

Abstract
Sepsis-induced immunosuppression increases the risk of chronic infection and reduces survival. Myeloid-derived suppressor cells (MDSCs) expand in the bone marrow and spleen during murine polymicrobial sepsis, contributing to immunosuppression. A better understanding of molecular controls of MDSC production is needed to identify treatment targets. We previously reported that miR-21 and miR-181b couple with transcription factor NFI-A to induce MDSCs during murine sepsis. Here, we expand upon these observations by showing that conditional deletion of the Nfia gene in the myeloid lineage precludes MDSC development. NFI-A-deficient Gr1+CD11b+ myeloid cells are not immunosuppressive and differentiate normally into macrophages and dendritic cells. In contrast, ectopically expressed NFI-A prevents differentiation of these immature Gr1+CD11b+ cells, while converting them into MDSCs. In addition, NFI-A-deficient Gr1+CD11b+ cells decreased, and cells transfected with NFI-A increase expression of miR-21 and miR181b. Our results support a myeloid cell loop in which NFI-A and miR-21 and miR-181b sustain Gr1+CD11b+ MDSC-dependent immunosuppression during sepsis.
Keywords: Sepsis, immune suppression, myeloid-derived suppressor cell, NFI-A
Introduction
Myeloid-derived suppressor cells (MDSCs) are generated during infection with systemic inflammation in animals and humans.1,2 Although phenotypically similar to the immature myeloid cells that are generated under normal conditions, MDSC cannot differentiate into mature innate immune cells.3,4 Mouse MDSCs are phenotyped by the same myeloid cell surface markers, Gr1 and CD11b, as found on normal immature myeloid cells.5 However, MDSCs repress both innate and adaptive immune responses by producing immunosuppressive mediators, such as IL-10 and TGF-β.3,6 Because infection and inflammation promote Gr1+CD11b+ MDSC expansion, they appear to be pathologically activated immature myeloid cells.7 In addition, myeloid progenitor pool expansion into Gr1+CD11b+ MDSCs comes at the expense of the effector innate immune cells. We and others have previously reported dramatic increases in the number of Gr1+CD11b+ MDSCs in the bone marrow and spleens of mice during polymicrobial sepsis.8,9 This becomes increasingly prominent during the late/chronic phase of sepsis,8 during which time immunosuppression correlates with increased mortality rates.8 Because Gr1+CD11b+ myeloid progenitors are generated as the immune system needs to repopulate lost immune cells, their expansion and re-programming into MDSCs may result from dysregulated or ‘emergency’ myelopoiesis.1,10
The transcription factor NFI-A has been implicated in regulation of myeloid cell differentiation, since it supports the undifferentiated state.11–13 Overexpression of NFI-A in human myeloid progenitor cells attenuates monocytic and granulocytic differentiation.12,14 We reported that NFI-A expression is induced during murine sepsis, and that Gr1+CD11b+ MDSCs from septic mice have high levels of NFI-A protein.15 Our previous loss- and gain-of-function studies revealed that NFI-A promotes Gr1+CD11b+ MDSC expansion during sepsis.15,16 We further showed that NFI-A knockdown in Gr1+CD11b+ MDSCs facilitates their differentiation and maturation to macrophages and dendritic cells, whereas its overexpression in Gr1+CD11b+ immature myeloid progenitors from naive mice switches them into the Gr1+CD11b+ MDSC phenotype.15 Those studies also revealed that NFI-A promotes late/chronic immunosuppression, which affects sepsis mortality in animals and humans.17–21
To further support our previous studies and probe the mechanisms by which NFI-A induces and sustains Gr1+CD11b+ MDSCs, we generated a mouse model with conditional, myeloid cell-specific deletion of the Nfia allele, where Nfia expression is inactivated only in the myeloid lineage. These mice have no gross phenotypic abnormalities and have a normal myeloid cell repertoire. Here, we show that NFI-A-deficient myeloid progenitors do not generate Gr1+CD11b+ MDSCs and differentiate normally during murine sepsis. We identify a loop between NFI-A and miR-21 and miR-181b that sustains Gr1+CD11b+ MDSC generation and limits differentiation of monocytes and dendritic cells. We further show that NFI-A decreases growth factor receptors that support normal myeloid differentiation. Findings from this study further endorse molecular targeting of Gr1+CD11b+ MDSC generation as potential treatment for prolonged sepsis immunosuppression.
Materials and methods
Mice
Generation of BALB/c Nfia conditional, myeloid cell-specific knockout mice has been described previously.22 The Nfiaflox/flox;Lyz2Cre/+ mice, where the expression of the Cre recombinase inactivates the floxed Nfia allele in the myeloid lineage cells, served as our myeloid-specific knockout. The Nfiaflox/flox;Lyz2+/+ mice, which do not express the Cre recombinase and thus the floxed Nfia allele is still expressed in the myeloid lineage cells, served as controls. The mice were bred and housed in a pathogen-free facility in the Division of Laboratory Animal Resources. Male mice, 8–10 wk old, were used in this study. All experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the East Tennessee State University Animal Care and Use Committee.
Polymicrobial sepsis
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) using a 23-G needle as described previously.23 Mice received (i.p.) 1 ml lactated Ringers solution plus 5% dextrose for fluid resuscitation. This model creates a prolonged infection with 100% mortality over 4 wk. To generate late sepsis, mice were subcutaneously administered antibiotic (imipenem; 25 mg/kg body mass) or an equivalent volume of 0.9% saline. To establish intra-abdominal infection and approximate the clinical situation of early human sepsis where there often is a delay between the onset of sepsis and the delivery of therapy,24 injections of imipenem were given at 8 and 16 h after CLP, which results in high mortality (∼70%) during the late/chronic phase, i.e., the time after d 5 of sepsis induction.23
Gr1+CD11b+ cells
Gr1+CD11b+ cells were isolated from the bone marrow by use of magnetically assisted cell sorting according to the manufacturer's protocol (Miltenyi Biotech, Auburn, CA, USA). The bone marrow cells were flushed out of the femurs with RPMI-1640 medium (without serum) under aseptic conditions.23 A single cell suspension of the bone marrow was made by pipetting up and down and filtering through a 70-µm nylon strainer, followed by incubation with erythrocyte lysis buffer. After washing, total Gr1+CD11b+ cells were purified by subjecting the single cell suspension to positive selection of the Gr1+CD11b+ cells by incubating with biotin-coupled mouse anti-Gr1 Ab (Clone RB6-8C5; eBioscience, San Diego, CA, USA) for 15 min at 4 ºC. Cells were then incubated with anti-biotin magnetic beads for 20 min at 4 ºC and subsequently passed over a MS column. Purified Gr1+CD11b+ cells were then washed and resuspended in sterile saline. The cell purity was determined by flow cytometry and was typically ∼90%.
Gr1+CD11b+ cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine (all from Hyclone Laboratories, Logan, UT, USA) and 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA) at 37℃ and 5% CO2. In some experiments, cells were stimulated for 12 h with 1 µg/ml of LPS, and culture supernatants were used for cytokine measurements by ELISA.
Gr1+CD11b+ cells differentiation
Gr1+CD11b+ cells were cultured for 6 d with complete RPMI 1640 medium in the presence of 10 ng/ml of M-CSF (PeproTech, Rocky Hill, NJ, USA) and 10 ng/ml rIL-4 (eBioscience). The cell phenotypes were analyzed by flow cytometry.
Flow cytometry
Cells were labeled by incubation for 30 min on ice in staining buffer (PBS plus 2% FBS) with the appropriate fluorochrome-conjugated Abs. After washing, the samples were analyzed by a FACSCaliber flow cytometer (BD Biosciences, Sparks, MD, USA). About 25,000 events were acquired and analyzed using the CellQuest Pro software (BD Biosciences). The following Abs were used: anti-Gr1 conjugated to FITC, anti-CD11b conjugated to phycoerythrin (PE), anti-F4/80 conjugated to allophycocyanin, anti-CD11c conjugated to PE, anti-MHC II conjugated to FITC and anti-CD4 conjugated to PE. An appropriate isotype-matched control was used for each Ab.
NFI-A expression construct
Full length mouse Nfia cDNA was cloned in a pEZ-M07 plasmid expression vector downstream of the CMV promoter, and NFI-A protein expression was verified by Western blot. An empty pEZ-M07 vector served as a negative control.
Gr1+CD11b+ cell transfection
For NFI-A transfection, plasmid DNA was suspended in HiPerFect reagent at a 0.5 µg/ml final concentration (Qiagen, Valencia, CA, USA). For Rb knockdown, pools of Rb-specific or scrambled (control) siRNAs were suspended in HiPerFect reagent at a 0.5 µM final concentration. Cells were transfected using the Gene Pulser MXCell system (Bio-Rad, Hercules, CA, USA). After 24 h, cells were differentiated for 6 d with M-CSF plus rIL-4. In some experiments, differentiated cells were stimulated for 12 h with Gram-negative bacterial LPS (Escherichia coli serotype 0111:B4; Sigma-Aldrich, St. Louis, MO, USA). Cell viability was assessed by a MTT Cell Viability Assay Kit (Biotium, Fremont, CA, USA).
CD4+ T cell proliferation assay
A co-culture of CD4+ T cells and Gr1+CD11b+ cells was used to determine the suppressive effect of Gr1+CD11b+ cells on T-cell proliferation. Briefly, spleen CD4+ T cells were isolated from normal (naive) mice by positive selection using magnetic beads (Miltenyi Biotech). Cells were fluorescently labeled with carboxy-fluorosceindiacetate, succinimidyl ester (CFSE) dye using the Vybrant CFDA SE Cell Tacer Kit (Invitrogen Molecular Probes, Eugene, OR, USA). Cells were incubated for 10 min at room temperature (20–25 ℃) with 10 µM CFSE dye and then co-cultured (at 1:1 ratio) with Gr1+CD11b+ cells, which were isolated from the bone marrow of late septic mice. T-Cell proliferation was induced by the stimulation with an anti-CD3 plus anti-CD28 Ab (1 µg/ml/each). After 3 d, cells were harvested and CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing, CD3-gated CD4+ T cells using flow cytometry.
Measurement of miRNA expression
Real-time qPCR (RT-qPCR) was used to determine levels of miR-21, miR-181b and miR-223 in control and NFI-A-deficient Gr1+CD11b+ cells. Cells were isolated from the bone marrow of sham and septic mice. miRNA-enriched RNA was isolated and measured using miScript SYBR Green PCR kit with miScript Primer Assays specific to miR-21 and miR-181b according to the manufacturer's protocol (Qiagen). The relative expression of each miRNA was calculated using the 2–ΔΔCt cycle threshold method after normalization to the endogenous U6 RNA as an internal control.
For M-CSFr and G-CSFr expression, RNA was measured using QuantiNova SYBR Green RT-PCR kit and QuantiTect Primer Assays specific to M-CSFr and G-CSFr (Qiagen). Sample data were normalized to GAPDH mRNA levels and are presented as fold change relative to RNA from sham cells.
Western blot
Equal amounts of whole cell lysate were mixed with 5 × Laemmeli sample buffer, separated by a SDS 10% polyacrylamide gel (Bio-Rad) and subsequently transferred to nitrocellulose membranes (Thermo Fisher Scientific, Waltham, MA, USA). After blocking with 5% milk in Tris-buffered saline/Tween-20 for 1 h at room temperature, membranes were probed for 16 h at 4 ºC with the appropriate primary Abs (Santa Cruz Biotechnology, Dallas, CA, USA). After washing, blots were incubated with appropriate HRP-conjugated secondary Ab (Life Technologies, Grand Island, NY, USA) for 2 h at room temperature. Proteins were detected with the enhanced chemiluminescence detection system (Thermo Fisher Scientific). The developed bands were visualized using the ChemiDoc XRS System (Bio-Rad) and the images were captured with the Image Lab Software V3.0. Membranes were stripped and re-probed with β-actin Ab (Sigma-Aldrich) as a loading control.
ELISA
Cytokine levels were determined using specific ELISA kits (eBioscience) according to the manufacturer's instructions. Each sample was run in duplicate.
Statistical analysis
Data are expressed as mean ± SD and were analyzed by Microsoft Excel, V3.0. Differences among groups were analyzed by a two-tailed Student's t-test for two groups and by ANOVA for multiple groups. P < 0.05 was considered statistically significant.
Results
NFI-A is responsible for the Gr1+CD11b+ MDSC expansion during sepsis
Targeted deletion of the Nfia allele in the myeloid lineage in mice improves sepsis survival by about 78%.25 In this conditional knockout model, NFI-A expression, which is induced following sepsis initiation,15 is disrupted only in the myeloid lineage cells, and these mice have normal innate and adaptive immune cell numbers.25 Because the immunosuppressive Gr1+CD11b+ cells generated during sepsis and the naive/normal Gr1+CD11b+ cells are phenotypically similar, we herein refer to sepsis Gr1+CD11b+ cells as ‘Gr1+CD11b+ MDSCs’. As shown in Figure 1, the Gr1+CD11b+ MDSC population did not expand in NFI-A knockout mice during sepsis. Compared with the control group, these mice still generate normal Gr1+CD11b+ cells at rates similar to sham mice. Thus, NFI-A drives Gr1+CD11b+ MDSC expansion during sepsis but does not affect generation of normal Gr1+CD11b+ cells.
Figure 1.
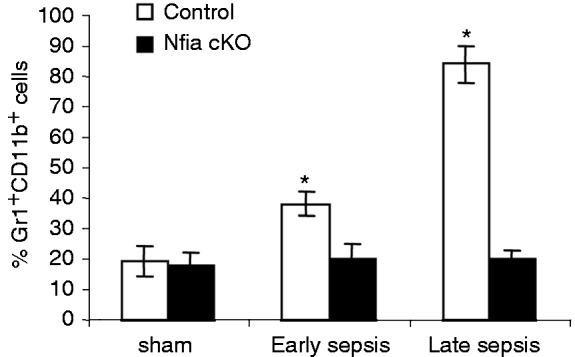
Myeloid-specific deletion of the Nfia gene attenuates Gr1+CD11b+ cell expansion during sepsis. Sepsis was induced by CLP using a 23-G needle, and mice were given antibiotics (imipenem) with fluid resuscitation. With this injury and treatment, sepsis develops into an early phase (defined as d 1–5) and a late phase (d 6 thereafter). Mice that were moribund (control group) during early or late sepsis were subjected to euthanasia. A corresponding number of surviving, healthy-appearing mice (Nfia KO group) were euthanized and analyzed at the same time and are reported here as “surviving.” Bone marrow cells were harvested, stained with anti-Gr1 and anti-CD11b Abs, and analyzed by flow cytometry. Percentages of the Gr1+CD11b+ in the bone marrow are shown. Data are expressed as mean ± SD (*P < 0.05) of five mice per group and represent one of three experiments. *Compared with conditional knockout (cKO).
NFI-A sustains expression of miR-21 and miR-181b during late sepsis
We have shown that the miR-21 and miR-181b are induced in early sepsis Gr1+CD11b+ MDSCs and are further increased during late sepsis.16 Inhibition of miR-21 and miR-181b in vivo using antagomiRs markedly reduced Gr1+CD11b+ MDSC expansion.16 Those studies also revealed that NFI-A is a downstream effector of miR-21 and miR-181b. To further investigate the effect of the Nfia myeloid-specific deletion on the miR-21 and miR-181b expression during sepsis, we measured their levels in the control and NFI-A conditional knockout mice during sepsis. Expression of miR-21 and miR-181b was induced in the Gr1+CD11b+ MDSCs in the control mice (expressing NFI-A) during early sepsis and was further increased during late sepsis (Figure 2a). Importantly, expression of miR-21 and miR-181b was diminished in the normal functioning Gr1+CD11b+ cells in NFI-A conditional knockout mice.
Figure 2.

Expression of miR-21 and miR-181b is lost in Gr1+CD11b+ cells from late, but not early, septic NFI-A knockout mice. Bone marrow Gr1+CD11b+ cells were isolated from sham and septic mice. (a) miRNA-enriched RNA was isolated, and levels of miR-21 and miR-181b were measured by RT-qPCR using miR-21 and miR-181b specific assay primer sets. Values were normalized to U6 RNA as an internal control. Data are expressed as mean ± SD (*P < 0.05) of five mice per group. Values from sepsis mice are presented relative to sham values (set at onefold) and represent one of two experiments. *Compared with sham. cKO: conditional knockout. (b) Knockout of NFI-A does not affect Stat3 phosphorylation or C/EBPβ protein levels in Gr1+CD11b+ cells during sepsis. Whole protein extracts from Gr1+CD11b+ cells were subjected to Western blotting of the indicated proteins. Lower panels show densitometry of the Stat3 and C/EBPβ protein bands. Values were normalized to β-actin and are presented relative to sham. The results represent one of two experiments.
Stat3 phosphorylation and C/EBPβ protein expression are induced in Gr1+CD11b+ MDSCs during sepsis in mice, where they bind to and activate the miR-21 and miR-181b promoters.26 We investigated whether NFI-A deficiency affects phosphorylated Stat3 and/or C/EBPβ protein levels in the normal Gr1+CD11b+ cells and thus may be responsible for the inhibition of miR-21 and miR-181b expression. Western blot analysis showed no differences in Stat3 phosphorylation or C/EBPβ protein between the control and NFI-A conditional knockout mice throughout sepsis response (Figure 2b). These results suggest that NFI-A may act in a feedback manner to sustain expression of miR-21 and miR-181b in the Gr1+CD11b+ MDSCs during sepsis.
NFI-A knockout reverses expression of the proteins that support development of Gr1+CD11b+ MDSCs
The NFI-A negatively regulates p21 gene expression in proliferating human cells.27 P21 is a cyclin-dependent kinase (cdk) inhibitor involved in cell differentiation.28 We reported previously that NFI-A and p21 proteins are reciprocally expressed in sepsis Gr1+CD11b+ MDSCs.15 We first determined p21 protein levels in the Gr1+CD11b+ cells from the control and NFI-A conditional knockout mice. As shown in Figure 3(a), p21 was expressed in sham Gr1+CD11b+ cells from the control mice, where NFI-A was not expressed, but sepsis induced NFI-A expression and diminished p21 expression. In contrast, p21 expression was maintained in the Gr1+CD11b+ cells in the conditional knockout mice during sepsis.
Figure 3.
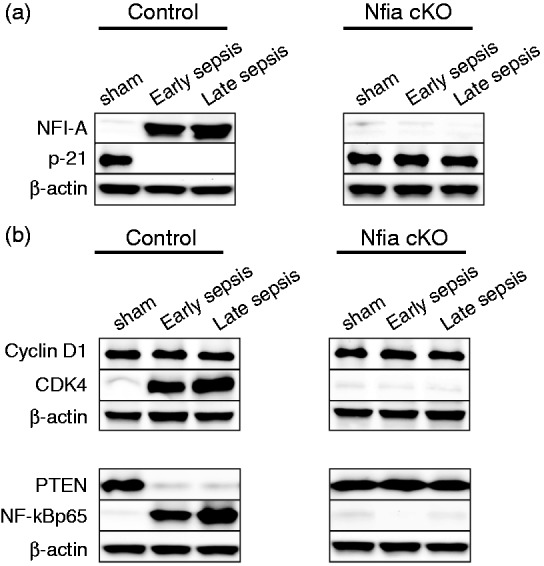
Measurements of proteins involved in Gr1+CD11b+ cell expansion in sepsis. Gr1+CD11b+ cells were isolated from the bone marrows of sham and septic mice (n = 6 mice per group). (a) p21 is expressed in sepsis Gr1+CD11b+ cells from Nfia knockout mice. Gr1+CD11b+ cell extracts were prepared, and levels of NFI-A and p21 proteins were determined by Western blot. (b) Reversal of CDK4, PTEN and NF-κBp65 protein expression patterns in sepsis Gr1+CD11b+ cells from NFI-A knockout mice. Levels of CDK4, PTEN and NF-κBp65 proteins in Gr1+CD11b+ cells were determined by Western blot. The results represent one of three experiments. cKO: conditional knockout.
The cdk4 protein is induced in sepsis Gr1+CD11b+ MDSCs owing to lack of p21 and forms a protein complex with cyclin D1, which inhibits PTEN, leading to NF-κB activation and thus expansion of the Gr1+CD11b+ MDSCs.15 We then investigated the effects of NFI-A conditional knockout on the expression of cyclin D1, cdk4, PTEN and NF-κB p65. Western blot analysis showed that levels of cdk4 protein and NF-κB activation were diminished, whereas PTEN expression was maintained in the normal Gr1+CD11b+ cells from the NFI-A conditional knockout mice (Figure 3b).
To test the inducing effect of NFI-A on NF-κB activation and its support of Gr1+CD11b+ MDSC expansion, we introduced NFI-A into late sepsis Gr1+CD11b+ cells from the NFI-A conditional knockout mice, which are functionally normal. Ectopic expression of NFI-A diminished p21 protein levels and activated NF-κB (Figure 4a). Moreover, differentiation into macrophages and dendritic cells in the presence of growth factors was reduced significantly compared with cells transfected with vector alone, and was similar to Gr1+CD11b+ MDSCs from the control mice expressing NFI-A (Figure 4b). The decrease in cell differentiation after NFI-A transfection was not due to increase in cell death, because cell survival analysis showed nearly equal numbers of cells in the control and transfected cells (Figure 4c). In addition, transfection efficiency of Gr1+CD11b+ cells with HiPerFect reagent using Gene Pulser system was about 65%, as demonstrated by GFP expression (Figure 4d). Collectively, these results support that NFI-A induction during sepsis is sufficient to trigger the molecular pathway leading to the expansion of Gr1+CD11b+ MDSCs.
Figure 4.

Ectopic expression of NFI-A in Gr1+CD11b+ from septic NFI-A knockout mice attenuates their differentiation into macrophages and dendritic cells. Gr1+CD11b+ cells were isolated from the bone marrow of late septic NFI-A conditional knockout or control mice and transfected with an NFI-A expression plasmid or an empty vector. (a) After 36 h, portions of the cells were harvested, cell extracts were prepared, and levels of NFI-A and p21 proteins were determined by Western blot. (b) The remainder of the cells was differentiated for 6 d with M-CSF plus rIL-4 (10 ng/ml/each). Flow cytometry analysis of the differentiated cells gated on F4/80+ CD11b+ or CD11c+ MHC II+ staining is shown. Data are expressed as mean ± SD (*/#P < 0.05) of five mice per group and represent one of three experiments. (c) Percentages of cell survival 36 h after transfection was determined by MTT cell viability assay. (d) Flow cytometry dot plot showing GFP transfection efficiency in Gr1+CD11b+ cells using HiPerFect reagent and Gene Pulser. Values are the mean ± SD of three transfections. *Compared with cells from NFI-A control mice; #Compared with cells isolated from NFI-A cKO mice and transfected with vector. cKO: conditional knockout.
Ectopic expression of NFI-A in sepsis Gr1+CD11b+ cells from the NFI-A knockout mice reactivates expression of miR-21 and miR-181b
Induction of miR-21 and miR-181b expression in sepsis Gr1+CD11b+ MDSCs is mediated by Rb phosphorylation by the cyclin D1–cdk4 protein complex.26 In these cells, phosphorylated Rb binds to and sequesters the C/EBPα protein, leading to Stat3 and C/EBPβ binding to and activating of the miR-21 and miR-181b promoters.26 The Gr1+CD11b+ cells from the NFI-A conditional knockout mice did not express miR-21 and miR-181b (Figure 2a). To confirm that NFI-A deficiency in Gr1+CD11b+ cells prevents miR-21 and miR-181b expression via modulating Rb phosphorylation, we expressed NFI-A in sepsis Gr1+CD11b+ cells from the NFI-A knockout mice and determined the miRNA levels. NFI-A induced cdk4 expression (Figure 5a), which is required for Rb phosphorylation as described above. Expression of cdk did not affect Rb protein levels but induced its phosphorylation. The phosphorylation of Rb resulted in reactivation of miR-21 and miR-181b expression (Figure 5b).
Figure 5.

Phosphorylated Rb protein promotes the expression of miR-21 and miR-181b in Gr1+CD11b+ cells during sepsis. Gr1+CD11b+ cells were isolated from the bone marrow of late septic NFI-A conditional knockout mice and transfected with an NFI-A expression plasmid or an empty vector for 36 h. (a) Ectopic expression of NFI-A restores RB phosphorylation and the miRNA expression. Levels of CDK4, Rb and p-Rb (Ser780) proteins were determined by Western blot. The results are representative of three experiments. (b) miRNA-enriched RNA was isolated, and levels of miR-21 and miR-181b expression were measured by RT-qPCR using miR-21 and miR-181b specific assay primer sets. Values were normalized to U6 RNA as an internal control. Data are expressed as mean ± SD (*P < 0.05) of five mice per group and represent one of two experiments. *Compared with vector. (c) Ectopic expression of NFI-A in the absence of RB does not restore the miRNA expression. Gr1+CD11b+ cells, isolated from the bone marrows of late septic conditional NFI-A knockout mice, were transfected with Rb-specific or scramble (control) siRNAs. After 12 h, cells were washed and transfected with an NFI-A expression plasmid or an empty vector for 24 h. Cell extracts were prepared from portion of the cells and used to determine Rb protein levels by Western blot. The results are representative of two Western blots. The remainder of the cells was used to isolated miRNA-enriched RNA, and levels of miR-21 and miR-181b were analyzed by real-time PCR as in (b). Data are expressed as mean ± SD (*P < 0.05) of five mice per group and represent one of three experiments. cKO: conditional knockout.
To further investigate the role of Rb phosphorylation in the induction of miR-21 and miR-181b expression, we expressed NFI-A in the sepsis, NFI-A-deficient Gr1+CD11b+ cells, in which Rb has been knocked down with siRNAs. As shown in Figure 5(c), lack of Rb diminished miR-21 and miR-181b expression despite the presence of ectopically expressed NFI-A protein. These results demonstrate that NFI-A sustains the miR-21 and miR-181 expression in Gr1+CD11b+ MDSCs during sepsis via promoting Rb phosphorylation.
NFI-A-deficient Gr1+CD11b+ cells from septic mice are not immunosuppressive
Gr1+CD11b+ MDSCs contribute to late sepsis immunosuppression by producing immunosuppressive mediators such as IL-10, and also by inhibiting T-cell activation and proliferation.8,15 We next examined the functional phenotypes of the Gr1+CD11b+ cells generated in the NFI-A conditional knockout mice during sepsis. We expressed NFI-A in late sepsis Gr1+CD11b+ cells from the NFI-A conditional knockout mice. As expected, upon stimulation with the Gram-negative bacterial LPS, the Gr1+CD11b+ MDSCs from control, septic mice produced significantly lower and higher levels, respectively, of TNF-α and IL-10 compared with cells from the NFI-A conditional knockout mice that were transfected with vector alone (Figure 6a). The introduction of NFI-A into the NFI-A-deficient cells from septic mice diminished TNF-α, while significantly increasing IL-10 production compared with the same cells that were transfected with vector alone. When co-cultured with CD4+ T cells, sepsis NFI-A-deficient Gr1+CD11b+ cells transfected with NFI-A plasmid significantly inhibited T-cell proliferation compared with cells transfected with vector alone (Figure 6b). This effect was similar to the Gr1+CD11b+ MDSCs from the control, NFI-A-expressing mice. These results show that Gr1+CD11b+ cells generated during sepsis in the absence of NFI-A are not immunosuppressive.
Figure 6.

Ectopic expression of NFI-A in Gr1+CD11b+ from septic NFI-A knockout mice switches their phenotype into immunosuppressive similar to those from control mice. Gr1+CD11b+ cells were isolated from the bone marrows of late septic NFI-A knockout mice and transfected with an NFI-A expression plasmid or an empty vector for 36 h. (a) NFI-A reconstitution inhibits TNF-α expression while inducing IL-10 expression in NFI-A conditional knockout cells. Cells (1 × 106) were cultured with 1 µg/ml LPS for 12 h. Supernatants were collected and levels of TNF-α and IL-10 were determined by ELISA. Cells from wild type, septic mice served as a control. (b) Gr1+CD11b+ NFI-A-deficient cells reconstituted with NFI-A suppress T cell proliferation. Gr1+CD11b+ cells from late septic NFI-A conditional knockout mice were transfected with an NFI-A expression plasmid. After 36 h, Gr1+CD11b+ cells were co-cultured (at 1:1 ratio) with spleen CD4+ T cells that have been isolated from normal (naive) mice and labeled with the fluorescent dye CFSE for 10 min at room temperature. The culture was incubated in the presence of anti-CD3 plus anti-CD28 Abs (1 µg/ml/each). After 3 d, CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing CD4+ T cells by flow cytometry. Percentages of cell proliferation were calculated as follow: % cell proliferation = 100 × (count from T cell + Gr1+CD11b+ cell culture/count from T cell culture). Data are expressed as mean ± SD (*/#P < 0.05) of five mice per group and represent one of three experiments. Cells from wild type septic mice serve as a control. *Compared with cells from NFI-A control mice; #compared with cells isolated from NFI-A cKO mice and transfected with vector. cKO: conditional knockout.
NFI-A diminishes expression of miR-223 and the receptors for colony-stimulating factors
To further investigate the mechanism of the Gr1+CD11b+ MDSC expansion by NFI-A during sepsis, we measured expression of miR-223, as well as the receptors for macrophage and granulocyte colony-stimulating factor (M-CSFr and G-CSFr). NFI-A targets miR-223, the expression of which is increased with myeloid cell differentiation and maturation.11 Our previous studies showed that miR-223 expression was not increased in sepsis Gr1+CD11b+ MDSCs.16 Real-time PCR analysis revealed that miR-223 was expressed at very low levels in the control, NFI-A-expressing Gr1+CD11b+ MDSCs, whereas it levels were significantly increased in NFI-A-deficient Gr1+CD11b+ cells (Figure 7a). In addition, NFI-A down-regulates expression of M-CSF receptor and G-CSF receptor on human hematopoietic progenitors,12,14 which attenuates their monocytic and granulocytic differentiation, respectively. During early sepsis, expression of M-CSFr and G-CSFr was significantly increased, with higher levels in the Gr1+CD11b+ MDSCs from the control compared with the Gr1+CD11b+ cells from the NFI-A conditional knockout mice (Figure 7b). Importantly, both M-CSFr and G-CSFr expressions diminished during late sepsis in Gr1+CD11b+ MDSCs from the control mice but remained significantly higher in the Gr1+CD11b+ cells from NFI-A knockout mice. Next, we measured miR-223, M-CSFr and G-CSFr levels after introducing NFI-A expression into late sepsis Gr1+CD11b+ cells from the NFI-A conditional knockout mice. As shown in Figure 7(c, d), ectopic expression of NFI-A significantly reduced miR-223, M-CSFr and G-CSFr expression levels (compared with late sepsis cells in Figure 7a and b). These results suggest that the induction of NFI-A expression in Gr1+CD11b+ MDSCs during sepsis is responsible for the downregulation of miR-223, M-CSFr and G-CSFr, thereby limiting normal differentiation and maturation of myeloid cells.
Figure 7.

Effects of NFI-A knockout on the expression of miR-223, M-CSFr and G-CSFr. (a) miR-223 is expressed in late sepsis Gr1+CD11b+ cells from NFI-A conditional knockout mice. Bone marrow Gr1+CD11b+ cells were isolated from sham, early and late septic mice. miRNA-enriched RNA was isolated, and levels of miR-21 and miR-181b were measured by RT-qPCR. Values were normalized to U6 RNA as an internal control. (b) Expression of M-CSFr and G-CSFr is increased in late sepsis Gr1+CD11b+ cells from NFI-A conditional knockout mice. Portion of the late sepsis Gr1+CD11b+ cells described in (a) was used for RNA extraction and determination of M-CSFr and G-CSFr mRNA levels by RT-qPCR. Values were normalized to GAPDH as an internal control. Data in (a) and (b) are expressed as mean ± SD (*/#P < 0.05) of five mice per group and are presented relative the values from sham mice (set at onefold). Data represent one of two experiments. *Compared with control mice; #compared with early sepsis mice. (c) Ectopic expression of NFI-A in late sepsis Gr1+CD11b+ cells from knockout mice abolishes miR-223 expression. Late sepsis Gr1+CD11b+ cells from NFI-A conditional knockout mice were transfected with empty vector or NFI-A expression plasmid for 36 h. RNA was isolated, and levels of miR-223 were measured as in (a). (d) Ectopic expression of NFI-A in late sepsis Gr1+CD11b+ cells from knockout mice inhibits M-CSFr and G-CSFr. Portion of the late sepsis Gr1+CD11b+ cells described in (c) was used to determine M-CSFr and G-CSFr mRNA levels by RT-qPCR as in (b). Data in (c) and (d) are expressed as mean ± SD (*P < 0.05) of five mice per group and are presented relative the values from control, sham mice as in (a) and (b) (set at onefold). Data represent one of two experiments. *Compared with vector. cKO: conditional knockout; r: receptor.
Discussion
While different molecular pathways promote Gr1+CD11b+ MDSC expansion under distinct pathological conditions,4,29 their immature phenotype supports the hypothesis that dysregulated myeloid cell differentiation and maturation is responsible for their accumulation.1 This study uses cell-type-specific genetics to show that NFI-A controls molecular pathways that generate Gr1+CD11b+ MDSCs during murine sepsis. It also identifies that NFI-A acts in a feedforward loop to sustain miR-21 and miR-181b expression and perpetuates its own expression and expansion of Gr1+CD11b+ MDSCs while disrupting normal myeloid cell differentiation. These molecular paths enlighten how chronic immunosuppression may persist during sepsis.
Accelerated or ‘emergency’ myelopoiesis coupled with attenuated differentiation and maturation of myeloid cells generates and expands Gr1+CD11b+ MDSCs during chronic inflammation and immunosuppression.1,4,30 We previously implicated NFI-A expression in supporting Gr1+CD11b+ MDSC accumulation during sepsis, especially in the late phases where immunosuppression predominates.15,16 The NFI-A myeloid cell-specific knockout used in the current study enabled us to further investigate the molecular processes of the NFI-A-mediated generation of Gr1+CD11b+ MDSCs in sepsis. These mice have normal immune cell repertoire and are immunocompetent, as demonstrated by mounting pro-inflammatory immune responses similar to naive mice upon challenge with bacterial LPS.25 Our finding that targeted deletion of Nfia in the myeloid lineage prevented generation of Gr1+CD11b+ MDSCs without affecting normal Gr1+CD11b+ cell development is particularly important. NFI-A is not involved in the steady-state myelopoiesis that generates normal Gr1+CD11b+ cells, but rather in the accelerated or “emergency” myelopoiesis that generates Gr1+CD11b+ MDSCs.
NFI-A promotes Gr1+CD11b+ MDSC expansion during sepsis by interacting with protein mediators involved in myeloid cell differentiation.15 Mechanistically, NFI-A blocks expression of the cdk inhibitor p21, which facilitates cdk4 protein complex formation and NF-κB control over differentiation.15 Here, we show that this pathway is reversed in Gr1+CD11b+ cells generated during sepsis in the NFI-A myeloid-specific knockout mice. Although Gr1+CD11b+ myeloid cells from these mice differentiate normally in the basal state, exogenous expression of NFI-A reverses the path of NF-κB control over Gr1+CD11b+ cell differentiation and maturation (Figure 4). Most strikingly, exogenous expression of NFI-A in Gr1+CD11b+ cells from mice with NFI-A conditional deletion switches them to the immunosuppressive Gr1+CD11b+ MDSC phenotype, which is disrupted in the NFI-A conditional knockout. Thus, this study clearly demonstrates that NFI-A induction during murine sepsis is necessary and sufficient for generating and expanding Gr1+CD11b+ MDSCs.
Sepsis induces expression of miR-21 and miR-181b in Gr1+CD11b+ MDSCs to support the initial increase in NFI-A expression,16 and in vivo inhibition of miR-21 and miR-181b simultaneously in septic mice using miRNA inhibitors (antagomiRs) diminishes NFI-A expression and limits Gr1+CD11b+ MDSC generation. Surprisingly, the current study supports that miR-21 and miR-181b expression is lost in the NFI-A conditional knockout mice during sepsis and restored with genetic knock-in conditions (Figures 2 and 5). C/EBPβ and Stat3 phosphorylation are induced in sepsis Gr1+CD11b+ MDSC, where they activate miR-21 and miR-181b promoters and expression levels required for initial NFI-A induction.26 Here, we found that C/EBPβ protein levels and Stat3 phosphorylation in the sepsis Gr1+CD11b+ cells from NFI-A conditional knockout mice simulate levels from the cells obtained from control mice that express miR-21 and miR-181b. The activation of miR-21 and miR-181b expression by C/EBPβ and Stat3 is regulated by phosphorylation of the Rb protein, which is induced by cdk4 downstream of NFI-A.26 In this study, Rb was not phosphorylated in Gr1+CD11b+ cells from the NFI-A conditional knockout mice during sepsis, owing to lack of cdk4, thus providing a plausible explanation for reduced miR-21 and miR-181b expression. In contrast, exogenous expression of NFI-A increased cdk4 expression and Rb phosphorylation, and reactivated miR-21 and miR-181b expression (Figure 5). A role of NFI-A coupled loop to miR-21 and miR-181b was confirmed by the simultaneous knockdown of Rb and the expression of NFI-A in NFI-A-deficient cells, which diminished the two miRNAs despite the presence of NFI-A.
Our studies showed that miR-21 and miR-181b play an anti-inflammatory/immunosuppressive role in the later phases of sepsis-induced immunosuppression.8,16 Although both miRNAs increase during the early/pro-inflammatory sepsis phase, antagomir blockage only affected the late/anti-inflammatory phenotype.15,16 These findings suggest that miR-21 and miR-181b may combine with other mediators during the late sepsis phase to exert their anti-inflammatory and immune repressor effect. Other studies have also described anti-inflammatory roles of miR-21 and miR-181b under different conditions.31,32 Barnett et al.31 reported miR-21 deficiency increases mortality in a mouse model of LPS-induced peritonitis. They also found that miR-21 attenuated production of pro-inflammatory cytokines TNF-α and IL-6 in LPS-stimulated peritoneal macrophages and reduced activation of the NF-κB p65 protein in bone marrow-derived macrophages. In contrast, miR-21 has been shown to promote inflammation in diseases associated with chronic inflammation such as colitis and type 2 diabetes, as well as in some cancer models where it promotes NF-κB activation in non-hematopoietic cells.33 This dual role of miR-21 may be influenced by the cell type affected and the inducing signal.33 Moreover, in vivo administration of miR-181b can attenuate vascular inflammation in a mouse model of endotoxemia via limiting NF-κB signaling, leukocyte influx and lung injury.32 These differences emphasize re-programming sepsis inflammation distinctly differs from pathways that may drive chronic inflammation.
We also found that M-CSFr and G-CSFr significantly increase during sepsis in the NFI-A conditional myeloid specific knockout mice (Figure 7). This shows that NFI-A induction and Gr1+CD11b+ MDSC increases are concomitant with attenuation of normal differentiation of myeloid precursors. Moreover, we show here that NFI-A attenuates Gr1+CD11b+ cell differentiation during sepsis by limiting miR-223, M-CSFr and G-CSFr expression. This is compatible with the concept that resistance mechanisms reciprocally give way to tolerance mechanisms, which may underlie profound immunosuppression associated with many septic deaths.
In summary, NFI-A access a key checkpoint to promote Gr1+CD11b+ MDSC generation and concomitantly limit growth factor dependent differentiation of normal myeloid monocytes and dendritic cells needed for competent innate and adaptive immunity. Moreover, sustained NFI-A expression continues to drive Gr1+CD11b+ MDSC repressor cells during chronic sepsis, and likely underlying continued high mortality. Targeting NFI-A may provide a novelist treatment approach for restoring competent immunity during the more chronic stage of sepsis.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a National Institutes of Health Grant R01GM103887 (to M.E.).
References
- 1.Cuenca AG, Delano MJ, Kelly-Scumpia KM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med 2011; 17: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong YY, Fuchsberger M, Xiang SD, et al. Myeloid derived suppressor cells and their role in diseases. Curr Med Chem 2013; 20: 1437–1444. [DOI] [PubMed] [Google Scholar]
- 4.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182: 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 2012; 22: 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun 2012; 80: 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delano MJ, Scumpia PO, Weinstein JS, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 2007; 204: 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda Y, Cain DW, Kuraoka M, et al. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J Immunol 2009; 182: 6477–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fazi F, Rosa A, Fatica A, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819–831. [DOI] [PubMed] [Google Scholar]
- 12.Rosa A, Ballarino M, Sorrentino A, et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci U S A 2007; 104: 19849–19854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laslo P, Pongubala JM, Lancki DW, Singh H. Gene regulatory networks directing myeloid and lymphoid cell fates within the immune system. Semin Immunol 2008; 20: 228–235. [DOI] [PubMed] [Google Scholar]
- 14.Starnes LM, Sorrentino A, Pelosi E, et al. NFI-A directs the fate of hematopoietic progenitors to the erythroid or granulocytic lineage and controls beta-globin and G-CSF receptor expression. Blood 2009; 114: 1753–1763. [DOI] [PubMed] [Google Scholar]
- 15.McClure C, Ali E, Youssef D, et al. NFI-A disrupts myeloid cell differentiation and maturation in septic mice. J Leukoc Biol 2016; 99: 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClure C, Brudecki L, Ferguson DA, et al. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun 2014; 82: 3816–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013; 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Remick DG. Pathophysiology of sepsis. Am J Pathol 2007; 170: 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCall CE, El Gazzar M, Liu T, et al. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J Leukoc Biol 2011; 90: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011; 306: 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013; 13: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPeak MB, Youssef D, Williams DA, et al. Frontline science: myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J Leukoc Biol 2017; 102: 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brudecki L, Ferguson DA, Yin D, et al. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect Immun 2012; 80: 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazuski JE, Sawyer RG, Nathens AB, et al. The Surgical Infection Society guidelines on antimicrobial therapy for intra-abdominal infections: an executive summary. Surg Infect (Larchmt ) 2002; 3: 161–173. [DOI] [PubMed] [Google Scholar]
- 25.McPeak MB, Youssef D, Williams DA, et al. Myeloid cell-specific knockout of NFI-A improves sepsis survival. Infect Immun 2017; 85. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClure C, McPeak MB, Youssef D, et al. Stat3 and C/EBPbeta synergize to induce miR-21 and miR-181b expression during sepsis. Immunol Cell Biol 2017; 95: 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouellet S, Vigneault F, Lessard M, et al. Transcriptional regulation of the cyclin-dependent kinase inhibitor 1A (p21) gene by NFI in proliferating human cells. Nucleic Acids Res 2006; 34: 6472–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000; 287: 1804–1808. [DOI] [PubMed] [Google Scholar]
- 29.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 2011; 32: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusmartsev S, Su Z, Heiser A, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res 2008; 14: 8270–8278. [DOI] [PubMed] [Google Scholar]
- 31.Barnett RE, Conklin DJ, Ryan L, et al. Anti-inflammatory effects of miR-21 in the macrophage response to peritonitis. J Leukoc Biol 2016; 99: 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun X, Icli B, Wara AK, et al. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J Clin Invest 2012; 122: 1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheedy FJ. Turning 21: induction of miR-21 as a key switch in the inflammatory response. Front Immunol 2015; 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]