

 The absence of the thyroid hormone (TH) could impair testicular function, but its mechanism is still rudimentary.
The absence of the thyroid hormone (TH) could impair testicular function, but its mechanism is still rudimentary.
Abstract
The absence of the thyroid hormone (TH) could impair testicular function, but its mechanism is still rudimentary. This study aims to explore the roles of estrogen receptor (ER α, β) and G protein-coupled receptor 30 (GPR30), extracellular signal regulated kinase (ERK1/2) and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways in apoptosis in testes of hypothyroidism rats. Male Wistar rats were randomly divided into control (C), low-(L) and high-hypothyroidism (H) groups [1 mL per 100 g BW per day normal saline, 0.001% and 0.1% propylthiouracil (PTU), respectively] by intragastrical gavage for 60 days. The levels of triiodothyronine (T3), thyroxine (T4) and thyroid stimulating hormone (TSH) in serum were measured. Expressions of ERα, ERβ and GPR30, pathway related protein expressions of ERK1/2 and PI3 K/AKT and apoptosis were detected in testicular homogenates. The results showed that T3 and T4 levels were decreased, and the TSH level was increased significantly in the H group. Protein expressions of ERα, ERβ and GPR30 decreased significantly in the H group. Significantly decreased protein expressions of p-ERK1/2, p-PI3K p85, p-AKT Ser473, Ras, p-Raf-1 Ser259, p-Raf-1 Ser338 and cyclin D1 in L and H groups as well PI3K p85, p-AKT and Thr308 in the H group were observed. Moreover, there was a significant increase in the Bad protein expression in L and H groups. In addition, there was a significant increase in the expression of Bax/Bcl-2, caspase 9 and cleaved caspase 3 and a significant decrease in the total caspase 3 protein expression in the H group. These results suggested that ERK1/2 and PI3K/AKT signaling pathways could be suppressed by hypothyroidism via inhibiting the expressions of ERs and could finally induce apoptosis in testes.
Introduction
Thyroid hormone (TH) plays an important role in the hypothalamo-hypophyseal testicular axis, and influences the sexual and spermatogenic function in men.1 As a physiological modulator, TH mediates the process of oxidative stress caused by reactive oxygen species (ROS).2 A deficiency of TH induces hypothyroidism and oxidative stress, and finally leads to testicular dysfunction and infertility.3,4
Research has reported that persistent hypothyroidism could downregulate the bioavailability of 17β-estradiol (E2).5 It is well established that E2 influences male spermatogenesis, sexual behavior and reproductive function, and the change in E2 expression might impair male fertility.6 In our previous study, we found that a significantly elevated E2 serum level changed the sperm quality in testes of hypothyroidism rats.7 However, the possible mechanisms for E2's influence on male fertility in hypothyroidism remain uncertain. Human and animal models have demonstrated that estrogen receptors (ERs), including estrogen receptor alpha (ERα) and beta (ERβ), mediate the biological effects of estrogen (particularly E2).8 It is well known that E2 interacts with ERα and ERβ to exert genomic (nuclear) effects.9 In addition to these established transcriptional effects, a transmembrane intracellular estrogen receptor, termed G-protein-coupled receptor 30 (GPR30), as well as ERs have been proved to be able to mediate the non-genomic effects of E2.9 After binding to ERs and GPR30, E2 activates multiple signaling pathways, influences cell proliferation, differentiation, survival and spermatogenesis, and induces apoptosis.8,9 It is well known that mitochondria are not only the major physiological source and the crucial targets of ROS, but also one of the major targets for the direct actions of steroid hormones and hormone receptors.10–12 Moreover, the ATP production, mitochondrial membrane potential and the regulation of calcium concentrations in mitochondria could be regulated by E2, and the ER-dependent mechanisms are the major ways by which E2 exerts action.13,14 The loss of ERs and GPR30 induces mitochondrial dysfunction by activating rapid survival signaling pathways and causes adverse effects on steroidogenesis and spermatogenesis.15–18 Studies have reported that extracellular signal regulated kinase (ERK1/2) and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways as well as the expressions of ERs and GPR30 could be affected by E2.19–23 Furthermore, a crosstalk regulation between ERK1/2 and PI3K/AKT signaling pathways has been found. The expression of ERK1/2 could be affected directly by PI3K or by AKT through Raf-1 Ser259.24,25 Additionally, Ras, as the common upstream of both signaling pathways, directly regulates the expression of ERK1/2 and PI3K/AKT signaling pathways.26
Moreover, ERK1/2 and PI3K/AKT signaling pathways play crucial roles in the cell cycle progression during the G1/S transition, and their activation or inhibition could promote or arrest the cell cycle progression.27,28 Cell cycle is an important regulator involved in the processes of cell proliferation, growth and survival. In our previous study, a changed cell cycle and an increased apoptotic level were observed in hypothyroidism rats.7 Cyclin D1, as one of the important proteins in the cell cycle, could drive the transition from the G1 phase to the S phase of the cell cycle and promote cell proliferation.29 In addition, the activation of ERK1/2 and PI3K/AKT signaling pathways induce the upregulation of Bcl-2 protein, which is a vital protein involved in the mitochondria apoptotic pathway.30 Furthermore, the release of cytochrome c, which is the biomarker of mitochondrial dysfunction, is under the regulation of Bcl-2 family proteins including pro-apoptotic proteins (such as Bax and Bad) and anti-apoptotic proteins (such as Bcl-2).31,32 Subsequently, caspase 9 and caspase 3, which are indicators of apoptosis could be activated.33 However, the roles of ERK1/2 and PI3K/AKT signaling pathways in apoptosis in testes of hypothyroidism rats are still unclear.
PTU (propylthiouracil) is used for treating hyperthyroid conditions.7 PTU can decrease the conversion of peripheral T4 to T3 and reduce the serum T3 concentration. Hence, it is usually applied to chemically induce a hypothyroid state.34
In our previous research, we found that the oxidative stress, arrested cell cycle and increased apoptosis in testes of hypothyroidism rats as well as all the changes mentioned above might be mediated by activating p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun NH2-terminal kinases(JNK) signaling pathways.7 In the present study, the hypothyroid rat-model induced by PTU was established to further explore the mRNA and protein expressions of ERs and GPR30, pathway related protein expressions of ERK1/2, PI3K/AKT and mitochondria-mediated apoptosis in testes to better understand the potential molecular mechanism of testicular dysfunction in hypothyroidism rats.
Materials and methods
Chemicals and reagents
PTU was obtained from Shanghai Chaohui Pharmaceutical Co., Ltd (Shanghai, China). Radioimmunoprecipitation assay (RIPA) buffer, phenyl methane sulfonyl fluoride (PMSF) and phosphatase inhibitors were obtained from Solarbio Sciences and Technology Co., Ltd (Beijing, China). Antibodies against ERα, ERβ, GPR30, Ras, ERK1/2, cyclin D1, Bax, Bcl-2, caspase 9 and caspase 3 were provided by Abcam Biotechnology (Cambridge, USA). Antibodies against Raf-1, p-Raf-1 Ser259, p-Raf-1 Ser338, p-ERK1/2, PI3K p85, p-PI3K p85, AKT, p-AKT Ser473, p-AKT Thr308, Bad and cytochrome c were provided by cell signaling technology (Boston, USA). β-Actin was chosen as the endogenous standard and purchased from Zhongshan Golden Bridge Biotech (Beijing, China). Peroxidase-conjugated secondary antibodies were purchased from Elabscience Biotechnology Co., Ltd (Wuhan, China). Bicinchoninic acid (BCA) and the enhanced chemiluminescence (ECL) reagent were provided by Thermo Scientific Biotechnology (Massachusetts, USA). TRIzol reagent was obtained from Life Technologies (Massachusetts, USA).
Animals and treatment
Male Wistar rats (6 weeks,35 240–270 g) were obtained from the Center for Experimental Animals at Gansu University of Chinese Medicine (Gansu, China) with the National Animal Use License number of SCXK (Gan 2015-0002), and acclimated to the laboratory conditions for 1 week before the experiment. The experimental rats were housed in plastic cages maintained in an air-conditioned animal room (temperature: 20 ± 2 °C, relative humidity: 60 ± 10%) with a 12 h/12 h light/dark cycle and received a standard chow diet with ad libitum access to tap water.
Then, the experimental rats were randomly divided into control (C), low dose (L) and high dose (H) groups, and were administered with 1 mL per 100 g BW per day normal saline, 0.001% and 0.1% PTU, respectively, by intragastrical gavage for 60 days.7,36 The body weights of the rats were measured every three days.
All experiments and treatments were approved by the Institutional Animal Ethical Clearance (IAEC) of Lanzhou University (ethical clearance number: IRB160218-1). All efforts were made to minimize the number of rats used in the study and reduce their suffering.
Sample preparation and hormone estimations
All rats were sacrificed after 60 days by cervical dislocation following a standard protocol and ethical procedures. Blood samples were rapidly obtained by cardiac puncture, and serum was obtained by centrifugation at 1204g for 10 min. After pretreatment of the serum sample according to the manufacturer's instructions, T3, T4 and TSH were analyzed in the γ-immuno counter by the method of radioimmunoassay.7 The bilateral testes were removed, washed with cold PBS, weighed and prepared for testicular homogenization to measure the mRNA and protein expressions.
RNA isolation and real time RT-PCR analysis
The mRNA expressions of ERα, ERβ and GPR30 were determined by real-time reverse-transcription polymerase chain reaction (real time RT-PCR) to quantify the relative expression levels of mRNA. Briefly, total RNA was extracted from approximately 95 mg of testis tissue using TRIzol reagent according to the manufacturer's protocol. Then, 1.5 μg of total RNA was reverse-transcribed with a PrimeScript™ RT Master Mix cDNA Synthesis Kit.
According to the manufacturer's instructions, real time PCR was performed using a SYBR® Premix Ex Tap™ II Kit in iQ™ 5 Real-Time PCR Detection System (Bio-Rad, USA) under the conditions of heating at 95 °C for 30 s, 50 cycles of denaturing at 95 °C for 5 s, annealing at 55–60 °C for 30 s and extension of annealing at 72 °C for 30 s. β-Actin was used as the internal reference, and the relative expression levels of mRNA were analyzed using the Pfaffl method.37 The primer sequences are shown in Table 1 and the relative mRNA expression of the target genes is expressed as a fold change compared with the control group.
Table 1. Primer sequences and theoretical amplification length.
| Genes | Orientation | Primer sequences (5′ to 3′) | Product size (bp) |
| β-Actin | Forward | GGAGATTACTGCCCTGGCTCCTA | 136 |
| Reverse | GACTCATCGTACTCCTGCTTGCTG | ||
| ERα | Forward | TTGCTCCTAACTTGCTCTTGG | 225 |
| Reverse | GGACTCGGTGGATGTGGT | ||
| ERβ | Forward | GTACCATAGACAAGAACCGGCGTA | 120 |
| Reverse | ATCCGCACTATACGGTACCCACA | ||
| GPR30 | Forward | AGGCTGTATGTGGCGCAGAA | 84 |
| Reverse | GCTGTCTGGTATGACTGCCTTGA |
Western blotting
Approximately 100 mg of testis tissue was homogenized in 1.0 mL RIPA buffer containing 10 μL protease inhibitor and 10 μL phosphatase inhibitor, followed by centrifugation at 14 000g at 4 °C for 5 min. Protein concentrations were quantified with the BCA kit. The protein samples were denatured at 100 °C for 10 min after mixing with 5× sodium dodecyl sulfate (SDS) buffer.
Protein samples (20 μg) and 2.5 μL rainbow prestained protein marker (10–170 kDa) were separated for western blot using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Initially, 6%–15% SDS-PAGE was used to separate the target proteins according to their molecular weights. Then, the proteins samples were transferred to polyvinylidene membranes (PVDF) in a transfer buffer, and the membranes were blocked for 2 hours at room temperature in Tris-saline-Tween 20 buffer (TBST) containing 5% bovine serum albumin (BSA). Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies. The primary antibodies were all diluted in TBST containing 5% BSA. The dilution ratio of anti-ERα, ERβ, PI3K p85, p-PI3K p85, AKT, Raf-1, p-Raf-1 ser259, p-Raf-1 ser338, ERK1/2, p-ERK1/2, Bad, Bax, Bcl-2, cytochrome c, caspase 9 and caspase 3 was 1 : 1000. The dilution ratio of anti-GPR30, p-AKT Thr308 and p-AKT Ser473 was 1 : 500. The dilution ratio of anti-cyclin D1 was 1 : 5000. After rinsing in TBST for three 10 min-periods, the membranes were incubated with secondary anti-mouse or anti-rabbit IgG antibodies diluted in TBST (1 : 4000 dilution) for 2 h at room temperature. Finally, the membranes were exposed in a Molecular Imager ChemiDoc XRS System (Bio-Rad, USA) after rinsing in TBST for three 10 min-periods and incubating with the enhanced chemiluminescence reagent. The intensity of each chemiluminescent band was quantified using Image ProPlus 6.0 software (Media Cybernetics, USA).
Statistical analysis
All the data were analyzed using SPSS 22.0 software (Chicago, USA) and presented as the mean ± standard deviation (SD). Comparisons among multiple groups were performed with the one-way analysis of variance (ANOVA). The significant difference between two groups was judged according to the least significant difference (LSD) test. Differences were considered to be significant when p < 0.05.
Results
The influence of hypothyroidism on serum hormone levels
To confirm whether the hypothyroidism model was established successfully by PTU, the serum levels of T3, T4 and TSH were detected. The results showed that T3 and T4 levels decreased significantly, and the TSH level increased significantly in the H group compared with those in C and L groups (p < 0.05, Table 2), suggesting that PTU induced a hypothyroid state in the H group.
Table 2. The influence of hypothyroidism on serum hormone levels.
| Groups | T3 (nmol L–1) | T4 (nmol L–1) | TSH (μIU mL–1) |
| C | 0.86 ± 0.08 | 45.56 ± 1.52 | 0.18 ± 0.06 |
| L | 0.79 ± 0.06 | 39.02 ± 1.33 | 0.27 ± 0.05 |
| H | 0.39 ± 0.01*# | 15.47 ± 1.21*# | 0.65 ± 0.09*# |
Influence of hypothyroidism on the mRNA and protein expressions of ERα, ERβ and GPR30
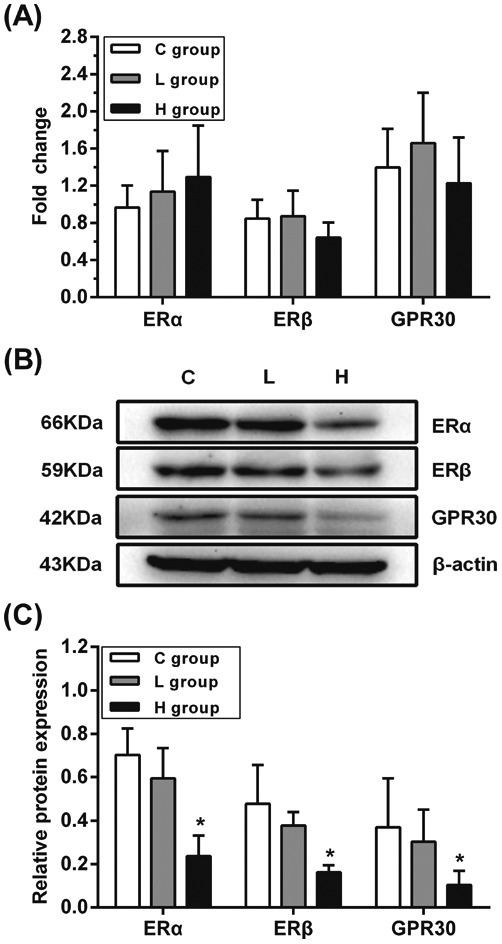
The mRNA and protein levels of ERα, ERβ and GPR30 in testes were examined to verify whether they were affected by hypothyroidism (Fig. 1). There were no significant differences in the mRNA expressions of ERα, ERβ and GPR30 in L and H groups (p > 0.05), while there was a significant decrease in the expressions of ERα, ERβ and GPR30 proteins in the H group compared with those in the C group (p < 0.05). These results showed that the decline in the expressions of ERα, ERβ and GPR30 proteins might be induced by hypothyroidism.
Fig. 1. Influence of hypothyroidism on mRNA and protein expression of ERα, ERβ and GPR30. (A) mRNA expression levels (B) protein expression levels and (C) semiquantitative analysis of ERα, ERβ and GPR30, n = 5. Significant difference from the C group, *p < 0.05.

Influence of hypothyroidism on the protein expression of ERK1/2 signaling pathway
To verify the role of ERK1/2 signaling pathway in testes, the relative protein expressions of ERK1/2 and phosphorylated ERK1/2 (p-ERK1/2) were analyzed (Fig. 2). Compared with the C group, ERK1/2 protein expression was not changed significantly (p > 0.05), while p-ERK1/2 protein expression was decreased significantly in L and H groups (p < 0.05), suggesting that ERK1/2 might have been inhibited.
Fig. 2. Influence of hypothyroidism on the protein expression of ERK1/2 signaling pathway. (A) Protein expression levels and (B) semiquantitative analysis of ERK1/2 and p-ERK1/2, n = 5. (C) The protein ratio of p-ERK1/2 to ERK1/2, n = 5. Significant difference from the C group, *p < 0.05.

Influence of hypothyroidism on the protein expression of PI3K/AKT signaling pathway
In order to determine whether the PI3K/AKT signaling pathway was suppressed in testes, the protein expression of PI3K p85, phosphorylated PI3K p85 (p-PI3K p85), total AKT, phosphorylated AKT Thr308 and Ser473 (p-AKT Thr308 and p-AKT Ser473) was detected (Fig. 3). There was no significant difference in the total AKT protein expression between L and H groups compared with that of the C group (p > 0.05). Moreover, decreased PI3K p85 and p-AKT Thr308 protein expressions in the H group and decreased p-PI3K p85 and p-AKT Ser473 protein expressions in L and H groups were observed when compared with those in the C group (p < 0.05), hinting that PI3K/AKT might have been suppressed.
Fig. 3. Influence of hypothyroidism on the protein expression of PI3K/AKT signaling pathway. (A), (D) Protein expression levels and (B), (E) semiquantitative analysis of PI3K p85, p-PI3K p85, AKT, p-AKT Thr308 and p-AKT Ser473, n = 5. (C), (F) The protein ratios of p-PI3K p85/PI3K p85, p-AKT Thr308/AKT and p-AKT Ser473/AKT, n = 5. Significant difference from the C group, *p < 0.05.

Influence of hypothyroidism on the protein expression of Ras and Raf-1
To explore the crosstalk between ERK1/2 and PI3K/AKT signaling pathways in testes, the protein expression of Ras, Raf-1, and phosphorylated Raf-1 Ser259 and Raf-1 Ser338 (p-Raf-1 Ser259 and p-Raf-1 Ser338) was detected. As shown in Fig. 4, the protein expression of Ras, p-Raf-1 Ser259, and p-Raf-1 Ser338 was significantly decreased in L and H groups when compared with that in the C group (p < 0.05). These results indicated that the downregulated Ras might have induced the inhibition of p-Raf-1 Ser338 and p-ERK.
Fig. 4. Influence of hypothyroidism on the protein expression of Ras and Raf-1. (A) Protein expression levels and (B) semiquantitative analysis of Ras, Raf-1, p-Raf-1 Ser259 and p-Raf-1 Ser338, n = 5. (C) The protein ratios of p-Raf-1 Ser259/Raf-1 and p-Raf-1 Ser338/Raf-1, n = 5. Significant difference from the C group, *p < 0.05.

Influence of hypothyroidism on the protein expression of cyclin D1
To confirm whether the cell cycle was arrested by ERK1/2 and PI3K/AKT signaling pathways, cyclin D1 protein expression was detected in testes. As shown in Fig. 5, there was a downregulated protein expression of cyclin D1 in L and H groups when compared with that in the C group (p < 0.05). This result implied that the cell cycle might have been arrested.
Fig. 5. Influence of hypothyroidism on the protein expression of cyclin D1. (A) Protein expression level and (B) semiquantitative analysis of cyclin D1, n = 5. Significant difference from the C group, *p < 0.05.

Influence of hypothyroidism on the protein expression of mitochondria-mediated apoptosis
In order to verify whether the mitochondria-mediated apoptosis was activated by ERK1/2 and PI3K/AKT signaling pathways, the protein expression of Bad, Bax, Bcl-2, cytochrome c, caspase 9 and caspase 3 was detected. As shown in Fig. 6, Bad protein expression was significantly upregulated in L and H groups when compared with that in the C group (p < 0.05). Moreover, the ratio of Bax/Bcl-2 and the protein expression of cytochrome c, caspase 9 and cleaved caspase 3 increased significantly, and the total caspase 3 protein expression decreased significantly in the H group when compared with the C group (p < 0.05). These results indicated that the mitochondria-mediated apoptosis might have been activated.
Fig. 6. Influence of hypothyroidism on the protein expressions of mitochondria-mediated apoptosis. (A), (C) Protein expression levels and (B), (D) semiquantitative analysis of Bad, Bax, Bcl-2, cytochrome c, caspase 9, cleaved caspase 3 and caspase 3, n = 5. Significant difference from the C group, *p < 0.05.

Discussion
In this study, a significant decrease in T3 and T4, and an increase in TSH levels were detected in the H group. These results showed that a PTU-induced hypothyroidism model has been established in the H group. This finding was consistent with the other previous studies, including our study.7,34
E2 is a typical steroid hormone associated with reproductive functions and male infertility, and could be affected by hypothyroidism.5,8,38 Some studies have indicated that an elevated E2 level induced a diminished expression of ERα, which led to spermatogenesis injury and male infertility.38 Other studies have also reported that testicular toxicity might be influenced by ERβ, and downregulated ERβ could promote cell apoptosis.39,40 Moreover, Sbert-Roig et al.41 demonstrated that E2 stimulated the mitochondrial oxidative capacity and reduced the cardiac oxidative stress through GPR30 in rat cardiac muscle. In addition, GPR30 could mimic the effects of E2 in enhancing the mitochondrial function while GPR30 inhibitor would weaken these effects. In our previous study, an increased E2 level has been observed in hypothyroidism rats,7 which was consistent with the study of Campo et al.42 In the present study, decreased protein expression of ERs and GPR30 was observed, whereas there was no significant difference in the mRNA expression. However, Campo et al.42 found that the mRNA expression of ERα decreased and the mRNA expression of ERβ as well as the protein expression of ERα increased in female rats. In their study, mammary tissue was used to extract RNA and protein, and S16 and total protein (Ponceau) were selected as the reference gene and the protein, respectively. β-Actin, which is commonly considered as a constitutive protein and used as the loading control for western blot, was strongly affected in tissue remodeling events following a strongly increased protein expression in the hypothyroidism group.42 In this study, β-actin was chosen as the reference gene and the protein, and there were no significant differences among all groups. This might be because the different sexes, tissues, reference genes and the proteins of the rats led to the differences between the results in these two studies. Moreover, Alarid et al.43 reported that TH prevented degradation of ERα protein induced by E2 while not influencing the mRNA expression; the results of the present study were consistent with that report. These results indicated that hypothyroidism might only induce the degradation of ERs and GPR30 proteins while not affecting the mRNA expression of ERs and GPR30 in testes of hypothyroidism rats.
ERK1/2 and PI3K/AKT signaling pathways are involved in the regulation of proliferation, differentiation, metabolism and apoptosis in response to diverse extracellular signals, including hormones, growth factors, and cytokines.44,45 Moreover, ERK1/2 and PI3K/AKT signaling pathways could be triggered by E2via binding to ERs and GPR30.9 However, the particular ER subtypes that exert the function in sperm survival signaling are still unclear.22,46 Zhang et al.19 found that ERK1/2 could be activated by the E2 treatment in human prostate stromal cells, and this effect was weakened by the absence of ERα but not GPR30. Kim et al.21 reported that ERβ could influence the ERK1/2 signaling pathway and downstream signals; the presence of ERβ increased the ERK1/2 expression, while the absence of ERβ exerted the opposite function. In addition, Lucas et al.47 also proved that even when the expression of ERα and ERβ was downregulated, the short-term treatment of E2 and GPR30 agonist could rapidly activate ERK1/2. In testes of congenital hypothyroidism rats, it has been demonstrated that the ERK1/2 signaling pathway was suppressed.48,49 Similarly, our results showed that hypothyroidism could decrease the protein expression of p-ERK1/2 while maintaining total protein expression, hinting that ERK1/2 signaling pathway might have been inactivated. According to the studies mentioned above, we speculated that the suppression of ERK1/2 might have been induced by the inhibition of ERs and GPR30 in testes of hypothyroidism rats.
ERα and ERβ could interact with the regulatory subunit of PI3K p85, and the absence of ERα and ERβ reduced the PI3K p85 expression, followed by the suppressed activation of downstream kinase AKT.22 AKT activation involves the phosphorylation of two residues: threonine 308 (Thr308) and serine 473 (Ser473).50 Stabile et al. proved that E2 induced the AKT activity via ERβ, and the decreased ERβ protein expression could suppress the p-AKT Ser473 activity in germ cells.51 Another study also indicated that GPR30 mediated the nontranscriptional effect of E2via PI3K/AKT signaling pathway, while downregulated GPR30 decreased the phosphorylation of AKT and directly reduced the cell proliferation without binding to PI3K in endometrial cancer cells.23 In the present study, we demonstrated that hypothyroidism inhibited the protein expression of p-PI3K p85, p-Akt Thr308 and p-Akt Ser473. This also suggested that the reason for the inhibited PI3K/AKT signaling pathway might be the decrease in expression of ERs and GPR30 in testes of hypothyroidism rats.
According to a previous report, there are crosstalks at different stages between Raf/ERK1/2 and PI3K/AKT signaling pathways.24 Downregulated AKT suppresses the expression of the inhibitory phosphorylation site of Raf-1 at Ser259, increases the expression of its stimulatory phosphorylation site at Ser338, and finally activates ERK1/2.24,25,52 In this study, decreased protein expression of p-AKT Thr308, p-AKT Ser473 and p-Raf-1 Ser259 was found, which indicates that both AKT and p-Raf-1 Ser259 were inactivated in testes of hypothyroidism rats. The current results were consistent with those obtained in previous studies mentioned above. However, the protein expression of p-Raf-1 Ser338 and ERK1/2 was inhibited. The reason might be that the expression of ERK1/2 could be directly mediated by Ras or PI3K protein.22,53–55
First, both Raf/ERK1/2 and PI3K/AKT signaling pathways are Ras-effector signaling pathways, and activated Ras could induce the phosphorylation of ERK1/2 and PI3K.26 Raf/ERK1/2 signaling pathway was identified to be the first Ras-effector signaling pathway.26 Raf-1 could bind to the effective region of Ras to exert the function. Then, the phosphorylation status of Raf-1 could promote the phosphorylation of MEK and subsequently activate ERK1/2.26 A study has shown that the inhibition of Ras blocked the expression of Raf-1 and finally decreased the expression of phosphorylated ERK1/2.56 Illario et al. also reported that Ras/Raf-1/Mek signal was needed by ERK1/2, and the activated Ras could promote the phosphorylation of ERK in thyroid cells.57 Moreover, ERK1/2 could be directly inhibited by the downregulation of PI3K p85.22 In the present study, protein expression of Ras and PI3K p85 were both inhibited. Therefore, we speculated that the expression of p-Raf-1 Ser338 and ERK1/2 might have been directly suppressed by the downregulation of Ras and PI3K. However, the exact mechanism requires further study.
According to a previous report, E2 stimulates the growth via a mechanism that involves the induction of the G1 to S phase transition.47 This induction is associated with the expression of cyclin D1, which is a vital cyclin-dependent kinase in the cell cycle.29,47 Moreover, a study has evidenced that cyclin D1 was regulated by several upstream signaling pathways, including ERK1/2 and PI3K/AKT signaling pathways.58 Sirianni et al.59 found that cyclin D1 expression could be upregulated by ERs via activating the ERK1/2 signaling pathway, whereas GPR30 only activated the ERK1/2 signaling pathway without affecting the expression of cyclin D1. Villanueva et al.60 reported that the activated ERK1/2 was dispensable for the induction of cyclin D1 at G1 phase. In addition, the inhibition of ERK1/2 suppresses the expression of cyclin D1.61 Moreover, the expression of cyclin D1 could be repressed by a downregulated PI3K/AKT signaling pathway and then, cell proliferation would be inhibited.62 In our previous study, an increase in ratio of G0/G1 and decrease in ratio of S and G2/M have been observed, suggesting that the cell cycle was arrested.7 In this study, the result showed that the protein expression of cyclin D1 was inhibited, hinting that the cell cycle arrest might have been caused by the suppression of ERK1/2 and PI3K/AKT signaling pathways.
Mitochondria are the target of sexual hormones to exert function, where sexual hormones could modulate the programmed cell death.63 In addition, it is well known that the mitochondria-mediated apoptosis pathway is regulated by the Bcl-2 protein family.64 Among the Bcl-2 proteins’ family, Bad and Bax promote apoptosis, whereas Bcl-2 inhibits apoptosis. The imbalanced ratios of anti- and pro-apoptotic Bcl-2 proteins family could affect the release of cytochrome c from mitochondria and the activation of apoptotic proteins caspase 9 and caspase 3,65 and would finally impair the stabilization of mitochondrial homeostasis and induce apoptosis.58 ERK1/2 and PI3K/AKT signaling pathways play important roles in the mitochondria-mediated apoptosis pathway and mediate the expression of Bcl-2 proteins family.28,65 Studies have found that mitochondria-mediated cellular apoptosis could be induced by the inhibition of ERK1/2 and AKT signaling pathways; the up-regulation of the pro-apoptotic protein Bad, an increase in the Bax/Bcl-2 ratio, and the activation of caspase 9 and caspase 3 had been observed in the apoptosis process.58,66 Similarly, the increase in ratio of Bax/Bcl-2 together with the upregulations of Bad, cytochrome c, caspase 9, cleaved caspase 3, and decreased total caspase 3 were observed in this study. Moreover, a significant increase in testicular cell apoptosis was observed in the hypothyroidism group in our previous study.7 All the results suggested that the mitochondria-mediated apoptosis pathway could be activated by suppressing the expression of ERK1/2 and PI3K/AKT signaling pathways in testes of hypothyroidism rats.
In summary, the results of this study indicated that hypothyroidism could lead to testicular dysfunction, and the possible molecular mechanisms are the changes in ERs and GPR30 expression, suppression of ERK1/2 and PI3K/AKT signaling pathways and cyclin D1, and the induction of the mitochondria-mediated apoptosis pathway. Further molecular mechanisms involved in these processes need to be determined in the future.
Funding
This study was supported by the National Natural Science Foundation of China (8157120143) and the Fundamental Research Funds for the Central Universities of China (lzujbky-2017-it34).
Conflicts of interest
The authors declare that there is no conflict of interest.
References
- Kumar A., Shekhar S., Dhole B. Indian J. Endocrinol. Metab. 2014;18(1):23–31. doi: 10.4103/2230-8210.126523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannini E. A., Ulisse S., Darmiento M. Endocr. Rev. 1995;16(4):443–459. doi: 10.1210/edrv-16-4-443. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S., Choudhury S., Roy A., Chainy G. B., Samanta L. Gen. Comp. Endocrinol. 2010;169(1):39–47. doi: 10.1016/j.ygcen.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Krassas G. E., Poppe K., Glinoer D. Endocr. Rev. 2010;31(5):702–755. doi: 10.1210/er.2009-0041. [DOI] [PubMed] [Google Scholar]
- Kala N., Ravisankar B., Govindarajulu P., Aruldhas M. M. Int. J. Androl. 2002;25(3):139–148. doi: 10.1046/j.1365-2605.2002.00338.x. [DOI] [PubMed] [Google Scholar]
- O'Donnell L., Robertson K. M., Jones M. E., Simpson E. R. Endocr. Rev. 2001:22289–22318. doi: 10.1210/edrv.22.3.0431. [DOI] [PubMed] [Google Scholar]
- Wang J. L., Zhang H. J., Wang H. L., Wang J. W., Gou P. H., Ye Z. H., Wang Y. L. Toxicol. Environ. Chem. 2015;97(10):1394–1407. [Google Scholar]
- Carreau S., Bois C., Zanatta L., Silva F. R., Bouraima-Lelong H., Delalande C. Life Sci. 2011;89(15–16):584–587. doi: 10.1016/j.lfs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Lazari M. F. M., Lucas T. F. G., Yasuhara F., Gomes G. R. O., Siu E. R., Royer C., Fernandes S. A. F., Porto C. S. Arq. Bras. Endocrinol. Metabol. 2009;53(8):923–933. doi: 10.1590/s0004-27302009000800005. [DOI] [PubMed] [Google Scholar]
- Psarra A. M., Sekeris C.E. IUBMB Life. 2008;60(4):210–223. doi: 10.1002/iub.37. [DOI] [PubMed] [Google Scholar]
- Psarra A. M., Solakidi S., Sekeris C. E. Mol. Cell. Endocrinol. 2006;246(1–2):21–33. doi: 10.1016/j.mce.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Borras C., Gambini J., Lopez-Grueso R., Pallardo F. V., Vina J. Biochim. Biophys. Acta. 2010;1802(1):205–211. doi: 10.1016/j.bbadis.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Wang J., Green P. S., Simpkins J. W. J. Neurochem. 2001;77(3):804–811. doi: 10.1046/j.1471-4159.2001.00271.x. [DOI] [PubMed] [Google Scholar]
- Duckles S. P., Krause D. N., Stirone C., Procaccio V. Mol. Interventions. 2006;6(1):26–35. doi: 10.1124/mi.6.1.6. [DOI] [PubMed] [Google Scholar]
- Pedram A., Razandi M., Wallace D. C., Levin E. R. Mol. Biol. Cell. 2006;17(5):2125–2137. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Q., Russo P. A., Cooke C., Russo I. H., Russo J. Biochim. Biophys. Acta. 2007;1773(12):1732–1746. doi: 10.1016/j.bbamcr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Pereira-Simon S., Xia X., Catanuto P., Elliot S. Endocrinology. 2012;153(11):5491–5499. doi: 10.1210/en.2012-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akingbemi B. T. Reprod. Biol. Endocrinol. 2005;3(1):1–13. doi: 10.1186/1477-7827-3-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Duan L., Du X., Ma H., Park I., Lee C., Zhang J., Shi J. Prostate. 2008;68(5):508–516. doi: 10.1002/pros.20722. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Maier B., Santen R. J., Song X. D. Biochem. Biophys. Res. Commun. 2002;294(5):926–933. doi: 10.1016/S0006-291X(02)00348-0. [DOI] [PubMed] [Google Scholar]
- Kim J. H., Jeong I. Y., Lim Y., Lee Y. H., Shin S. Y. BMB Rep. 2011;44(7):452–457. doi: 10.5483/BMBRep.2011.44.7.452. [DOI] [PubMed] [Google Scholar]
- Mannella P., Brinton R. D. J. Neurosci. 2006;26(37):9439–9447. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X., Guo R., Qiao Y., Zhang Y., Lei J., Wang X., Li L., Hu D. Int. J. Gynecol. Cancer. 2013;23(1):52–59. doi: 10.1097/IGC.0b013e31827912b8. [DOI] [PubMed] [Google Scholar]
- Zhou J., Du T., Li B., Rong Y., Verkhratsky A., Peng L. ASN Neuro. 2015;7(5):1–16. doi: 10.1177/1759091415602463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C., Clarke B. A., Zimmermann S., Nunez L., Rossman R., Reid K., Moelling K., Yancopoulos G. D., Glass D. J. Science. 1999;286(5445):1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- Castellano E., Downward J. Curr. Top. Microbiol. Immunol. 2010:346143–346169. doi: 10.1007/82_2010_56. [DOI] [PubMed] [Google Scholar]
- Xu G., Li Y., Yoshimoto K., Wu Q., Chen G., Iwata T., Mizusawa N., Wan C., Nie X. Toxicol. Lett. 2014;224(3):362–370. doi: 10.1016/j.toxlet.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Li L., Wang X., Sharvan R., Gao J., Qu S. Biomed. Pharmacother. 2017:951225–951231. doi: 10.1016/j.biopha.2017.09.010. [DOI] [PubMed] [Google Scholar]
- Baldin V., Lukas J., Marcote M. J., Pagano M., Draetta G. Genes Dev. 1993;7(5):812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Cawley N. X., Loh Y. P. PLoS One. 2013;8(8):e71578. doi: 10.1371/journal.pone.0071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L., Korsmeyer S. J. Biochem. Biophys. Res. Commun. 2003;304(3):437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- Er E., Oliver L., Cartron P. F., Juin P., Manon S., Vallette F. M. Biochim. Biophys. Acta. 2006;1757(9–10):1301–1311. doi: 10.1016/j.bbabio.2006.05.032. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S., Ahmad M. Cell. 1997:91479–91489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Sahoo D. K., Roy A., Bhanja S., Chainy G. B. N. Gen. Comp. Endocrinol. 2008;156(1):63–70. doi: 10.1016/j.ygcen.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Sengupta P. Int. J. Prev. Med. 2013;4(6):624–630. [PMC free article] [PubMed] [Google Scholar]
- Shiraki A., Saito F., Akane H., Akahori Y., Imatanaka N., Itahashi M., Yoshida T., Shibutani M. J. Appl. Toxicol. 2016;36(1):24–34. doi: 10.1002/jat.3140. [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W. Nucleic Acids Res. 2001:292002–292007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavy M., Trottmann M., Liedl B., Reese S., Stief C., Freitag B., Baugh J., Spagnoli G., Kolle S. Sci. Rep. 2017;7(39931):1–11. doi: 10.1038/srep39931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J. L., Wang C. G., Wu T., Moreno J. M. L., Zhong Y. F., Huang X., Chen Y. X., Zuo Z. H. Toxicology. 2011;287(1):21–28. doi: 10.1016/j.tox.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Liang J., Xie Q., Li P., Zhong X., Chen Y. Mol. Cell. Biochem. 2015;401(1–2):71–86. doi: 10.1007/s11010-014-2293-y. [DOI] [PubMed] [Google Scholar]
- Sbert-Roig M., Bauza-Thorbrugge M., Galmes-Pascual B. M., Capllonch-Amer G., Garcia-Palmer F. J., Llado I., Proenza A. M., Gianotti M. Mol. Cell. Endocrinol. 2016:420116–420124. doi: 10.1016/j.mce.2015.11.027. [DOI] [PubMed] [Google Scholar]
- Campo V. A. F., Sasso C. V., Actis E. A., Caron R. W., Hapon M. B., Jahn G. A. Mol. Cell. Endocrinol. 2016:41918–41928. doi: 10.1016/j.mce.2015.09.023. [DOI] [PubMed] [Google Scholar]
- Alarid E. T., Preisler-Mashek M. T., Solodin N. M. Endocrinology. 2003;144(8):3469–3476. doi: 10.1210/en.2002-0092. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C., Eychène A. Biol. Cell. 2001;93(1–2):53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- Cantley L. C. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Aquila S., Sisci D., Gentile M., Middea E., Catalano S., Carpino A., Rago V., Ando S. J. Clin. Endocrinol. Metab. 2004;89(3):1443–1451. doi: 10.1210/jc.2003-031681. [DOI] [PubMed] [Google Scholar]
- Lucas T. F., Royer C., Siu E. R., Lazari M. F., Porto C. S. Biol. Reprod. 2010;83(2):307–317. doi: 10.1095/biolreprod.110.084160. [DOI] [PubMed] [Google Scholar]
- Zamoner A., Heimfarth L., Pessoa-Pureur R. Neurotoxicology. 2008;29(6):1092–1099. doi: 10.1016/j.neuro.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Zamoner A., Barreto K. P., Filho D. W., Sell F., Woehl V. M., Guma F. C., Pessoa-Pureur R., Silva F. R. J. Mol. Endocrinol. 2008;40(3):125–135. doi: 10.1677/JME-07-0089. [DOI] [PubMed] [Google Scholar]
- Vincent E. E., Elder D. J. E., Thomas E. C., Phillips L., Morgan C., Pawade J., Sohail M., May M. T., Hetzel M. R., Tavaré J. M. Br. J. Cancer. 2011;104(11):1755–1761. doi: 10.1038/bjc.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabile V., Russo M., Chieffi P. Reproduction. 2006;132(3):477–484. doi: 10.1530/rep.1.01107. [DOI] [PubMed] [Google Scholar]
- Moelling K., Schad K., Bosse M., Zimmermann S., Schweneker M. J. Biol. Chem. 2002;277(34):31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- Ebi H., Costa C., Faber A. C., Nishtala M., Kotani H., Juric D., Della Pelle P., Song Y., Yano S., Mino-Kenudson M., Benes C. H., Engelman J. A. Proc. Natl. Acad. Sci. U. S. A. 2013;110(52):21124–21129. doi: 10.1073/pnas.1314124110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G., Khanin R., Carvajal R. D., Schwartz G. K. Mol. Cancer Ther. 2014;13(8):2073–2080. doi: 10.1158/1535-7163.MCT-14-0095. [DOI] [PubMed] [Google Scholar]
- Minano A., Xifro X., Perez V., Barneda-Zahonero B., Saura C. A., Rodriguez-Alvarez J. Mol. Cell. Neurosci. 2008;39(2):143–151. doi: 10.1016/j.mcn.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Li D., Wu L. J., Tashiro S., Onodera S., Ikejima T. J. Pharmacol. Sci. 2007;103(1):56–66. doi: 10.1254/jphs.fpj06016x. [DOI] [PubMed] [Google Scholar]
- Illario M., Cavallo A. L., Monaco S., Di V. E., Mueller F., Marzano L. A., Troncone G., Fenzi G., Rossi G., Vitale M. J. Clin. Endocrinol. Metab. 2005;90(5):2865–2873. doi: 10.1210/jc.2004-1520. [DOI] [PubMed] [Google Scholar]
- Liu J., Li Q., Liu Z., Lin L., Zhang X., Cao M., Jiang J. Oncol. Rep. 2016;35(6):3363–3370. doi: 10.3892/or.2016.4695. [DOI] [PubMed] [Google Scholar]
- Sirianni R., Chimento A., Ruggiero C., De L. A., Lappano R., Andò S., Maggiolini M., Pezzi V. Endocrinology. 2008;149(10):5043. doi: 10.1210/en.2007-1593. [DOI] [PubMed] [Google Scholar]
- Villanueva J., Yung Y., Walker J. L., Assoian R. K. Mol. Biol. Cell. 2007;18(4):1457–1463. doi: 10.1091/mbc.E06-10-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P. L., Chen L. P., Hess R. A., Müller R., Gonzalez F. J., Peters J. M. J. Biol. Chem. 2015;290(38):23416–23431. doi: 10.1074/jbc.M115.664508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Guo M., Zhuang Y., Chu J., Zhang S. Mol. Cell. Biochem. 2014;393(1–2):155–164. doi: 10.1007/s11010-014-2055-x. [DOI] [PubMed] [Google Scholar]
- Psarra A. M., Solakidi S., Sekeris C. E. Mol. Cell. Endocrinol. 2006;246(1–2):21–33. doi: 10.1016/j.mce.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Tait S. W., Green D. R. Nat. Rev. Mol. Cell Biol. 2010;11(9):621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Ding D., Wei S., Song Y., Li L., Du G., Zhan H., Cao Y. Cell. Physiol. Biochem. 2013;32(6):1751–1760. doi: 10.1159/000356609. [DOI] [PubMed] [Google Scholar]
- Wang A. S., Xu C. W., Xie H. Y., Yao A. J., Shen Y. Z., Wan J. J., Zhang H. Q., Fu J. F., Chen Z. M., Zou Z. Q. J. Funct. Foods. 2016:21517–21524. [Google Scholar]