

Abstract
The 20S proteasome is the main protease that directly targets intrinsically disordered proteins (IDPs) for proteolytic degradation. Mutations, oxidative stress, or aging can induce the buildup of IDPs resulting in incorrect signaling or aggregation, associated with the pathogenesis of many cancers and neurodegenerative diseases. Drugs that facilitate 20S-mediated proteolysis therefore have many potential therapeutic applications. We report herein the modulation of proteasome assembly by the small molecule TCH-165, resulting in an increase in 20S levels. The increase in the level of free 20S corresponds to enhanced proteolysis of IDPs, including α-synuclein, tau, ornithine decarboxylase, and c-Fos, but not structured proteins. Clearance of ubiquitinated protein was largely maintained by single capped proteasome complexes (19S–20S), but accumulation occurs when all 19S capped proteasome complexes are depleted. This study illustrates the first example of a small molecule capable of targeting disordered proteins for degradation by regulating the dynamic equilibrium between different proteasome complexes.
Graphical Abstract
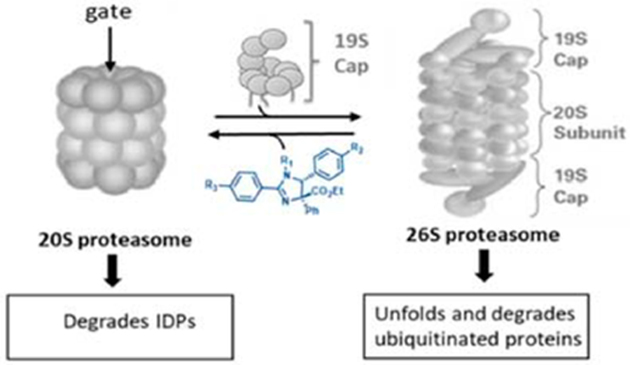
Intrinsically disordered proteins (IDPs) make up a class of proteins that lack a stable tertiary structure.1 IDPs are involved in multiple regulatory and signaling events as their disordered structures permit interactions with multiple proteins and signaling pathways.2 Not surprisingly, accumulation of IDPs can lead to harmful signaling events directly associated with the pathogenesis of many human diseases.3–7Unfortunately, the lack of a defined three-dimensional structure has impeded traditional small molecule-binding pocket drug design approaches to block the many detrimental effects of amassed IDPs.
The proteasome is the main protease responsible for the degradation of misfolded, oxidatively damaged, and redundant proteins.8–11 The proteasome can form different complexes that include active complexes (such as the 26S proteasome) and a weakly active/latent complex, called the 20S proteasome. In contrast to the 26S proteasome, which requires proteins to be ubiquitinated prior to their degradation, IDPs are directly targeted by the 20S proteasome for degradation.8,12 IDPs are therefore typically short-lived and present in only small-to-undetectable quantity because of unremitted degradation by the 20S proteasome.8,12 In most cells, the 20S:26S ratio is approximately 3:1,13 but when IDPs accumulate, the 26S proteasome complex will further disassemble to form more 20S proteasome particles to prevent toxic signaling.14–16 Considering the pathological role of accumulated IDPs in protein aggregation and amyloid formation (notable examples include α-synuclein and tau),17–23 cell growth (i.e., ornithine decarboxylase),12,24–27 and survival and inflammation (i.e., c-Fos),28 the reduction of the level of IDP buildup has great potential for the treatment of multiple human diseases.3–7
Enhancing proteasome activity is a relatively new approach, but despite its enormous therapeutic potential, very few molecules have been identified as direct29–31 or indirect32–34 enhancers of the proteasome degradation pathway.30,35 Herein, we report a new approach that uses small molecules to modulate the dynamic equilibrium between different protea-some complexes, favoring the 20S proteasome, thus mimicking the natural defense response of cells to reduce high levels of IDPs.
The 26S proteasome is comprised of a barrel-shaped 20S core particle (CP) capped by two regulatory particles (RP) or “caps”.36 The 20S CP is a threonine protease that consists of four stacked rings. The two inner β-rings contain three catalytic subunits (β5, β2, and β1) that exhibit chymotrypsin-like (CT-L), trypsin-like (Tryp-L), and caspase-like (Casp-L) proteolytic activity, respectively.37 The outer α-rings do not exhibit proteolytic activity but control access to the proteolytic core chamber via a gate opening and closing mechanism.38,39 The free 20S proteasome exists primarily in its latent (closed-gate) conformation but can accept peptides or IDP substrates when in its intermittent open-gate conformation.8,40
Better access to the inner 20S core particle (CP) is achieved when additional modules, such as a regulatory particle (RP), or PA28 or PA200 activators dock onto the α-ring and induce conformational changes promoting gate opening.41–43 The 26S proteasome is assembled by the docking of two 19S (or PA700) RPs onto the α-rings of the 20S CP.44 During the docking stage, six ATPases (Rpt 1–6) on the base of the 19S lid insert C-terminal hydrophobic peptides containing an Hb-Y-X (hydrophobic amino acid-tyrosine-any amino acid) motif into the intersubunit pockets in the α-rings.45,46 This docking opens the gate to the 20S CP; however, details of this 20S gate opening mechanism remain ambiguous.38,45,47–49 The RPs contain receptors for ubiquitin recognition and will subsequently deubiquitinate, unfold, and transfer the substrate protein into the CP following ATP hydrolysis, as it proceeds through at least three major distinct conformational stages.50,51 The degradation of ubiquitinated proteins requires one (19S–20S) or two regulatory particles (19S–20S–19S) to be docked onto the CP.38,52 Not all additional modules require ATP hydrolysis to activate the 20S CP. PA28αβ, PA28γ, and Blm10 are examples of activators of the 20S that do not have ATPase activity and do not recognize ubiquitinated substrates.43,53PA28αβ and PA28γ have been shown to compete with the 19S RP for 20S binding to provide hybrid proteasomes (19S–20S–PA28).52,54,55 Other 20S activators, such as Blm10, allosterically regulate the 20S gate to provide a clear path for IDPs to enter the proteolytic 20S core.56,57
We previously reported that noncompetitive modulation of a proteasome by imidazolines could overcome bortezomib resistance and block tumor growth in vivo.58 The parent compound of that study was found not to interact with the catalytic sites of the 20S proteasome but to modulate protein proteolysis through an unknown mechanism. Our efforts to optimize the activity of the imidazoline scaffold generated the small molecule TCH-165 (Figure 1).59 Intrigued by conflicting signs of activation and inhibition of proteasome activity in various assays, we pursued a full investigation of its mechanism of action. Here we report that TCH-165 regulates the dynamic equilibrium between the 20S and 26S proteasome complexes, favoring 20S-mediated protein degradation.
Figure 1.

Structure and activities of imidazoline scaffolds.
MATERIALS AND METHODS
Materials and Reagents.
Human proteasomes (20S, i20S, and 26S) and fluorogenic substrates N-succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC), carboxyl benzyl-Leu-Leu-Glu-7-amido-4-methylcoumarin (Z-LLEAMC), tert-butyloxycarbonyl-Leu-Arg-Arg-7-amido-4-methylcoumarin (Boc-LRR-AMC), acetyl-Pro-Ala-Leu-7-amido-4-methylcoumarin (Ac-PAL-AMC), and bortezomib were obtained from Boston Biochem, Inc. (Cambridge, MA). Epoxomicin was purchased from Cayman Chemical (Ann Arbor, MI). The PVDF membrane, Clarity western ECL reagent, blocking grade milk, and precast sodium dodecyl sulfate (SDS) gels were from Bio-Rad (Hercules, CA). The DCFDA/H2DCFDA cellular reactive oxygen species detection assay kit, recombinant wild type α-synuclein, tau441, and GAPDH were obtained from Abcam (Cambridge, MA). Rabbit polyclonal anti-α-synuclein (C-20), anti-tau, goat anti-rabbit HRP, rabbit polyclonal GAPDH-HRP, GFP-HRP, and mouse monoclonal anti-β5 proteasome subunit were purchased from Santa Cruz Biotechnologies. Mouse monoclonal anti-Rpt1 and the yeast (Saccharomyces cerevisiae) proteasome were obtained from Enzo Life Sciences Inc. Rabbit polyclonal anti-ubiquitin, anti-K-48 ubiquitin, rabbit polyclonal anti c-Fos, and goat anti-mouse antibodies were purchased from Cell Signaling Technology Inc. AQueous One Solution Cell Proliferation Reagent (MTS) was obtained from Promega. The human ornithine decarboxylase/ODC1 gene ORF cDNA clone expression plasmid, N-GFPSpark, and sinofection transfection reagent were purchased from Sino Biological Inc. Embryonic kidney cells (HEK293T) were a gift from B. Sjögren (Department of Pharmacology & Toxicology, Michigan State University), while glioblastoma astrocytoma cells (U-87MG) was obtained from ATCC. The plasmid from which the Mtb proteasome was expressed and purified by the method of Lin et al.60 was a gift from the Abramovitch lab (Department of Microbiology & Molecular Genetics, Michigan State University).
Cell Culture.
RPMI-8226 cells were maintained in RPMI media. Human embryonic kidney cells (HEK293T) or glioblastoma cells (U-87MG) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin, at 37 °C with 5% CO2.
Proteasome Activity in a Purified Protein Assay.
Activity assays were performed in a 200 μL reaction volume. Different concentrations of test compounds were added to a black flat/clear bottom 96-well plate containing 1 nM human constitutive 20S proteasome, immunoproteasome (i20S), 26S proteasome, Mtb 20S proteasome, or yeast proteasome in 50 mM Tris-HCl (pH 7.5) and allowed to sit for 10 min at room temperature (RT). Fluorogenic substrates were then added, and the enzymatic activity was measured at 37 °C on a SpectraMax M5e spectrometer by measuring the increase in fluorescence units per minute for 1 h at 380/460 nm. The fluorescence unit for the vehicle control was set at 100%, and the ratio of drug-treated sample to that of vehicle control was used to calculate the fold change in enzymatic activity. Fold activity was plotted as a function of drug concentration, using GraphPad Prism 5. The fluorogenic substrates Suc-LLVYAMC (CT-L activity, 10 μM, and 40 μM for Mtb), Z-LLEAMC (Casp-L activity, 10 μM), and Boc-LRR-AMC (T-L activity, 20 μM) were used. Magnesium chloride (5 mM) and ATP (2.5 mM) were included in assays containing 26S proteasome.
Atomic Force Microscopy (AFM) Imaging.
AFM imaging of proteasome complexes was performed in tapping mode in liquid using the MultiMode Nanoscope IIIa microscope and oxide-sharpened silicon nitride probes with a nominal spring constant 0.32 N/m (Bruker Corp., Billerica, MA), as previously described.40,61 To assess the dynamics of the 20S proteasomes, the purified core particles were diluted with 5 mM Tris-HCl (pH 7.5) to 3–5 ng/μL, deposited on muscovite mica, and briefly incubated at RT (≤5 min) to allow for electrostatic binding to mica. After a few fields had been scanned, the particles were treated with dimethyl sulfoxide (DMSO) (control) or TCH-165 dissolved in DMSO directly injected into the AFM chamber. The scanning was continued to collect 6–10 images; some fields were scanned continuously and others distinctly. The titration procedure was repeated until the desired concentration of the drug was achieved. The DMSO concentration in imaging buffer was always kept <3% (v/v). Fields of 0.5–1 μm2, with a digital pixel size of 1–2 nm, were scanned with a rate of 2.65 or 3.05 Hz, with a drive voltage of 200–500 mV and a set point of 1.5–2.0 V. A majority of the particles were imaged in “standing” (top-view) position with the α-ring exposed. The topography of proteasome particles was analyzed in plane-fitted and flattened raw height mode images. Distinguishing between “closed” and “open-gate” particles was based on analysis of sections through the center of the α-ring, with the sections presenting a smooth concave surface or a central dip.
Proteasome Native Gel.
HEK293Tor U-87MG cells were grown to 80% confluency in a T-75 flask. Cells were treated with either vehicle, TCH165, TCH-023, or bortezomib at the concentrations indicated under each figure. Cells were trypsinized and washed three times with chilled phosphate-buffered saline (PBS). Cell pellets were homogenized in native lysis buffer [50 mM Tris-HCl (pH 8.0), 2 mM Na2ATP, 5 Mm MgCl2, 0.5 mM EDTA, and 10% glycerol] using acid-washed glass beads. The total protein was quantified by a bicinchoninic acid (BCA) assay, and equal amounts of lysates (30 μg) were resolved on a 3%–4%–5% (stacking–resolving–resolving) Tris-borate-EDTA (TBE) gel supplemented with Rhinohide polyacrylamide gel strengthener, for 3 h at 150 V, at 4 °C. Gels were blotted onto a PVDF membrane for 3 h at 100 V at 4 °C. Proteasome subcomplexes were probed with an anti-β5 or Rpt1 antibody.
Docking Studies.
In silico docking was performed as previously reported.29
In Vitro Degradation of α-Synuclein.
Digestion of α-synuclein was performed in a 50 μL reaction volume of 20 mM HEPES (pH 7.4), 2 mM EDTA, 1 mM EGTA, 0.5 μM purified α-synuclein, 0.5 μM GAPDH, and 15 nM purified human 20S proteasome as previously described.29 Briefly, 20S proteasome was diluted to 17 nM in reaction buffer. Test compounds or the vehicle (1 μL of a 50× stock) was added to 44 μL of 17 nM 20S and incubated at RT for 20 min. The substrate (5 μL of a 5 μM GAPDH/synuclein mixture) was then added to the reaction mixture and incubated at 37 °C for 1 h. The reactions were quenched with concentrated SDS loading buffer. After being boiled for 5 min, samples were resolved on 4 to 20% Tris-glycine sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotted with rabbit polyclonal anti-α-synuclein IgG (1:4000) and goat anti-rabbit HRP (1:5000)/anti-GAPDH-HRP. Blots were developed with ECL Western reagent and imaged with X-ray film. EDTA and EGTA were excluded from 26S degradation buffer and supplemented with magnesium chloride (5 mM) and ATP (2.5 mM).
In Vitro Degradation of Tau441.
Tau degradation was performed in the same way as that of α-synuclein with the exceptions that tau and GAPDH were used at final concentrations of 0.1 μM.
Generation of HEK293T Cells Stably Expressing Human Ornithine Decarboxylase with N-Terminal Spark Green Fluorescent Protein (GFPSpark-ODC).
HEK293T cells expressing GFPSpark-ODC were generated as previously reported.29 Briefly, HEK293T cells were seeded at a density of 1 × 105 cells/mL in a 24-well plate overnight. DNA (1 μg of GFPSpark-ODC plasmid) was mixed with 250 μL of serum free DMEM medium. Sinofection transfection reagent (5 μL) was also mixed with 250 μL of serum free medium in a separate vial. The separate mixtures were combined and allowed to sit at RT for 15 min. The mixture was then added to HEK293 cells in a 24-well plate and allowed to incubate for 4 h at 37 °C and 5% CO2 in a tissue culture incubator. The transfection medium was replaced with fresh complete culture medium (with 10% FBS). Three days later, cells were trypsinized and resuspended in hygromycin (100 μg/mL) selection medium. Surviving clones were picked and expanded in hygromycin selection medium for 6 weeks. After three passages, stable expression was confirmed by confocal fluorescent imaging, using standard filters for GFP.
GFPSpark-ODC Degradation in HEK293T Cells.
HEK293T cells stably expressing GFPSpark-ODC were seeded in a T-75 flask, in hygromycin selection medium 2 days prior to treatment, such that cells were ~80% confluent at the time of treatment. Cells were incubated with fresh medium (no hygromycin) with or without 50 μg/mL cycloheximide, in combination with either the vehicle, bortezomib (3 μM), or TCH165 (3 and 10 μM) for 24 h. GFPSpark-ODC degradation was monitored by immunoblotting of cell lysates with the GFP antibody.
c-Fos Degradation in U-87MG Cells.
U-87MG cells were grown to approximately 80% confluency in a 100 mm dish. Cells were treated with the vehicle, TCH-165 (3, 10, or 30 μM), or epoxomicin (100 nM) without or with cycloheximide (50 μg/mL) for 8 h. Cells were washed twice with warm PBS and scraped into chilled RIPA buffer supplemented with sigmafast protease inhibitor cocktail. Total protein was quantified by a BCA assay and total protein normalized to 2 mg/mL and boiled with 5× SDS loading buffer. Equal amounts of lysates were resolved on a 4 to 20% Tris-glycine gel and transferred to a PVDF membrane. The membrane was probed with anti c-Fos and anti-GAPDH antibodies.
Proteasome Activity in HEK293T Cell Lysates.
HEK293T cells were grown in a T-75 flask to ~80% confluency. Cells were treated with the vehicle, TCH-165 (10 μM), and epoxomycin (1 μM) for 12 h. Cells were trypsinized and washed twice with PBS (pH7.4). Cells were resuspended in 500 μL of lysis buffer (20 mM Tris-HCl, 5 mM MgCl2, 1 mM ATP, 0.5 mM EDTA, 1 mM DTT, and 10% glycerol) and lysed by sonication. Samples were clarified for 20 min at 14000g. The supernatant was assayed for total protein using the BCA assay and normalized to 1 mg/mL. Samples were diluted to 0.036 μg/μL in assay buffer (20 mM Tris-HCl, 5 mM MgCl2, 1 mM ATP, and 1 mM DTT), and 140 μL (5 μg of total protein) of the diluted samples was transferred to three wells of a black clear bottom 96-well plate. The substrate (10 μL of Suc-LLVY-AMC) in assay buffer was added to a final concentration of 25 μM. Kinetic readings were taken every 5 min at 37 °C, at 380/460 nm for 1 h. Epoxomicin treatment was set to zero, and the vehicle control was set to 100%.
Ubiquitin Immunoblot.
Lysates of HEK293T cells and U-87MG cells used for 26S assembly were also blotted for total ubiquitin and K-48-linked ubiquitin following SDS–PAGE.
Cell Viability Assay (MTS assay).
HEK293T cells or U-87MG cells were seeded in a clear 96 well plate at a density of 1.0 × 104 cells/well and 5.0 × 104 cells/well for RPMI-8226 cells. Cells were treated with different concentrations of TCH-165 for 24 h or 72 h. A MTS solution (20 μL) was then added and incubated under cell culture conditions for 2 h. The absorbance was read at 490 nm and expressed as a percentage of the vehicle control
ROS Detection Assay.
Reactive oxygen species were detected with DCFDA as specified by the manufacturer. Briefly, U-87MG cells were seeded in a black clear bottom 96-well plate at a density of 5.0 × 104 cells/well in 100 μL of medium overnight. Cells were treated with the vehicle, TCH-165 (10 μM), TCH-023 (10 μM), or menadione (50 μM) for 12 h. DCFDA at a 2× final concentration (50 μM final concentration) was added, and cells were incubated for a further 45 min and fluorescence readings taken at 485/535 nm. DCFDA and medium only wells were used as blanks. An additional control included cells without DCFDA (called unstained cells).
Statistical Analyses.
Data are presented as means ± the standard deviation of at least three independent experiments (for data with statistical analysis). Western blots were quantified with ImageJ, and statistical analysis was performed with GraphPad Prism 5. An unpaired Student’s t test was used for two group comparisons, while one-way analysis of variance with a post hoc Bonferroni test was used for multiple comparisons of group means. A p value of <0.05 vas considered significant.
RESULTS
TCH-165 Induces an Active 20S Conformation.
The induction of a proteolytically active 20S proteasome conformation can be achieved in vitro by pretreating the 20S proteasome with detergents such as sodium dodecyl sulfate, heat, or certain ionic conditions.62 Proteolysis of the fluorogenic chymotrypsin-Like (CT-L) peptide substrate (Suc-LLVY-AMC)29,30 occurs if an open-gate conformation of the α-ring allows the substrate to enter the core particle, resulting in the release of 7-amino-4-methylcoumarin (AMC).63 The human 20S proteasome was pretreated with various concentrations of TCH-165 and proteasome activity quantified by measuring the release of AMC from the fluorogenic chymotrypsin-like (CT-L) peptide substrate (Suc-LLVY-AMC) over time. The concentration of the drug required for induction of 50% maximum activity (EC50) was determined by fitting the relative fluorescence units and concentrations into a four-parameter dose–response curve. No fluorescence was detected in the absence of the AMC-labeled substrate, indicating that TCH-165 does not show intrinsic fluorescence at the AMC wavelength. The EC50 of TCH-165 was 4.2 μM and saturates at concentrations of >7 μM (tested up to the limits of its solubility of 70 μM). It should be noted that saturation of activity readily distinguishes these agents from detergents such as SDS, which display a sharp drop in activity at higher concentrations (Figure 2A, left).30 The imidazoline analogue TCH-023 (Figure 1) did not exhibit significant activity and was used as a negative control in many of our studies. We tested each of the three proteolytic activities of the proteasome using the standard AMC-labeled peptide substrates63,64 and found that TCH-165 enhances the chymotrypsin-like (CT-L; EC50 = 4.2 μM), trypsin-like (Tryp-L; EC50 = 3.2 μM), and caspase-like (Casp-L; EC50 = 4.7 μM) activities (Figures 1 and 2B). This observation suggests enrichment in the open-gate 20S conformation that allows access of each of the substrates to the catalytic chamber.65 Under similar assay conditions, TCH-165 had no effect on the proteolytic activity of the fully assembled 26S proteasome (Figure 2A, right).
Figure 2.

TCH-165 enhances 20S-mediated degradation of peptides, α-syn, and tau by inducing an open-gate 20S conformation. (A) Concentration–response curve of TCH-165 for 20S (left) and 26S (right) proteasome-mediated proteolysis of the fluorogenic substrates Suc-LLVY-AMC for chymotrypsin-like (CT-L), Boc-LRR-AMC for trypsin-like (Tryp-L), and Z-LLE-AMC for caspase-like (Casp-L) activities. (B) EC50 values of TCH-165 and maximum fold enhancement of 20S activities. (C) Immunoblot and quantification of GAPDH and α-synuclein digestion with the 20S proteasome pretreated with TCH-165 or the proteasome inhibitor bortezomib (BTZ, 2 μM). (D) Immunoblot and quantification of GAPDH and tau441 digestion with the 20S proteasome pretreated with TCH-165 or the proteasome inhibitor bortezomib (BTZ, 2 μM). (E) Tilted top-view AFM images of standing particles of closed- and open-gate 20S proteasome particles and content of open-gate conformers in populations of the 20S proteasome treated with various concentrations of TCH-165. The bar graph shows means ± the standard deviation (SD) of at least four fields with 120–260 particles per field. Western blots were quantified with ImageJ and are presented as means ± SD of at least three independent experiments. (F) Top view of the α-ring showing the preferred docking site utilizing Autodock Vina of TCH-165 in the α1/α2 intersubunit-binding pocket of the α-ring of the 20S proteasome. (G) The Rpt-3 peptide (2 μM) inhibits TCH-165 (1 μM) enhancement of 20S proteolysis (n = 3). One-way analysis of variance with the post hoc Bonferroni test was used for multiple comparisons of means in GraphPad Prism 5 (ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001).
To determine whether the enhanced 20S activity translated to more physiologically relevant targets, we investigated the ability of TCH-165 to enhance 20S-mediated degradation of the intrinsically disordered proteins α-synuclein (α-syn) and tau (tau441).29,30 For these studies, the disordered proteins α-syn (Figure 2C) and tau (Figure 2D) were mixed with the structured protein, GAPDH, and treated with the purified 20S proteasome in the presence of various concentrations of TCH-165 or bortezomib (BTZ, proteasome inhibitor as a negative control)66 for 1 h. The mixture was analyzed for protein degradation by Western blotting. Panels C and D of Figure 2 clearly demonstrate that TCH-165 enhanced the degradation of both α-syn and tau over the vehicle control (Figure 2C,D; p < 0.01). Importantly, TCH-165 did not induce the degradation of GAPDH (Figure 2C,D, bottom panels). These data demonstrate two important findings. (1) TCH-165 enhances 20S-mediated degradation of IDPs, α-syn, and tau in vitro, and (2) TCH-165 does not induce the degradation of structured proteins such as GAPDH.
To gain insight into the mechanism of imidazoline-induced activation of 20S in a single-molecule fashion, AFM was used to study TCH-165-mediated gate-switching dynamics of the 20S proteasome. Control native eukaryotic proteasome particles repeatedly scanned and imaged by oscillating (tapping) mode AFM in liquid constantly alternate between open and closed forms, with the more stable closed-gate form in ~3:1 excess (Figure 2E).40,61 Analysis of >100 single 20S particles in the top-view position revealed a concentration-dependent increase in the abundance of open-gate conformers upon treatment with TCH-165, from 28 ± 4% in the control to 59 ± 3% at 2 μM TCH-165 (Figure 2E; p < 0.001 for ≥400 nM). The TCH-165-treated particles continuously switched between open and closed forms, similar to control particles. However, unlike the control, the population of particles in the open-gate state constituted the majority of conformers. We speculate that the open-gate form represents particles with an unobscured or just-clearing path for substrates to enter the catalytic chamber. Thus, treatment with TCH-165 increases the population of substrate-receptive, activated 20S proteasomes. This notion is consistent with our agglomerated data demonstrating enhanced activities of TCH-165-treated 20S core proteasomes.
The Collective Data Suggest an Interaction of TCH-165 with the α-Ring of the 20S Proteasome.
In silico docking studies were performed to determine possible binding sites of TCH-165 that may explain its gate regulation seen in the AFM studies. We used Autodock Vina67 run through PyRx68 to manage the workflow. For these studies, TCH-165 was geometrically optimized with the MM2 force field. Autodock Vina identified molecular conformations with the best fit and strongest binding affinity (global minima). These unbiased docking studies found that one pocket, the α1/α2 intersubunit pocket, was primarily targeted by TCH-165 (Figure 2F and Figure S1). Binding affinities at this binding pocket were >9 kcal/mol for the active compound. Binding affinities for the inactive analogue TCH-023 (Figure 1) were <6 kcal/mol. Interestingly, this pocket was also primarily targeted by the phenothiazine-based 20S agonists.29
The α1/α2 intersubunit pocket is known to bind the C-terminal Hb-Y-X-motif of the Rpt-3 subunit of the 19S cap.69 Thus, we prepared the peptide based on the C-terminatl sequence of the Rpt-3 subunit (KDEQEHEFYK) and performed a competition experiment with the Rpt-3 peptide and TCH-165. At 1 μM TCH-165, 20S activity is enhanced 2-fold. Treatment with the Rpt-3 peptide (2 μM) alone had no effect on 20S-mediated proteolysis. However, the presence of the Rpt-3 peptide inhibited TCH-165 enhancement of 20S activity by approximately 30% (Figure 2G). The Rpt-5 peptide indicated no signs of competition with TCH-165 (data not shown). Although these data indicate that the interaction of the Rpt-3 peptide with the α-ring prevents 20S activation by TCH-165 (Figure 2G), it does not necessarily dictate pocket selectivity as higher concentrations of Rpt3 could not overcome TCH-165 activity any further. To further examine the role of the α-ring, we tested the immuno-proteasome (i20S), the yeast proteasome, and the Mycobacterium tuberculosis proteasome (Mtb 20S) for activation by TCH-165. The i20S has an α-ring identical to that of the constitutive 20S CP but incorporates the structurally different catalytic subunits LMP7, MECL1, and LMP2, instead of β5, β2, and β1, into its core.70,71 The yeast proteasome is similar to the human 20S proteasome but has significant topological differences in the subunits of the α-ring.72 In contrast, the Mtb 20S proteasome has an α-ring comprised of seven identical subunits (α1–α7) and displays a very different α-ring topology.73 Similar to the case for the constitutive 20S, TCH-165 was capable of enhancing the proteolytic activities of the i20S by >3-fold (Figure S2); however, TCH-165 was unable to enhance the activity of the yeast or Mtb 20S proteasome (Figure S3). These data confirm the significance of α-ring topology for TCH-165 activity. Consistent with these observations, TCH-165 was unable to enhance 26S proteasome-mediated proteolysis of fluorescent peptides (Figure 2A, right) or disordered protein α-syn (Figure S4). This suggests that blocking the α-rings with two 19S RPs (i.e., in the fully assembled 26S proteasome) prevented TCH-165 from binding on the α-ring and enhancing proteolysis. The activity of the TCH-165-treated 20S was higher than that of the equimolar 26S proteasome in these assays. This suggests that the lack of enhancement of 26S activity by TCH-165 was not due to the maxium activity of the core enzyme of 26S, but rather a consequence of the TCH-165-binding site being occupied by the 19S caps. Collectively, these data are consistent with an interaction of TCH-165 with the α-ring of the 20S proteasome.
TCH-165 Enhances the Degradation of Intrinsically Disordered Proteins in Cell Cultures.
Next, we assessed if the in vitro activation of the 20S translated in the enhancement of 20S-mediated degradation of IDPs in cells. Given the antitumor efficacy of this molecule class, we turned our attention to proteins that are overexpressed in certain cancers.58,59 For these studies, we tested TCH-165 in HEK293T cells expressing ornithine decarboxylase (ODC), labeled with GFPSpark at its N-terminus.30 GFPSpark is a structured 28 kDa protein and is not degraded by the 20S, whereas the disordered C-terminal of ODC is a well-known 20S substrate (Figure 3A).12,24,30 Thus, 20S agonists will enhance the C-terminal degradation of the GFPSpark-ODC fusion protein (78 kDa), but not GFP, resulting in a band shift that can be detected by Western blotting using an anti-GFP antibody. Cells were cultured with TCH-165 for 24 h, and then the lysates were immunoblotted with anti-GFP (Figure 3B,C). Cycloheximide was used to ensure changes were occurring at the post-translational level. Figure 3B shows that TCH-165 enhanced the cleavage of ODC, but not the structured GFP, resulting in the presence of free GFPSpark (28 kDa) and the GFPSpark-ODC fragments (~50 kDa). Importantly, TCH-165-enhanced proteolytic degradation was blocked by bortezomib (Figure 3B, BTZ, 3 μM). The fact that TCH-165-enhanced ODC degradation is blocked by BTZ indicated that this event is proteasome-mediated. Figure 3C shows that the enhanced proteolytic degradation is also concentration-dependent. In addition, no changes in levels of other structured proteins were detected, as illustrated by the levels of GAPDH and β-actin. In the absence of cycloheximide, a continuous production of the fusion protein was seen (i.e., no decrease in the level of the GFPSpark-ODC fusion protein), but a concentration-dependent increase in the level of 20S-mediated cleavage products of ODC was maintained (Figure S5).
Figure 3.

TCH-165 enhances the degradation of intrinsically disordered proteins in cell cultures. (A) Schematic of ODC-GFPSpark degradation by the 20S proteasome. (B) HEK cells stably expressing GFPSpark-ODC were treated with cycloheximide (50 μg/mL) and either the vehicle, TCH-165 (10 μM), BTZ (3 μM), or a combination of TCH-165 and BTZ for 24 h. Cell lysates were immunoblotted with anti-GFP. (C) HEK cells stably expressing GFPSpark-ODC were treated with TCH-165 (0, 3, and 10 μM) or bortezomib (BTZ, 3 μM) with cycloheximide (50 μg/mL) for 24 h. Cell lysates were immunoblotted with anti GFP. (D) Glioblastoma cells (U-87MG) were treated with the vehicle or TCH-165 (10 μM) in combination with or without epoxomicin (100 nM) for 8 h and immunoblotted with anti c-Fos. GAPDH and β-actin were used as loading controls. (E) Glioblastoma cells (U-87MG) were treated with TCH-165 (0, 3, 10, or 30 μM) and epoxomicin (100 nM) in combination with either the vehicle or TCH-165 (10 μM) for 8 h (with 50 μg/mL) and immunoblotted with anti c-Fos. GAPDH and β-actin were used as loading controls.
The proto-oncoprotein c-Fos is also intrinsically disordered in its C-terminal transactivation domain.74 Similar to ODC, c-Fos is targeted for degradation by the 26S proteasome in a ubiquitin-dependent manner, but the bulk of its degradation is mediated by the 20S in a ubiquitin-independent fashion.75–79 Because of its involvement in c-Fos/c-Jun AP-1-mediated expression of pro-inflammatory cytokines and neoplastic cellular transformation, the pharmacological regulation of c-Fos may have therapeutic implications in the treatments of intervertebral disk degeneration, rheumatoid arthritis, and several types of cancer, including malignant gliomas.80–83 Considering that the brain expresses one of the highest levels of disordered proteins,84,85 we evaluated the effects of TCH-165 on c-Fos degradation in the human glioblastoma cells (U-87MG). Treatment of U-87MG cells with TCH-165 for 8 h resulted in a significant reduction in c-Fos levels compared to the vehicle control (Figure 3D). Cells treated with TCH-165 also exhibited a concentration-dependent decrease in c-Fos protein levels in cycloheximide-treated cells (Figure 3E), which confirms that the c-Fos reduction is at the post-translation level and not due to changes in protein synthesis. Importantly, the enhanced degradation of c-Fos by TCH-165 was blocked by the selective proteasome inhibitor epoxomicin (100 nM), confirming the proteasome as the main target for TCH-165-enhanced degradation of c-Fos (Figure 3D,E). Again, the structured protein GAPDH was not affected by TCH-165 treatment. These data show that TCH-165 induces the proteasomal degradation of IDPs, such as ODC and c-Fos, over structured proteins in cell cultures.
TCH-165-Treated Cells Display a Decrease in the Assembled 26S and an Increase in the 20S Proteasome.
Endogenous proteasome activators, including PA28αβ, PA28γ, and Blm10, compete with the 19S RP for binding to the 20S CP.52,54,55 Enhanced degradation of intrinsically disordered proteins by TCH-165 can be a result of the increased enzymatic activity of the 20S, the increased amount of free 20S via competition with the 19S caps, or a combination of both effects. To examine the role of small molecule activators on 19S RP-CP binding, we examined possible changes in the distribution of proteasome subcomplexes. HEK293T cells were treated with the vehicle and various concentrations of TCH-165 (3, 10, and 30 μM) for 24 h, and then cell lysates were resolved on a native gel and immunoblotted for proteasome subcomplexes (using anti-β5 and anti-Rpt1). Remarkably, the immunoblot revealed a concentration-dependent decrease in the amount of fully assembled 26S proteasome (19S–20S–19S) and singly capped proteasome [19S–20S (Figure 4A)]. This was accompanied by a concentration-dependent increase in the levels of free 19S and 20S proteasomes (Figure 4B). No changes were observed in the levels of individual subunits (β5 of the 20S core and Rpt1 of the 19S cap) (Figure 4A). This suggests that changes in the level of 26S and 20S were not occurring at the transcription/translation level but are due to a shift in the equilibrium between free 20S CP and assembled 26S proteasome particles (Figure 4B). Furthermore, treatment of HEK293T cells with TCH-165 (30 μM) resulted in a time-dependent decrease in assembled 26S (singly and doubly capped) and an increase in the free 20S proteasome (Figure 4C,D). The inactive analogue TCH-023 had no effect on the assembled complexes (Figure 4E). Endogenous disassembly of the 26S proteasome can also occur following oxidative stress. Therefore, we checked whether TCH-165 induced reactive oxygen species (ROS) via an alternative mechanism, but no significant difference was observed in ROS levels for vehicle- or TCH-165-treated cells (Figure S7). Additionally, no significant cell death was observed after treatment for 24 h using concentrations of ≤10 μM TCH-165 (<20% cell death) and some (22%) at higher concentrations (30 μM), perhaps because of the accumulation of ubiquitinated proteins (Figure S8).
Figure 4.

TCH-165 induces a concentration- and time-dependent decrease of assembled 26S, favoring an increase of free 20S proteasome. (A) HEK293T cells treated with the vehicle or TCH-165 (3, 10, and 30 μM) for 24 h were immunoblotted for proteasome subcomplexes following native PAGE. Proteasome subunits (β5 and Rpt1) and GAPDH (loading control) were also immunoblotted after SDS–PAGE. (B) The data in panel A were quantified with ImageJ (n = 4). (C) HEK293T cells were treated with TCH-165 (30 μM) for 0, 2, 4, 8, 12, and 24 h and immunoblotted for proteasome subcomplexes following native PAGE. (D) The data in panel C were quantified with ImageJ (n = 3). (E) HEK293T cells were treated with the vehicle, TCH-165 or TCH-023 (0, 10, and 30 μM), or bortezomib (BTZ, 50 nM) for 24 h and immunoblotted for proteasome subcomplexes following native PAGE. (F) Hydrolysis of Suc-LLVY-AMC in HEK cell lysates. HEK293T cells were treated with the vehicle, TCH-165 (10 μM), or epoxomycin (1 μM) for 12 h followed by cell lysis and subsequent exposure to the CT-L peptide substrate Suc-LLVY-AMC. Bar graphs are presented as means ± SD of four independent experiments.
Next, we investigated whether TCH-165 enhances the proteasome activity in cells by measuring the chymotryptic-like activity in HEK293T cells. For this, we exposed the cells to the vehicle, TCH-165, and the nonreversible selective proteasome inhibitor, epoxomycin, and evaluated CT-L activity in the cell lysate after exposure for 12 h, using the CT-L probe Suc-LLVY-AMC. Figure 4F shows that the CT-L activity is increased significantly (p < 0.05) after TCH-165 treatment for 12 h compared to the vehicle control. Whether the observed enhanced CT-L proteolysis is due to the total increase in 20S particles or to an increase in 20S open-gate particles will be difficult to assess in cells.
Considering the decrease in the amount of fully assembled 26S proteasome, we investigated possible changes in the clearance of ubiquitinated proteins. For these studies, we treated HEK293T cells with the vehicle or TCH-165 for 2, 4, 8, 12, and 24 h, and then the lysates were probed for ubiquitinated proteins. Panels A and B of Figure 5 show that the level of total ubiquitinated and K-48 ubiquitinated proteins (Figure 5C) remains largely the same. As anticipated, only when there is no 19S-capped proteasome present (i.e., 19S–20S–19S and 19S–20S) do ubiquitinated substrates start to accumulate (Figure 5D). These data illustrate that the singly capped (i.e., 19S–20S) or hybrid proteasome is still fully capable of processing ubiquitinated proteins, and perhaps even more efficient than the doubly capped form. Thus, at low or intermediate concentrations, TCH-165 does not induce significant accumulation of ubiquitinated proteins.
Figure 5.

Accumulation of ubiquitinated proteins correlates to the levels of singly and doubly capped 20S. (A) HEK293T cells were treated TCH-165 (30 μM) for 0, 2, 4, 8, 12, and 24 h and immunoblotted for proteasome subcomplexes following native PAGE and ubiquitin and proteasome subunit β−5 following SDS–PAGE. GAPDH was used as a loading control. (B) Quantification of ubiquitinated proteins was performed with ImageJ, and bar graphs are presented as means ± SD of three independent experiments. (C) Quantification of K-48-ubiquitinated proteins was performed with ImageJ, and bar graphs are presented as means ± SD of three independent experiments. (D) HEK cells were treated with the vehicle or TCH-165 (3, 10, and 30 μM) for 24 h and immunoblotted with anti-β5 (top panel, native gel) and anti-K-48 ubiquitin (bottom panel, denaturing gel). (C) U-87MG cells treated with the vehicle or TCH-165 (3, 10, and 30 μM) for 24 h and cell lysates were immunoblotted for proteasome subcomplexes (top panel, native gel) and K-48-linked ubiquitin (bottom panel, SDS−PAGE).
Similarly, when U-87MG cells were probed for TCH-165 modulation of proteasome assembly, a concentration response decrease of the fully assembled 26S proteasome was also detected (Figure 5E, top panel). Here, the singly capped, hybrid proteasome (i.e., 19S−20S) persisted at tested concentrations of ≤30 μM, resulting in no accumulation of ubiquitinated proteins (Figure 5E, bottom panel). The significance of this finding is that as long as hybrid proteasome complexes (19S–20S) remain in the cells, cells are capable of processing ubiquitinated substrates. However, the amount of cleared ubiquitinated substrates varies among cell lines and may depend on several additional factors. For example, many E3 ubiquitin ligases are also IDPs, and the enhanced degradation of disordered E3 ligases by these molecules may prevent substantial accumulation of polyubiquitinated proteins.86,87 These contributing factors that regulate ubiquitinated substrates are under further investigation in our lab.
In conclusion, we report here the unique finding of a small molecule that is capable of enhancing the clearance of intrinsically disordered proteins by modulating the dynamic equilibrium between proteasome complexes, favoring the free 20S. These findings represent a new mechanism of proteasome regulation, consistent with our previous findings that these agents effectively block growth in bortezomib resistant THP-1 cells.59 A similar efficacy was observed among various additional cancer cell lines tested with CC50 values of 1.6 and 2.4 μM for multiple myeloma cell line RPMI 8226 and glioblastoma cell line U-87MG, respectively (Figure S8). These studies provide support for the possibility of enhancing 20S selective degradation of intrinsically disordered proteins by modulation of 26S proteasome assembly.
DISCUSSION
Intrinsically disordered proteins are unique in that they do not adopt a defined three-dimensional structure, which allows them to interact with multiple protein partners and control multiple cellular events. When IDP production outpaces its removal, the subsequent accumulation can lead to harmful signaling directly associated with the pathogenesis of many different human diseases. Targeting IDPs for therapeutic intervention is a daunting challenge because of the lack of a defined three-dimensional structure, which has impeded classic “small molecule-binding pocket” drug design. Here, we describe the identification of a small molecule that modulates 26S assembly favoring an increase in 20S proteasome particles, resulting in an enhancement of 20S-mediated degradation of IDPs. Proteolytically active singly capped proteasome complexes (19S–20S) remain in the cells, still capable of degrading ubiquitinated substrates at effective concentrations at which enhancement of 20S proteolysis of IDP is observed. However, at high concentrations, the decrease in 19S-capped proteasome complexes will prevent the efficient degradation of ubiquitinated substrates. These effects may be cell type specific and will likely induce apoptosis in proteasome reliant cells, including multiple myeloma cells. These findings illustrate that regulation of the dynamic equilibrium between different proteasome complexes by small molecules represents a new approach to controlling the proteolytic degradation of specific protein classes. Modulation of proteasome assembly by small molecules can provide a new therapeutic strategy for targeting dysregulation of IDPs such as α-synuclein (most notably associated with Parkinson’s disease), tau (most notably associated with Alzheimer’s disease), and the proto-oncogenes ornithine decarboxylase and c-Fos, and likely many other IDPs associated with the pathogenesis of different human diseases.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank T. A. Lansdell for helpful discussions.
Funding
Financial support for this work was provided in part by the National Institute of Allergy and Infectious Diseases (1R21AI117018–01A1) and the National Institute of General Medical Sciences (T32GM092715) of the National Institutes of Health. The authors also gratefully acknowledge financial support from the SPG of Michigan State University (J.J.T.), the Clinical and Translational Science Institute (J.J.T) and the William and Ella Owens Foundation for Medical Research (P.A.O. and M.G).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00579.
In silico docking studies using Autodock Vina, showing binding of TCH-165 in the α1/α2 intersubunit pocket (Figure S1), figure showing that TCH-165 also enhances the proteolytic activity of the immunoproteasome core particle (i20S) (Figure S2), figure showing that TCH-165 does not enhance the proteolytic activity of the M. tuberculosis or yeast proteasome core particle (Figure S3), figure showing that TCH-165 does not enhance 26S-mediated proteolysis of α-synuclein (Figure S4), figure showing that TCH-165 enhances the degradation of the intrinsically disordered protein, ODC, in cell cultures (Figure S5), evaluation of reactive oxygen species in U-87MG cells treated with the vehicle (negative control), TCH-165, TCH-023, and menadione (positive control) (Figure S6), viability of HEK293T cells treated with various concentrations of TCH-165 for 24 h (Figure S7), and cytotoxicity in RPMI-8226 and U-87MG cells treated with various concentrations of TCH-165 for 72 h (Figure S8) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, et al. (2001) Intrinsically disordered protein. J. Mol. Graphics Modell 19, 26–59. [DOI] [PubMed] [Google Scholar]
- (2).DeForte S, and Uversky VN (2016) Order, Disorder, and Everything in Between. Molecules 21, e1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Babu MM, van der Lee R, de Groot NS, and Gsponer J (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol 21, 432–440. [DOI] [PubMed] [Google Scholar]
- (4).Berrocal R, Vasquez V, Krs SR, Gadad BS, and Rao KS (2015) alpha-Synuclein Misfolding Versus Aggregation Relevance to Parkinson’s Disease: Critical Assessment and Modeling. Mol. Neurobiol 51, 1417–1431. [DOI] [PubMed] [Google Scholar]
- (5).Korsak M, and Kozyreva T (2015) Beta Amyloid Hallmarks: From Intrinsically Disordered Proteins to Alzheimer’s Disease. Adv. Exp. Med. Biol 870, 401–421. [DOI] [PubMed] [Google Scholar]
- (6).Uversky VN, Na I, Landau KS, and Schenck RO (2017) Highly Disordered Proteins in Prostate Cancer. Curr. Protein Pept. Sci 18, 453–481. [DOI] [PubMed] [Google Scholar]
- (7).Uversky VN, Oldfield CJ, and Dunker AK (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys 37, 215–246. [DOI] [PubMed] [Google Scholar]
- (8).Ben-Nissan G, and Sharon M (2014) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jung T, Hohn A, and Grune T (2014) The proteasome and the degradation of oxidized proteins: part III-Redox regulation of the proteasomal system. Redox Biol. 2, 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jung T, Hohn A, and Grune T (2014) The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol. 2, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tanaka K, Mizushima T, and Saeki Y (2012) The proteasome: molecular machinery and pathophysiological roles. Biol. Chem 393, 217–234. [DOI] [PubMed] [Google Scholar]
- (12).Erales J, and Coffino P (2014) Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta, Mol. Cell Res 1843, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fabre B, Lambour T, Delobel J, Amalric F, Monsarrat B, Burlet-Schiltz O, and Bousquet-Dubouch MP (2013) Subcellular distribution and dynamics of active proteasome complexes unraveled by a workflow combining in vivo complex cross-linking and quantitative proteomics. Mol. Cell. Proteomics 12, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grune T, Catalgol B, Licht A, Ermak G, Pickering AM, Ngo JK, and Davies KJ (2011) HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radical Biol. Med 51, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Huang Q, Wang H, Perry SW, and Figueiredo-Pereira ME (2013) Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J. Biol. Chem 288, 12161–12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang X, Yen J, Kaiser P, and Huang L (2010) Regulation of the 26S proteasome complex during oxidative stress. Sci. Signaling 3, ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alvarez-Castelao B, Goethals M, Vandekerckhove J, and Castano JG (2014) Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim. Biophys. Acta, Mol. Cell Res 1843, 352–365. [DOI] [PubMed] [Google Scholar]
- (18).David DC, Layfield R, Serpell L, Narain Y, Goedert M, and Spillantini MG (2002) Proteasomal degradation of tau protein. J. Neurochem 83, 176–185. [DOI] [PubMed] [Google Scholar]
- (19).Grune T, Botzen D, Engels M, Voss P, Kaiser B, Jung T, Grimm S, Ermak G, and Davies KJ (2010) Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys 500, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Radwan M, Wood RJ, Sui X, and Hatters DM (2017) When proteostasis goes bad: Protein aggregation in the cell. IUBMB Life 69, 49–54. [DOI] [PubMed] [Google Scholar]
- (21).Tofaris GK, Layfield R, and Spillantini MG (2001) alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26. [DOI] [PubMed] [Google Scholar]
- (22).Yen SS (2011) Proteasome degradation of brain cytosolic tau in Alzheimer’s disease. Int. J. Clin. Exp. Pathol 4, 385–402. [PMC free article] [PubMed] [Google Scholar]
- (23).Zouambia M, Fischer DF, Hobo B, De Vos RA, Hol EM, Varndell IM, Sheppard PW, and Van Leeuwen FW (2008) Proteasome subunit proteins and neuropathology in tauopathies and synucleinopathies: Consequences for proteomic analyses. Proteomics 8, 1221–1236. [DOI] [PubMed] [Google Scholar]
- (24).Asher G, Bercovich Z, Tsvetkov P, Shaul Y, and Kahana C (2005) 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 17, 645–655. [DOI] [PubMed] [Google Scholar]
- (25).Asher G, Reuven N, and Shaul Y (2006) 20S proteasomes and protein degradation ″by default″. BioEssays 28, 844–849. [DOI] [PubMed] [Google Scholar]
- (26).Mohan RR, Challa A, Gupta S, Bostwick DG, Ahmad N, Agarwal R, Marengo SR, Amini SB, Paras F, MacLennan GT, et al. (1999) Overexpression of ornithine decarboxylase in prostate cancer and prostatic fluid in humans. Clin. Cancer Res. 5, 143–147. [PubMed] [Google Scholar]
- (27).Shukla-Dave A, Castillo-Martin M, Chen M, Lobo J, Gladoun N, Collazo-Lorduy A, Khan FM, Ponomarev V, Yi Z, Zhang W, et al. (2016) Ornithine Decarboxylase Is Sufficient for Prostate Tumorigenesis via Androgen Receptor Signaling. Am. J. Pathol 186, 3131–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, Kenner L, Tschachler E, and Wagner EF (2008) Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther 10, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jones CL, Njomen E, Sjogren B, Dexheimer TS, and Tepe JJ (2017) Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol 12, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Trader DJ, Simanski S, Dickson P, and Kodadek T (2017) Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim. Biophys. Acta, Gen. Subj 1861, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Trippier PC, Zhao KT, Fox SG, Schiefer IT, Benmohamed R, Moran J, Kirsch DR, Morimoto RI, and Silverman RB (2014) Proteasome Activation is a Mechanism for Pyrazolone Small Molecules Displaying Therapeutic Potential in Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci 5, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B, Zaal EA, van de Kooij B, Laufer S, Heck AJR, et al. (2017) Proteasome Activation by Small Molecules. Cell Chem. Biol 24, 725–736. [DOI] [PubMed] [Google Scholar]
- (34).Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, and Duff KE (2016) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med 22, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Krahn JH, Kaschani F, and Kaiser M (2017) Turning-ON Proteasomes. Cell Chem. Biol 24, 653–655. [DOI] [PubMed] [Google Scholar]
- (36).Budenholzer L, Cheng CL, Li Y, and Hochstrasser M (2017) Proteasome Structure and Assembly. J. Mol. Biol 429, 3500–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, and Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386, 463–471. [DOI] [PubMed] [Google Scholar]
- (38).Finley D, Chen X, and Walters KJ (2016) Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci 41, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, and Finley D (2000) A gated channel into the proteasome core particle. Nat. Struct. Biol 7, 1062–1067. [DOI] [PubMed] [Google Scholar]
- (40).Osmulski PA, Hochstrasser M, and Gaczynska M (2009) A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the alpha-ring channel. Structure 17, 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Forster A, Masters EI, Whitby FG, Robinson H, and Hill CP (2005) The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol. Cell 18, 589–599. [DOI] [PubMed] [Google Scholar]
- (42).Stadtmueller BM, Ferrell K, Whitby FG, Heroux A, Robinson H, Myszka DG, and Hill CP (2010) Structural models for interactions between the 20S proteasome and its PAN/19S activators. J. Biol. Chem 285, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, and Hill CP (2000) Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 408, 115–120. [DOI] [PubMed] [Google Scholar]
- (44).Tomko RJ Jr., and Hochstrasser M (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem 82, 415–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chen S, Wu J, Lu Y, Ma YB, Lee BH, Yu Z, Ouyang Q, Finley DJ, Kirschner MW, and Mao Y (2016) Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U. S. A 113, 12991–12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Smith DM, Chang SC, Park S, Finley D, Cheng Y, and Goldberg AL (2007) Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 27, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ding Z, Fu Z, Xu C, Wang Y, Wang Y, Li J, Kong L, Chen J, Li N, Zhang R, et al. (2017) High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res. 27, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Huang X, Luan B, Wu J, and Shi Y (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol 23, 778–785. [DOI] [PubMed] [Google Scholar]
- (49).Latham MP, Sekhar A, and Kay LE (2014) Understanding the mechanism of proteasome 20S core particle gating. Proc. Natl. Acad. Sci. U. S. A 111, 5532–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, and Groll M (2008) Ubiquitin docking at the proteasome through a novel pleckstrinhomology domain interaction. Nature 453, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wehmer M, and Sakata E (2016) Recent advances in the structural biology of the 26S proteasome. Int. J. Biochem. Cell Biol 79, 437–442. [DOI] [PubMed] [Google Scholar]
- (52).Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, and Tanaka K (2000) Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J. Biol. Chem 275, 14336–14345. [DOI] [PubMed] [Google Scholar]
- (53).Wilk S, Chen WE, and Magnusson RP (1999) Modulators of the activation of the proteasome by PA28 (11S reg). Mol. Biol. Rep 26, 39–44. [DOI] [PubMed] [Google Scholar]
- (54).Dahlmann B, Ruppert T, Kuehn L, Merforth S, and Kloetzel PM (2000) Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J. Mol. Biol 303, 643–653. [DOI] [PubMed] [Google Scholar]
- (55).Pickart CM, and VanDemark AP (2000) Opening doors into the proteasome. Nat. Struct. Biol 7, 999–1001. [DOI] [PubMed] [Google Scholar]
- (56).Dange T, Smith D, Noy T, Rommel PC, Jurzitza L, Cordero RJ, Legendre A, Finley D, Goldberg AL, and Schmidt M (2011) Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem 286, 42830–42839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sadre-Bazzaz K, Whitby FG, Robinson H, Formosa T, and Hill CP (2010) Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 37, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Lansdell TA, Hurchla MA, Xiang J, Hovde S, Weilbaecher KN, Henry RW, and Tepe JJ (2013) Noncompetitive Modulation of the Proteasome by Imidazoline Scaffolds Overcomes Bortezomib Resistance and Delays MM Tumor Growth in Vivo. ACS Chem. Biol 8, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Azevedo LM, Lansdell TA, Ludwig JR, Mosey RA, Woloch DK, Cogan DP, Patten GP, Kuszpit MR, Fisk JS, and Tepe JJ (2013) Inhibition of the human proteasome by imidazoline scaffolds. J. Med. Chem 56, 5974–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Lin G, Hu G, Tsu C, Kunes YZ, Li H, Dick L, Parsons T, Li P, Chen Z, Zwickl P, et al. (2006) Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol 59, 1405–1416. [DOI] [PubMed] [Google Scholar]
- (61).Gaczynska M, and Osmulski PA (2011) Atomic force microscopy of proteasome assemblies. Methods Mol. Biol 736, 117–132. [DOI] [PubMed] [Google Scholar]
- (62).DeMartino GN, and Slaughter CA (1999) The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem 274, 22123–22126. [DOI] [PubMed] [Google Scholar]
- (63).Gaczynska M, and Osmulski PA (2005) Characterization of noncompetitive regulators of proteasome activity. Methods Enzymol. 398, 425–438. [DOI] [PubMed] [Google Scholar]
- (64).Lansdell TA, Hewlett NM, Skoumbourdis AP, Fodor MD, Seiple IB, Su S, Baran PS, Feldman KS, and Tepe JJ (2012) Palau’amine and Related Oroidin Alkaloids Dibromophakellin and Dibromophakellstatin Inhibit the Human 20S Proteasome. J. Nat. Prod 75, 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Choi WH, de Poot SA, Lee JH, Kim JH, Han DH, Kim YK, Finley D, and Lee MJ (2016) Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun 7, 10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Groll M, Berkers CR, Ploegh HL, and Ovaa H (2006) Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 14, 451–456. [DOI] [PubMed] [Google Scholar]
- (67).Trott O, and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Dallakyan S, and Olson AJ (2015) Small-molecule library screening by docking with PyRx. Methods Mol. Biol 1263, 243–250. [DOI] [PubMed] [Google Scholar]
- (69).Smith DM, Chang SC, Park S, Finley D, Cheng Y, and Goldberg AL (2007) Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 27, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ferrington DA, and Gregerson DS (2012) Immunoproteasomes: structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci 109, 75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, and Groll M (2012) Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–738. [DOI] [PubMed] [Google Scholar]
- (72).Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Forster F, and Baumeister W (2016) Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U. S. A 113, 7816–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Li D, Li H, Wang T, Pan H, Lin G, and Li H (2010) Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 29, 2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Campbell KM, Terrell AR, Laybourn PJ, and Lumb KJ (2000) Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry 39, 2708–2713. [DOI] [PubMed] [Google Scholar]
- (75).Adler J, Reuven N, Kahana C, and Shaul Y (2010) c-Fos proteasomal degradation is activated by a default mechanism, and its regulation by NAD(P)H:quinone oxidoreductase 1 determines c-Fos serum response kinetics. Mol. Cell. Biol 30, 3767–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Basbous J, Jariel-Encontre I, Gomard T, Bossis G, and Piechaczyk M (2008) Ubiquitin-independent- versus ubiquitin-dependent proteasomal degradation of the c-Fos and Fra-1 transcription factors: is there a unique answer? Biochimie 90, 296–305. [DOI] [PubMed] [Google Scholar]
- (77).Bossis G, Ferrara P, Acquaviva C, Jariel-Encontre I, and Piechaczyk M (2003) c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol. Cell. Biol 23, 7425–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Gomard T, Jariel-Encontre I, Basbous J, Bossis G, Mocquet-Torcy G, and Piechaczyk M (2008) Fos family protein degradation by the proteasome. Biochem. Soc. Trans 36, 858–863. [DOI] [PubMed] [Google Scholar]
- (79).Jariel-Encontre I, Bossis G, and Piechaczyk M (2008) Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta, Rev. Cancer 1786, 153–177. [DOI] [PubMed] [Google Scholar]
- (80).Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S, and Shiozawa S (2008) Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol 26, 817–823. [DOI] [PubMed] [Google Scholar]
- (81).Liu ZG, Jiang G, Tang J, Wang H, Feng G, Chen F, Tu Z, Liu G, Zhao Y, Peng MJ, et al. (2016) c-Fos over-expression promotes radioresistance and predicts poor prognosis in malignant glioma. Oncotarget 7, 65946–65956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Makino H, Seki S, Yahara Y, Shiozawa S, Aikawa Y, Motomura H, Nogami M, Watanabe K, Sainoh T, Ito H, et al. (2017) A selective inhibition of c-Fos/activator protein-1 as a potential therapeutic target for intervertebral disc degeneration and associated pain. Sci. Rep 7, 16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Motomura H, Seki S, Shiozawa S, Aikawa Y, Nogami M, and Kimura T (2018) A selective c-Fos/AP-1 inhibitor prevents cartilage destruction and subsequent osteophyte formation. Biochem. Biophys. Res. Commun 497, 756–761. [DOI] [PubMed] [Google Scholar]
- (84).Panda A, Acharya D, and Chandra Ghosh T (2017) Insights into human intrinsically disordered proteins from their gene expression profile. Mol. BioSyst 13, 2521–2530. [DOI] [PubMed] [Google Scholar]
- (85).Uversky VN (2014) Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Front. Mol. Biosci 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Bhowmick P, Pancsa R, Guharoy M, and Tompa P (2013) Functional diversity and structural disorder in the human ubiquiti-nation pathway. PLoS One 8, e65443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Guharoy M, Bhowmick P, and Tompa P (2016) Design Principles Involving Protein Disorder Facilitate Specific Substrate Selection and Degradation by the Ubiquitin-Proteasome System. J. Biol. Chem 291, 6723–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.