

Abstract
Background
Epithelial ovarian cancer is one of the most severe public health threats in women. Since it is still challenging to screen in early stages, identification of core genes that play an essential role in epithelial ovarian cancer initiation and progression is of vital importance.
Results
Seven gene expression datasets (GSE6008, GSE18520, GSE26712, GSE27651, GSE29450, GSE36668, and GSE52037) containing 396 ovarian cancer samples and 54 healthy control samples were analyzed to identify the significant differentially expressed genes (DEGs). We identified 563 DEGs, including 245 upregulated and 318 downregulated genes. Enrichment analysis based on the gene ontology (GO) functions and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways showed that the upregulated genes were significantly enriched in cell division, cell cycle, tight junction, and oocyte meiosis, while the downregulated genes were associated with response to endogenous stimuli, complement and coagulation cascades, the cGMP-PKG signaling pathway, and serotonergic synapse. Two significant modules were identified from a protein-protein interaction network by using the Molecular Complex Detection (MCODE) software. Moreover, 12 hub genes with degree centrality more than 29 were selected from the protein-protein interaction network, and module analysis illustrated that these 12 hub genes belong to module 1. Furthermore, Kaplan-Meier analysis for overall survival indicated that 9 of these hub genes was correlated with poor prognosis of epithelial ovarian cancer patients.
Conclusion
The present study systematically validates the results of previous studies and fills the gap regarding a large-scale meta-analysis in the field of epithelial ovarian cancer. Furthermore, hub genes that could be used as a novel biomarkers to facilitate early diagnosis and therapeutic approaches are evaluated, providing compelling evidence for future genomic-based individualized treatment of epithelial ovarian cancer.
Electronic supplementary material
The online version of this article (10.1186/s13048-018-0467-z) contains supplementary material, which is available to authorized users.
Keywords: Ovarian cancer, Meta-analysis, Differentially expressed genes
Introduction
Ovarian cancer is the most lethal gynecological cancer in the world and a general term which contains some cancers derived from various tissues [1]. Epithelial ovarian cancer is the most common and representative histological types in primary ovarian cancer and is the primary cause of deaths in female cancer patients in North America and over 100,000 deaths every year worldwide [2]. High-grade serous carcinoma (HGSC) is the most lethal subtype in the epithelial ovarian cancer, and most of them are diagnosed in an advanced stage [3]. The standard treatment for ovarian cancer is maximal cytoreductive surgery and platinum-based chemotherapy [4]. Although ovarian cancer actively responds to the initial anticancer therapy, nearly 75% of patients may relapse within two years and cannot be treated with the available chemotherapy drugs [5, 6]. Meanwhile, metastasis within the peritoneal cavity and resistance to chemotherapy are the leading causes of the high mortality rate associated with ovarian cancer, because this cancer is often diagnosed in late clinical stages as what has been mentioned above [5]. Thus, identifying new targets for treatment and seeking effective chemotherapy drugs are crucial for overcoming drug resistance in advanced ovarian cancer [7].
Thus far, many genetic factors, such as BRCA1, BRCA2, P53 (TP53), KRAS, PIK3CA, CTNNB1, and PTEN, have been correlated with ovarian cancer [8]. In recent years, many studies have shown promise for gene-targeted therapies in ovarian cancer [9–11]. The poly-ADP-ribose polymerase (PARP) inhibitor olaparib, a targeted therapeutic drug approved by the Food and Drug Administration, is used to treat ovarian cancer patients with BRCA1 and BRCA2 mutations. Olaparib has also been used as maintenance therapy for patients with platinum-sensitive recurrent BRCA-mutated ovarian cancer [12]. Therefore, gene-targeted therapies provide a new possibility for the personalized treatment of ovarian cancer patients.
However, the lack of large-scale studies for the identification of differentially expressed genes (DEGs) in ovarian cancer limits the reliability of previous results and makes it difficult to screen potential targets. DNA microarray analysis is a systematic and global approach to analyze genomic expression profiles and physiological mechanisms in diseases [13, 14]. High-throughput microarray experiments have been used to analyze gene expression patterns and identify potential target genes in ovarian cancer [15].
To fill the gap regarding the identification of DEGs in ovarian cancer, we performed this meta-analysis to identify DEGs between ovarian cancer and healthy control tissues and aimed to provide a powerful tool to investigate microarray datasets by integrating data from multiple studies. An important advantage of this large-scale analysis lies in the reduction of discrepancies among different study conditions; additionally, this analysis combines the results from previous studies to assess an existing problem from a novel perspective [16]. it is worth noting that although ovarian cancer is known as a series of different molecular and histological diseases [17], we aim to uncover those common genes across different molecular subtypes of epithelial ovarian cancer. Genes discovered in meta-analyses generally overlap with genes identified in various studies, indicating increased reliability [18]. The present meta-analysis aimed to identify DEGs between ovarian tissues and control tissues. In addition, we attempted to identify potential core genes associated with epithelial ovarian cancer and to investigate some possible related mechanisms.
Materials and methods
Selection of microarray datasets for the meta-analysis
According to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines published in 2009, we performed a comprehensive search of Gene Expression Omnibus (GEO) databases from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/geo/). The keywords “ovarian neoplasms” and “ovarian cancer” were used in our search. The datasets were required to meet the following criteria: (1) the samples had to be from the Affymetrix Human Genome U133A Array platform or Affymetrix Human Genome U133 Plus 2.0 Array platform; (2) the study organisms had to be Homo sapiens; (3) the datasets had to contain ovarian cancer and normal ovarian tissue samples; and (4) the number of normal ovarian tissue samples had to be greater than three. Studies were excluded if (1) they were cell line studies; (2) they involved dual-channel arrays; (3) they did not have literature traceability; (4) they were DNA methylation studies; (5) they were miRNA-based studies; and (6) they lacked cases and controls. The data from the original studies were selected by two independent analysts. Any disagreement between the two analysts was solved by consultation with a third analyst.
Meta-analysis of multiple microarray datasets
From the GEO database, we downloaded Ovarian files (.CEL) of microarray datasets that met the inclusion criteria. A total of 13,294 common genes were obtained by integrating the genes from seven datasets using the R statistical software (http://www.r-project.org/). Then, we performed a meta-analysis of gene expression profiles of the 13,294 common genes according to combined p-values and Z scores using R statistical software. Combined p-values (pval_test) included the test-statistic and p-value. The meta-analysis of common genes was conducted by the MAMA, mataMA, affyPLM, CLL and RankProd packages. In performing two meta-analysis with R statistical software, combined p-values method (where the threshold was an absolute value more than 5) and Z-scored meta-analysis (where the threshold was an absolute value more than 7) were used as the cutoff criteria, and a list of DEGs (up- or downregulated) was identified.
GO annotations and KEGG pathway enrichment analysis
Identifying the biological characteristics of DEGs is vital. Based on the results of the meta-analysis, the most significant DEGs were evaluated by enrichment analyses. Then, gene ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted to identify the most significant DEGs using the WEB-based GEne SeT AnaLysis Toolkit (http://www.webgestalt.org/option.php) with a significance threshold of false-discovery rate (FDR) less than 0.1.
PPI network construction
The Search Tool for the Retrieval of Interacting Genes (STRING) database (http://string-db.org) displays information on protein-protein interactions (PPIs) [19]. We charted a PPI network of DEGs using STRING with confidence score more than 0.7 as the significance cutoff criterion to acquire an in-depth understanding and predict the cellular functions and biological behaviors of the identified DEGs. Further, PPI networks were visualized utilizing the Cytoscape software [19].
Selection of hub genes and modules
CentiScaPe 2.1 was used to calculate the degree, closeness, and betweenness of the PPI network. The degree of a node is the average number of edges (interactions) incident on the node [20]. According to the degree of a node, we identified the hub genes. The Molecular Complex Detection (MCODE) software was employed to select the most important clustering modules of PPI networks in Cytoscape with degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max. Depth = 100. Furthermore, KEGG pathway enrichment analysis was conducted for DEGs in every module using the WEB-based GEne SeT AnaLysis Toolkit with a significance threshold of FDR less than 0.1.
Survival analysis using hub genes
The Kaplan-Meier plotter (KM plotter, http://kmplot.com/analysis/) was used to display the relevance of the identified genes regarding patient survival using 1816 ovarian cancer samples. Gene expression data and relapse-free and overall survival (OS) information were downloaded from the GEO (Affymetrix microarrays only), the European Genome-phenome Archive (EGA) and the Cancer Genome Atlas (TCGA) databases. Hazard ratios (HRs) with 95% confidence intervals and log-rank p-value were calculated and displayed on the plot.
Results
Identification of upregulated or downregulated DEGs through the meta-analysis
According to the inclusion criteria, the following seven GEO datasets from the NCBI were obtained: GSE6008, GSE18520, GSE26712, GSE27651, GSE29450, GSE36668, and GSE52037 (see “Materials and Methods”, Fig. 1). A total of 396 ovarian cancer samples and 54 normal ovarian tissue samples were analyzed. The GEO Platform Files (GPLs) from the seven datasets were obtained using Affymetrix gene chips (Table 1).
Fig. 1.
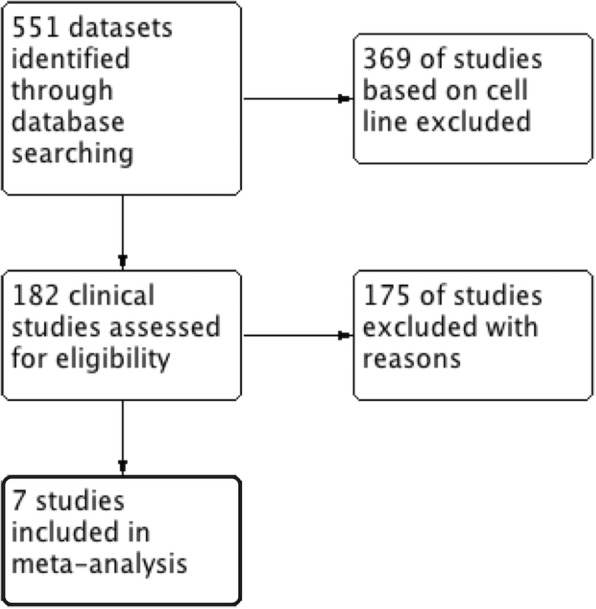
Selection procession of microarray datasets for meta-analysis
Table 1.
Characteristic of individual studies retrieved from Gene Expression Omnibus for meta-analysis
| Dataset | Samples | Case/Control | Country | PMID | Platforms | Gene# | Gene chip |
|---|---|---|---|---|---|---|---|
| GSE6008 | 103 | 99/4 | USA | 16,452,189/ 19,843,521/ 17,418,409/ 27538791 |
GPL96 | 13,909 | Affymetrix Human Genome U133A Array |
| GSE18520 | 63 | 53/10 | USA | 19,962,670 | GPL570 | 22,838 | Affymetrix Human Genome U133 Plus 2.0 Array |
| GSE26712 | 195 | 185/10 | USA | 18,593,951/25944803 | GPL96 | 13,909 | Affymetrix Human Genome U133A Array |
| GSE27651 | 41 | 35/6 | USA | 21,451,362 | GPL570 | 22,838 | Affymetrix Human Genome U133 Plus 2.0 Array |
| GSE29450 | 20 | 10/10 | USA | 21,754,983 | GPL570 | 22,838 | Affymetrix Human Genome U133 Plus 2.0 Array |
| GSE36668 | 8 | 4/4 | Norway | 23,029,477 | GPL570 | 22,838 | Affymetrix Human Genome U133 Plus 2.0 Array |
| GSE52037 | 20 | 10/10 | USA | 24,666,724 | GPL570 | 19,257 | Affymetrix Human Genome U133 Plus 2.0 Array |
We identified common genes across all datasets and performed a meta-analysis of multiple gene expression profiles using two platforms according to combined p-values and Z scores. According to the combined p-values (the threshold was 5) and Z scores (the limit was 7), 563 DEGs including 245 upregulated and 318 downregulated genes were identified (Additional file 1: Table S1 and Additional file 2: Table S2). The overlapping DEGs based on the combined p-values and Z scores are shown in Fig. 2.
Fig. 2.

The 563 overlapping DEGs based on |pval_test| < 5 and |Z| > 7 were detected using Venny 2.1.0
GO term and KEGG pathway enrichment analyses
To further investigate the functions of the DEGs, we separately classified the upregulated and downregulated genes into functional GO and KEGG categories and then performed pathway enrichment analyses with a significance threshold less than 0.05. The top five terms enriched in each category were selected according to the p-value.
For biological processes, GO analysis results showed that the upregulated DEGs were separately enriched in ‘cell division’ (GO:0051301), ‘cell-cell junction’ (GO:0005911) and ‘enzyme binding’ (GO:0019899), whereas and the downregulated DEGs were enriched in ‘response to endogenous stimulus’ (GO:0009719), ‘extracellular space’ (GO:0005615) and ‘RNA polymerase II transcription factor activity, and ‘sequence-specific DNA binding’ (GO:0000981, Table 2).
Table 2.
GO analysis of differentially expressed genes
| Category | Term | O | C | E | R | P Value |
|---|---|---|---|---|---|---|
| Up-regulated | ||||||
| GOTERM_BP_DIRECT | GO:0051301~cell division | 32 | 553 | 7.28 | 4.40 | 1.64E-12 |
| GO:0051276~chromosome organization | 33 | 597 | 7.86 | 4.20 | 2.48E-12 | |
| GO:0000819~sister chromatid segregation | 19 | 215 | 2.83 | 6.71 | 6.47E-11 | |
| GO:0007059~chromosome segregation | 23 | 329 | 4.33 | 5.31 | 7.00E-11 | |
| GO:0000278~mitotic cell cycle | 40 | 972 | 12.79 | 3.13 | 9.53E-11 | |
| GOTERM_CC_DIRECT | GO:0005911~cell-cell junction | 28 | 626 | 7.05 | 3.97 | 4.55E-10 |
| GO:0030054~cell junction | 39 | 1357 | 15.28 | 2.55 | 4.45E-08 | |
| GO:0000777~condensed chromosome kinetochore | 10 | 101 | 1.14 | 8.79 | 2.05E-07 | |
| GO:0000776~kinetochore | 11 | 130 | 1.46 | 7.52 | 2.52E-07 | |
| GO:0043296~apical junction complex | 11 | 130 | 1.46 | 7.52 | 2.52E-07 | |
| GOTERM_MF_DIRECT | GO:0019899~enzyme binding | 44 | 1756 | 22.14 | 1.99 | 6.09E-06 |
| GO:0098632~protein binding involved in cell-cell adhesion | 14 | 288 | 3.63 | 3.86 | 1.79E-05 | |
| GO:0098631~protein binding involved in cell adhesion | 14 | 293 | 3.69 | 3.79 | 2.17E-05 | |
| GO:0042802~identical protein binding | 35 | 1330 | 16.77 | 2.09 | 2.39E-05 | |
| GO:0098641~cadherin binding involved in cell-cell adhesion | 13 | 277 | 3.49 | 3.72 | 5.15E-05 | |
| Down-regulated | ||||||
| GOTERM_BP_DIRECT | GO:0009719~response to endogenous stimulus | 62 | 1536 | 25.89 | 2.40 | 5.06E-11 |
| GO:0071495~cellular response to endogenous stimulus | 53 | 1190 | 20.06 | 2.64 | 5.06E-11 | |
| GO:0009725~response to hormone | 42 | 832 | 14.02 | 3.00 | 1.65E-10 | |
| GO:0032870~cellular response to hormone stimulus | 34 | 608 | 10.25 | 3.32 | 8.17E-10 | |
| GO:1901700~response to oxygen-containing compound | 55 | 1445 | 24.35 | 2.26 | 6.68E-09 | |
| GOTERM_CC_DIRECT | GO:0005615~extracellular space | 49 | 1385 | 17.58 | 2.79 | 3.25E-11 |
| GO:0005578~proteinaceous extracellular matrix | 21 | 347 | 4.40 | 4.77 | 3.59E-09 | |
| GO:0031012~extracellular matrix | 25 | 503 | 6.39 | 3.92 | 6.13E-09 | |
| GO:0042995~cell projection | 42 | 1806 | 22.93 | 1.83 | 7.75E-05 | |
| GO:0005925~focal adhesion | 15 | 390 | 4.95 | 3.03 | 1.44E-04 | |
| GOTERM_MF_DIRECT | GO:0000981~RNA polymerase II transcription factor activity, sequence-specific DNA binding | 31 | 652 | 10.49 | 2.95 | 6.92E-08 |
| GO:0003700~transcription factor activity, sequence-specific DNA binding | 42 | 1203 | 19.36 | 2.17 | 1.53E-06 | |
| GO:0001071~nucleic acid binding transcription factor activity | 42 | 1204 | 19.38 | 2.17 | 1.56E-06 | |
| GO:0000982~transcription factor activity, RNA polymerase II core promoter proximal region sequence-specific binding | 19 | 342 | 5.50 | 3.45 | 2.96E-06 | |
| GO:0001228~transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding | 17 | 323 | 5.20 | 3.27 | 2.00E-05 | |
If there are more than five pathways in this category, the top five are selected according to the P value
O number of genes in this category in the user gene list, E expected number of genes in this category, R concentration ratio
The most enriched KEGG pathway term for the upregulated DEGs was ‘cell cycle’ (KEGG:04110) and for the downregulated DEGs was ‘complement and coagulation cascades’ (KEGG:04610, Table 3).
Table 3.
KEGG enrichment analysis of differentially expressed genes
| Term | C | O | E | R | P Value | Genes |
|---|---|---|---|---|---|---|
| Up-regulated | ||||||
| hsa04110: Cell cycle | 124 | 11 | 1.90 | 5.79 | 2.64E-06 | E2F3, SFN, MCM4, BUB1, BUB1B, TTK, YWHAZ, CCNE1, CCNB2, CDK1, CDC20 |
| hsa04530: Tight junction | 139 | 9 | 2.13 | 4.22 | 0.00026240 | CLDN4, CLDN3, CLDN7, TJP3, KRAS, LLGL2, PRKCI, CLDN10, MAGI1 |
| hsa04114: Oocyte meiosis | 124 | 8 | 1.90 | 4.21 | 0.00059620 | ITPR3, BUB1, YWHAZ, CCNE1, CCNB2, CALML4, CDK1, CDC20 |
| hsa01230: Biosynthesis of amino acids | 75 | 6 | 1.15 | 5.22 | 0.00096638 | PSAT1, IDH2, PFKP, PKM, PYCR1, TPI1 |
| hsa00051: Fructose and mannose metabolism | 33 | 4 | 0.51 | 7.91 | 0.00151933 | PFKP, SORD, TPI1, TSTA3 |
| Down-regulated | ||||||
| hsa04610: Complement and coagulation cascades | 79 | 7 | 1.42 | 4.93 | 0.00051053 | PROCR, CFH, TFPI, THBD, C1S, C4BPB, C7 |
| hsa04022: cGMP-PKG signaling pathway | 168 | 10 | 3.02 | 3.31 | 0.00084616 | AKT3, ADCY2, EDNRA, AKT2, GATA4, ITPR1, KCNJ8, MEF2C, PLN, RGS2 |
| hsa04726: Serotonergic synapse | 113 | 8 | 2.03 | 3.94 | 0.00092536 | DUSP1, GNG4, GNG11, HTR2B, ITPR1, MAOA, MAOB, TRPC1 |
| hsa04550: Signaling pathways regulating pluripotency of stem cells | 142 | 9 | 2.55 | 3.53 | 0.00098921 | AKT3, AKT2, APC, ID3, IL6ST, WNT4, TBX3, LEFTY2, KLF4 |
| hsa04270: Vascular smooth muscle contraction | 121 | 8 | 2.18 | 3.68 | 0.00144356 | CALCRL, ADCY2, EDNRA, ITPR1, PPP1R12B, PLA2G1B, PLA2G5, CALD1 |
If there are more than five pathways in this category, the top five are selected according to the P value
O number of genes in this category in the user gene list, E expected number of genes in this category, R concentration ratio
Hub gene and module screening from the PPI network
First, we determined the PPI network of the DEGs. The PPI network consisted of 275 nodes and 770 edges with a confidence score of more than 0.7 based on the STRING database. The top 12 hubs with degree centrality more than 29 were screened from the PPI network as hub genes. These hub genes included Cyclin-dependent kinase 1 (CDK1), DNA topoisomerase 2-alpha (TOP2A), cell-division cycle protein 2 (CDC20), G2/mitotic-specific cyclin-B2 (CCNB2), baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), ubiquitin-conjugating enzyme E2 C2 (UBE2C), budding uninhibited by benzimidazoles 1 (BUB1), non-SMC condensin I complex subunit G (NCAPG), Ribonucleoside-diphosphate reductase subunit M2 (RRM2), Kinesin-like protein (KIF2C), centromere protein A (CENPA), and maternal embryonic leucine zipper kinase (MELK).
Moreover, the top 2 significant modules were obtained from the PPI network of DEGs using the MCODE software (Fig. 3). Then, KEGG pathway enrichment analyses of the genes in these two modules were performed using the WEB-based GEne SeT AnaLysis Toolkit (Additional file 3: Table S3). The results demonstrated that the genes in module 1 were mainly associated with cell cycle, oocyte meiosis and the p53 signaling pathway, while the genes in module 2 were primarily in tight junction proteins, leukocyte transendo thelial migration, hepatitis C, and cell adhesion molecules (CAMs). The top 12 hub genes belonged to module 1 and confirmed the critical pathways associated with ovarian cancer. In conclusion, these essential genes provide new ideas for the treatment of ovarian cancer.
Fig. 3.
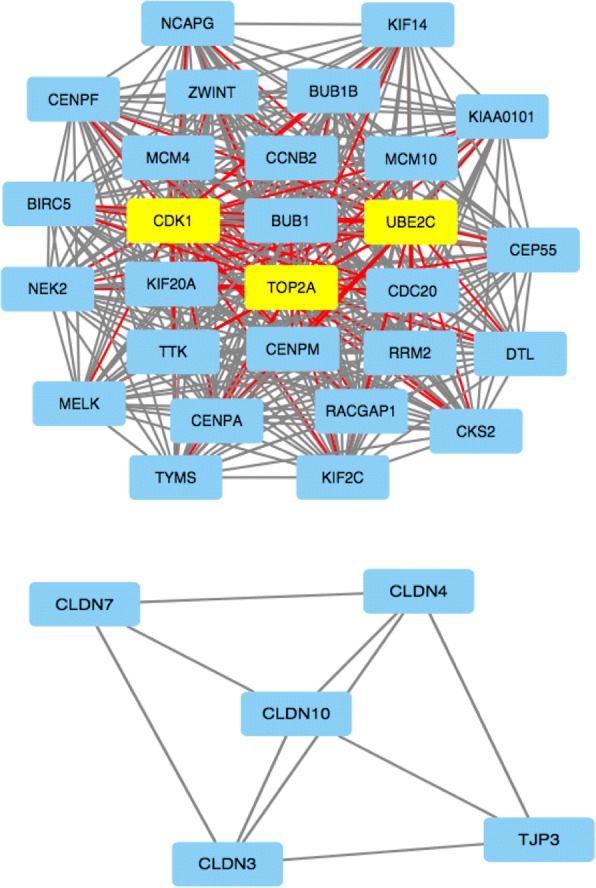
2 modules obtained from PPI network of DEGs using the MCODE software. The top 12 hub genes were all in the module 1
KM plots for hub genes
The prognostic information of the 12 hub genes is freely available in http://kmplot.com/analysis/. The results demonstrated that the expression of CDK1 (203213_at, HR 1.27 (1.11–1.46), p = 6 × 10− 4), TOP2A (201291_s_at, HR 1.27 (1.11–1.44), p = 3.9 × 10− 4), CCNB2 (202705_at, HR 1.15 (1.22–1.81), p = 0.049), UBE2C (202954_at, HR 1.28 (1.12–1.47), p = 3.8 × 10− 4), BUB1 (209642_at, HR 1.26 (1.08–1.46), p = 2.9 × 10− 3), NCAPG (218663_at, HR 1.26 (1.09–1.46), p = 1.9 × 10− 3), RRM2 (201890_at, HR 1.17 (1.03–1.34), p = 1.7 × 10− 2), KIF2C (209408_at, HR 1.15 (1.01–1.32), p = 3.8 × 10− 2) and CENPA (204962_s_at, HR 1.23 (1.08–1.41), p = 2.4 × 10− 3) was negatively associated with the OS of epithelial ovarian cancer patients (Fig. 4 and Additional file 4: Figure S1).
Fig. 4.

Top 3 genes (CDK1, TOP2A and UBE2C) significantly correlates with poor OS of ovarian cancer
Discussion
The problematic diagnosis in an early stage and recurrence and resistance to current chemotherapeutic agents are the leading causes of high mortality in ovarian cancer based on data from The Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute [21]. Therefore, the development of novel therapies for ovarian cancer is of great urgency. Previous research has proven that ovarian cancer is caused by the activation of oncogenes and the inactivation of cancer suppressor gene [22]. With continued advancements in high-throughput technologies, some genetic alterations associated with ovarian cancer, such as specific mutations in KRAS, loss-of-function mutations in PTEN, mutations in TP53, modifications in BRCA1/2, and changes in homologous recombination genes, have been uncovered [23]. Although a significant amount of data were produced by microarray studies, the sample sizes of most studies are small and may affect the identification of DEGs. However, meta-analysis of multiple microarray datasets makes the identification of DEGs more reliable by increasing the sample size.
In the present study, we performed a meta-analysis to determine the DEGs between ovarian cancer and normal ovarian tissues. We identified 563 DEGs, including 245 upregulated and 318 downregulated DEGs, in ovarian tissues by combining p-values (cutoff value of 5) and Z scores (cutoff value of 7). We classified the DEGs into functional categories based on their GO functions and KEGG pathways. Furthermore, we screened the following top 12 hub nodes with degree centrality more than 29 from the PPI network as hub genes: CDK1, TOP2A, CDC20, CCNB2, BIRC5, UBE2C, BUB1, NCAPG, RRM2, KIF2C, CENPA, and MELK. Additionally, we obtained two top significant modules from PPI networks of DEGs using MCODE analysis. Genes in module 1 were mainly associated with cell cycle, oocyte meiosis and the p53 signaling pathway, while genes in module 2 were primarily enriched in tight junction proteins, leukocyte transendothelial migration, hepatitis C, and CAMs.
Among the top 12 hub genes, nine hub genes were associated with poor OS in epithelial ovarian cancer patients. Based on GO functional analysis, KEGG pathway analysis, and survival analysis, we found that CDK1, TOP2A, and UBE2C might be the core genes contributing to the development of epithelial ovarian cancer at the molecular level.
CDK1 plays a vital role in the regulation of the cell cycle by modulating the centrosome. CDK1 not only promotes G2-M transition but also regulates G1 progression and G1-S transition by binding with multiple interphase cyclins [24, 25] In ovarian cancer, the expression of CDK1 is significantly associated with survival status, histological grade, FIGO stage, lymph node metastasis, and metastasis in epithelial ovarian cancer patients [26].
TOP2A is a nuclear enzyme involved involved in cell division and the cell cycle. TOP2A controls topological states of DNA by transiently breaking and subsequently rejoining of DNA strands [27]. Additionally, TOP2A is a direct molecular target of topoisomerase inhibitor, and its upregulation has been reported in several cancers including lung, nasopharyngeal, esophageal, gallbladder, hepatocellular, colorectal, breast, endometrial, pancreatic and ovarian cancer [28–31].
UBE2C, an essential factor of the anaphase-promoting complex/cyclosome (APC/C), is required for the destruction of mitotic cyclins and cell cycle progression [32]. The N-terminal extension of UBE2C contributes to the regulation of APC/C activity for substrate selection and checkpoint control [33]. UBE2C, the exclusive partner of APC/C, participates in the degradation of the APC/C target protein family by initiating the formation of a Lys11-linked ubiquitin chain. Thus, UBE2C plays a vital role in the destruction of mitotic cyclins and other mitosis-related substrates. During early mitosis, the APC is activated through cyclin B/Cdk1-dependent phosphorylation and binding of its activator CDC20. During metaphase, UBE2C degrades securin and cyclin B by APC/CCDC20 to promote progression to anaphase [34]. UBE2C is significantly upregulated in several types of cancer including bladder, breast, brain, cervical, esophageal, colorectal, liver, lung, nasopharyngeal, prostate (late-stage), pancreatic, thyroid, stomach, and ovarian cancer [33]. UBE2C is associated with tumor progression. I. van Ree et al. identified UBE2C as a prominent proto-oncogene that contributes to whole chromosome instability and tumor formation over a wide range of overexpression levels [35].
In our study, in addition to UBE2C upregulation, CDC20, CDK1, and CCNB2 are overexpressed. Combined with the above results, it is logic to assume that the interaction among UBE2C, CDC20, CDK1, and CCNB2 may play a vital role in the formation and development of ovarian cancer. Overexpression of UBE2C was associated with poor OS for ovarian cancer patients, and thus UBE2C might be a promising prognostic molecular biomarker and therapeutic target for ovarian cancer. It is worth to emphasize that though a series of distinct molecular and histologic subtypes of ovarian cancer exists and each subtype has different tumor microenvironment. The present research mainly focuses on those common pathways of epithelial ovarian cancer. Yet we have analyzed the corresponding gene expression data acquired from GSE9891 and the results shows that 11 out of 12 hub genes (expression data of CDK1 cannot be found in the datasets GSE9891) are significantly up-regulated (Additional file 5: Figure S2, Additional file 6: Figure S3, Additional file 7: Figure S4, Additional file 8: Figure S5, Additional file 9: Figure S6, Additional file 10: Figure S7, Additional file 11: Figure S8, Additional file 12: Figure S9, Additional file 13: Figure S10, Additional file 14: Figure S11 and Additional file 15: S12, Additional file 16: Table S4) in all of the molecular subtypes (differential, immunoreacted, proliferation and mesenchymal) comparing to control group. Subsequent researches will be conducted to investigate the role each hub gene played in each subtype of ovarian cancer,
Besides, we consider the protein expression of these hub genes might be instructive to the further study. The protein expression data of hub genes is acquired from the Human Protein Atlas for evaluation. The protein expressions of 5 hub genes (CDK1, TOP2A, CDC20, NCAPG, and MELK) are significantly up-regulated in ovarian cancer compared to normal tissues (Additional file 17: Table S5.). Also, we have done a chi-square analysis to explore the relationship between the expression of 12 hub genes and the metastasis of ovarian cancer. The results show none of these genes has connections to the cancer metastasis (P > 0.05). Overall, the present study was designed to identify DEGs through integrated bioinformatics analysis to find potential biomarkers and predict the development and prognosis of ovarian cancer. However, to obtain more accurate correlation results, we need to performed a series of validation experiments. In conclusion, this study provides robust evidence for future genomic-based individualized treatment of ovarian cancer.
Conclusion
Until now, a large-scale meta-analysis identifying DEGs was absent from the ovarian cancer literature. Our study systematically validated previous studies and filled the gap regarding a large-scale meta-analysis in the field of ovarian cancer. Moreover, our meta-analysis identified three specific genes, namely, CDK1, TOP2A and UBE2C, which may be potential targets of ovarian cancer. Thus, this study provides convincing evidence for future genomic individualized treatment of epithelial ovarian cancer.
Additional files
Table S1. Information of 245 upregulated DEGs. (XLSX 49 kb)
Table S2. Information of 318 downregulated DEGs. (XLSX 59 kb)
Table S3. KEGG pathway enrichment analyses of the genes in the two modules using the WEB-based GEne SeT AnaLysis Toolkit. (XLSX 13 kb)
Figure S1. The Kaplan-Meier plots of 9 hub gene selected from PPI network are associated with poor prognosis of ovarian cancer patients. (TIF 893 kb)
Figure S2. The difference of BIRC5 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S3. The difference of BUB1 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S4. The difference of CCNB2 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 4 kb)
Figure S5. The difference of CDC20 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S6. The difference of CENPA expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S7. The difference of KIF2C expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S8. The difference of MELK expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 4 kb)
Figure S9. The difference of NCAPG expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S10. The difference of RRM2 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S11. The difference of TOP2A expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S12. The difference of UBE2C expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Table S4. The difference of 11 hub genes expression changes in four molecular subtypes of epithelial ovarian cancer to normal tissues. (XLSX 9 kb)
Table S5. The protein expression changes of hub genes in the ovarian cancer. (XLSX 9 kb)
Acknowledgements
Thanks to Prof. Deng Libin for providing the technical help and writing assistance.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. #81760504 to F Fu) and the Natural Science Foundation of Jiangxi Province (Grant No. #20171BAB205109 to X. Tang).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional file.
Abbreviations
- APC/C
Anaphase-promoting complex/cyclosome
- BIRC5
Baculoviral inhibitor of apoptosis repeat-containing 5
- BUB1
Budding uninhibited by benzimidazoles 1
- CAMs
Cell adhesion molecules
- CCNB2
G2/mitotic-specific cyclin-B2
- CDC20
Cell-division cycle protein 2
- CDK1
Cyclin-dependent kinase 1
- CENPA
Centromere protein A
- DEGs
Differentially expressed genes
- EGA
The European Genome-phenome Archive
- FDR
False-discovery rate
- GEO
Gene Expression Omnibus
- GO
Gene ontology
- GPLs
The GEO Platform Files
- HGSC
High-grade serous carcinoma
- HRs
Hazard ratios
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KIF2C
Kinesin-like protein
- KM plotter
Kaplan-Meier plotter
- MCODE
Molecular Complex Detection
- MELK
Maternal embryonic leucine zipper kinase
- NCAPG
Non-SMC condensin I complex subunit G
- NCBI
National Center for Biotechnology Information
- OS
Overall survival
- PARP
Poly-ADP-ribose polymerase
- PPIs
Protein-protein interactions
- PRISMA
Systematic Reviews and Meta-Analyses
- RRM2
Ribonucleoside-diphosphate reductase subunit M2
- SEER
The Surveillance, Epidemiology, and End Results
- TCGA
The Cancer Genome Atlas
- TOP2A
DNA topoisomerase 2-alpha
- UBE2C
Ubiquitin-conjugating enzyme E2 C2
Authors’ contributions
FF and XT conceived of the concept. WLi, ZL and BL made substantial contributions to data collection and interpretation of results; WLou, SC, XZ, XT and LL analyzed the data; ZL and BL wrote the manuscript. WLi assisted with manuscript preparation. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xiaoli Tang, Phone: +8615960195936, Email: xltangmail@163.com.
Fen Fu, Phone: +8683827169, Email: FF.NCU@hotmail.com.
References
- 1.Desai Arpita. Epithelial ovarian cancer: An overview. World Journal of Translational Medicine. 2014;3(1):1. doi: 10.5528/wjtm.v3.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6):415. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortez AJ, Tudrej P, Kujawa KA, Lisowska KM. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol. 2018;81:17–38. doi: 10.1007/s00280-017-3501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banno Kouji, Yanokura Megumi, Iida Miho, Adachi Masataka, Nakamura Kanako, Nogami Yuya, Umene Kiyoko, Masuda Kenta, Kisu Iori, Nomura Hiroyuki, Kataoka Fumio, Tominaga Eiichiro, Aoki Daisuke. Application of MicroRNA in Diagnosis and Treatment of Ovarian Cancer. BioMed Research International. 2014;2014:1–6. doi: 10.1155/2014/232817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal R, Kaye SB. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 7.Norouzi-Barough L, Sarookhani MR, Sharifi M, Moghbelinejad S, Jangjoo S, Salehi R. Molecular mechanisms of drug resistance in ovarian cancer. J Cell Physiol. 2018;233:4546–4562. doi: 10.1002/jcp.26289. [DOI] [PubMed] [Google Scholar]
- 8.Lech A, Daneva T, Pashova S, Gagov H, Crayton R, Kukwa W, et al. Ovarian cancer as a genetic disease. Front Biosci. 2013;18:543–563. doi: 10.2741/4119. [DOI] [PubMed] [Google Scholar]
- 9.An J, Lv W, Zhang Y. LncRNA NEATI contributes to paclitaxel resistance of ovarian cancer cells by regulating ZEBI expression via miR-194. Onco Targets Ther. 2017;10:5377–5390. doi: 10.2147/OTT.S147586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother Pharmacol. 2018;81:647–658. doi: 10.1007/s00280-018-3532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetter MH, Hays JL. Use of targeted therapeutics in epithelial ovarian cancer: a review of current literature and future directions. Clin Ther. 2018;40:361–371. doi: 10.1016/j.clinthera.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Johnson N, Liao JB. Novel therapeutics for ovarian cancer: the 11th Biennial Rivkin Center Ovarian cancer Research Symposium. Int J Gynecol Cancer. 2017;27(9S):S14–S19. doi: 10.1097/IGC.0000000000001115. [DOI] [PubMed] [Google Scholar]
- 13.Spies M, Dasu MRK, Svrakic N, Nesic O, Barrow RE, Perez-Polo JR, et al. Gene expression analysis in burn wounds of rats. Am J Physiol Regul Integr Comp Physiol. 2002;283(4):R918–R930. doi: 10.1152/ajpregu.00170.2002. [DOI] [PubMed] [Google Scholar]
- 14.Guo QM. DNA microarray and cancer. Curr Opin Oncol. 2003;15:36–43. doi: 10.1097/00001622-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci. 2011;108(30):12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chee M, Yang R, Hubbell E, et al. Accessing genetic information with high-density DNA arrays. Science. 1996;274(5287):610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 17.Vaughan S, Coward JI, Bast RC, Jr, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22(22):2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
- 19.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scardoni G, Tosadori G, Faizan M, et al. Biological network analysis with CentiScaPe: centralities and experimental dataset integration. F1000Res. 2015;139(3):1-9. [DOI] [PMC free article] [PubMed]
- 21.Cronin Kathleen A., Lake Andrew J., Scott Susan, Sherman Recinda L., Noone Anne-Michelle, Howlader Nadia, Henley S. Jane, Anderson Robert N., Firth Albert U., Ma Jiemin, Kohler Betsy A., Jemal Ahmedin. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer. 2018;124(13):2785–2800. doi: 10.1002/cncr.31551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Men C, Liu QN, Ren Q. A prognostic 11 genes expression model for ovarian cancer. J Cell Biochem. 2018;119(2):1971–1978. doi: 10.1002/jcb.26358. [DOI] [PubMed] [Google Scholar]
- 23.Previs RA, Sood AK, Mills GB, Westin SN. The rise of genomic profiling in ovarian cancer. Expert Rev Mol Diagn. 2016;16:1337–1351. doi: 10.1080/14737159.2016.1259069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci. 2012;109(10):3826–3831. doi: 10.1073/pnas.1115201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xi Q, Huang M, Wang Y, Zhong J, Liu R, Xu G, et al. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumor Biol. 2015;36(7):4939–4948. doi: 10.1007/s13277-015-3141-8. [DOI] [PubMed] [Google Scholar]
- 27.Md AS, Md S, Mazharol HM, et al. Inhibition of DNA topoisomerase type IIα(TOP2A) by mitoxantrone and its halogenated derivatives: a combined density functional and molecular docking study. Biomed Res Int. 2016;2016(5):12. doi: 10.1155/2016/6817502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Z, Liu S, Zhang M, Zhou R, Liu J, Chang Y, et al. Overexpression of topoisomerase 2-alpha confers a poor prognosis i pancreatic adenocarcinoma identified by co-expression analysis. Dig Dis Sci. 2017;62(10):2790–2800. doi: 10.1007/s10620-017-4718-4. [DOI] [PubMed] [Google Scholar]
- 29.Ito F, Furukawa N, Nakai T. Evaluation of TOP2A as a predictive marker for endometrial cancer with taxane-containing adjuvant chemotherapy. Int J Gynecol Cancer. 2016;26(2):325–330. doi: 10.1097/IGC.0000000000000607. [DOI] [PubMed] [Google Scholar]
- 30.Erriquez J, Becco P, Olivero M, Ponzone R, Maggiorotto F, Ferrero A, et al. TOP2A gene copy gain predicts response of epithelial ovarian cancers to pegylated liposomal doxorubicin. Gynecol Oncol. 2015;138(3):627–633. doi: 10.1016/j.ygyno.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Chen T, Sun Y, Ji P, Kopetz S, Zhang W. Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene. 2015;34(31):4019–4031. doi: 10.1038/onc.2014.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi M, VanderLinden R, Weissmann F, Qiao R, Dube P, Brown NG, et al. Cryo-EM of mitotic checkpoint complex-bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. Mol Cell. 2016;63(4):593–607. doi: 10.1016/j.molcel.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie C, Powell C, Yao M, et al. Ubiquitin-conjugating enzyme E2C: a potential cancer biomarker. Int J Biochem Cell Biol. 2014;47:113–117. doi: 10.1016/j.biocel.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 34.Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 2006;124(1):89–103. doi: 10.1016/j.cell.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 35.Van Ree JH, Jeganathan KB, Malureanu L, Van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188(1):83–100. doi: 10.1083/jcb.200906147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information of 245 upregulated DEGs. (XLSX 49 kb)
Table S2. Information of 318 downregulated DEGs. (XLSX 59 kb)
Table S3. KEGG pathway enrichment analyses of the genes in the two modules using the WEB-based GEne SeT AnaLysis Toolkit. (XLSX 13 kb)
Figure S1. The Kaplan-Meier plots of 9 hub gene selected from PPI network are associated with poor prognosis of ovarian cancer patients. (TIF 893 kb)
Figure S2. The difference of BIRC5 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S3. The difference of BUB1 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S4. The difference of CCNB2 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 4 kb)
Figure S5. The difference of CDC20 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S6. The difference of CENPA expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S7. The difference of KIF2C expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S8. The difference of MELK expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 4 kb)
Figure S9. The difference of NCAPG expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S10. The difference of RRM2 expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S11. The difference of TOP2A expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Figure S12. The difference of UBE2C expression level in four molecular subtypes of epithelial ovarian cancer to control group. (PDF 5 kb)
Table S4. The difference of 11 hub genes expression changes in four molecular subtypes of epithelial ovarian cancer to normal tissues. (XLSX 9 kb)
Table S5. The protein expression changes of hub genes in the ovarian cancer. (XLSX 9 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional file.