

Abstract
Cancer cells undergo metabolic reprogramming such as enhanced aerobic glycolysis, mutations in the tricarboxylic acid cycle enzymes, and upregulation of de novo lipid synthesis and glutaminolysis. These alterations are pivotal to the development and maintenance of the malignant phenotype of cancer cells in unfavorable tumor microenvironment or metastatic sites. Although mitochondrial fatty acid β-oxidation (FAO) is a primary bioenergetic source, it has not been generally recognized as part of the metabolic landscape of cancer. The last few years, however, have seen a dramatic change in the view of cancer relevance of the FAO pathway. Many recent studies have provided significant evidence to support a “lipolytic phenotype” of cancer. FAO, like other well-defined metabolic pathways involved in cancer, is dysregulated in diverse human malignancies. Cancer cells rely on FAO for proliferation, survival, stemness, drug resistance, and metastatic progression. FAO is also reprogrammed in cancer-associated immune and other host cells, which may contribute to immune suppression and tumor-promoting microenvironment. This article reviews and puts into context our current understanding of multi-faceted roles of FAO in oncogenesis as well as anti-cancer therapeutic opportunities posed by the FAO pathway.
Keywords: Fatty acid β-oxidation, Cancer, Lipolytic phenotype, ATP, NADPH
1. Introduction
During the process of transformation from normal precursors, cancer cells acquire the ability of rewiring the metabolism of their own as well as diverse types of immune and stromal cells in the tumor microenvironment to meet the demands of uncontrolled growth and metastatic progression [1]. As early as in 1920s, Warburg observed that neoplastic cells prefer glycolysis to form lactate as the final product instead of mitochondrial oxidation, even in the presence of abundant oxygen [2]. Since then, much progress has been made in understanding metabolic reprogramming in cancer. Although limited in generating ATP, the Warburg effect of aerobic glycolysis offers an advantage in supplying cancer cells with quick ATP and biosynthetic intermediates for rapid proliferation [3]. In addition to the Warburg effect, increases in glutaminolysis and de novo fatty acid synthesis are also prominent hallmarks of cancer. Glutaminolysis feeds the tricarboxylic acid (TCA) cycle and contributes nitrogen and carbon skeletons to nucleotide and amino acid synthesis [4]. On the other hand, heightened de novo fatty acid synthesis from acetyl-CoA and reducing power (NADPH) is required for membrane biogenesis in rapidly dividing cancer cells [5].
In contrast to the lipogenic phenotype, the role of mitochondrial fatty acid β-oxidation (FAO) in cancer has not been well defined. Although FAO is one of the major sources of ATP production, most previous studies of cancer bioenergetics have focused on the Warburg effect [3,6,7]. In addition to ATP, FAO is involved in production of cytosolic NADPH [8–10], the reducing power to support biosynthesis and to counteract oxidative stress. However, there are other alternative or redundant routes to replenish cytosolic NADPH including the pentose phosphate pathway and the conversion of malate to pyruvate catalyzed by malic enzyme [10]. Another argument against the oncogenic involvement of FAO is that lipogenesis and FAO are mutually exclusive processes coordinated by the level of malonyl-CoA [10–12]. Malonyl-CoA, an intermediate of fatty acid biosynthesis, acts as an allosteric inhibitor of the FAO rate-setting enzyme carnitine palmitoyltransferase 1 (CPT1), presumably preventing FAO from occurring simultaneously with active lipogenesis [10–12]. Finally, in sharp contrast to the well-studied glycolysis, glutaminolysis, and lipogenesis cascades where key enzymes or their regulators are known to be overexpressed, mutated, or dysregulated in connection to oncoproteins or tumor suppressors [13–18], limited evidence suggests that the FAO pathway is “reprogrammed” in cancer or pathophysiologically linked to activation of specific oncogenes or loss of tumor suppressors. However, recent advances in the field have substantially changed the view on the relevance of FAO to cancer. Multiple lines of evidence now suggest that abnormal FAO activity is involved in diverse aspects of oncogenesis [19]. Many cancer cells rely on FAO for proliferation, survival, stemness, drug resistance or metastasis. The key enzymes or regulators of FAO have therefore emerged as promising targets for cancer therapy. The review aims to dissect the multi-faceted roles of FAO in cancer and non-cancerous cells in the tumor microenvironment.
2. FAO basics
FAO is a multi-step catabolic process that allows for the mitochondrial conversion of long chain fatty acids into acetyl-CoA which will be fully oxidized through the TCA cycle and the electron transport chain (ETC) to produce ATP (Fig. 1). Fatty acids enter mammalian cells through fatty acid transport proteins such as CD36 and fatty-acid-binding proteins that facilitate the transfer of fatty acids between extra-and intra-cellular membranes [20]. Before shuttling into the mitochondrion for oxidation, fatty acids are activated to fatty acyl-CoA by fatty acyl CoA synthetase. On the outer mitochondrial membrane, fatty acyl-CoA is converted to fatty acyl-carnitine by the action of CPT1. Carnitine/acylcarnitine translocase (CACT), located on the inner mitochondrial membrane shuttles acylcarnitine into the mitochondrial matrix. Carnitine palmitoyltransferase II (CPT2) on the matrix side of the inner membrane reconverts acylcarnitine to acyl-CoA. In the mitochondrion, acyl CoA is cleaved into acetyl CoA by a repeated 4-step cycle catalyzed sequentially by activities of acyl-CoA dehydrogenase, hydroxyacyl-CoA dehydrogenase, enoyl-CoA hydratase and 3-ketoacyl-CoA thiolase (3-KAT). The breakdown product acetyl-CoA enters the TCA cycle which is coupled to oxidative phosphorylation to generate ATP (Fig. 1).
Fig. 1. FAO basics.
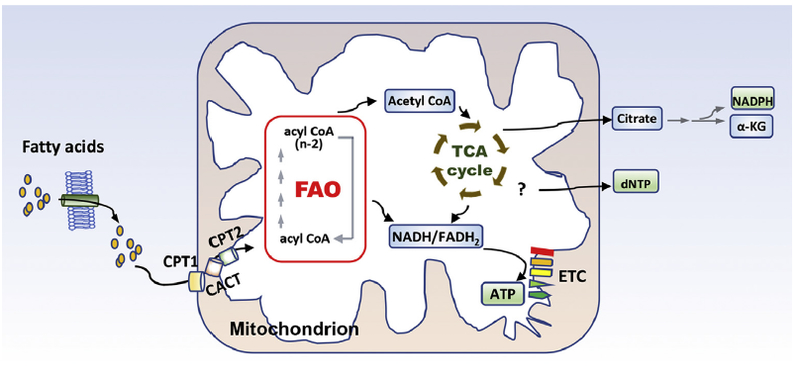
Long chain fatty acids enter cells via fatty acid transport proteins and then shuttled into the mitochondrion by the carnitine shuttle system. In the mitochondrion, fatty acids undergo oxidative removal of successive 2-carbon unit in the form of acetyl-CoA. Acetyl-CoA will be oxidized to CO2 through the TCA cycle. Both FAO and the TCA cycle produce reduced electron carriers (NADH/FADH2), which will pass electrons to ETC to yield ATP. Apart from bioenergetic production, the carbon and hydrogen sources of FAO-generated acetyl CoA can be exported out of the TCA cycle to the cytoplasm to engage in NADPH and dNTP production as detailed in the text.
In addition to bioenergetic production, FAO-generated acetyl CoA enters TCA to form citrate, which can be exported to the cytoplasm to engage NADPH-producing oxidation of isocitrate to α-ketoglutarate by isocitrate dehydrogenase [10] (Fig. 1). Indeed, as demonstrated by several groups, FAO is involved in regulation of cytosolic NADPH [8–10], the reducing agent to support biosynthesis and redox homeostasis. Several recent studies have identified FAO-generated acetyl CoA to be carbon source for incorporation into aspartate (a nucleotide precursor), uridine monophosphate (a precursor of pyrimidine nucleoside triphosphates) and subsequently cellular DNA in endothelial cells [21–23] (Fig. 1). However, this catabolic fate of fatty acid has not been extrapolated to other cell types or tissues yet.
The cellular FAO rate is regulated at multiple sites of fatty acid trafficking and the subsequent breakdown via transcriptional and post-transcriptional mechanisms. CPT1 is considered to be the rate-limiting enzyme that is allosterically inhibited by malonyl-CoA [21]. There are three members of the CPT1 family, CPT1A (liver form), CPT1B (muscle form) and CPT1C (brain form, enzymatic inactive) encoded by three paralogous genes. Compared to CPT1B, CPT1A is 30–100-fold more resistant to allosteric inhibition by malonyl CoA [12,24] and is therefore more likely to be enzymatically active in cancer cells exhibiting high activities of both lipogenesis and FAO. Although the atypical CPT1C has been reported to be expressed in various cell types and functions to cope with metabolic stresses [25,26], the location in endoplasmic reticulum (ER) of neurons and the lack of an enzymatic activity in mitochondria make it unlikely that CPT1C plays a direct role in mitochondrial FAO [21,27,28].
The most prominent transcriptional regulators of FAO are peroxisome proliferator-activated receptors (PPARs) of the ligand-activated nuclear receptor superfamily. They act as environmental fat sensors and transcriptional activators of FAO enzymes [29]. PPARα has been shown to stimulate CPT1A transcription in the liver by binding to the peroxisome proliferator response element on the CPT1A gene promoter [30]. PPARδ drives expression of various FAO enzymes to promote FAO in hematopoietic stem cells [31]. In addition, the FAO rate is tightly controlled by the activity of AMP-activated protein kinases (AMPKs). AMPKs regulate cellular metabolic states by shutting down energy-consuming anabolic processes and activating energy-yielding catabolic ones [32]. Activation of AMPK stimulates FAO primarily through phosphorylation and inactivation of its downstream target acetyl-CoA carboxylase (ACC) [33], the enzyme that synthesizes malonyl CoA to inhibit CPT1 and FAO in physiological conditions [10–12]. It seems that two isoforms of ACC (ACC1 and ACC2) differ in cellular locations and functions [34]. ACC1 is present in the cytosol and produces malonyl-CoA mainly as substrate for fatty acid synthesis. In contrast, ACC2 is located on the outer mitochondrial membrane responsible for synthesizing malonyl-CoA serving as a CPT1 inhibitor [35]. In cancer cells, ACC2 has been reported to be repressed by sirtuin-mediated histone deacetylation or loss of prolyl hydroxylase 3 [36,37], potentially explaining the simultaneous occurrence of active lipogenesis and FAO in neoplastic cells.
3. Dysregulation of FAO in cancer
A critical question concerning the role of FAO in cancer pathogenicity is whether the FAO enzymes or their regulators are dysregulated as those in the glycolysis and glutaminolysis pathways. Mutations of FAO enzymes have not been detected at significant rates in cancer. Substantial studies, however, have revealed overexpression of various FAO enzymes including CD36 [38], CPT1A [39,40], CPT1B [40,41], CPT1C [25], CPT-2 [40], carnitine transporter CT2 [42], and Acyl-CoA synthetase long chain 3 [43] in multiple malignancies relative to their normal counterparts. For instance, 3- to 4-fold increases in expression of CPT-1 A, CPT-1B, and CPT-2 were observed in chronic lymphocytic leukemia (CLL) cells compared to normal stromal cells [40]. Over-expression of some FAO enzymes such as CPT1A correlates strongly with poor patient outcomes of cancer including acute myeloid leukemia (AML) and ovarian cancer [39,44].
Consistent with cancer-associated overexpression of key FAO enzymes, many types of cancer exhibit a high activity of FAO such as KRas mutant lung cancer [43], triple negative breast cancer (TNBC) [45,46], AML [42], hepatitis B-induced hepatocellular carcinoma [47], glioma [48], and low-grade astrocytoma [48]. Most significantly, several groups have recently reported that expression of a number of FAO enzymes was activated by prominent oncoproteins. For instance, a metabolomics study revealed that FAO enzymes and metabolic intermediates were upregulated in a subset of TNBC that overexpress c-Myc [49], an oncogenic transcription factor disproportionately elevated in TNBC. Inhibition of FAO blocked Myc-driven tumorigenesis [49]. Such a role of c-Myc in activation of FAO has been further confirmed in MCF-10A-Ras cells where FAO enzymes were upregulated by the c-Myc/PGC-1β/ERRα signaling [50]. In addition, another group recently identified CPT1B as a downstream target of the JAK/STAT3 transcription factor in breast cancer. The JAK/STAT3-CPT1B axis was activated in response to mammary adipocyte-derived leptin [41]. Collectively these studies suggest that FAO is activated in cancer as a result of c-Myc over-expression or JAK/STAT3 activation. Furthermore, FAO can act upstream of oncoproteins to regulate their functions. In metastatic TNBC with elevated levels of FAO, the knocking down of CPT enzymes resulted in inhibition of Src and Src-mediated metastasis [51], suggesting that FAO activity may be instrumental in maintaining activated status of certain oncoproteins, another mode of FAO involvement in oncogenic processes.
4. Reliance on FAO for cancer cell growth and survival
Multiple types of cancer have been demonstrated to rely on FAO for cell growth and survival. Most previous observations implicating FAO in oncogenesis were based on pharmacological inhibition of CPT1. CPT1 inhibitors suppressed growth and/or viability of cell lines of myeloid leukemia [9,52], ovarian cancer [44], hepatocellular carcinoma [53], prostate cancer [54,55], glioma [8], and multiple myeloma [56]. The effects of these CPT1 inhibitors in ovarian [44] and prostate [54] cancer cells were validated by siRNA or shRNA silencing approach. In an integrated genomics study published in 2014, Gatza et al. identified 8 genes essential for the luminal subtype of breast cancer [57]. CPT1A, but not other CPT1 isoforms, was among this short panel of the 8 growth-dependent genes [57]. Mechanistically, many types of cancer seem to rely on FAO as an essential ATP source to fuel rapid growth [8,9,44,58]. This is particularly relevant in ovarian cancer wherein inhibition of CPT1 reduced cellular ATP levels and activated AMPK, which was associated with cell cycle arrest at G1/G0 [44]. The meta-static spread of ovarian cancer characteristically first involves the abdominal fatty tissue of the omentum. Upon interactions, fatty acids are hydrolyzed and released from omental adipocytes [58]. Ovarian tumor cells depend on these adipocyte-derived fatty acids to support rapid growth and continued peritoneal dissemination [58]. More recent studies from colon and breast cancer suggest that cancer cells have a clear predilection for spreading to adipocyte-rich tissues [59,60]. Uptake of fatty acids from surrounding adipocytes promoted FAO in cancer cells [59,61] (Fig. 2).
Fig. 2. FAO signaling pathways and multi-faceted roles in cancer.
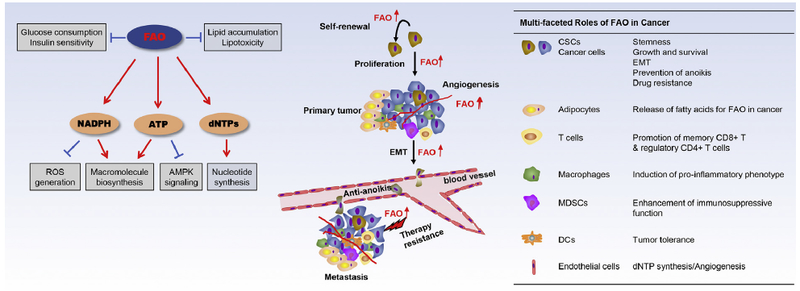
Shown on the left are signaling pathways regulated by FAO in physiological conditions. FAO is implicated in multiple aspects of tumorigenesis including cancer cell growth, survival, CSC maintenance, drug resistance, and metastasis. In addition to abnormal activation in cancer cells, FAO and related lipid metabolic processes are also reprogrammed in tumor-associated immune cells, adipocytes and endothelial cells, which may contribute to immune suppression and tumor-promoting microenvironment.
Compared to other malignancies, prostate cancer is less glycolytic [62,63]. The 18F-fluorodeoxyglucose with positron emission tomography (FDG-PET) has limited value in diagnostic imaging of prostate cancer [64–66]. Pharmacological interference with FAO led to not only growth inhibition but also metabolic switch to more glycolysis and therefore enhanced sensitivity to FDG-PET scan as demonstrated in mouse xenografts of human prostate cancer cell lines [54,55]. Glioma may be another example of cancer with limited glycolysis. Less than 50% of acetyl-CoA in glioma was derived from glucose as demonstrated in a study of human nuclear magnetic resonance (NMR) spectroscopy [67]. Pharmacological inhibition of FAO decreased cell proliferation, Ki-67 positive index, and S + G2/M phases of cell cycle, as well as delayed emergence and progression of glioma in vivo [48]. In glioma patient-derived tumor xenografts, FAO was demonstrated to support major respiration activity compared to glucose catabolism [48].
Besides the impact on cellular proliferation, inhibition of FAO was also linked to induction of apoptosis or reduced viability in cell lines of myeloid leukemia, glioma, and hepatocellular carcinoma [47,48,52]. The anti-survival effect of FAO inhibition was most likely resulted from perturbation of NADPH homeostasis, reactive oxidative species (ROS) production, oxidative stress, and/or mitochondrial damages [9,52]. There is also evidence that FAO flux or FAO enzymes regulated expression or functions of the Bcl-2 family proteins or other death/survival mediators [68,69]. Other studies suggested the involvement of FAO metabolic intermediates as functional effectors of cell survival [8,9,22]. In addition, FAO inhibition could lead to cytotoxic accumulation of long-chain fatty acids (lipotoxicity) and the subsequent ER stress, and ultimately cell death [70,71].
5. FAO and metastasis
Cancer metastasis encompasses multiple steps including local invasion of tumor cells into adjacent tissues, intravasation of cancer cells into blood or lymphatic vessels, survival of cancer cells in the circulatory and lymphatic systems, extravasation, and colonization of cancer cells in target organs. FAO has been implicated in promotion of many of these steps leading to a metastatic phenotype (Fig. 2) [22,23,38,59,72,73].
Local invasion of cancer cells is facilitated by epithelial-to-mesenchymal transition (EMT), a process critical for normal embryonic morphogenesis. A metabolomics profiling study of immortalized breast epithelial cell lines with stem cell properties and their EMT-derived mesenchymal phenotype revealed that FAO was more active, driven by PPAR, in the mesenchymal phenotype, whereas amino acid metabolism and glycolysis were at higher levels in the epithelial counterpart [74]. In line with this, FAO induction in colon cancer cells by co-cultivation with adipocytes was associated with induction of EMT as indicated by decreased E-cadherin but increased vimentin expression [59].
Once migration into the lumina of lymphatic or blood vessels, the first challenge for the survival of cancer cells is the detachment-triggered metabolic stress. When detached from the extracellular matrix, normal or non-metastatic cells undergo anoikis, a caspase-mediated apoptosis [72,73,75]. The detachment results in ATP deficiency as a result of decreased glucose uptake and catabolism, associated with a decrease in NADPH and increase in ROS [72]. Several independent studies showed that cancer cells upregulated FAO to overcome anoikis [73,75,76]. Upon loss of attachment, FAO in breast cancer cells was stimulated to increase ATP and cell survival by overexpression of the promyelocytic leukemia (PML) gene [75]. The PML protein is a driver of FAO as further elaborated below.
FAO may also contribute to the development of the metastatic phenotype through its potential role in regulation of cancer stem cells (CSCs). CSCs are capable of self-renewing and differentiating into the non-stem cancer cells. They are also responsible for tumor metastasis and resistance to therapies. Ito et al. reported that the PML-PPARδ-FAO axis was required for asymmetric division of hematopoietic stem cells [31]. Genetic interference with this pathway resulted in the exhaustion of the stem cell pool. Another work showed that a deficient mutation in trimethyllysine hydroxylase, a key enzyme in biosynthesis of the CPT1 substrate carnitine, reduced neural stem cells in the mouse embryonic neocortex, an abnormality shared by silencing CPT1A or limiting fatty acid availability [77]. These observations indicate that normal tissue stem cells benefit from active FAO for phenotypic maintenance.
Such a role for FAO in cancer stemness has gained some recent attention. The first observation alluding to the possibility was from Samudio et al. who reported that treatment with etomoxir decreased the number of quiescent leukemia progenitor cells in approximately 50% of primary human AML samples [52]. Another study reported that leukemic stem cells could be divided into two distinct populations based on the expression of the fatty acid transporter CD36 [78]. CD36-positive leukemic stem cells displayed a higher FAO rate and more resistance to drugs than CD36-negative leukemic stem cells, indicating that FAO activity was a determinant of cancer stem cell properties. In a most recent study, Wang et al. reported that the Leptin-JAK/STAT3 pathway upregulated expression of CPT1B, FAO activity and stem cell self-renewal in breast cancer [41].
6. FAO in drug resistance
Systemic treatment of cancer inevitably induces drug resistance of tumor cells, which prevents most cancer therapies from being curative [79]. Accumulating studies showed that FAO activation is an important mechanism employed by cancer cells to develop drug resistance (Fig. 2). FAO activity has been demonstrated to be elevated in cancer cells in response to treatment with the therapeutic reagents dexamethasone [80], L-asparaginase [81], imatinib or rapamycin [82,83], cytarabine [84], and tamoxifen [85].
Multiple mechanisms have been proposed to explain drug-induced FAO activation and FAO-mediated drug resistance. Dexamethasone induced FAO in CLL cells via upregulation of PPARα [80]. Treatment with L-asparaginase, a chemotherapeutic agent for childhood acute lymphoblastic leukemia, inhibited the RagB (a Ras-related GTPase)-mTORC1 pathway, leading to metabolic stress and FAO activation [81]. Intriguingly, although it has been previously shown to be enzymatically inactive, CPT1C is upregulated and involved in survival of breast, brain, and lung cancers, especially under conditions of hypoxia or nutrient stress [25,86]. Recently, CPT1C upregulation was found to be linked to increased FAO in BCR-ABL positive leukemia cells, responsible for imatinib or rapamycin resistance [82,83]. Although the functional relationship of CPT1C to FAO is yet to be elucidated, these studies make CPT1C, an atypical isoform of CPT1, a promising anti-cancer target. In AML cells, resistance to cytarabine was found to be mediated by increases in expression of the fatty acid transporter CD36 and FAO rates [84]. In addition, retinoblastoma (Rb) deficiency in breast cancer cells upregulated FAO-related genes, which might contribute to resistance to tamoxifen [85]. In these studies, addition of FAO inhibitors completely or partially inhibited the drug resistances of cancer cells [80,81,83,84,87,88].
7. Beyond cancer cells
While the crucial role for FAO in cancer cells has been recognized, the emerging correlative studies also suggest association of FAO with tumor-promoting functions of non-cancerous cells in the tumor micro-environment (Fig. 2). For example, it was recently shown that FAO was involved in supporting cellular DNA synthesis of endothelium [22,23], suggesting that blockade of FAO may prevent vessel sprouting and tumor angiogenesis through targeting endothelial cells within a tumor.
Depending on the cellular context, FAO activity may be immune promoting or inhibitory. Cellular immune responses against tumors are typically mediated by CD8 T cells. Tumor necrosis factor receptor-associated factor 6 (TRAF6) regulated long-lived memory CD8 T cell development by promoting FAO [89]. Similarly, activation of FAO was reported to be a mechanism by which IL-15 regulated the memory phenotype of CD8+ T cells [90]. Restoration of FAO increased CD8 memory cells and improved the potency of anticancer vaccination [89]. It will be interesting to determine whether FAO is repressed in CD8 memory cells in cancer.
On the other hand, activation of the FAO pathway in other immune cells, such as regulatory CD4+ T cells (Treg), M2 macrophages, myeloid-derived suppressor cells (MDSCs), and dendritic cells (DCs), may be a potential mechanism for cancer cells to escape from immune surveillance. Distinct subsets of helper CD4+ T cells require specific metabolic programs to meet their differing energetic and biosynthetic demands. Effector CD4+ T cells (Teff) relied on glycolytic metabolism, whereas Treg demonstrated higher levels of FAO [91]. Etomoxir suppressed Treg population while addition of exogenous fatty acids promoted their differentiation or survival [91]. High infiltration of Treg cells in tumors correlated with poor prognosis in multiple cancers [92]. Several groups reported that M2 macrophages used FAO to fuel mitochondrial oxidative phosphorylation. Inhibition of FAO could prevent M2 polarization of macrophages [93,94], Heightened FAO is believed to provide survival advantage to M2 macrophages over M0 and M1 phenotypes [94]. However, a recent report using a genetic model disrupting FAO in the myeloid lineage did not support such a function of FAO in M2 polarization and differentiation [95]. Intriguingly, the rate of FAO in MDSCs, a major component of tumor-associated immunosuppressive network [96,97], correlated positively with their T-cell inhibitory activity [98]. A recent study showed that FAO inhibition could delay tumor growth and enhance the antitumor effect of adoptive T-cell therapy [98]. Earlier studies indicated that abnormal lipid accumulation in tumor-associated DCs contributed to their tolerogenic phenotype [99,100]. The dysfunction of these DCs was thought to be due to the excessive lipid burden but the results could be equally explained by the role of the abundant intracellular fat store in supplying substrates for active FAO, which could metabolically and functionally alter tumor-associated DCs. Indeed, a more recent observation demonstrated that FAO increased the activity of indoleamine 2,3-dioxgenase-1 (IDO), culminating in enhanced tolerization of DCs and generation of Treg cells [101]. Blockade of FAO enhanced antitumor immunity and improved the efficacy of anti-PD-1 inhibitors [101].
8. FAO as a druggable metabolic pathway for cancer treatment
Physiologically, the FAO flux in normal tissues including the high energy-demanding skeletal muscle and heart as well as the liver, the central organ of lipid metabolism, is controlled by the availability of glucose in the circulation [102]. FAO is activated when the environmental glucose becomes limited. On the other hand, inhibition of FAO switches energy metabolism from fatty acid to glucose oxidation, a condition typically alleviating oxygen shortage and insulin resistance, as exemplified by therapeutic benefits of a number of FAO inhibitors in patients with type II diabetes or myocardial ischemia [103–108]. Apparently, normal tissues differ from cancer. FAO is required constitutively for rapid proliferation of malignant cells as recently shown in multiple types of cancer [19,63]. Differential dependence of cancerous and normal tissues on FAO could provide a sufficient therapeutic window to target cancer cells with little side effect on normal ones. The cancer-special dependence on FAO may be related to the glycolytic phenotype of neoplastic cells. Unlike mitochondrial glucose oxidation, glycolysis is limited in ATP generation [3,6] and can’t fully replace the bioenergetic function of FAO. This may explain why glycolysis and FAO occur simultaneously in cancer. The hyperactive catabolism of both glucose and fatty acids could be also a major cause of weight loss and cachexia associated with cancer progression in patients [109,110].
FAO inhibitors are generally safe and well tolerated in vivo [103–108] (Table 1). Some of them belong to a new class of drugs termed “metabolic modulators” which are already in clinical use for treatment of angina pectoris in USA or other parts of the world [10,104–108]. These include perhexiline (CPT1/CPT2 dual inhibitor) and trimetazidine or ranolazine [inhibitor of 3-KAT of the trifunctional protein TFP (hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase/3-KAT)] [106,107] (Table 1). The TFP complex catalyzes the last three steps of β-oxidation within the mitochondrion. Although the exact targets of these medicines remain controversial, substantial in vitro and in vivo data indicate that their clinical benefits are at least partially mediated through inhibiting FAO to improve myocardial glucose oxidation [10,104–108]. However, the activities of these metabolic modulators against malignant diseases have not been tested. If proved effective, these medicines could be readily repurposed to treat cancer or as adjuvants to enhance efficacies of other anti-cancer drugs.
Table 1.
FAO modulators in clinical and research uses.
| Compounds | Targeted Enzymes |
Clinical applications | Research applications | Anti-cancer effects |
|---|---|---|---|---|
| Etomoxir | CPT1 | Retired from phase II clinical trial for diabetes and heart failures |
Leukemia, breast, prostate cancer cell lines in culture and xenograft model |
Inhibition of cancer cell growth, survival, and tumorigenicity [49,52,54] |
| Oxfenicine | CPT1 | No | Melanoma cell line in culture | Growth inhibition [117] |
| Aminocarnitine | CPT2 | No | Glioma cell lines in culture | Growth inhibition [118] |
| Perhexiline | CPT1/CPT2 | Medicine for angina pectoris in Australia and Asia |
Prostate cancer cell lines, primary CLL cells, and CLL transgenic mice |
Inhibition of cancer cell survival, and CLL progression in mice [40,119] |
| Trimetazidine | 3-KAT of TFP | Medicine for angina pectoris in Europe and Asia |
Glioblastoma and breast cancer cell lines in culture |
Inhibition of cancer cell growth and induction of cell death [120,121] |
| Ranolazine | 3-KAT of TFP | Medicine for angina pectoris in Europe and USA |
Leukemia, lung, and colon cancer cell lines in culture and xenograft model |
Inhibition of cancer cell growth, survival, and tumorigenicity in mice [37,52,98] |
| ST1326 | CPT1A | No | Leukemia and lymphoma cell lines in culture and Eμ-myc transgenic mice |
Growth inhibition and cytotoxicity in cell lines, and prevention of lymphomagenesis in mice [115] |
| Avocatin B | CPT1 | No | Primary AML cells and cell lines in culture | ROS-dependent cell death [9] |
Of note, relatively high doses (high micro-to millimolar) are required for these FAO inhibitors to efficiently reduce FAO. This common feature of diverse FAO inhibitors seems to be related to the limited conversion of the compounds (pro-drugs) to their active metabolites after entering cells. For example, oxfenicine (4-Hydroxy-L-phenylglycine) is intracellularly transaminated to the active 4-hydro-xyphenylglyoxylate to inhibit CPT1 [111]. Etomoxir is attached to CoA to turn to an active CPT1 inhibitor [112]. Although etomoxir is well tolerated in experimental rodents and has been a most commonly used FAO inhibitor in scientific research, its clinical development for treatment of type II diabetes was terminated in phase II clinical trials because of adverse side effects in the liver and heart [113]. One possibility for the undesired side effects is that the high doses of etomoxir target all CPT1 isoforms including CPT1B in the heart.
An aminocarnitine derivative (ST1326) has been recently found to be a CPT1A-specific inhibitor [114]. ST1326 was shown to be effective in inducing apoptosis and growth arrest in lymphoma and leukemia cells including primary leukemia cells [114,115]. It also suppressed lymphomagenesis in mice [114,115]. In addition, certain natural compounds such as Avocatin B, an odd-numbered carbon lipid isolated from avocado fruit, has been recently shown to interfere with growth and viability of AML cells through inhibiting mitochondrial FAO and NADPH production leading to overproduction of ROS and oxidative stress [9].
9. Future perspectives
While recent advances in the field have generated considerable enthusiasm, the concept of “lipolytic phenotype” of cancer remains to be fully evaluated. As requirement for FAO may vary considerably among malignancies, it will not be surprising if the pathway turns out to be more critical to certain types of cancer than others. In particular, the less glycolytic prostate cancer and those that grow from adipocyte-rich environments such as breast and ovarian carcinomas are more likely to be FAO-addicted forms of cancers [58,60]. Furthermore, FAO might be a preferred fuel choice for cancer cells undergoing metastatic progression or during development of drug resistance. FAO activity in host cells may play roles of both friend and foe to cancer as discussed earlier.
Previous studies rely heavily on pharmacological approaches that are limited in potency and specificity, especially for in vivo applications [95,116]. Those clinically used for treatment of angina pectoris summarized in Table 1 are generally considered to be only partial FAO inhibitors. Although it is of interest to test these medicines in animals or patients for treatment or prevention of cancer, more potent and CPT1-isoform specific FAO inhibitors are needed to confirm the oncogenic function of FAO. Given the complexity and plasticity of cancer cell metabolism, inhibition of FAO together with glycolysis or glutaminolysis could be a more effective strategy to treat cancer. An obstacle in the field is the lack of appropriate genetic mouse models targeting a key enzyme of FAO in specific tissues or distinct immune lineages. The availability of relevant animal models should offer critical opportunities to elucidate exact roles of FAO in tumor initiation, progression and immunity.
Acknowledgements
The work was supported in part by the DoD Breast Cancer Research Award W81XWH-17-1-0317 (XF), Astar Biotechnology Research Award (XF), and the NIH grant P30 CA16059 to Massey Cancer Center of VCU School of Medicine. We apologize to colleagues whose work is not cited due to space constraints.
Abbreviations
- FAO
fatty acid β-oxidation
- TCA cycle
the tricarboxylic acid cycle
- CPT
carnitine palmitoyltransferase
- ETC
electron transport chain
- CACT
carnitine/acylcarnitine translocase
- 3-KAT
3-ketoacyl-CoA thiolase
- TFP
the trifunctional protein (hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase/3-KAT)
- ER
endoplasmic reticulum
- PPAR
peroxisome proliferator-activated receptor
- AMPK
AMP-activated protein kinase
- ACC
acetyl-CoA carboxylase
- FDG-PET
18F-fluorodeoxyglucose with positron emission tomography
- NMR
nuclear magnetic resonance
- ROS
reactive oxidative species
- CLL
chronic lymphocytic leukemia
- AML
acute myeloid leukemia
- TNBC
triple negative breast cancer
- EMT
epithelial-to-mesenchymal transition
- PML
promyelocytic leukemia
- CSC
cancer stem cell
- Rb
retinoblastoma
- TRAF6
tumor necrosis factor receptor-associated factor 6
- Treg
regulatory CD4+ T cell
- MDSC
Myeloid-Derived Suppressor Cell
- DC
dendritic cell
- Teff
Effector CD4+ T cell
- HCC
hepatocellular carcinoma
- IDO
indoleamine 2,3-dioxgenase-1
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- [1].Pavlova NN, Thompson CB, The emerging hallmarks of cancer metabolism, Cell Metabol. 23 (2016) 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Warburg O, Posener K, Negelein E, üeber den Stoffwechsel der Tumoren, Biochem. Z 152 (1924) 319–344. [Google Scholar]
- [3].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science 324 (2009) 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jin L, Alesi GN, Kang S, Glutaminolysis as a target for cancer therapy, Oncogene 35 (2016) 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rohrig F, Schulze A, The multifaceted roles of fatty acid synthesis in cancer, Nat. Rev. Canc 16 (2016) 732–749. [DOI] [PubMed] [Google Scholar]
- [6].Warburg O, On the origin of cancer cells, Science 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [7].Cairns RA, Harris IS, Mak TW, Regulation of cancer cell metabolism, Nat. Rev. Canc 11 (2011) 85–95. [DOI] [PubMed] [Google Scholar]
- [8].Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M, Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells, Biochim. Biophys. Acta 1807 (2011) 726–734. [DOI] [PubMed] [Google Scholar]
- [9].Lee EA, Angka L, Rota SG, Hanlon T, Mitchell A, Hurren R, Wang XM,Gronda M, Boyaci E, Bojko B, Minden M, Sriskanthadevan S, Datti A,Wrana JL, Edginton A, Pawliszyn J, Joseph JW, Quadrilatero J,Schimmer AD, Spagnuolo PA, Targeting mitochondria with Avocatin B induces selective leukemia cell death, Canc. Res 75 (2015) 2478–2488. [DOI] [PubMed] [Google Scholar]
- [10].Carracedo A, Cantley LC, Pandolfi PP, Cancer metabolism: fatty acid oxidation in the limelight, Nat. Rev. Canc 13 (2013) 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr., Cellular fatty acid metabolism and cancer, Cell Metabol. 18 (2013) 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qu Q, Zeng F, Liu X, Wang QJ, Deng F, Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer, Cell Death Dis. 7 (2016) e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Menendez JA, Lupu R, Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis, Nat. Rev. Canc 7 (2007) 763–777. [DOI] [PubMed] [Google Scholar]
- [14].Wang C, Rajput S, Watabe K, Liao DF, Cao D, Acetyl-CoA carboxylase-a as a novel target for cancer therapy, Front. Biosci. (Schol. Ed.) 2 (2010) 515–526. [DOI] [PubMed] [Google Scholar]
- [15].Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I,Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D,Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD, IDH1 and IDH2 mutations in gliomas, N. Engl. J. Med 360 (2009) 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cheong H, Lu C, Lindsten T, Thompson CB, Therapeutic targets in cancer cell metabolism and autophagy, Nat. Biotechnol 30 (2012) 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N,Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF,Robey RB, Hay N, Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer, Canc. Cell 24 (2013) 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ma Y, Yu C, Mohamed EM, Shao H, Wang L, Sundaresan G, Zweit J,Idowu M, Fang X, A causal link from ALK to hexokinase II overexpression and hyperactive glycolysis in EML4-ALK-positive lung cancer, Oncogene 35 (2016) 6132–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K,Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S,Marto JA, Shipp MA, Danial NN, Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma, Canc. Cell 22 (2012) 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schwenk RW, Holloway GP, Luiken JJ, Bonen A, Glatz JF, Fatty acid transport across the cell membrane: regulation by fatty acid transporters, Prostaglandins Leukot. Essent. Fatty Acids 82 (2010) 149–154. [DOI] [PubMed] [Google Scholar]
- [21].Zammit VA, Carnitine palmitoyltransferase 1: central to cell function, IUBMB Life 60 (2008) 347–354. [DOI] [PubMed] [Google Scholar]
- [22].Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Godde L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M,De Bock K, Ghesquiere B, Lunt SY, Fendt SM, Carmeliet P, Fatty acid carbon is essential for dNTP synthesis in endothelial cells, Nature 520 (2015) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J,Garcia-Caballero M, Missiaen R, Huang H, Bruning U, Blacher S, Vinckier S,Goveia J, Knobloch M, Zhao H, Dierkes C, Shi C, Hagerling R, Moral-Darde V,Wyns S, Lippens M, Jessberger S, Fendt SM, Luttun A, Noel A, Kiefer F,Ghesquiere B, Moons L, Schoonjans L, Dewerchin M, Eelen G, Lambrechts D,Carmeliet P, The role of fatty acid beta-oxidation in lymphangiogenesis, Nature 542 (2017) 49–54. [DOI] [PubMed] [Google Scholar]
- [24].Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J, Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects, Mol. Aspect. Med 25 (2004) 495–520. [DOI] [PubMed] [Google Scholar]
- [25].Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, Kalliomaki T, Elia A, Luo X, Nadeem V,Bungard D, Yalavarthi S, Growney JD, Wakeham A, Moolani Y, Silvester J, Ten AY, Bakker W, Tsuchihara K, Berger SL, Hill RP, Jones RG, Tsao M, Robinson MO, Thompson CB, Pan G, Mak TW, Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress, Genes Dev. 25 (2011) 1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Reilly PT, Mak TW, Molecular pathways: tumor cells Co-opt the brain-specific metabolism gene CPT1C to promote survival, Clin. Canc. Res 18 (2012) 5850–5855. [DOI] [PubMed] [Google Scholar]
- [27].Sierra AY, Gratacos E, Carrasco P, Clotet J, Urena J, Serra D, Asins G,Hegardt FG, Casals N, CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity, J. Biol. Chem 283 (2008) 6878–6885. [DOI] [PubMed] [Google Scholar]
- [28].Lee J, Wolfgang MJ, Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism, BMC Biochem. 13 (2012) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Poulsen L, Siersbaek M, Mandrup S, PPARs: fatty acid sensors controlling metabolism, Semin. Cell Dev. Biol 23 (2012) 631–639. [DOI] [PubMed] [Google Scholar]
- [30].Pirat C, Farce A, Lebegue N, Renault N, Furman C, Millet R, Yous S, Speca S,Berthelot P, Desreumaux P, Chavatte P, Targeting peroxisome proliferator-activated receptors (PPARs): development of modulators, J. Med. Chem 55 (2012) 4027–4061. [DOI] [PubMed] [Google Scholar]
- [31].Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT,Evans RM, Suda T, Lee CH, Pandolfi PP, A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance, Nat. Med 18 (2012) 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mihaylova MM, Shaw RJ, The AMPK signalling pathway coordinates cell growth, autophagy and metabolism, Nat. Cell Biol 13 (2011) 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hardie DG, Pan DA, Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase, Biochem. Soc. Trans 30 (2002) 1064–1070. [DOI] [PubMed] [Google Scholar]
- [34].Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ, The subcellular localization of acetyl-CoA carboxylase 2, Proc. Natl. Acad. Sci. U.S. A 97 (2000) 1444–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McGarry JD, Leatherman GF, Foster DW, Carnitine palmitoyltransferase I The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA, J. Biol. Chem 253 (1978) 4128–4136. [PubMed] [Google Scholar]
- [36].Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O, Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation, Cell Metabol. 24 (2016) 311–323. [DOI] [PubMed] [Google Scholar]
- [37].German NJ, Yoon H, Yusuf RZ, Murphy JP, Finley LW, Laurent G, Haas W, Satterstrom FK, Guarnerio J, Zaganjor E, Santos D, Pandolfi PP, Beck AH, Gygi SP, Scadden DT, Kaelin WG Jr., M.C. Haigis, PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2, Mol. Cell 63 (2016) 1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS,Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA, Targeting metastasis-initiating cells through the fatty acid receptor CD36, Nature 541 (2017) 41–45. [DOI] [PubMed] [Google Scholar]
- [39].Shi J, Fu H, Jia Z, He K, Fu L, Wang W, High expression of CPT1A predicts adverse outcomes: a potential therapeutic target for acute myeloid leukemia, EBioMedicine 14 (2016) 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu PP, Liu J, Jiang WQ, Carew JS, Ogasawara MA, Pelicano H, Croce CM,Estrov Z, Xu RH, Keating MJ, Huang P, Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline, Oncogene 35 (2016) 5663–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V,Song J, Yuan Y, Somlo G, Jandial R, Ann D, Hanash S, Jove R, Yu H, JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance, Cell Metabol. 27 (2017) 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu Y, Hurren R, MacLean N, Gronda M, Jitkova Y, Sukhai MA, Minden MD, Schimmer AD, Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells, Apoptosis 20 (2015) 1099–1108. [DOI] [PubMed] [Google Scholar]
- [43].Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S,Mitsche M, Yang C, Batten K, Huffman KE, Liu J, Tang X, Rodriguez-Canales J, Kalhor N, Shay JW, Minna JD, McDonald J, Wistuba II RJ DeBerardinis, P.P. Scaglioni, Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis, Cell Rep. 16 (2016) 1614–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, Zhang Y, Spiegel S, Idowu MO, Fang X, Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer, Oncotarget 7 (2016) 3832–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cook KL, Soto-Pantoja DR, Clarke PA, Cruz MI, Zwart A, Warri A,Hilakivi-Clarke L, Roberts DD, Clarke R, Endoplasmic reticulum stress protein GRP78 modulates lipid metabolism to control drug sensitivity and antitumor immunity in breast cancer, Canc. Res 76 (2016) 5657–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Linher-Melville K, Zantinge S, Sanli T, Gerstein H, Tsakiridis T, Singh G, Establishing a relationship between prolactin and altered fatty acid beta-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells, BMC Canc. 11 (2011) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang MD, Wu H, Huang S, Zhang HL, Qin CJ, Zhao LH, Fu GB, Zhou X,Wang XM, Tang L, Wen W, Yang W, Tang SH, Cao D, Guo LN, Zeng M, Wu MC, Yan HX, Wang HY, HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress, Oncotarget 7 (2016) 6711–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA, Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells, Neuro Oncol. 19 (2017) 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B,Eyob H, Kajimura S, Tward A, Krings G, Nomura DK, Goga A, Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer, Nat. Med 22 (2016) 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yan X, Zhang G, Bie F, Lv Y, Ma Y, Ma M, Wang Y, Hao X, Yuan N, Jiang X, Eugenol inhibits oxidative phosphorylation and fatty acid oxidation via down-regulation of c-Myc/PGC-1beta/ERRalpha signaling pathway in MCF10A-ras cells, Sci. Rep 7 (2017) 12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V,Bhat VB, Bhowmik SK, Gupta V, Arora K, Wu D, Tsouko E, Zhang Y, Maity S, Donti TR, Graham BH, Frigo DE, Coarfa C, Yotnda P, Putluri N, Sreekumar A, Lewis MT, Creighton CJ, Wong LJ, Kaipparettu BA, Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer, Cell Rep 14 (2016) 2154–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B,Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M, Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction, J. Clin. Invest 120 (2010) 142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Merrill CL, Ni H, Yoon LW, Tirmenstein MA, Narayanan P, Benavides GR, Easton MJ, Creech DR, Hu CX, McFarland DC, Hahn LM, Thomas HC, Morgan KT, Etomoxir-induced oxidative stress in HepG2 cells detected by differential gene expression is confirmed biochemically, Toxicol. Sci 68 (2002) 93–101. [DOI] [PubMed] [Google Scholar]
- [54].Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L,Cimic A, Sirintrapun SJ, Glode LM, Eckel RH, Cramer SD, Lipid catabolism via CPT1 as a therapeutic target for prostate cancer, Mol. Canc. Therapeut 13 (2014) 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schlaepfer IR, Glode LM, Hitz CA, Pac CT, Boyle KE, Maroni P, Deep G,Agarwal R, Lucia SM, Cramer SD, Serkova NJ, Eckel RH, Inhibition of lipid oxidation increases glucose metabolism and enhances 2-deoxy-2-[(18)F]fluoro-dglucose uptake in prostate cancer mouse xenografts, Mol. Imag. Biol 17 (2015) 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tirado-Velez JM, Joumady I, Saez-Benito A, Cozar-Castellano I, Perdomo G, Inhibition of fatty acid metabolism reduces human myeloma cells proliferation, PLoS One 7 (2012) e46484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gatza ML, Silva GO, Parker JS, Fan C, Perou CM, An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer, Nat. Genet 46 (2014) 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R,Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E, Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth, Nat. Med 17 (2011) 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, Weiss HL,Mark Evers B, Gao T, Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer, Cell Death Dis. 8 (2017) e2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J,Lazar I, Alet N, Laurent V, Le Gonidec S, Biard D, Herve C, Bost F, Ren GS,Bono F, Escourrou G, Prentki M, Nieto L, Valet P, Muller C, Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells, JCI Insight 2 (2017) e87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S,Moro C, Soldan V, Dalle S, Balor S, Golzio M, Burlet-Schiltz O, Valet P,Muller C, Nieto L, Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer, Canc. Res 76 (2016) 4051–4057. [DOI] [PubMed] [Google Scholar]
- [62].Liu Y, Zuckier LS, Ghesani NV, Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach, Anticancer Res. 30 (2010) 369–374. [PubMed] [Google Scholar]
- [63].Liu Y, Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer, Prostate Cancer Prostatic Dis. 9 (2006) 230–234. [DOI] [PubMed] [Google Scholar]
- [64].Hofer C, Laubenbacher C, Block T, Breul J, Hartung R, Schwaiger M, Fluorine-18-fluorodeoxyglucose positron emission tomography is useless for the detection of local recurrence after radical prostatectomy, Eur. Urol 36 (1999) 31–35. [DOI] [PubMed] [Google Scholar]
- [65].Effert PJ, Bares R, Handt S, Wolff JM, Bull U, Jakse G, Metabolic imaging of untreated prostate cancer by positron emission tomography with 18 fluorine-labeled deoxyglucose, J. Urol 155 (1996) 994–998. [PubMed] [Google Scholar]
- [66].Schuster DM, Nanni C, Fanti S, PET tracers beyond FDG in prostate cancer, Semin. Nucl. Med 46 (2016) 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J,Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C, Mathews D,Pascual JM, Mickey BE, Malloy CR, DeBerardinis RJ, Metabolism of [U-13 C] glucose in human brain tumors in vivo, NMR Biomed 25 (2012) 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Paumen MB, Ishida Y, Muramatsu M, Yamamoto M, Honjo T, Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis, J. Biol. Chem 272 (1997) 3324–3329. [DOI] [PubMed] [Google Scholar]
- [69].Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MA, Bonelli P,Calarco A, Peluso G, tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1, Cell Death Differ. 12 (2005) 603–613. [DOI] [PubMed] [Google Scholar]
- [70].Dobbins RL, Szczepaniak LS, Bentley B, Esser V, Myhill J, McGarry JD, Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats, Diabetes 50 (2001) 123–130. [DOI] [PubMed] [Google Scholar]
- [71].Leamy AK, Egnatchik RA, Young JD, Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease, Prog. Lipid Res. 52 (2013) 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S,Puigserver P, Brugge JS, Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment, Nature 461 (2009) 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G,Elstrom RL, Thompson CB, The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation, Oncogene 24 (2005) 4165–4173. [DOI] [PubMed] [Google Scholar]
- [74].Halldorsson S, Rohatgi N, Magnusdottir M, Choudhary KS, Gudjonsson T,Knutsen E, Barkovskaya A, Hilmarsdottir B, Perander M, Maelandsmo GM,Gudmundsson S, Rolfsson O, Metabolic re-wiring of isogenic breast epithelial cell lines following epithelial to mesenchymal transition, Canc. Lett 396 (2017) 117–129. [DOI] [PubMed] [Google Scholar]
- [75].Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K, Finley LS, Egia A, Libermann T,Gerhart-Hines Z, Puigserver P, Haigis MC, Maratos-Flier E, Richardson AL, Schafer ZT, Pandolfi PP, A metabolic prosurvival role for PML in breast cancer,J. Clin. Invest 122 (2012) 3088–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, Zhao Q, Wang ZX, Li T, Lu YX, Wu QN, Yu K, Wang F, Pu HY, Li B, Jia WH, Shi M, Xie D,Kang TB, Huang P, Ju HQ, Xu RH, CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis, Oncogene (2018),10.1038/s41388-018-0384-z. [DOI] [PubMed] [Google Scholar]
- [77].Xie Z, Jones A, Deeney JT, Hur SK, Bankaitis VA, Inborn errors of long-chain fatty acid beta-oxidation link neural stem cell self-renewal to autism, Cell Rep. 14 (2016) 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B,Pei S, Balys M, Ashton JM, Klemm DJ, Woolthuis CM, Stranahan AW, Park CY, Jordan CT, Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche, Cell Stem Cell 19 (2016) 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG, Cancer drug resistance: an evolving paradigm, Nat. Rev. Canc 13 (2013) 714–726. [DOI] [PubMed] [Google Scholar]
- [80].Tung S, Shi Y, Wong K, Zhu F, Gorczynski R, Laister RC, Minden M,Blechert AK, Genzel Y, Reichl U, Spaner DE, PPARalpha and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia, Blood 122 (2013) 969–980. [DOI] [PubMed] [Google Scholar]
- [81].Hermanova I, Arruabarrena-Aristorena A, Valis K, Nuskova H, Alberich-Jorda M, Fiser K, Fernandez-Ruiz S, Kavan D, Pecinova A, Niso-Santano M,Zaliova M, Novak P, Houstek J, Mracek T, Kroemer G, Carracedo A, Trka J,Starkova J, Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells, Leukemia 30 (2016) 209–218. [DOI] [PubMed] [Google Scholar]
- [82].Barger JF, Gallo CA, Tandon P, Liu H, Sullivan A, Grimes HL, Plas DR, S6K1 determines the metabolic requirements for BCR-ABL survival, Oncogene 32 (2013) 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, Kuranaga Y,Taniguchi K, Yamada N, Otsuki Y, Naoe T, Akao Y, Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells, Canc. Lett 371 (2016) 1–11. [DOI] [PubMed] [Google Scholar]
- [84].Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M,Stuani L, Fraisse M, Scotland S, Larrue C, Boutzen H, Feliu V, Nicolau-Travers ML, Cassant-Sourdy S, Broin N, David M, Serhan N, Sarry A, Tavitian S,Kaoma T, Vallar L, Iacovoni J, Linares LK, Montersino C, Castellano R,Griessinger E, Collette Y, Duchamp O, Barreira Y, Hirsch P, Palama T, Gales L,Delhommeau F, Garmy-Susini BH, Portais JC, Vergez F, Selak M, Danet-Desnoyers G, Carroll M, Recher C, Sarry JE, Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism, Canc. Discov 7 (2017) 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kitajima S, Yoshida A, Kohno S, Li F, Suzuki S, Nagatani N, Nishimoto Y,Sasaki N, Muranaka H, Wan Y, Thai TC, Okahashi N, Matsuda F, Shimizu H,Nishiuchi T, Suzuki Y, Tominaga K, Gotoh N, Suzuki M, Ewen ME,Barbie DA, Hirose O, Tanaka T, Takahashi C, The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity, Oncogene 36 (2017) 5145–5157. [DOI] [PubMed] [Google Scholar]
- [86].Casals N, Zammit V, Herrero L, Fado R, Rodriguez-Rodriguez R, Serra D, Carnitine palmitoyltransferase 1C: from cognition to cancer, Prog. Lipid Res 61 (2016) 134–148. [DOI] [PubMed] [Google Scholar]
- [87].Li J, Zhao S, Zhou X, Zhang T, Zhao L, Miao P, Song S, Sun X, Liu J, Zhao X,Huang G, Inhibition of lipolysis by mercaptoacetate and etomoxir specifically sensitize drug-resistant lung adenocarcinoma cell to paclitaxel, PLoS One 8 (2013) e74623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, Hess S,Machida K, NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism, Cell Metabol. 23 (2016) 206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, Jones RG,Choi Y, Enhancing CD8 T-cell memory by modulating fatty acid metabolism, Nature 460 (2009) 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Gerritje JW van der Windt, B. Everts, C.-H. Chang, Jonathan D. Curtis, ToriC. Freitas, E. Amiel, Edward J. Pearce, Erika L. Pearce, Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development, Immunity 36 (2012) 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ,Mason EF, Sullivan SA, Nichols AG, Rathmell JC, Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets, J. Immunol 186 (2011) 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fridman WH, Pages F, Sautes-Fridman C, Galon J, The immune contexture in human tumours: impact on clinical outcome, Nat. Rev. Canc 12 (2012) 298–306. [DOI] [PubMed] [Google Scholar]
- [93].Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR,Wagner RA, Greaves DR, Murray PJ, Chawla A, Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation, Cell Metabol. 4 (2006) 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM,Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr., Artyomov MN, Pearce EL, Pearce EJ, Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages, Nat. Immunol 15 (2014) 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nomura M, Liu J, Rovira II E Gonzalez-Hurtado J Lee MJ Wolfgang T Finkel, Fatty acid oxidation in macrophage polarization, Nat. Immunol 17 (2016) 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zou W, Immunosuppressive networks in the tumour environment and their therapeutic relevance, Nat. Rev. Canc 5 (2005) 263–274. [DOI] [PubMed] [Google Scholar]
- [97].Grivennikov SI, Greten FR, Karin M, Immunity, inflammation, and cancer, Cell 140 (2010) 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K,Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC, Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies, Cancer Immunol Res 3 (2015) 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA,Corzo A, Cho HI, Celis E, Lennox B, Knight SC, Padhya T, McCaffrey TV, McCaffrey JC, Antonia S, Fishman M, Ferris RL, Kagan VE, Gabrilovich DI, Lipid accumulation and dendritic cell dysfunction in cancer, Nat. Med 16 (2010) 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A,Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH, ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis, Cell 161 (2015) 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, Liu J,Liu X, Boczkowski D, Nair S, Locasale JW, Hanks BA, Paracrine Wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization, Immunity 48 (2018) 147–160 e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Houten SM, Violante S, Ventura FV, Wanders RJ, The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders, Annu. Rev. Physiol 78 (2016) 23–44. [DOI] [PubMed] [Google Scholar]
- [103].Bressler R, Gay R, Copeland JG, Bahl JJ, Bedotto J, Goldman S, Chronic inhibition of fatty acid oxidation: new model of diastolic dysfunction, Life Sci. 44 (1989) 1897–1906. [DOI] [PubMed] [Google Scholar]
- [104].Kennedy JA, Kiosoglous AJ, Murphy GA, Pelle MA, Horowitz JD, Effect of perhexiline and oxfenicine on myocardial function and metabolism during low-flow ischemia/reperfusion in the isolated rat heart, J. Cardiovasc. Pharmacol 36 (2000) 794–801. [DOI] [PubMed] [Google Scholar]
- [105].Ashrafian H, Horowitz JD, Frenneaux MP, Perhexiline, Cardiovasc. Drug Rev 25 (2007) 76–97. [DOI] [PubMed] [Google Scholar]
- [106].Kantor PF, Lucien A, Kozak R, Lopaschuk GD, The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase, Circ. Res 86 (2000) 580–588. [DOI] [PubMed] [Google Scholar]
- [107].Bagger JP, Botker HE, Thomassen A, Nielsen TT, Effects of ranolazine on ischemic threshold, coronary sinus blood flow, and myocardial metabolism in coronary artery disease, Cardiovasc. Drugs Ther 11 (1997) 479–484. [DOI] [PubMed] [Google Scholar]
- [108].Fragasso G, Spoladore R, Cuko A, Palloshi A, Modulation of fatty acids oxidation in heart failure by selective pharmacological inhibition of 3-ketoacyl coenzyme-A thiolase, Curr. Clin. Pharmacol 2 (2007) 190–196. [DOI] [PubMed] [Google Scholar]
- [109].Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN,Ong P, Li Z, Chen S, Mak SY, Lim WJ, Kanayama HO, Mohan RE, Wang RR, Lai JH, Chua C, Ong HS, Tan KK, Ho YS, Tan IB, Teh BT,Shyh-Chang N, Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia, Nat. Med 22 (2016) 666–671. [DOI] [PubMed] [Google Scholar]
- [110].Petruzzelli M, Wagner EF, Mechanisms of metabolic dysfunction in cancer-associated cachexia, Genes Dev. 30 (2016) 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Keung W, Ussher JR, Jaswal JS, Raubenheimer M, Lam VH, Wagg CS,Lopaschuk GD, Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice, Diabetes 62 (2013) 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Weis BC, Cowan AT, Brown N, Foster DW, McGarry JD, Use of a selective inhibitor of liver carnitine palmitoyltransferase I (CPT I) allows quantification of its contribution to total CPT I activity in rat heart. Evidence that the dominant cardiac CPT I isoform is identical to the skeletal muscle enzyme, J. Biol. Chem 269 (1994) 26443–26448. [PubMed] [Google Scholar]
- [113].Ceccarelli SM, Chomienne O, Gubler M, Arduini A, Carnitine palmitoyltransferase (CPT) modulators: a medicinal chemistry perspective on 35 years of research, J. Med. Chem 54 (2011) 3109–3152. [DOI] [PubMed] [Google Scholar]
- [114].Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR,Foa R, Nicolai R, Peluso G, Tafuri A, Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias, Blood 126 (2015) 1925–1929. [DOI] [PubMed] [Google Scholar]
- [115].Pacilli A, Calienni M, Margarucci S, D’Apolito M, Petillo O, Rocchi L,Pasquinelli G, Nicolai R, Koverech A, Calvani M, Peluso G, Montanaro L, Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis, J. Natl. Cancer Inst 105 (2013) 489–498. [DOI] [PubMed] [Google Scholar]
- [116].Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ, Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation, PLoS Biol. 16 (2018) e2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mascagna D, Ghanem G, Morandini R, d’Ischia M, Misuraca G, Lejeune F,Prota G, Synthesis and cytotoxic properties of new N-substituted 4-aminophenol derivatives with a potential as antimelanoma agents, Melanoma Res. 2 (1992) 25–32. [DOI] [PubMed] [Google Scholar]
- [118].Berge K, Tronstad KJ, Bohov P, Madsen L, Berge RK, Impact of mitochondrial beta-oxidation in fatty acid-mediated inhibition of glioma cell proliferation, J. Lipid Res 44 (2003) 118–127. [DOI] [PubMed] [Google Scholar]
- [119].Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, Hart C,Guldvik IJ, Takhar M, Heemers HV, Erho N, Bloch K, Davicioni E, Derua R,Waelkens E, Mohler JL, Clarke N, Swinnen JV, Keun HC, Rekvig OP,Mills IG, Lipid degradation promotes prostate cancer cell survival, Oncotarget 8 (2017) 38264–38275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE,Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL, Fatty acid uptake and lipid storage induced by HIF-1 alpha contribute to cell growth and survival after hypoxia-reoxygenation, Cell Rep. 9 (2014) 349–365. [DOI] [PubMed] [Google Scholar]
- [121].Halama A, Kulinski M, Dib SS, Zaghlool SB, Siveen KS, Iskandarani A,Zierer J, Prabhu KS, Satheesh NJ, Bhagwat AM, Uddin S, Kastenmuller G,Elemento O, Gross SS, Suhre K, Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis, Canc. Lett 430 (2018) 133–147. [DOI] [PubMed] [Google Scholar]