

Genome-wide RNA-seq and mutant analyses reveal the importance of alternative splicing in the mineral deficiency response and in mineral uptake and remobilization in rice.
Abstract
Alternative splicing (AS) of pre-mRNAs promotes transcriptome and proteome diversity and plays important roles in a wide range of biological processes. However, the role of AS in maintaining mineral nutrient homeostasis in plants is largely unknown. To clarify this role, we obtained whole transcriptome RNA sequencing data from rice (Oryza sativa) roots grown in the presence or absence of several mineral nutrients (Fe, Zn, Cu, Mn, and P). Our systematic analysis revealed 13,291 alternatively spliced genes, representing ∼53.3% of the multiexon genes in the rice genome. As the overlap between differentially expressed genes and differentially alternatively spliced genes is small, a molecular understanding of the plant’s response to mineral deficiency is limited by analyzing differentially expressed genes alone. We found that the targets of AS are highly nutrient-specific. To verify the role of AS in mineral nutrition, we characterized mutants in genes encoding Ser/Arg (SR) proteins that function in AS. We identified several SR proteins as critical regulators of Zn, Mn, and P nutrition and showed that three SR protein-encoding genes regulate P uptake and remobilization between leaves and shoots of rice, demonstrating that AS has a key role in regulating mineral nutrient homeostasis in rice.
INTRODUCTION
The uptake and utilization of minerals are crucial for plant growth and development (Marschner, 2012). In agriculture, the availability of certain required minerals is often inadequate for optimal growth, and this can be a severe limitation for crop productivity (Yadav et al., 2000). For over 150 years, the widespread use of fertilizers has vastly increased production, supporting the growing global population. However, the application of fertilizers has resulted in environmental and ecological problems worldwide and added substantially to production cost (Sutton et al., 2011). Breeding crops with a higher nutrient use efficiency, either by conventional or genetic engineering approaches, should lead to more sustainable agriculture (Ronald, 2011).
Plants have evolved flexible and complex regulatory systems that allow them to adapt to changes in soil nutrient availability. Much progress has been made in understanding the adaptive strategies used by plants to withstand a deficiency or excess of a specific nutrient (Aibara and Miwa, 2014). For example, under P-deficient conditions, the phosphate (Pi) starvation response pathway is activated, and this pathway upregulates phosphate transporters and other downstream Pi starvation-induced genes. This increases both the uptake of Pi from the soil and the utilization of internal Pi in planta and thereby promotes survival under P-deficient conditions (Chiou and Lin, 2011; Wu et al., 2013). To mediate Fe absorption under low Fe availability, nongrass plants such as thale cress (Arabidopsis thaliana) use a reduction-based mechanism that is regulated by Fe deficiency-induced transcription factor 1, a master regulator. By contrast, the graminaceous plant, rice (Oryza sativa) uses a chelation-based mechanism involving iron deficiency-responsive element binding factor 1 to mediate Fe(III) uptake (Kobayashi and Nishizawa, 2012).
Alternative splicing (AS) is the process by which multiple mRNA variants are produced from a single gene via differential intron removal and retention of different exon combinations from pre-mRNAs (Berget et al., 1977; Chow et al., 1977). This both increases protein diversity and is an additional regulatory point of gene expression. As shown in Figure 1A, transcript variations resulting from AS can be grouped into five types: intron retention (IR), alternative 3′ splice sites, alternative 5′ splice sites, exon skipping (ES), and mutually exclusive exons (Breitbart et al., 1987).
Figure 1.

RT-qPCR Validation of AS Events Associated with Nutrient Deficiency in Rice Roots.
(A) Overview of the five different types of AS and their frequency in the RNA-seq data sets of rice grown in the presence or absence of mineral nutrients. For each of the five types, two different AS events are shown. A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exons. Black boxes, exons flanking AS events; white boxes, exons differentially included; thin lines, sequences spliced together.
(B) to (F) The effect of mineral deprivation on the frequency of AS events. In each figure part, the sashimi plot on the top left shows the number of RNA-seq reads mapping to selected DASGs. The exon-intron structure for each of the two major transcript isoforms (bottom left) shows the 5′-UTR (green), coding exons (blue), and the 3′-UTR (red). The colored bar graph (right) represents the exclusion/inclusion ratio of the IR events as determined from RNA-seq and RT-qPCR results of DAS genes. For each mineral nutrient deficiency condition, one exemplary gene is shown: P-deficiency-regulated gene, Os04g48390 (SPX-MFS1) (B); Fe-deficiency-regulated gene Os03g09970 (C); Zn-deficiency-regulated gene Os04g40080 (D); Cu-deficiency-regulated gene Os01g01830 (E); and Mn-deficiency-regulated gene Os01g58290 (F). Upper numbers in the sashimi plots indicate inclusion junction count (IJC), which represents splicing junctions that connect the upstream and downstream exon to the retained intron. Lower numbers indicate skipping junction count (SJC), which represents splicing junctions that connect the upstream exon to downstream exon. For RNA-seq data, the exclusive/inclusive ratio was calculated using the following formula: exclusion/inclusion ratio = PSI/(1 − PSI). The PSI of AS event is calculated from the IC (inclusion count) and SC (skipping count) quantification model (see Methods). Data in bar graphs are the means ± se of four biological replicates.

IR is the most common AS event in plants, whereas ES is the most prevalent AS event in animals. This difference is proposed to be due to the presence of small introns in plants and the use of an intron-defined splicing mechanism in plant genomes (Zhang et al., 2010). Next-generation sequencing transcriptomic studies have revealed an extensive number of AS events in plant cells: ∼40 to 60% of intron-containing genes are estimated to undergo at least one type of AS (Zhang et al., 2010; Marquez et al., 2012; Li et al., 2014; Shen et al., 2014b; Thatcher et al., 2014). By contrast, up to 95% of intron-containing genes in humans (Homo sapiens) have been estimated to undergo AS (Wang et al., 2008). It is likely that more AS genes and events will be identified in plants as more data (such as tissue-specific or condition-specific transcriptomic sequence data) become available. Indeed, recent studies have shown that AS occurs in response both to environmental stresses, including heat, cold, salt, drought, and ions, and to biotic stresses (Staiger and Brown, 2013; Filichkin et al., 2015). However, the functional significance of stress-induced AS in plants is poorly understood.
AS is finely regulated by a group of RNA binding proteins that recognize sequence signals in RNA and regulate splicing (Reddy et al., 2013; Staiger and Brown, 2013). Among these proteins are the Ser/Arg (SR)-rich proteins, which contain one or two characteristic RNA recognition motifs (RRMs) in the N-terminal portion and an Arg/Ser-rich domain at the C-terminal end. These proteins interact with pre-mRNA sequences and splicing factors during spliceosome assembly in order to perform essential functions in constitutive and AS (Barta et al., 2010). Moreover, SR proteins play important roles in mediating responses to environmental stresses. The Arabidopsis arginine/serine-rich domain containing protein 40 and arginine/serine-rich domain containing protein 41 mutants are more sensitive than the wild type to abscisic acid (ABA) and salt treatment (Chen et al., 2013), and a mutant of a member of the SC35-like (SCL) subfamily, scl30a, is also hypersensitive to ABA during seed germination (Cruz et al., 2014). A loss-of-function sr45 mutant is hypersensitive to ABA (Carvalho et al., 2010), while mutation of SR34b leads to increased sensitivity to Cd stress (Zhang et al., 2014). Over 4000 target RNAs are associated with the SR45 protein, and SR45 is proposed to be involved in the AS of 30% of ABA responses in Arabidopsis (Xing et al., 2015). Thus, SR proteins appear to function in plant responses to environmental stress. However, their precise role in this process is unclear.
In this study, we investigated the role of AS in the nutrient deficiency responses of the model crop species, rice. Numerous AS genes and events were identified for plants grown in nutrient-limited (–P, –Fe, –Zn, –Cu, or –Mn) conditions. To experimentally verify the role of SR proteins in the response to mineral deficiency, we generated mutants of 11 SR genes (representing half of the rice SR family). Detailed analyses of three sr mutants revealed that SR proteins have a regulatory role in Pi uptake and mobilization in rice shoots.
RESULTS
AS Events Are Frequent under Mineral Deficiency
To profile the rice transcriptome and AS in response to mineral deficiency, RNA-seq was performed on 2-week-old rice seedlings grown under control (replete) or Fe-, Zn-, Cu-, or Mn-deficient (depleted) growth conditions for 10 d. Mineral deficiency was verified by a significantly lower concentration of each element in the root and shoot (Supplemental Figure 1). A deficiency in Fe, Zn, or Mn significantly induced the uptake of the other three microelements evaluated (Supplemental Figure 1). We then focused on the macronutrient P, analyzing publicly available transcriptomic data derived from a previous Pi starvation time course study that included root and shoot samples (Secco et al., 2013).
A total of 141 RNA-seq samples were obtained in this study. After filtering out low-abundance transcripts at TPM (transcripts per million) < 1 in all samples, the remaining 107,499 novel and 34,635 known assembled transcripts were merged with an additional 8190 known expressed transcripts annotated in the RAP-DB (International Rice Annotation Project Database) and MSU (Rice Genome Annotation Project) into a unified set of transcripts. This included 37,350 genes and 150,324 transcripts in total (Supplemental Table 1). Of the 24,957 intron-containing genes, 19,023 (76.2%) had more than one intron and 21,601 (86.6%) had fewer than 10 introns (Supplemental Figure 2A). The median and mean intron sizes were 156 and 412 bp, respectively (Supplemental Figure 2B).
A total of 81,969 AS events were identified from all of the nutrient deficiency data sets using rMATS software (Supplemental Data Set 1) (Shen et al., 2014a). These AS events were distributed across 13,291 genes, which accounted for ∼53.3% of the total 24,957 intron-containing genes in the rice genome (Supplemental Figure 3). The ribo-minus RNA-sequencing method used in this study commonly results in a significantly higher IR ratio (Cui et al., 2010; Zhao et al., 2014). Not surprisingly, a high frequency of IR (83%) was identified, followed by the alternative 3′ splice site, alternative 5′ splice site, ES, and mutually exclusive exon event types (Figure 1A). To verify the accuracy of our AS event identification, we compared our results with previously defined AS events deposited in the RAPDB and MSU databases.
The total number of novel AS events was about 14 times that of known AS events (Supplemental Figure 2C and Supplemental Data Set 1). For the novel AS events defined in this study, the canonical dinucleotides GT and AG for donor and acceptor sites represented the highest proportion of all sites (98.6%), GC-AG splice pairs were the second most common splice site (1.2%), and the remaining 0.2% represented a variety of small groups, consistent with the GT-AG (96.7%), GC-AG (2.1%), and other (1.2%) patterns observed in previous studies examining AS (Supplemental Figure 2C) (Shen et al., 2014b).
To identify rice genes that had an increased frequency of AS events under nutrient deficiency, all intron-containing genes were classified into four groups: the high-frequency AS gene group (2204) with ≥11 AS events, the medium-frequency AS gene group (9064) with 2 to 10 AS events, the low-frequency AS gene group (2023) with only 1 AS event, and the no AS gene group (11,666), which lacked AS events. In our data sets, AS frequency was positively correlated with exon/intron number, exon/intron length, and transcriptional level, and negatively associated with the GC content (Supplemental Figure 2D), a pattern similar to that described previously in soybean (Glycine max) during normal development (Shen et al., 2014b).
Verification of Differential AS under Mineral Limitation
To validate the differential AS events, 11 differentially alternatively spliced genes (DASGs; criteria for genes being DASGs were that if the difference in the percentage splicing index [PSI] of AS event between two contrast conditions exceeded a stringent threshold [FDR ≤ 0.05, ΔPSI ≥10%]; see Methods) were selected and the existence of differential AS for these genes under mineral nutrient deficiency was confirmed using RT-qPCR. The RT-qPCR results showed the same trend as the RNA-seq data (Figures 1B to 1F; Supplemental Figure 4), demonstrating that our statistical method was reliable for AS event identification. SPX-MFS1 is a key regulator of phosphate homeostasis in rice (Wang et al., 2012). Compared with the control, the exclusion/inclusion ratio for the IR events of SPX-MFS1 was reduced more than 6-fold under P-deficient conditions in the root (Figure 1B). Significant differences were also observed in other genes involved in rice phosphate homeostasis, such as SPX1 and PHO2 (Supplemental Figure 4A and Supplemental Data Set 1). These results suggest that AS is directly involved in the plant’s response to mineral deficiency.
Differentially Expressed Genes and DASGs Display Little Overlap under Mineral Deficiency
To define genes that were differentially alternatively spliced in response to each type of mineral deficiency, DASGs were detected between each nutrient deficiency and control condition using rMATS software using a FDR cutoff of ≤0.05 and ΔPSI of ≥10% (Supplemental Data Sets 1 and 2) (Shen et al., 2014a). Under different nutrient limitations, 2109, 1783, 1513, and 1903 DASGs were defined from 12,927, 12,962, 12,965, and 12,895 alternatively spliced genes (ASGs; criteria for genes being ASGs were that it contains AS events supported by two sets of transcripts with different splicing patterns; see Methods) under –Fe, –Zn, –Cu, and –Mn stress conditions in roots (Figure 2; Supplemental Data Set 2). After 21 d of Pi deficiency, 766 DASGs were defined from 12,831 ASGs in the roots (Figure 2; Supplemental Data Set 2).
Figure 2.

Analysis of Overlaps in DEGs, DASGs, and ASGs under Mineral Deficiency in Rice Roots.
The Venn diagrams on the left indicate overlaps among DEGs, DASGs, and ASGs under Cu-, Zn-, Mn-, Fe-, or Pi-deficient conditions. The colored bar charts in the right panels represent the total number of DEGs, DASGs, and alternatively spliced genes (ASGs), respectively, for each of the nutrient treatments. The black bar charts on top show the number of overlapping genes between the treatments as indicated by connected black dots.
To investigate the relationship between differentially expressed genes (DEGs) and DASGs, DEGs were also defined as those with a FDR of ≤0.05 and a 2-fold change for each mineral deficiency using DESeq2 (Supplemental Data Set 3) (Love et al., 2014). The overlap between DEGs and DASGs in roots was low: 1.84% for Fe-deficient conditions, 0.05% for zinc-deficient conditions, 0% for Cu-deficient conditions, 0.81% for Mn-deficient conditions, and 2.03% for P-deficient conditions (Figure 2). Whereas only 36 and 45 genes were differentially expressed under Cu- and Zn-deficient conditions, over 1500 genes were differentially alternatively spliced (Figure 2). Likewise, for Mn limitation, the number of DASGs was twice that of DEGs (1903 versus 961). For Fe and P limitation, there were more DEGs than DASGs, with 2109 and 766 genes being differentially alternatively spliced under Fe and P limitation, respectively (Figure 2; Supplemental Data Sets 2 and 3).
We then examined the functions of genes enriched within the identified DASGs and DEGs using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (P value ≤ 0.05) (Supplemental Data Sets 4 to 7). The significantly enriched functional categories differed between the above two groups (Figure 3; Supplemental Figures 5 to 7). Under –Fe stress conditions, GO categories, such as “sulfate transport,” “nucleic acid metabolic,” “nucleobase-containing compound metabolic,” and “RNA processing” were only overrepresented in the DASGs of rice roots, while some GO categories related to oxidation reduction processes were only overrepresented in the DEGs (Figure 3A; Supplemental Data Set 6). In –P roots, three photosynthesis GO categories, “photosynthesis,” “photosynthesis, light harvesting,” and “photosynthesis, light reaction” were only overrepresented in the DEGs, whereas GO categories of “nucleotide salvage,” “NAD biosynthetic process,” and “organic cyclic compound metabolic” were only overrepresented in the DASGs (Figure 3B; Supplemental Data Set 6). Few GO categories were overrepresented in both the DEGs and DASGs; this phenomenon was also observed for Zn, Cu, and Mn deficiency stress samples (Supplemental Figures 5 to 7 and Supplemental Data Set 6). Thus, the gene targets for DEGs and DASGs were different under different nutrient deficiencies, and the latter represents an unknown but possibly important part of the molecular response to mineral deficiency.
Figure 3.

Functional GO Analysis of DAS and DE Genes.
Graphs show functional categorization (GO) of DASGs (red) and DEGs (blue) under either Fe deficiency (A) or P deficiency (B) conditions in rice roots. The log10(1/P) values represents the enrichment ratio of members of the specific GO terms are shown.
Under –Fe stress, the top GO enrichment terms for the DASGs were “inorganic anion transport” and “sulfate transport” and included several sulfate transporter genes (Figure 3A; Supplemental Data Set 6). To evaluate the potential consequence of Fe-deficiency-regulated AS on Fe homeostasis, we analyzed one sulfate transporter gene, SULFATE TRANSPORTER 3;2 (SULTR3;2 or OsSultr3;2) (Os03g06520; Zhao et al., 2016). Under +Fe conditions, less than one-fourth of the reads retained the third intron, resulting in expression of the major isoform of the functional sulfate transporter protein (Figure 4A). By contrast, under –Fe conditions, more than half of the expressed transcripts retained the third intron. This introduces a premature termination codon (PTC), either resulting in a truncated protein or leading to nonsense-mediated mRNA decay (NMD) (Figure 4A). Alternative splicing of this sulfate transporter gene would potentially affect sulfate homeostasis under –Fe. Furthermore, we found that lack of Fe induced AS using an alternative 3′ acceptor site in exon 4 of a MYB transcription factor gene, Os07g30130. This results in a shorter 3′-untranslated region (UTR) that may affect the stability or efficiency of translation of its mRNA (Figure 4B; Supplemental Data Set 6), thus regulating downstream gene expression.
Figure 4.

AS of Four Genes under Fe Deficiency or P Deficiency Conditions in Rice Roots.
Visualization of the exon-intron structure of representative transcripts for four alternatively spliced genes in roots under Fe deficiency conditions ([A] and [B]) or under P deficiency conditions ([C] and [D]). Sashimi plots show the number of RNA-seq reads mapping to loci associated with AS events in +Fe and –Fe, +P and –P samples. The heights of the bars represent overall read coverage. Structure diagrams below the sashimi plots show the 5′-UTRs (green), coding exons (blue), 3′-UTRs (red), and specific protein domains (purple, yellow) in major transcript isoforms. Upper numbers in the sashimi plots indicate the IJC and lower numbers indicate the SJC, as described in Figure 1.
Previous studies have reported that the galactolipid synthetic pathway is upregulated under P stress (Nakamura et al., 2009). The “liposaccharide metabolic” group, which includes the galactolipid synthetic pathway, is overrepresented in the DASGs (Figure 3B; Supplemental Data Set 6). Thus, we analyzed the AS isoforms and structure of the encoded protein of one MGD gene (Os02g55910). Phosphorus deficiency significantly induced the ratio of IR of the second intron and resulted in a shorter protein that either contained only a truncated partial MGDG synthase domain or led to NMD (Figure 4C). The MGD gene is included in both the DEG and DASG groups (Supplemental Data Sets 2 and 3), and under –P conditions, the functional significance of AS regulation of MGD was underestimated. The additional layer of AS regulation may better adapt the plant to mineral nutrient starvation conditions.
Moreover, nine P-signaling associated genes (PHO2, PHR2, PHR3, PHF1, PHF1L, SPX1, SPX4, SPX-MFS1, and SPX-MFS2) had differential IR events in the root and shoot (Supplemental Data Set 8) (Secco et al., 2013; Wu et al., 2013). Although previous studies have indicated that PHO2 transcript splicing was differentially regulated under –P conditions (Secco et al., 2013), a total of seven IR events with significantly increased levels of intron inclusion after 7 d of Pi deficiency were identified in both the root and shoot. The positions of the six retained introns were evenly distributed within the coding regions of the gene, and one retained intron was located in the 5′-UTR (Figure 5D; Supplemental Data Set 8). Depending on location, this may lead to removal of the region encoding the UBC domain of the protein. Long-term P deficiency may alter PHO2 pre-mRNA splicing by decreasing the rate of intron removal, leading to an increase in the accumulation of unspliced RNA.
Figure 5.

Differential AS of SR Genes under Mineral Deficiency.
(A) The 20 most significantly enriched Pfam domains in AS genes having more than two AS events are ranked by the hypergeometric test, with log10(1/P value) indicated by a line connecting the dots above each domain name. Both observed (red) and expected (blue) numbers are shown.
(B) AS of 20 two rice SR genes as related to the indicated mineral nutrient deficiency. Red boxes represent corresponding transcripts that had significantly altered AS patterns, and white boxes indicate transcripts with unaltered AS patterns.
(C) The exon-intron structure of two representative transcripts for two alternatively spliced SR genes in roots after 21 d in Pi-deficient conditions. Sashimi plots show the number of RNA-seq reads mapping to a locus associated with differentially alternatively spliced events in +Pi and –Pi samples. The heights of the bars represent the overall read coverage. The 5′-UTRs (green), coding exons (blue), 3′-UTRs (red), and protein domains (purple for the RRM domain and yellow for the zinc knuckle domain) are indicated for each of the two major transcript isoforms shown. Upper numbers in the sashimi plot indicate the IJC and lower numbers indicate the SJC, as described in Figure 1.
A hypergeometric test was performed to evaluate statistical specific protein family overrepresentation in each AS gene group compared with all rice genes annotated by Pfam domain genes. The results showed that four enriched protein families, including RRM, WD domain G-β repeat, helicase conserved C-terminal domain, and DEAD/DEAH box helicase, had the most AS events during nutrient deficiency (Figure 5A). The top-ranked protein family (RRM) contains the most widely spread motif of RNA binding proteins and is involved in biological functions such as mRNA splicing and regulation. Autoregulatory feedback mechanisms of RNA binding proteins play a major role in maintaining protein homeostasis as proteins bind to their own pre-mRNA to activate AS events, which may trigger NMD (Thomas et al., 2012; Reddy et al., 2013; Laloum et al., 2018). Thus, AS variations are common in RNA binding proteins.
Functional Significance of AS in Response to Mineral Nutrient Limitation
Because many splicing factors and RNA binding proteins regulate splicing and AS (Fu and Ares, 2014; Meyer et al., 2015), we focused on SR proteins in this study. We examined the expression and AS patterns of the 22 SR genes in the rice genome (Barta et al., 2010) under mineral nutrient limitation. Only one gene (SC34, Os08g37960) was significantly downregulated at 21 d + 6 h of Pi deficiency in the root using the 2-fold change and FDR of ≤0.05 as the cutoff. This showed that the transcription levels of SR genes were not strongly regulated by mineral deficiency, either in the root or the shoot (Supplemental Data Set 9).
In total, 15 SR genes (∼68.2%) underwent extensive AS, with one or more AS event detected (Supplemental Data Set 10). For example, 13 SR genes were significantly differentially alternatively spliced during micronutrient deficiency, and 14 SR genes during Pi deficiency (Figure 5B). Furthermore, AS of nine SR genes (∼40.9%) was common under at least three different nutrient deficiencies (Figure 5B). A comparison of two alternatively spliced transcripts of SR revealed changes in putative protein sequences after 21 d of Pi deficiency in the root (Figure 5C). The AS transcript of RS2Z36 (Os05g02880) with a fourth retained intron under –P conditions induced a PTC, resulting in a truncated protein containing the RRM domain or leading to NMD (Figure 5C), likely altering the function of this splicing factor. For another SR gene, RS33 (Os02g03040), the longer transcript (RS33.2), which includes an alternative 3′ acceptor site in exon 3, led either to the production of a PTC that resulted in a shorter protein lacking the RRM domain or to NMD (Figure 5C). These results show that although nutrient limitation might not lead to changes in the transcript abundance of some genes, changes in AS might lead to altered protein function or protein abundance by regulating transcript levels via NMD, which might regulate downstream processes.
To provide functional evidence that SR proteins are involved in regulating mineral homeostasis, 11 T-DNA insertion mutants were obtained from various rice mutant depositories (http://cbi.khu.ac.kr/RISD_DB.html; http://rmd.ncpgr.cn/) (Jeong et al., 2006; Zhang et al., 2006) (Supplemental Figure 8). Homozygous lines were identified and used to analyze mineral accumulation. Two types of analyses were undertaken: First, we examined if T-DNA inactivation of SR genes affects mineral accumulation under replete conditions; second, we evaluated whether inactivation of SR genes that are alternatively spliced under mineral limitation affects the response to that specific nutrient limitation.
For Mn and Zn limitations, 10-d-old seedlings were transferred to either a mineral-deficient solution or a control solution for 7 d, and the concentrations of Mn and Zn in the sr mutants were measured (Figures 6A and 6B). The rs29 mutant accumulated significantly less Mn in the shoot than did the wild type under control (+Mn) conditions (Figure 6A). RS33 was differentially alternatively spliced when grown in –Zn conditions. Also, a higher Zn concentration in roots, but a lower concentration in shoots, was observed in the rs33 background (Figure 6B), suggesting that RS33 functions in root-to-shoot Zn allocation under Zn-replete conditions.
Figure 6.

Nutrient Concentrations in Rice sr Mutants and Their Corresponding Wild-Type Controls.
(A) Root and shoot Mn concentrations of the rs29 mutant and wild-type plants under +Mn and –Mn conditions. (B) Root and shoot Zn concentrations of the rs33 mutant and the wild type under +Zn and –Zn conditions.
(C) Root and shoot P concentrations of eleven sr mutants and their wild types under +Pi and –Pi conditions.
Ten-day-old rice seedlings of sr mutants and wild-type rice were transferred to a control solution (half-strength Kimura B) or a –Mn, –Zn, or –P solution for 7 d. Roots and shoots were sampled and Mn, Zn, and P concentrations were determined by ICP-MS. Data are means ± sd of three or four biological replicates. Asterisks indicate significant differences (*P ≤ 0.05 and **P ≤ 0.01; Student’s t test) between the mutant and the corresponding wild type. DW, dry weight.
To investigate the accumulation of Pi in sr mutants, 10-d-old seedlings were transferred to a –P solution for 8 d, and the Pi concentrations in shoots and roots were measured. In shoots, the P concentration was higher in the sr40 (41.3% higher), scl57 (43.1% higher), and rs2z38 (11.6% higher) mutants than in the wild type under P-replete conditions (Figure 6C). Under P-limited growing conditions, a higher P concentration in at least one organ was observed in all but one (rs29) of the 11 mutants examined. The four mutants with the highest P concentration in shoots were sr40 (46.1% higher than the wild type), scl57 (43.3% higher), rs2z36 (49.5% higher), and sr33 (48.9% higher) (Figure 6C). Two mutants (sr40 [17.3% higher] and rs2z36 [25.3% higher]) had a higher P concentration in the root compared with the wild-type control (Figure 6C). Under –P conditions, rs2z36 (25.7% higher) and sr33 (16.2% higher) had a higher P concentration in the root compared with the wild type (Figure 6C). Overall, these results show that SR proteins play important roles in maintaining mineral homeostasis in rice and that P homeostasis is a target of many SR proteins.
Phosphate Uptake and Accumulation in the sr40, scl57, and scl25 Mutants
To establish the function of SR genes in P homeostasis in rice, we selected SR40, SCL57, and SCL25 for further investigation. The sr40 and scl57 mutants had a significantly higher P concentration in shoots than did the wild type, whereas scl25 exhibited only slight relative differences in P concentration (Figure 6C). SR40 were differentially expressed (less than a 2-fold change) and were alternatively spliced in the shoot under –Pi conditions. By contrast, SCL57 showed differential expression (less than a twofold change) in both the root and shoot under –Pi conditions (Supplemental Data Set 9). A null mutant of SCL25 (scl25-1) was generated by CRISPR/Cas9 (Xie and Yang, 2013). When the seedlings were grown in control conditions (91 µM Pi), all three mutants showed a significantly lower shoot height and biomass, and leaf tip necrosis (Figures 7A to 7E) when compared with the wild type. The growth of sr40, scl57, and scl25-1 plants was also reduced under excess P conditions (637 μM Pi = 7 times that of the control Pi level) (Supplemental Figures 9A to 9E). Under –P conditions, the scl25-1 mutant showed decreased root and shoot biomass compared with the wild type, while the growth of sr40 and scl57 was better than that of the wild type under Pi-depleted conditions (Figures 8A to 8E).
Figure 7.

Phenotypic Analysis of The sr40, scl57, and scl25 Mutants.
(A) Photographs of 24-d-old sr40, scl57, and scl25 rice seedlings and their corresponding wild-type plants grown on a +P solution. Bar = 5 cm.
(B) Phenotypes and quantification of P distribution in leaf 2 by micro x-ray fluorescence . Tomographs of leaf 2 showing the relative abundance of P concentration according to the color scale at the right. The dotted lines indicate that the images are different parts of the same photograph. WT1, Dongjing; WT4, Nipponbare. Bar = 1 cm.
(C) to (E) Growth of sr40, scl57, and scl25. Root length (C), shoot height (D), biomass (E), and Pi concentration (F) in roots and shoots of sr40, scl57, and scl25 rice mutants. Data are means ± sd of four biological replicates. Asterisks indicate significant differences (*P ≤ 0.05 and **P ≤ 0.01; Student’s t test) between the mutant and corresponding wild type. FW, fresh weight; DW, dry weight.
Figure 8.

Phenotypic Analysis of The sr40, scl57, and scl25 Mutants under −Pi Conditions.
(A) Photographs of 24-d-old sr40, scl57, and scl25 rice seedlings and their corresponding wild types grown on a −Pi (0 mM) solution. Bar = 5 cm.
(B) Phenotypes of leaf 2. The dotted lines indicate that the images are different parts of the same photograph. WT1, Dongjing; WT4, Nipponbare. Bar = 1 cm.
(C) to (E) Growth parameters of sr40, scl57, and scl25. Root length (C), shoot height (D), biomass (E), and Pi concentration (F) in roots and shoots of sr40, scl57, and scl25 rice mutants are shown.
Ten-day-old rice seedlings were grown in −Pi solution for 14 d. In the –Pi solution, KH2PO4 (0.09 mM) was replaced with KCl (0.09 mM). Data are means ± sd of four biological replicates. Asterisks indicate significant differences (*P ≤ 0.05 and **P ≤ 0.01; Student’s t test) between the mutant and corresponding wild type. FW, fresh weight; DW, dry weight.
We then determined the P concentrations of the mutant and wild-type rice. When grown under normal (replete) solution, the scl25-1 mutant showed a significantly higher Pi concentration compared with the wild type in the shoot, but not in the root (Figure 7F). However, under –P and excess P conditions, there was no difference in Pi concentration in the shoot between the scl25-1 and the wild type (Figure 8F; Supplemental Figure 9F). The sr40 mutant exhibited a higher Pi concentration in both the shoot and root compared with the wild type under all Pi growth conditions, i.e., deplete, replete, and excess conditions (Figures 7F and 8F; Supplemental Figure 9F). Similarly, the scl57 mutant exhibited a higher Pi concentration in both the shoot and root compared with the wild type under Pi-replete conditions, whereas it showed a significantly higher Pi concentration compared with the wild type in the shoot, but not in the root under Pi-deplete and excess conditions (Figures 7F and 8F; Supplemental Figure 9F). These results indicate that the loss of the three SR genes results in Pi accumulation in rice, and it appears that different regulatory pathways are involved.
When seedlings of sr40, scl57, and scl25-1 were grown in normal replete solution, necrotic spots, a symptom of Pi-excess toxicity, occurred on the tips of leaf 2. This was even more pronounced under excess P conditions (Figures 7A and 7B; Supplemental Figures 9A and 9B). Under –P conditions, this symptom of P excess was not observed (Figures 8A and 8B). As necrotic spots only arise under excessive accumulation of P, we next determined the P distribution in leaf 2 by micro x-ray fluorescence scanning at higher resolution and found that the sr40, scl57, and scl25-1 mutants accumulated more P, especially in the region of the necrotic spots (Figure 7B), than did the wild type. This confirmed that the necrotic spots in leaves of the mutants were caused by the accumulation of excess P.
We then performed a time-course experiment to identify the role of SR proteins in regulating P distribution in leaves. Phosphorus concentration and mobility were analyzed in sr40 and scl57. Four time points (10-, 16-, 22-, and 28-d-old seedlings) were selected, and the Pi concentration in the root and each leaf was analyzed beginning on day 10. We found that the Pi concentration in older leaves (leaf 2 to 6) of sr40 and scl57 was significantly higher than in those of the wild type. However, no significant difference was observed in the youngest leaf (leaf 7) (Figures 9A and 9B). In addition, there was a gradual increase in Pi concentration from the youngest (leaf 7) to the oldest (leaf 2) leaf (Figures 9A and 9B). The highest Pi concentration was found in leaf 2 of sr40 and scl57, which had a 3.5- and 3.0-fold difference compared with the wild type, respectively, when the seedlings were 28 d old (Figures 9A and 9B).
Figure 9.

Distribution and Accumulation of Pi in Rice sr40 and scl57 Mutants over a 28-d Period.
(A) and (B) Pi distribution in the indicated leaves of the sr40 (A) and scl57 (B) mutant at 28 d.
(C) to (F) Time-course of Pi accumulation in the roots and leaf 2 of sr40, scl57, and wild-type rice plants.
Rice seedlings were grown in half-strength Kimura B nutrient solution, and roots and the indicated leaves were sampled at 10, 16, 22, and 28 d for Pi concentration measurements. Data are means ± sd of three biological replicates. Asterisks indicate significant differences (*P ≤ 0.05 and **P ≤ 0.01; Student’s t test) between the mutant and wild type. FW, fresh weight.
The Pi concentration in roots increased slightly in the wild type over time, with roots of sr40 almost doubling in Pi concentration over 18 d. The Pi concentration of sr40 was 504.9 μg/g at day 28 compared with 311.6 μg/g for the wild type. The scl57 mutant showed a similar trend, with a Pi concentration at day 28 of 380.8 μg/g compared with 288.1 μg/g in the wild type (Figures 9C and 9D). In wild-type rice, the Pi concentration of leaf 2 constantly declined, likely due to remobilization to younger leaves, but it increased slightly in sr40 and scl57 (Figures 9E and 9F). These results suggest that SR40 and SCL57 participate in regulating P uptake as concentrations increase, and also in remobilization, as no decrease was observed in older leaves of the mutants compared with the wild type.
DISCUSSION
AS Is Common and Is Regulated by Specific Nutrient Deficiencies in Rice
Plants have evolved tightly regulated systems to regulate growth and development in response to variable mineral nutrient availability in the environment. The availability of mineral nutrients is crucial for crop plants because the nutrient-use efficiency directly affects the yield and quality of crops (Yadav et al., 2000). Many previous studies have focused on how transcript levels are adjusted under nutrient-limited conditions, with the aim of identifying the signal transduction pathways and ultimately the transcription factors that control these changes. Our study reveals a previously uncharacterized mode of regulation under nutrient-limited conditions. Traditionally, the biological significance of AS in higher plants has been underestimated (Reddy et al., 2013; Staiger and Brown, 2013). With the advent of high-throughput next-generation sequencing, large amounts of transcriptomic data have allowed the identification of more AS events in plants. Recent studies have shown that AS is involved in ∼60% of intron-containing genes in Arabidopsis and soybean, and ∼40% in maize (Zea mays) and cotton (Gossypium hirsutum) (Marquez et al., 2012; Li et al., 2014; Shen et al., 2014b; Thatcher et al., 2014). This study reveals large numbers of previously unidentified AS events, increasing the proportion of alternatively spliced genes in rice from 42% as revealed by Zhang et al. (2010) to 53% of all expressed multiexon genes, a percentage comparable to that of other model plants (Marquez et al., 2012; Li et al., 2014; Shen et al., 2014b; Thatcher et al., 2014).
Our analyses of the overlap in gene sets demonstrated that most of the ASGs among all the nutrient treatments were shared. Pfam enrichment analysis showed that the common ASGs were most highly enriched in RRM domain-containing proteins, which are usually contained in splicing factors and directly involved in alternative pre-mRNA splicing. This is consistent with previous studies showing that AS has a regulatory function and that splicing factors regulate AS (Shen et al., 2014b; Meyer et al., 2015; Xing et al., 2015).
By contrast, the DASGs are largely nutrient-specific. Analysis of the list of genes with DASGs under P deficiency reveals that several P-related genes that play roles in transcriptional and posttranslational regulation are alternatively spliced during P starvation, including PHR2, PHF1, and PHO2, and genes encoding the SPX domain-containing proteins SPX1, SPX4, SPX-MFS1, SPX-MFS2, and SPX-MFS3. This finding suggests that AS has an important role in regulating P deficiency responses and P uptake and distribution in plants.
Several transporter genes that may be involved in Fe transport or homeostasis were found to be differentially alternatively spliced under Fe deficiency, including ENA1, ZIP2, ZIP7, NRAMP3, and YSL5 (Kobayashi and Nishizawa, 2012). The differential AS of the gene encoding sulfate transporters under –Fe may play an important role in the interaction between Fe and sulfate, an interaction that is of crucial importance because most of the metabolically active Fe is bound to S in Fe–S clusters (Forieri et al., 2013). Auxin-related genes, including IAA15, IAA16, IAA25, and ARF16, and two genes (Os09g31478 and Os09g38130) encoding proteins that contain the auxin efflux carrier domain were among the DASGs under –Zn stress, pointing to the significant role of AS in the effect of Zn deficiency on the auxin biosynthesis and signaling pathway. All of the above examples suggest that nutrient-specific regulation of AS contributes to the homeostasis of important nutrients in rice.
AS Is an Additional Layer of Regulation in Response to Nutrient Stress in Rice
Transcriptome analysis has been widely used to investigate the response of plants to Fe and P deficiency stresses (Zheng et al., 2009; Secco et al., 2013). The DEGs identified in this study are highly consistent with those identified in these previous studies. Few studies have examined changes in transcriptome in response to –Zn, –Cu, and –Mn. In this study, only 45 and 36 DEGs have been identified in rice for –Zn and –Cu stress, suggesting that 10-d starvation from these two elements is not long enough to observe the full response. Nonetheless, we identified the well-established –Zn marker gene, ZIP4 (Ishimaru et al., 2005), among the DEGs in rice grown under –Zn conditions. There is no established –Cu marker gene in rice; however, we found that a homolog of Arabidopsis ZIP2 (Wintz et al., 2003) in rice, ZIP1, was significantly induced by –Cu, as were several metallothionein genes. The DEGs identified in plants grown under –Cu were enriched in photosynthesis-related processes. The transcriptional regulation of Mn deficiency is largely unknown. As only two studies on Arabidopsis (Yang et al., 2008; Rodríguez-Celma et al., 2016) are available and no –Mn transcriptomic analysis has been reported in rice, we compared our –Mn DEGs with Arabidopsis –Mn genes. We found that, similar to Arabidopsis, the glucosinolate metabolism pathway was enriched in rice DEGs under –Mn, as were several Fe-marker genes such as TOM1 and ENA1 that were antagonistically regulated by deficiencies of Fe and Mn, suggesting that crosstalk occurs between the acquisition responses of the two nutrients, as proposed previously for Arabidopsis (Rodríguez-Celma et al., 2016). In our GO enrichment of DEG analyses, we found a significant enrichment in genes associated with the response to biotic stimulus and cellular detoxification among DEGs identified for plants grown under –Mn conditions. Further studies should examine what role these DEGs have in plants subjected to a nutrient deficiency.
This study indicated that in addition to DEGs, DASGs are an important part of the regulatory mechanism of the plant’s response to stress. This is consistent with a recent study showing that changes in transcript structure contribute to the transcriptome complexity in Arabidopsis plants under nutrient stress (Nishida et al., 2017). Our comparative analysis of DEGs and DASGs under nutrient deficiency conditions revealed that there was little overlap between DASGs and DEGs, and similarly between functional groups (GO analysis). A similar finding was reported in two AS studies of Arabidopsis (Li et al., 2013; Nishida et al., 2017). Interestingly, our GO enrichment analysis of the DASGs that were identified under nutrient deficiency commonly include nucleic acid metabolic pathways, mRNA splicing, and protein kinases that regulate gene/protein expression and function, similar to the situation in Arabidopsis (Nishida et al., 2017). This suggests that the regulation of transcription and mRNA splicing is controlled by an independent regulatory system that is conserved among different plant species and supports the hypothesis that environmental and developmental cues affect gene expression at the level of transcription and AS, which are mediated by transcription factors and splicing factors, respectively (Staiger and Brown, 2013).
It is well established that stress can cause changes at the epigenomic and transcriptomic level (Secco et al., 2013, 2015); AS adds a layer of complexity to the regulation under nutrient limitation that may directly or indirectly impact both the epigenetic and transcriptomic regulation of nutrient availability. As a variety of key factors in nutrient limitation, such as PHO2 in Pi homeostasis (Aung et al., 2006; Bari et al., 2006), are not regulated at the transcript abundance level, the mechanism of action is unclear. However, as PHO2 is a negative regulator of Pi accumulation, it likely prevents Pi toxicity, and the AS of PHO2 under Pi limitation yields a transcript that does not encode a functional protein. Thus, a negative regulator of Pi accumulation is essentially removed under limitation by AS. Therefore, the lack of overlap between DEGs and DASGs indicates that expression and AS need to be analyzed in conjunction to fully capture the response to nutrient limitation.
SR Proteins Play Key Roles in Regulating Mineral Homeostasis in Rice
Although the key role of SR proteins in the plant’s response to environmental stress has long been recognized, few studies have shown direct evidence of their function (Reddy et al., 2013; Staiger and Brown, 2013). There are more SR genes in plant than in animal genomes: Arabidopsis contains 18 SR genes and rice 22, whereas roundworm (Caenorhabditis elegans) contains 7 and humans 12 (Barta et al., 2010; Manley and Krainer, 2010). It has been proposed that the greater number of SR genes in plants provides additional SR proteins that help them survive environmental challenges that they cannot avoid.
In plants, our understanding of the function of SR proteins is largely derived from studies in Arabidopsis (Cruz et al., 2014; Xing et al., 2015; Laloum et al., 2018). Several Arabidopsis mutants defective in SR genes have shown to be severely impaired under certain abiotic stresses, including salt, drought, ABA, and heavy metal treatment (Kim et al., 2007; Cruz et al., 2014; Zhang et al., 2014; Laloum et al., 2018). However, in rice, only one study has reported that overexpression of RSZ36 and SRp33b results in changes in AS patterns of their own pre-mRNAs and of other SR genes (Isshiki et al., 2006). The 22 SR genes in rice provide the capacity to respond to a variety of environmental challenges. Our analysis revealed that the RRM domain-containing protein family, of which SR proteins are key members, is the most overrepresented family among AS gene groups with more than two events. Moreover, as has already been reported, SR transcripts themselves undergo AS and regulate downstream AS genes (Barta et al., 2010; Filichkin et al., 2015).
A recent study (Nishida et al., 2017) identified several SR genes that were transcriptionally regulated by N and K deficiency stress, but AS of SR genes was not investigated under nutrient stress. By contrast, a genome-wide analysis in Arabidopsis found that the abundance of several SRs was altered at the protein level but not at the transcriptional level under Mn deficiency (Rodríguez-Celma et al., 2016). Our analysis showed that most rice SR genes (15 out of 22) were differentially alternatively spliced, but not differentially expressed under mineral deficiency. Our mutant phenotype analysis showed that SR proteins are required for mineral accumulation in rice. Mutants of rs29 and rs33 resulted in impaired Mn and Zn accumulation in rice shoots. Among our 11 rice sr mutants, 10 showed changed P accumulation compared with the wild type, supporting the essential role of rice SR genes in mineral nutrient homeostasis, specifically for P.
To determine how rice SR proteins regulate P accumulation in rice, we performed a time-course P accumulation analysis in the sr40 and scl57 mutants. The impaired remobilization of P from old to young leaves suggests that SR40 and SCL57 regulate P transporters that are involved in Pi remobilization via phloem transport in rice. Phosphate uptake and allocation are tightly regulated in plants with highly efficient Pi remobilization (Nagarajan et al., 2011; Nussaume et al., 2011). In rice, studies have shown that several PHT family transporters are involved in redistributing Pi from older leaves to younger tissues (Młodzińska and Zboińska, 2016). Further work should examine whether these PHT transporter genes are posttranscriptionally regulated by rice SR40 and SCL57.
In conclusion, our study both provides a comprehensive analysis of genes that undergo AS in rice and demonstrates that rice transcripts are subjected to AS in a nutrient-dependent manner, which does not necessarily lead to changes in transcript abundance. Moreover, key splicing factors such as SR proteins play a central role in the response to nutrient limitation in rice and are directly involved in mineral homeostasis. Our findings have potential applications for generating crops with increased P use efficiency.
METHODS
Plant Materials and Growth Conditions
Rice (Oryza sativa) T-DNA insertion mutants in either the cv Dongjing background [sr40 (PFG_3A-01681), rs33 (PFG_1B-04705), rs2z38 (PFG_4A-00839), and scl57 (PFG_1A-11441)]; the cv Hwayoung background [scl26 (PFG_1D-05056) and rs2z36 (PFG_2D-30710)]; or the cv Zhonghua 11 background [sr33 (RMD_04Z11MX35), rsz21a (RMD_04Z11OK81), scl25 (RMD_03Z11LS20), scl30 (RMD_04Z11GP01), and rs29 (RMD_04Z11HY43)] were obtained from rice mutant databases (http://cbi.khu.ac.kr/RISD_DB.html; http://rmd.ncpgr.cn/) (Jeong et al., 2006; Zhang et al., 2006).
Homozygosity of lines was confirmed by PCR using a primer flanking the T-DNA insertion site and a primer representing the T-DNA border (primer pairs are listed in Supplemental Table 2). O. sativa cvs Nipponbare, Zhonghua 11, Dongjing, and Hwayoung were used as wild types. The scl25-1 mutation was created in the SCL25 target sequence (Supplemental Table 2) by CRISPR/Cas9 (Xie and Yang, 2013), and homozygous lines were confirmed by sequencing.
Hydroponic experiments were performed in a greenhouse at 25 to 30°C. Rice seeds were pregerminated in water for 2 d before being transferred to a 0.5 mM CaCl2 solution for 4 d. Seedlings were then transferred into half-strength Kimura B solution [(NH4)2SO4 (0.18 mM), MgSO4·7H2O (0.27 mM), KNO3 (0.09 mM), Ca(NO3)2·4H2O (0.18 mM), KH2PO4 (0.09 mM), MnCl2·4H2O (0.5 μM), H3BO3 (3 μM), (NH4)6Mo7O24·4H2O (1 μM), ZnSO4·7H2O (0.4 μM), CuSO4·5H2O (0.2 μM), and FeSO4·7H2O (2 μM)]. The pH was adjusted to 5.5, and the nutrient solution was replaced every 2 d. All experiments were repeated at least three times with three replicates each and representative results of one experiment are shown.
For RNA-seq, 14-d-old seedlings of O. sativa cv Nipponbare were transferred to the –Fe, –Zn, –Cu, –Mn, and control solution. After 10 d under mineral deficiency conditions, root samples were harvested for total RNA extraction with three replications.
Phenotypic Analysis of Rice sr Mutants
To select for nutrient-related SR genes, 11 sr T-DNA insertion mutants and three corresponding wild-type rice varieties were used. To investigate the effects of SR genes in micronutrient homeostasis, 10-d-old rice seedlings of sr mutants and wild-type rice were transferred to the –Fe, –Zn, –Cu, and –Mn solution for 7 d. Seedlings grown in half-strength Kimura B solution were used as controls. At harvest, shoots and roots were sampled to determine the concentrations of Fe, Zn, Cu, and Mn. For P-related phenotype analysis, half of the 10-d-old seedlings were transferred into a –P solution, while the other half remained in a +P solution (0.09 mM KH2PO4). After 8 d of treatment (mutants in the Dongjing background were treated for 6 d), shoots and roots were sampled and total P and/or Pi concentration was measured.
sr40, scl57, and scl25-1 were analyzed further. Ten-day-old seedlings were transferred to either –P or excess Pi (0.64 mM KH2PO4) solutions, with seedlings grown in normal nutrient solution (+P) used as controls. Phenotypes were analyzed after 14 d of treatment. Different tissues were sampled for the Pi time-course analysis when the seedlings were 10, 16, 22, or 28 d old. In the –P solution, KH2PO4 (0.09 mM) was replaced with KCl (0.09 mM), whereas NaH2PO4 (0.54 mM) was added to the solutions for excess P treatments.
Measurement of Total Element and Pi Concentration and Distribution of P
Rice tissues were dried at 80°C for 3 d, and samples were digested with HNO3:HClO4 (87:13 v/v) at 180°C. After dissolving samples in 2% HNO3, the concentrations of Fe, Zn, Cu, Mn, and P were determined by ICP-MS (Perkin-Elmer NexION 300X). The high-resolution distribution analysis of P in the leaf was analyzed using a micro x-ray fluorescence spectrometer (M4 Tornado). Pi was measured using the molybdate blue method (Nanamori et al., 2004).
RNA Isolation, RNA-Seq Library Preparation, and Sequencing
Total RNA was extracted from tissues using a MiniBEST plant RNA extraction kit (TaKaRa), and genomic DNA was removed by DNase I (TaKaRa) treatment. RNA was then quantitatively analyzed using either an ND-8000 spectrophotometer (Nanodrop Technologies) after purification by agarose gel electrophoresis or a 2100-Bioanalyzer (Agilent Technologies). RNA samples that did not produce a smear on agarose gels and had a 260/280 ratio of above 2.0 and an RNA integrity number (RIN; Agilent Technologies) of >8.0 were used. The RIN was a RNA quality assessment tool, which assigns a RIN score in the range from 1 (highly degraded RNA) to 10 (completely intact RNA) (Schroeder et al., 2006).
RNA samples (three biological replicates per condition and four plants per replicate) were sent to Genergy Biotechnology for sequencing. This was done by first depleting total RNA of rRNA using the Ribo-Zero rRNA removal kit (Plant Leaf and Plant Seed/Root kits; Epicentre). Sequencing libraries were generated using the TruSeq RNA Sample Prep Kit (Illumina). The libraries were sequenced as 151-bp paired-end reads using Illumina HiSeq X Ten according to the manufacturer’s instructions. Illumina reads of all samples were deposited in the Sequence Read Archive at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/sra) under accession number SRP117202.
Read Mapping and Transcript Assembly
The coordinates of mRNAs, UTRs, exons, and coding sequences of rice protein-coding genes were extracted from the RAPDB (http://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_representative_2016-08-05.tar.gz, IRGSP-1.0_predicted_2016-08-05.tar.gz) and MSU 7.0 (http://rice.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/all.gff3) databases. The MSU transcript models that had the same 5′ and 3′ ends, exons, and introns as RAPDB transcripts were discarded. Overlapping transcripts from the two databases were merged into a nonredundant reference gene GTF file with the same gene locus for downstream alignment and assembly using Bedtools version 2.17.0. For each sample, raw paired-end reads were quality- and adaptor-trimmed using Trim Galore version 0.4.2 (-q 25, -length 30). Next, clean reads were aligned to the Os-Nipponbare-Reference-IRGSP-1.0 reference genome (http://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-.0_genome.fasta.gz) using STAR version 2.5.2b (Dobin et al., 2013) and the above merged reference gene GTF file. STAR was run with default settings.
Mapped reads were assembled into putative transcripts based on a reference guided assembly strategy using the single-sample transcript assembly tool StringTie version 1.3.1 (Pertea et al., 2015). Multiple putative transcripts were merged into a unified set of transcripts using the meta-assembly tool TACO version 0.7.3 (Niknafs et al., 2017). TACO employs a change-point detection model to break read-through transcripts apart and accurately delineate transcript start and end sites. This set of transcripts was then compared with the above merged reference gene GTF file using gffcompare version 0.9.9c (Trapnell et al., 2012).
Based on transcript classification codes, “j” transcripts with at least one splice junction were shared with a reference transcript, “o” transcripts had generic exonic overlap with a reference transcript, “u” (unknown) intergenic transcripts were potentially regarded as novel isoforms from known or novel gene loci, and other assembled transcripts including “=” transcripts, which completely matched all introns in the reference transcript, were filtered. The closest reference transcripts in three class codes (=, j, o) were extracted from the above merged reference gene GTF file as candidate known isoforms detected in the data set. To reduce the number of potentially novel misassembled isoforms, each splice junction of each isoform was required to be supported by at least 40 unique spliced reads totally mapped by STAR for all samples. Therefore, novel isoforms with a single exon or poorly supported by splice junction reads were removed. These resulting novel transcripts and known transcripts deposited in RAPDB and MSU databases were then merged into nonredundant transcripts.
Finally, transcript expression was quantified using Salmon (version 0.8.2), and only the transcripts with a TPM of ≥1 in at least one sample were used for downstream DEG and DASG analysis. To identify the protein-coding region of each novel transcript, protein-coding potential was evaluated using CPC2 (version 0.1) and its putative longest open reading frame (ORF) was identified using Transdecoder version 3.0.1.
Differential Expression Analysis
For all novel and known genes in the above high-quality transcript GTF file, the gene-level count matrices were generated by summing the transcript counts estimated using Salmon within genes. The median-of-ratios method was used to normalize for sequencing depth and RNA composition using DESeq2 available as an R/Bioconductor package (Gentleman et al., 2004; Anders and Huber, 2010; Love et al., 2014).
To identify DEGs under each nutrient deficiency condition with respect to its control at each time point after filtering out genes that were expressed at low levels (<30 total read counts in three replicate samples per condition), a negative binomial generalized linear model was fit for each gene, then the Wald significance test allowed testing against the null hypothesis of zero logarithmic fold change and the moderated estimation of fold change and P values are calculated in DESeq2 (Love et al., 2014). Differentially expressed genes exhibiting twofold changes and Benjamini and Hochberg-adjusted P values (FDR) of ≤0.05 were selected. The matrix data of expression value or percentage splicing index were visualized using pheatmap available as an R/cran package (http://cran.r-project.org/web/packages/pheatmap/index.html). Significantly enriched GO terms and KEGG pathways compared with the genome-wide background were detected using the hypergeometric test in our in-house pipeline. The hypergeometric test, in which sampling occurs without replacement, calculates the P value as the probability to determine whether any GO term or KEGG pathway annotates a specified list of genes at a frequency greater than that would be expected by chance using hypergeometric distribution. Significantly enriched GO terms and KEGG pathways were also selected using a threshold P value of ≤0.05.
AS Event and Gene Detection
All putative AS events were extracted from the above high-quality transcript GTF file using rMATS (Shen et al., 2014a). Each putative AS event was supported by two sets of transcripts with different splicing patterns. For example, an intron-retention event was identified if both transcripts (with or without a retained intron) were observed. Two quantification models of AS events were provided by rMATS based on different effective lengths of isoform-specific exon or intron regions and read counting. Inclusive and skipping reads satisfied all of the following criteria: (1) spanned the splicing junction, or occurred in the alternative exon or intron target (inclusion count [IC] and skipping count [SC]); and (2) only spanned splicing junctions (inclusion junction count [IJC] and skipping junction count [SJC]). Expressed AS events and the associated AS genes were declared if a total IJC of ≥1 and total SJC ≥1 in all samples (including replicates). Based on the distribution of the number of AS events detected per gene, the high-frequency cutoff was equal to or greater than one sd above the mean and the low-frequency cutoff was less than or equal to one sd below the mean. These AS genes were then classified into four groups: high-frequency (≥11 AS events), medium-frequency (≥2 AS events), low-frequency (1 AS event), and no AS intron-containing genes. Pfam domains of all known rice proteins were screened for using the Interproscan tool (Finn et al., 2017). Similar to GO or KEGG enrichment analysis, a hypergeometric test was used to detect the significantly enriched Pfam domains for each AS gene group. For genes that had multiple assembled transcript variants, the representative transcript with the most exons was used to calculate the exon/intron number, exon/intron length, and GC content. Comparisons of each genomic feature and of expression levels between two AS gene groups were conducted using box plot visualization and a Wilcoxon rank sum test using stats package in R (Bauer, 1972).
Differential AS Gene Detection and AS Function
Effective length normalization was performed before the PSI of an AS event was robustly estimated using the inclusion count and skipping count quantification model. A hierarchical statistical framework based on binomial distribution and logit-normal models was used to simultaneously account for the estimation uncertainty of PSI individual replicates and variability among replicates. After weakly expressed AS events were filtered below 40% samples with total IJC+SJC ≥ 30, differentially AS events between control and each nutrient deficiency condition were identified using rMATS if the difference in the PSI of AS event between two conditions exceeds a stringent threshold (FDR ≤ 0.05, ΔPSI ≥10%). Multiple transcripts per DASG were classified into either an inclusive transcript group or an exclusive transcript group based on whether or not they contained the alternative exon (or intron) within an AS event. Two representative transcripts selected from each group based on the highest mean expression value (TPM) among all samples were used to predict the impact of AS on transcript structure and protein function. For the alternative exon (or intron), different mapped regions (translated or nontranslated) from two representative isoforms were compared manually. The putative longest ORF identified by Transdecoder may predict false-positive ORFs of alternative spliced transcripts based on the AUG position located downstream of the authentic translation start site. To accurately compare ORFs of two representative transcripts, the authentic translation start AUG, which did not lead to an unlikely long 5′-UTR or to the same sequence between the two transcripts, was manually selected (Brown et al., 2015). If a PTC was located >55 nucleotides upstream of the 3′-most exon-exon junction, AS produced nonsense transcripts as a result of NMD. AS events were predicted to change the N or C terminus of proteins or to result in the insertion/deletion of amino acids within proteins by introducing an in-frame start codon or stop codon. Two completely different proteins or the loss of protein-coding capacity were produced by frame shift AS. For a differential AS event, an in-house visualization plot showed the normalized mapped read density across exons and the number of junction reads spanning the exon-intron boundary affected in an AS event in each sample, color-coded by condition.
An overview of our workflow for RNA-seq data analysis is illustrated in Supplemental Figure 10.
Validation of Differential AS Events by RT-qPCR
cDNA was synthesized using a PrimeScript RT reagent kit with gDNA Eraser (TaKaRa). RT-qPCR reactions were done using TaKaRa SYBR Premix Ex Taq (Tli RNaseH Plus) on a Mastercycler ep Realplex (Eppendorf). The relative expression data was calculated by the ΔΔCt (cycle threshold) method. Four biological replications were performed. AS events were validated by RT-qPCR analysis, according to previous reports (Zhou et al., 2014; Yan et al., 2017). The exclusion/inclusion ratio was determined from the level of exon exclusion RNA normalized to the level of exon inclusion RNA.
For RNA-seq data, the ratio was calculated using the following formula: exclusion/inclusion ratio = 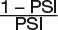 . PCR primer sets were designed to ensure that either spliced RNA or unspliced RNA was amplified. The first primer pair was designed to exactly cover the retained intron or alternative exon region for alternative exon/intron inclusive transcripts, and the second primer pair amplified the splice-junction region connecting the upstream flanking exon and the downstream flanking exon of alternative exon/intron exclusive transcripts. Sequences for primer pairs used in this study are listed in Supplemental Table 2.
. PCR primer sets were designed to ensure that either spliced RNA or unspliced RNA was amplified. The first primer pair was designed to exactly cover the retained intron or alternative exon region for alternative exon/intron inclusive transcripts, and the second primer pair amplified the splice-junction region connecting the upstream flanking exon and the downstream flanking exon of alternative exon/intron exclusive transcripts. Sequences for primer pairs used in this study are listed in Supplemental Table 2.
Accession Numbers
RNA-seq raw sequence data of the 15 samples for micronutrient deficiency roots used in this article can be found in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession number SRP117202. All the RNA-seq data of the time course of Pi-starved root and shoot samples are available under the accession number SRA097415 (Secco et al., 2013). The code underlying the AS analyses of RNA-seq data in this study is available at https://github.com/feihestudy/Alternative-Splicing-Study-Under-Mineral-Nutrient-Deficiency. Accession numbers for T-DNA mutants are given in the Plant Materials and Growth Conditions section of Methods.
Supplemental Data
Supplemental Figure 1. Fe, Zn, Cu, and Mn concentrations in roots and shoots of rice under mineral deficiency.
Supplemental Figure 2. Overview of alternative splicing identified in rice mineral deficiency RNA-seq data sets.
Supplemental Figure 3. Number of detected AS genes.
Supplemental Figure 4. RT-qPCR validation of AS regulated by nutrient deficiency in rice.
Supplemental Figure 5. Functional categorization (biological process) of DASGs and DEGs under Zn deficiency conditions of rice roots.
Supplemental Figure 6. Functional categorization (biological process) of DASGs and DEGs under Cu deficiency conditions of rice roots.
Supplemental Figure 7. Functional categorization (biological process) of DASGs and DEGs under Mn deficiency conditions of rice roots.
Supplemental Figure 8. Locations and verification of T-DNA insertion sites in rice sr mutants.
Supplemental Figure 9. Phenotypic analysis of rice sr40, scl57, and scl25 mutants under excess Pi conditions.
Supplemental Figure 10. Overview of our RNA-seq data analysis workflow.
Supplemental Table 1. A set of high-quality transcripts from TACO multisample assembly in our data set and known databases using stringent filters.
Supplemental Table 2. Primers used in this research.
Supplemental Data Set 1. All putative AS events from the transcript GTF file by rMATS.
Supplemental Data Set 2. DASGs in response to mineral nutrient deficiencies.
Supplemental Data Set 3. DEGs in response to mineral nutrient deficiencies.
Supplemental Data Set 4. Gene Ontology enrichment analysis results for mineral nutrient deficiency regulated DEGs.
Supplemental Data Set 5. KEGG pathway enrichment analysis results for mineral nutrient deficiency regulated DEGs.
Supplemental Data Set 6. Gene Ontology enrichment analysis results for mineral nutrient deficiency regulated DASGs.
Supplemental Data Set 7. KEGG pathway enrichment analysis results for mineral nutrient deficiency regulated DASGs.
Supplemental Data Set 8. Differential alternative splicing patterns of Pi signaling-associated genes in response to Pi deficiency.
Supplemental Data Set 9. Differential expression patterns of 22 SR genes in response to mineral nutrient deficiency.
Supplemental Data Set 10. Differential alternative splicing patterns of 22 SR genes in response to nutrient deficiency.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
NMD Gramene: nonsense-mediated mRNA decay
NMD Araport: nonsense-mediated mRNA decay
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2016YFD0100700); the National Natural Science Foundation of China (31770269); the Fundamental Research Funds for the Central Universities (KYTZ201402); and the PAPD project of Jiangsu Higher Education Institutions. C.D. was supported by the NAU creative project for doctoral dissertations. J.W. and O.B. are supported by the Australian Research Council Centre of Excellence in Plant Energy Biology (CE140100008). We thank the National Center of Plant Gene Research (Wuhan, China), Huazhong Agricultural University, and Gynheung An (Kyung Hee University, Korea) for providing the T-DNA insertion lines of rice used in this study. We thank Paula Duque (Instituto Gulbenkian de Ciência, Portugal) and Yufeng Wu (Nanjing Agricultural University, China) for helpful comments.
AUTHOR CONTRIBUTIONS
L.Z. conceived and supervised the project. L.Z. and J.W. designed the study. F.H. and O.B. performed the bioinformatics analyses. C.D. generated and screened SR mutants. C.D., P.C., J.L., M.T., and W.W. performed mutant phenotype analyses. C.D., Q.L., and H.S. carried out the RT-qPCR validation. C.D., Z.S., and L.Z. analyzed the data. L.Z., C.D., F.H., and J.W. wrote the manuscript. All authors read and approved the final manuscript.
References
- Aibara I., Miwa K. (2014). Strategies for optimization of mineral nutrient transport in plants: multilevel regulation of nutrient-dependent dynamics of root architecture and transporter activity. Plant Cell Physiol. 55: 2027–2036. [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung K., Lin S.I., Wu C.C., Huang Y.T., Su C.L., Chiou T.J. (2006). pho2, a phosphate overaccumulator, is caused by a nonsense mutation in a microRNA399 target gene. Plant Physiol. 141: 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari R., Datt Pant B., Stitt M., Scheible W.R. (2006). PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 141: 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barta A., Kalyna M., Reddy A.S. (2010). Implementing a rational and consistent nomenclature for serine/arginine-rich protein splicing factors (SR proteins) in plants. Plant Cell 22: 2926–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer D.F. (1972). Constructing confidence sets using rank statistics. J. Am. Stat. Assoc. 67: 687–690. [Google Scholar]
- Berget S.M., Moore C., Sharp P.A. (1977). Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 74: 3171–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart R.E., Andreadis A., Nadal-Ginard B. (1987). Alternative splicing: a ubiquitous mechanism for the generation of multiple protein isoforms from single genes. Annu. Rev. Biochem. 56: 467–495. [DOI] [PubMed] [Google Scholar]
- Brown J.W., Simpson C.G., Marquez Y., Gadd G.M., Barta A., Kalyna M. (2015). Lost in translation: pitfalls in deciphering plant alternative splicing transcripts. Plant Cell 27: 2083–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho R.F., Carvalho S.D., Duque P. (2010). The plant-specific SR45 protein negatively regulates glucose and ABA signaling during early seedling development in Arabidopsis. Plant Physiol. 154: 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Cui P., Chen H., Ali S., Zhang S., Xiong L. (2013). A KH-domain RNA-binding protein interacts with FIERY2/CTD phosphatase-like 1 and splicing factors and is important for pre-mRNA splicing in Arabidopsis. PLoS Genet. 9: e1003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou T.J., Lin S.I. (2011). Signaling network in sensing phosphate availability in plants. Annu. Rev. Plant Biol. 62: 185–206. [DOI] [PubMed] [Google Scholar]
- Chow L.T., Gelinas R.E., Broker T.R., Roberts R.J. (1977). An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12: 1–8. [DOI] [PubMed] [Google Scholar]
- Cruz T.M., Carvalho R.F., Richardson D.N., Duque P. (2014). Abscisic acid (ABA) regulation of Arabidopsis SR protein gene expression. Int. J. Mol. Sci. 15: 17541–17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P., Lin Q., Ding F., Xin C., Gong W., Zhang L., Geng J., Zhang B., Yu X., Yang J., Hu S., Yu J. (2010). A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 96: 259–265. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichkin S., Priest H.D., Megraw M., Mockler T.C. (2015). Alternative splicing in plants: directing traffic at the crossroads of adaptation and environmental stress. Curr. Opin. Plant Biol. 24: 125–135. [DOI] [PubMed] [Google Scholar]
- Finn R.D., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45: D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forieri I., Wirtz M., Hell R. (2013). Toward new perspectives on the interaction of iron and sulfur metabolism in plants. Front. Plant Sci. 4: 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.D., Ares M. Jr (2014). Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru Y., Suzuki M., Kobayashi T., Takahashi M., Nakanishi H., Mori S., Nishizawa N.K. (2005). OsZIP4, a novel zinc-regulated zinc transporter in rice. J. Exp. Bot. 56: 3207–3214. [DOI] [PubMed] [Google Scholar]
- Isshiki M., Tsumoto A., Shimamoto K. (2006). The serine/arginine-rich protein family in rice plays important roles in constitutive and alternative splicing of pre-mRNA. Plant Cell 18: 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong D.-H., et al. (2006). Generation of a flanking sequence-tag database for activation-tagging lines in japonica rice. Plant J. 45: 123–132. [DOI] [PubMed] [Google Scholar]
- Kim Y.O., Pan S., Jung C.H., Kang H. (2007). A zinc finger-containing glycine-rich RNA-binding protein, atRZ-1a, has a negative impact on seed germination and seedling growth of Arabidopsis thaliana under salt or drought stress conditions. Plant Cell Physiol. 48: 1170–1181. [DOI] [PubMed] [Google Scholar]
- Kobayashi T., Nishizawa N.K. (2012). Iron uptake, translocation, and regulation in higher plants. Annu. Rev. Plant Biol. 63: 131–152. [DOI] [PubMed] [Google Scholar]
- Laloum T., Martín G., Duque P. (2018). Alternative splicing control of abiotic stress responses. Trends Plant Sci. 23: 140–150. [DOI] [PubMed] [Google Scholar]
- Li Q., Xiao G., Zhu Y.X. (2014). Single-nucleotide resolution mapping of the Gossypium raimondii transcriptome reveals a new mechanism for alternative splicing of introns. Mol. Plant 7: 829–840. [DOI] [PubMed] [Google Scholar]
- Li W., Lin W.D., Ray P., Lan P., Schmidt W. (2013). Genome-wide detection of condition-sensitive alternative splicing in Arabidopsis roots. Plant Physiol. 162: 1750–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J.L., Krainer A.R. (2010). A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 24: 1073–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez Y., Brown J.W., Simpson C., Barta A., Kalyna M. (2012). Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 22: 1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschner P. (2012). Mineral Nutrition of Higher Plants, 3rd ed. (London: Academic Press; ). [Google Scholar]
- Meyer K., Koester T., Staiger D. (2015). Pre-mRNA splicing in plants: in vivo functions of RNA-binding proteins implicated in the splicing process. Biomolecules 5: 1717–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Młodzińska E., Zboińska M. (2016). Phosphate uptake and allocation: A closer look at Arabidopsis thaliana L. and Oryza sativa L. Front. Plant Sci. 7: 1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan V.K., Jain A., Poling M.D., Lewis A.J., Raghothama K.G., Smith A.P. (2011). Arabidopsis Pht1;5 mobilizes phosphate between source and sink organs and influences the interaction between phosphate homeostasis and ethylene signaling. Plant Physiol. 156: 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y., Koizumi R., Shui G., Shimojima M., Wenk M.R., Ito T., Ohta H. (2009). Arabidopsis lipins mediate eukaryotic pathway of lipid metabolism and cope critically with phosphate starvation. Proc. Natl. Acad. Sci. USA 106: 20978–20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanamori M., Shinano T., Wasaki J., Yamamura T., Rao I.M., Osaki M. (2004). Low phosphorus tolerance mechanisms: phosphorus recycling and photosynthate partitioning in the tropical forage grass, Brachiaria hybrid cultivar Mulato compared with rice. Plant Cell Physiol. 45: 460–469. [DOI] [PubMed] [Google Scholar]
- Niknafs Y.S., Pandian B., Iyer H.K., Chinnaiyan A.M., Iyer M.K. (2017). TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. Methods 14: 68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida S., Kakei Y., Shimada Y., Fujiwara T. (2017). Genome-wide analysis of specific alterations in transcript structure and accumulation caused by nutrient deficiencies in Arabidopsis thaliana. Plant J. 91: 741–753. [DOI] [PubMed] [Google Scholar]
- Nussaume L., Kanno S., Javot H., Marin E., Pochon N., Ayadi A., Nakanishi T.M., Thibaud M.C. (2011). Phosphate import in plants: Focus on the PHT1 transporters. Front. Plant Sci. 2: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A.S., Marquez Y., Kalyna M., Barta A. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25: 3657–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Celma J., Tsai Y.H., Wen T.N., Wu Y.C., Curie C., Schmidt W. (2016). Systems-wide analysis of manganese deficiency-induced changes in gene activity of Arabidopsis roots. Sci. Rep. 6: 35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald P. (2011). Plant genetics, sustainable agriculture and global food security. Genetics 188: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A., Mueller O., Stocker S., Salowsky R., Leiber M., Gassmann M., Lightfoot S., Menzel W., Granzow M., Ragg T. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secco D., Jabnoune M., Walker H., Shou H., Wu P., Poirier Y., Whelan J. (2013). Spatio-temporal transcript profiling of rice roots and shoots in response to phosphate starvation and recovery. Plant Cell 25: 4285–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secco D., Wang C., Shou H., Schultz M.D., Chiarenza S., Nussaume L., Ecker J.R., Whelan J., Lister R. (2015). Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 4: 09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. (2014a). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl. Acad. Sci. USA 111: E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Zhou Z., Wang Z., Li W., Fang C., Wu M., Ma Y., Liu T., Kong L.A., Peng D.L., Tian Z. (2014b). Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26: 996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D., Brown J.W. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25: 3640–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton M.A., Oenema O., Erisman J.W., Leip A., van Grinsven H., Winiwarter W. (2011). Too much of a good thing. Nature 472: 159–161. [DOI] [PubMed] [Google Scholar]
- Thatcher S.R., Zhou W., Leonard A., Wang B.B., Beatty M., Zastrow-Hayes G., Zhao X., Baumgarten A., Li B. (2014). Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell 26: 3472–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J., Palusa S.G., Prasad K.V., Ali G.S., Surabhi G.K., Ben-Hur A., Abdel-Ghany S.E., Reddy A.S. (2012). Identification of an intronic splicing regulatory element involved in auto-regulation of alternative splicing of SCL33 pre-mRNA. Plant J. 72: 935–946. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Huang W., Ying Y., Li S., Secco D., Tyerman S., Whelan J., Shou H. (2012). Functional characterization of the rice SPX-MFS family reveals a key role of OsSPX-MFS1 in controlling phosphate homeostasis in leaves. New Phytol. 196: 139–148. [DOI] [PubMed] [Google Scholar]
- Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintz H., Fox T., Wu Y.Y., Feng V., Chen W., Chang H.S., Zhu T., Vulpe C. (2003). Expression profiles of Arabidopsis thaliana in mineral deficiencies reveal novel transporters involved in metal homeostasis. J. Biol. Chem. 278: 47644–47653. [DOI] [PubMed] [Google Scholar]
- Wu P., Shou H., Xu G., Lian X. (2013). Improvement of phosphorus efficiency in rice on the basis of understanding phosphate signaling and homeostasis. Curr. Opin. Plant Biol. 16: 205–212. [DOI] [PubMed] [Google Scholar]
- Xie K., Yang Y. (2013). RNA-guided genome editing in plants using a CRISPR-Cas system. Mol. Plant 6: 1975–1983. [DOI] [PubMed] [Google Scholar]
- Xing D., Wang Y., Hamilton M., Ben-Hur A., Reddy A.S. (2015). Transcriptome-wide identification of RNA targets of Arabidopsis SERINE/ARGININE-RICH45 uncovers the unexpected roles of this RNA binding protein in RNA processing. Plant Cell 27: 3294–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R.L., Dwivedi B.S., Prasad K., Tomar O.K., Shurpali N.J., Pandey P.S. (2000). Yield trends, and changes in soil organic-C and available NPK in a long-term rice-wheat system under integrated use of manures and fertilisers. Field Crops Res. 68: 219–246. [Google Scholar]
- Yan Q., Xia X., Sun Z., Fang Y. (2017). Depletion of Arabidopsis SC35 and SC35-like serine/arginine-rich proteins affects the transcription and splicing of a subset of genes. PLoS Genet. 13: e1006663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.J., Perry P.J., Ciani S., Pandian S., Schmidt W. (2008). Manganese deficiency alters the patterning and development of root hairs in Arabidopsis. J. Exp. Bot. 59: 3453–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., et al. (2010). Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 20: 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Li C., Wu C., Xiong L., Chen G., Zhang Q., Wang S. (2006). RMD: a rice mutant database for functional analysis of the rice genome. Nucleic Acids Res. 34: D745–D748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Du B., Liu D., Qi X. (2014). Splicing factor SR34b mutation reduces cadmium tolerance in Arabidopsis by regulating iron-regulated transporter 1 gene. Biochem. Biophys. Res. Commun. 455: 312–317. [DOI] [PubMed] [Google Scholar]
- Zhao H., Frank T., Tan Y., Zhou C., Jabnoune M., Arpat A.B., Cui H., Huang J., He Z., Poirier Y., Engel K.H., Shu Q. (2016). Disruption of OsSULTR3;3 reduces phytate and phosphorus concentrations and alters the metabolite profile in rice grains. New Phytol. 211: 926–939. [DOI] [PubMed] [Google Scholar]
- Zhao W., He X., Hoadley K.A., Parker J.S., Hayes D.N., Perou C.M. (2014). Comparison of RNA-Seq by poly(A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics 15: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Huang F., Narsai R., Wu J., Giraud E., He F., Cheng L., Wang F., Wu P., Whelan J., Shou H. (2009). Physiological and transcriptome analysis of iron and phosphorus interaction in rice seedlings. Plant Physiol. 151: 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Wu W., Li H., Cheng Y., Wei N., Zong J., Feng X., Xie Z., Chen D., Manley J.L., Wang H., Feng Y. (2014). Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 42: 4019–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]