplemented with kanamycin (30 μg ml−1), or in LB medium at 16 °C and supplemented with kanamycin (20 μg/ml) and chloromycetin (17 μg/ml). After induction with isopropyl β-d-thiogalactoside (final solution: 1 mm) for 16 h, cells were centrifuged at 12,000 × g for 3 min at 4 °C, washed twice with water, and stored at −20 °C until use.
Cell pellets of His-tagged proteins were disrupted by sonication and purified via Ni2+-affinity chromatography on chelating Sepharose columns (GE Healthcare, Rydalmere, Australia), respectively. The tagged proteins were eluted with the start buffer supplemented with a high concentration of imidazole (0.5 m). Pellets of cells of GST-tagged proteins were resuspended in ice-cold lysis buffer (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.3) and purified by affinity chromatography on a GSH-Sepharose column (GE Healthcare). The GST-tagged proteins were eluted with elution buffer (50 mm Tris-HCl, 10 mm reduced GSH, pH 8.0). PCB was isolated from Spirulina platensis as described (39); its concentration was determined spectroscopically using an extinction coefficient ϵ690 = 37,900 m−1 cm−1 in methanol, 2% HCl (40).
Bacterial two-hybrid assay
For the bacterial two-hybrid approach (Stratagene, USA), full-length slr1393, slr1393g3, and ntrB were cloned into pBT bait plasmid, and slr2111 was cloned into pTRG target plasmid (Table S3). Positive clones could be obtained (following the manufacturer's protocol) using the reporter of HIS3-aadA, after two cycles of differential screening in the absence of 3-amino-1,2,4-triazole (3-AT), or in the presence of 3-AT and streptomycin, respectively.
Pulldown assay
Proteins were expressed as above. For the His6-tag affinity pulldown assay, 0.4 μmol of His-tagged proteins were immobilized on a Ni2+ affinity column, and incubated independently with (0.8 μmol) GST-tagged proteins, respectively, in the start buffer (pH 7.4) overnight at 4 °C, and then purified as described above. For GST-affinity pulldown: 0.4 μmol of GST-Slr2111 in lysis buffer (pH 7.3) was immobilized on a GST-affinity column, incubated with 0.8 μmol of PCB-His6tag-Slr1393g3 and His6tag-Slr1393g3 in lysis buffer (pH 7.3) overnight at 4 °C, respectively, and then purified as described above. For testing effects of nucleotides, after addition of 1 μmol of ATP, ADP, or AMP to the column, the chromatography was done similarly. For GST-affinity pulldown with PCB: after addition of 20 μl of PCB (20 mm stock in DMSO) or 20 μl of DMSO (as the negative control) were added to the column, the chromatography was done similarly. All fractions were analyzed by SDS-PAGE with the buffer system of Laemmli (41). Proteins were stained with Coomassie Brilliant Blue, and those covalently binding bilin chromophores were identified by Zn2+-induced fluorescence (42).
Co-immunoprecipitation and Western blotting
Synechocystis sp. PCC 6803 cells were grown in BG-11 liquid medium to an optical density of 0.2 at 730 nm (A730). Cells were collected by centrifugation and then lysed into extraction buffer (150 mmol/liter of NaCl, 50 mmol/liter of Tris-HCl (pH 7.8), 1% Triton X-100 (v/v), 5 mmol/liter of EDTA, 10 mmol/liter of NaF, 1 mmol/liter of NaVO3, 10% glycerin). After 30 min on ice and centrifugation in a microcentrifuge (14,000 × g for 10 min), the supernatants were collected and used for immunoprecipitation. Lysates were prepared and and mixed with affinity-purified rabbit polyclonal antibody (anti-Slr2111, 0.5–1 μg) for immunoprecipitation (at 4 °C for 6 h). Immunoglobulin G (IgG) was also added in other lysates as control. After incubation with antibody, 30 μl (settled volume) of protein A-Sepharose beads (pre-blocked overnight with 10% BSA in PBS) were added to each tube for 3 h. The beads were washed three times with extraction buffer, and resuspended in 50 μl of SDS sample buffer without reducing agents. Eluted proteins were analyzed by Western blotting through anti-Slr1393.
Oligomerization analysis
To determine the oligomeric states of the Slr2111 and Slr1393g3 complexes, the proteins His6tag-Slr1393g3, PCB-His6tag-Slr1393g3, and His6tag-Slr2111 were purified as described above. The samples were then dialyzed twice against KPB (20 mm, pH 7.4) containing 200 mm NaCl and adjusted to a concentration of 10 μm. His6tag-Slr1393g3 was mixed with His6tag-Slr2111, and PCB-His6tag-Slr1393g3 with His6tag-Slr2111 at equal molarity, and 1.0 ml of the mixture was loaded onto a Superdex 200 preparative grade column (60 × 1.6 cm) and developed (1.0 ml/min) with KPB (20 mm, pH 7.4) containing NaCl (0.2 m). The apparent molecular mass was determined by comparison to a marker set (12.4–66 kDa). Protein concentrations were determined by the Bradford assay (43), calibrated with BSA.
Fluorescence titrations
The fluorescence emission spectra and the fluorescence intensities from titrations of Slr2111 were measured with a fluorophotometer (Horiba Jobin Yvon, Edison, NJ). The emission spectra were recorded over a wavelength of 280–400 nm with an excitation wavelength at 260 nm. The excitation and emission slits were 5 nm, and the data acquisition interval was 1 nm. Binding experiments were performed at 25 °C, in 50 mm sodium phosphate buffer (pH 7.4) using ATP, AMP, and ADP adenosine as ligands. Increasing amounts of the ligands, in the 0–120 μm range, were added to a solution of Slr2111 at a fixed protein concentration of 8 μm. Fluorescence of the resulting samples was measured after overnight incubation at 4 °C to ensure equilibration. The binding of ATP, ADP, or AMP to Slr2111 resulted in quenching of the phenylalanine fluorescence. Each presented spectrum was an average of three scans. The fluorescence intensities (F) were plotted against the total concentration of quencher (Q) and the data were analyzed according to the modified Stern-Volmer equations (44). In the case where there is a heterogeneous population of fluorophores, the modified Stern-Volmer relationship is used,
| (Eq. 1) |
where fa is the fractional number of fluorophores accessible to the quencher and KQ is the quenching constant. The dissociation constants were calculated graphically using the modified Stern-Volmer plot (plot of Fo/(Fo − F) versus 1/[Q]), where KQ = 1/Kd (45). Inner-filter effects at 260 nm were corrected for the absorbance of the corresponding ligand (46). Absorbance measurements were carried out via a UV-visible absorption spectrometer (Beckman-Coulter DU 800), using a 1-cm path length quartz cell (Hellma). Typically, every fluorescence titration was repeated three times with freshly prepared samples.
Synchronous fluorescence spectra
Synchronous fluorescence spectra were recorded by a fluorophotometer (Horiba Jobin Yvon, Edison, NJ). This method allows studying the environment of amino acid residues by measuring the possible shift in wavelength emission maximum, λmax, the shift in position of emission maximum corresponding to the changes of the polarity around the chromophore molecule (47). In the case of fluorescence synchronous scans, the initial excitation wavelength was set at 250 nm and scanned up to 500 nm, whereas the wavelength shift Δλ was equal to 15 (for tyrosine residues) and 60 nm (for tryptophan residues) (48).
Circular dichroism (CD)
CD measurements were performed on a MOS-500 CD spectrometer (Bio-Logic, Claix, France, bandwidth: 2 nm, scan range: 190–250 nm, scan speed: 1 nm s−1) at room temperature. Measurements were performed in a 0.1-cm path length quartz cuvette (Hellma) with 10 μm protein in sodium phosphate buffer (50 mm, pH 7.4, supplemented with 100 mm NaCl), to which the same amount (10 μm) of the adenosine ligands (ATP, ADP, or AMP) was added. Experiments were also performed in the presence of 500 mm NaCl to ascertain the influence of the NaCl on the ligand binding. Each experiment was scanned for three times with new samples.
Assay of enzyme activity
The kinetic of PCB binding by Slr1393g3 was monitored by a fluorophotometer (Horiba Jobin Yvon) with autoscanning at 670 nm/s after PCB and Slr1393g3 was mixed. Before reaction, Slr2111 was mixed with ATP at a molar ratio of 1:6, and with AMP at 1:2, respectively. Chromophorylation reactions were carried out at 25 °C by addition of 2% PCB (v/v, stock PCB in DMSO, 200 μm) in buffer (20 mm Tris, 20 mm KPB, 150 mm NaCl, pH 7.4) containing 10 μm Slr1393g3 in the presence of different amounts of Slr2111 (0–8 μm), Slr2111/ATP (0–8 μm), or Slr2111/AMP (0–8 μm). The reaction velocity was monitored by following the fluorescence increase of the reaction product (PCB-Slr1393g3) at 670 nm (10). The initial velocity data at different substrate concentrations were fitted by the Michaelis-Menten equation,
| (Eq. 2) |
where v is the initial velocity, Vmax is the maximum reaction velocity, [S] is the substrate concentration, and Km is the Km. A Lineweaver-Burk plot with double reciprocals (1/V versus 1/[S]) was plotted and Km was determined by linear fitting.
For inhibition tests, the inhibitors were added simultaneously. Ki values was calculated from the sigmoidal dose-response curves by using nonlinear regression analysis with the program GraphPad (GraphPad, San Diego, CA).
Model building and sequence alignment
Images of protein structures were created using PyMOL. The model of Slr2111 was built on the SWISS-MODEL server (49, 50), using the crystal structure (PDB code 4fry) of BamMC 406_4587 (UniProtKB/Swiss-Prot entry B1YXI0, a putative signal-transduction protein with CBS domains from B. ambifaria strain MC40–6) as template (26). The interaction model of Slr1393g3 (PDB code 5dfx) and Slr2111 was built using Hex 8.0 (http://hex.loria.fr/). 3 Alignments were edited on the GeneDoc sequence editor (51). CBS domains used to sequence alignment were the CBS pair of putative d-arabinose 5-phosphate isomerase (Yrbh) with AMP from E. coli CTF073 (PDB code 3fna), the CBS domain containing protein ATU1752 (UniProtKB/Swiss-Prot entry A9CIP4) bound AMP from Agrobacterium tumefaciens (PDB code 3fhm), MJ1225 (UniProtKB/Swiss-Prot entry Q58622) from Methanocaldococcus jannaschii complexed to ATP, ADP, and AMP (PDB codes 3kh5 and 3lfz) (52), the single CBS domain-containing protein CBSX2 from A. thaliana (UniProtKB/Swiss-Prot entry Q9C5D0), which binds AMP (PDB code 4gqy) (27), AMPKγ1 (UniProtKB/Swiss-Prot entry Q80385) of the energy sensor AMPK from Rattus norvegicus binding AMP or ATP in five different combinations (PDB codes 4eag, 4eai, 4eak, 4eaj, and 4eal) (53).
Author contributions
Q. H., W. G., and K.-H. Z. formal analysis; Q. H., Q.-Y. T., Y.-F. S., and M. Z. investigation; Q. H., W. G., and K.-H. Z. writing-original draft; M. Z. and K.-H. Z. supervision; M. Z. and K.-H. Z. funding acquisition; W. G. and K.-H. Z. writing-review and editing.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grants 31370777 (to M. Z.) and 21472055 and 31770822 (to K. H. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CBCR
- cyanobacteriochrome
- CBS
- cystathionine β-synthase
- GAF
- cGMP phosphodiesterase, adenylyl cyclase, and FhlA protein domain
- PAS
- Per, ARNT, and SIM protein domain
- PCB
- phycocyanobilin
- AAT
- aspartate aminotransferase
- AMPK
- AMP kinase
- 3-AT
- 3-amino-1,2,4-triazole
- aa
- amino acids
- HK
- histidine kinase
- GST
- glutathione S-transferase
- PDB
- Protein Data Bank
- AXP
- ATP, ADP, and AMP.
References
- 1. Ikeuchi M., and Ishizuka T. (2008) Cyanobacteriochromes: a new superfamily of tetrapyrrole-binding photoreceptors in cyanobacteria. Photochem. Photobiol. Sci. 7, 1159–1167 10.1039/b802660m [DOI] [PubMed] [Google Scholar]
- 2. Rockwell N. C., Martin S. S., Feoktistova K., and Lagarias J. C. (2011) Diverse two-cysteine photocycles in phytochromes and cyanobacteriochromes. Proc. Natl. Acad. Sci. U.S.A. 108, 11854–11859 10.1073/pnas.1107844108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fushimi K., Nakajima T., Aono Y., Yamamoto T., Ni-Ni W., Ikeuchi M., Sato M., and Narikawa R. (2016) Photoconversion and fluorescence properties of a red/green-type cyanobacteriochrome AM1_C0023g2 that binds not only phycocyanobilin but also biliverdin. Front. Microbiol. 7, 588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narikawa R., Fukushima Y., Ishizuka T., Itoh S., and Ikeuchi M. (2008) A novel photoactive GAF domain of cyanobacteriochrome AnPixJ that shows reversible green/red photoconversion. J. Mol. Biol. 380, 844–855 10.1016/j.jmb.2008.05.035 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Zhang J., Luo J., Tu J. M., Zeng X. L., Xie J., Zhou M., Zhao J. Q., Scheer H., and Zhao K. H. (2012) Photophysical diversity of two novel cyanobacteriochromes with phycocyanobilin chromophores: photochemistry and dark reversion kinetics. FEBS J. 279, 40–54 10.1111/j.1742-4658.2011.08397.x [DOI] [PubMed] [Google Scholar]
- 6. Rockwell N. C., Martin S. S., and Lagarias J. C. (2012) Red/green cyanobacteriochromes: sensors of color and power. Biochemistry 51, 9667–9677 10.1021/bi3013565 [DOI] [PubMed] [Google Scholar]
- 7. Ulijasz A. T., Cornilescu G., von Stetten D., Cornilescu C., Velazquez Escobar F., Zhang J., Stankey R. J., Rivera M., Hildebrandt P., and Vierstra R. D. (2009) Cyanochromes are blue/green light photoreversible photoreceptors defined by a stable double cysteine linkage to a phycoviolobilin-type chromophore. J. Biol. Chem. 284, 29757–29772 10.1074/jbc.M109.038513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma Q., Hua H. H., Chen Y., Liu B. B., Krämer A. L., Scheer H., Zhao K. H., and Zhou M. (2012) A rising tide of blue-absorbing biliprotein photoreceptors: characterization of seven such bilin-binding GAF domains in Nostoc sp. PCC7120. FEBS J. 279, 4095–4108 10.1111/febs.12003 [DOI] [PubMed] [Google Scholar]
- 9. Bussell A. N., and Kehoe D. M. (2013) Control of a four-color sensing photoreceptor by a two-color sensing photoreceptor reveals complex light regulation in cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 12834–12839 10.1073/pnas.1303371110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J., Wu X. J., Wang Z. B., Chen Y., Wang X., Zhou M., Scheer H., and Zhao K. H. (2010) Fused-gene approach to photoswitchable and fluorescent biliproteins. Angew. Chem. Int. Ed. Engl. 49, 5456–5458 10.1002/anie.201001094 [DOI] [PubMed] [Google Scholar]
- 11. Kemp B. E. (2004) Bateman domains and adenosine derivatives form a binding contract. J. Clin. Invest. 113, 182–184 10.1172/JCI200420846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bateman A. (1997) The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22, 12–13 10.1016/S0968-0004(96)30046-7 [DOI] [PubMed] [Google Scholar]
- 13. Ignoul S., and Eggermont J. (2005) CBS domains: structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 289, C1369–C1378 10.1152/ajpcell.00282.2005 [DOI] [PubMed] [Google Scholar]
- 14. Kushwaha H. R., Singh A. K., Sopory S. K., Singla-Pareek S. L., and Pareek A. (2009) Genome wide expression analysis of CBS domain containing proteins in Arabidopsis thaliana (L.) Heynh and Oryza sativa L. reveals their developmental and stress regulation. BMC Genomics 10, 200 10.1186/1471-2164-10-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott J. W., Hawley S. A., Green K. A., Anis M., Stewart G., Scullion G. A., Norman D. G., and Hardie D. G. (2004) CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 113, 274–284 10.1172/JCI19874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baykov A. A., Tuominen H. K., and Lahti R. (2011) The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem. Biol. 6, 1156–1163 10.1021/cb200231c [DOI] [PubMed] [Google Scholar]
- 17. Jämsen J., Tuominen H., Salminen A., Belogurov G. A., Magretova N. N., Baykov A. A., and Lahti R. (2007) A CBS domain-containing pyrophosphatase of Moorella thermoacetica is regulated by adenine nucleotides. Biochem. J. 408, 327–333 10.1042/BJ20071017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lucas M., Encinar J. A., Arribas E. A., Oyenarte I., García I. G., Kortazar D., Fernández J. A., Mato J. M., Martínez-Chantar M. L., and Martínez-Cruz L. A. (2010) Binding of S-methyl-5′-thioadenosine and S-adenosyl-l-methionine to protein MJ0100 triggers an open-to-closed conformational change in its CBS motif pair. J. Mol. Biol. 396, 800–820 10.1016/j.jmb.2009.12.012 [DOI] [PubMed] [Google Scholar]
- 19. Pimkin M., and Markham G. D. (2008) The CBS subdomain of inosine 5′-monophosphate dehydrogenase regulates purine nucleotide turnover. Mol. Microbiol. 68, 342–359 10.1111/j.1365-2958.2008.06153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oakhill J. S., Steel R., Chen Z. P., Scott J. W., Ling N., Tam S., and Kemp B. E. (2011) AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 10.1126/science.1200094 [DOI] [PubMed] [Google Scholar]
- 21. De Angeli A., Moran O., Wege S., Filleur S., Ephritikhine G., Thomine S., Barbier-Brygoo H., and Gambale F. (2009) ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J. Biol. Chem. 284, 26526–26532 10.1074/jbc.M109.005132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W., Papov V. V., Minakawa N., Matsuda A., Biemann K., and Hedstrom L. (1996) Inactivation of inosine 5′-monophosphate dehydrogenase by the antiviral agent 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboxamide 5′-monophosphate. Biochemistry 35, 95–101 10.1021/bi951499q [DOI] [PubMed] [Google Scholar]
- 23. Sato S., Shimoda Y., Muraki A., Kohara M., Nakamura Y., and Tabata S. (2007) A large-scale protein-protein interaction analysis in Synechocystis sp. PCC6803. DNA Res. 14, 207–216 10.1093/dnares/dsm021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu X. L., Gutt A., Mechelke J., Raffelberg S., Tang K., Miao D., Valle L., Borsarelli C. D., Zhao K. H., and Gärtner W. (2014) Combined mutagenesis and kinetics characterization of the bilin-binding GAF domain of the protein Slr1393 from the Cyanobacterium Synechocystis PCC6803. ChemBioChem 15, 1190–1199 10.1002/cbic.201400053 [DOI] [PubMed] [Google Scholar]
- 25. Ereño-Orbea J., Oyenarte I., and Martínez-Cruz L. A. (2013) CBS domains: ligand binding sites and conformational variability. Arch. Biochem. Biophys. 540, 70–81 10.1016/j.abb.2013.10.008 [DOI] [PubMed] [Google Scholar]
- 26. Baugh L., Gallagher L. A., Patrapuvich R., Clifton M. C., Gardberg A. S., Edwards T. E., Armour B., Begley D. W., Dieterich S. H., Dranow D. M., Abendroth J., Fairman J. W., Fox D. 3rd, Staker B. L., Phan I., et al. (2013) Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS ONE 8, e53851 10.1371/journal.pone.0053851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeong B. C., Park S. H., Yoo K. S., Shin J. S., and Song H. K. (2013) Change in single cystathionine β-synthase domain-containing protein from a bent to flat conformation upon adenosine monophosphate binding. J. Struct. Biol. 183, 40–46 10.1016/j.jsb.2013.04.013 [DOI] [PubMed] [Google Scholar]
- 28. Townley R., and Shapiro L. (2007) Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science 315, 1726–1729 10.1126/science.1137503 [DOI] [PubMed] [Google Scholar]
- 29. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., and Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 10.1038/nature09932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meyer S., Savaresi S., Forster I. C., and Dutzler R. (2007) Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 14, 60–67 10.1038/nsmb1188 [DOI] [PubMed] [Google Scholar]
- 31. Huynh T. N., Choi P. H., Sureka K., Ledvina H. E., Campillo J., Tong L., and Woodward J. J. (2016) Cyclic di-AMP targets the cystathionine β-synthase domain of the osmolyte transporter OpuC. Mol. Microbiol. 102, 233–243 10.1111/mmi.13456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bilwes A. M., Quezada C. M., Croal L. R., Crane B. R., and Simon M. I. (2001) Nucleotide binding by the histidine kinase CheA. Nat. Struct. Biol. 8, 353–360 10.1038/86243 [DOI] [PubMed] [Google Scholar]
- 33. Stewart R. C., VanBruggen R., Ellefson D. D., and Wolfe A. J. (1998) TNP-ATP and TNP-ADP as probes of the nucleotide binding site of CheA, the histidine protein kinase in the chemotaxis signal transduction pathway of Escherichia coli. Biochemistry 37, 12269–12279 10.1021/bi980970n [DOI] [PubMed] [Google Scholar]
- 34. Tawa P., and Stewart R. C. (1994) Kinetics of CheA autophosphorylation and dephosphorylation reactions. Biochemistry 33, 7917–7924 10.1021/bi00191a019 [DOI] [PubMed] [Google Scholar]
- 35. Shrivastava R., Ghosh A. K., and Das A. K. (2007) Probing the nucleotide binding and phosphorylation by the histidine kinase of a novel three-protein two-component system from Mycobacterium tuberculosis. FEBS Lett. 581, 1903–1909 10.1016/j.febslet.2007.03.089 [DOI] [PubMed] [Google Scholar]
- 36. Surette M. G., Levit M., Liu Y., Lukat G., Ninfa E. G., Ninfa A., and Stock J. B. (1996) Dimerization is required for the activity of the protein histidine kinase CheA that mediates signal transduction in bacterial chemotaxis. J. Biol. Chem. 271, 939–945 10.1074/jbc.271.2.939 [DOI] [PubMed] [Google Scholar]
- 37. Sambrook J., Fritsch E., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 38. Zhao K. H., Su P., Li J., Tu J. M., Zhou M., Bubenzer C., and Scheer H. (2006) Chromophore attachment to phycobiliprotein ββ-subunits: phycocyanobilin:cysteine-ββ84 phycobiliprotein lyase activity of CpeS-like protein from Anabaena sp. PCC7120. J. Biol. Chem. 281, 8573–8581 10.1074/jbc.M513796200 [DOI] [PubMed] [Google Scholar]
- 39. Storf M., Parbel A., Meyer M., Strohmann B., Scheer H., Deng M. G., Zheng M., Zhou M., and Zhao K. H. (2001) Chromophore attachment to biliproteins: specificity of PecE/PecF, a lyase-isomerase for the photoactive 31-Cys-α84-phycoviolobilin chromophore of Phycoerythrocyanin. Biochemistry 40, 12444–12456 10.1021/bi010776s [DOI] [PubMed] [Google Scholar]
- 40. Cole W. J., Chapman D. J., and Siegelman H. W. (1968) The structure and properties of phycocyanobilin and related bilatrienes. Biochemistry 7, 2929–2935 10.1021/bi00848a033 [DOI] [PubMed] [Google Scholar]
- 41. Laemmli U. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 42. Berkelman T. R., and Lagarias J. C. (1986) Visualization of bilin-linked peptides and proteins in polyacrylamide gels. Anal. Biochem. 156, 194–201 10.1016/0003-2697(86)90173-9 [DOI] [PubMed] [Google Scholar]
- 43. Bradford M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 44. Lehrer S. S. (1971) Solute perturbation of protein fluorescence: the quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10, 3254–3263 10.1021/bi00793a015 [DOI] [PubMed] [Google Scholar]
- 45. Samworth C. M., Degli Esposti M., and Lenaz G. (1988) Quenching of the intrinsic tryptophan fluorescence of mitochondrial ubiquinol–cytochrome-c reductase by the binding of ubiquinone. Eur. J. Biochem. 171, 81–86 10.1111/j.1432-1033.1988.tb13761.x [DOI] [PubMed] [Google Scholar]
- 46. Birdsall B., King R. W., Wheeler M. R., Lewis C. A. Jr, Goode S. R., Dunlap R. B., and Roberts G. C. (1983) Correction for light absorption in fluorescence studies of protein-ligand interactions. Anal. Biochem. 132, 353–361 10.1016/0003-2697(83)90020-9 [DOI] [PubMed] [Google Scholar]
- 47. Paul B. K., Bhattacharjee K., Bose S., and Guchhait N. (2012) A spectroscopic investigation on the interaction of a magnetic ferrofluid with a model plasma protein: effect on the conformation and activity of the protein. Phys. Chem. Chem. Phys. 14, 15482–15493 10.1039/c2cp42415k [DOI] [PubMed] [Google Scholar]
- 48. Miller J. N. (1979) Recent advances in molecular luminescence analysis. Proc. Anal. Div. Chem. Soc. 16, 203–208 [Google Scholar]
- 49. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- 50. Guex N., Peitsch M. C., and Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30, S162–S173 10.1002/elps.200900140 [DOI] [PubMed] [Google Scholar]
- 51. Nicholas K. B., Nicholas H. B. Jr., and Deerfield D. W. II (1997) GeneDoc: analysis and visualization of genetic variation. EMBnet News 4, 1–4 [Google Scholar]
- 52. Gómez-García I., Oyenarte I., and Martínez-Cruz L. A. (2010) The crystal structure of protein MJ1225 from Methanocaldococcus jannaschii shows strong conservation of key structural features seen in the eukaryal γ-AMPK. J. Mol. Biol. 399, 53–70 10.1016/j.jmb.2010.03.045 [DOI] [PubMed] [Google Scholar]
- 53. Chen L., Wang J., Zhang Y. Y., Yan S. F., Neumann D., Schlattner U., Wang Z. X., and Wu J. W. (2012) AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 19, 716–718 10.1038/nsmb.2319 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.