

Abstract
Alkylations of proline-based imidazolidinones are described based on the principle of self-regeneration of stereocenters (SRS), affording high levels of either the cis or trans configured products. Stereoselectivity is dictated solely on the nature of the “temporary” group, where isobutyraldehyde-derived imidazolidinones provide the cis configured products and 1-naphthaldehyde-derived imidazolidinones afford the complementary trans configured products. These stereodivergent products can be readily cleaved to afford both α-alkylated proline enantiomers from readily available L-proline. A series of imidazolidinones were alkylated to investigate the origin of the anti-selectivity. Potential contributions toward the observed anti-selectivity are discussed on the basis of these experiments, suggesting a refined hypothesis for selectivity may be in order.
Keywords: α-quaternary amino amides, self-regeneration of stereochemistry, imidazolidinones, alkylation, enolate
Graphical abstract
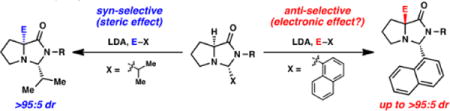
Introduction
The self-regeneration of stereocenters (SRS), pioneered by Seebach, has been well-established as a sound method for generating compounds with extremely high levels of stereoenrichment.1 The general conceit of this method is to conserve the existing stereogenicity in a conveniently accessed starting material via a sequence of diastereoselective processes. The overall sequence, illustrated for α-alkylation in Fig. 1, can be considered as follows: (1) diastereoselectively attach a “temporary” group to the starting material based on the prevailing stereochemistry; (2) execute a second transformation in a diastereoselective fashion, steered by the stereochemistry associated with that temporary group; and (3) remove the temporary group. This net process accomplishes a derivatization that, if executed differently, would destroy the originating stereochemistry. This generalized process has been applied in a multitude of contexts based on amino acids, α-hydroxy acids, among other motifs.
Fig. 1.
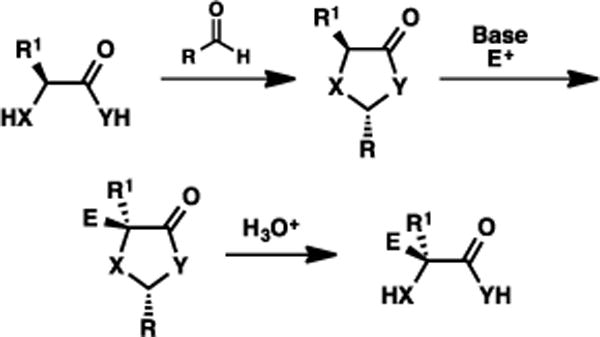
General concept for Self-Regeneration of Stereocenters (SRS).
As part of an overarching strategy for novel catalytic directed C-H functionalizations based on temporary molecular scaffolds,2 we had discovered compounds reminiscent of the structural classes that are synthesized via SRS chemistry (e.g., amide 1, Fig. 2). These compounds were designed to covalently attach to carbonyl substrates, and when attached (as imidazolidinones), could induce site-specific Pd-catalyzed acetoxylations and olefinations on both sp2- and sp3-hybridized bonds. Given the observed ability of these molecules to impart this reactivity, we desired a simple and direct approach to this class of scaffolds, ideally with sufficient modularity to accommodate variations in both amide and ligating group. More generally, the proposed approach could provide ready access to a range of α-quaternary proline derviatives.3,4 Last year, we reported the complementary generation of α-quaternary proline-based amino amides based on SRS chemistry, where we found that the structure of the “temporary” substituent had a direct impact on the stereochemical outcome of enolate alkylations (Fig. 2).5 Herein, we report our overall observations in this alkylation chemistry, including a more thorough analysis of this “temporary” substituent that suggests the need for refinement of our original hypothesis of the stereochemical rationale.
Fig. 2.
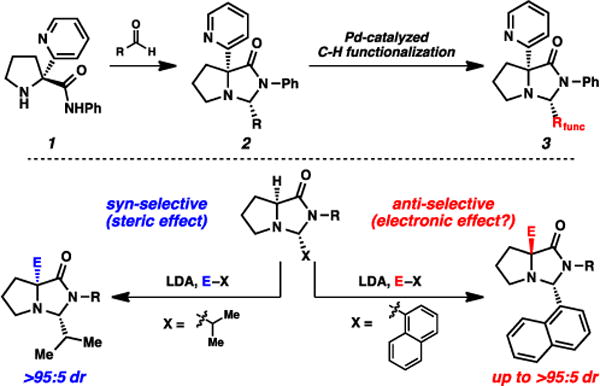
Molecular scaffolds for C–H functionalizations: motivation for complementary α-quaternary amino amide synthesis.
Background
In Seebach’s original self-regeneration of stereocenters with proline, an oxazolidinone based on pivalaldehyde is generated, and alkylation occurs from lithium enolate 5 to form oxazolidinone cis-6 (Fig. 3).6,7 The use of pivalaldehyde is common in SRS chemistry; the bulkiness and inertness of the tert-butyl group reliably imparts steric-driven selectivities. Diastereoselection in this particular process have been reported to be consistently excellent. A 1,3-syn relationship between the tert-butyl group and the electrophile is observed, originating from the preference of the bulky t-Bu group to be positioned on the convex face of the bicyclic system. A noted oxidative sensitivity of this molecule was addressed by Wang and Germanas, where they applied the more robust chloral-based oxazolidinone exo-7 in similar alkylative processes.8,9 This overall method has been widely employed to access α-quaternary proline-based amino acids.3,4
Fig. 3.
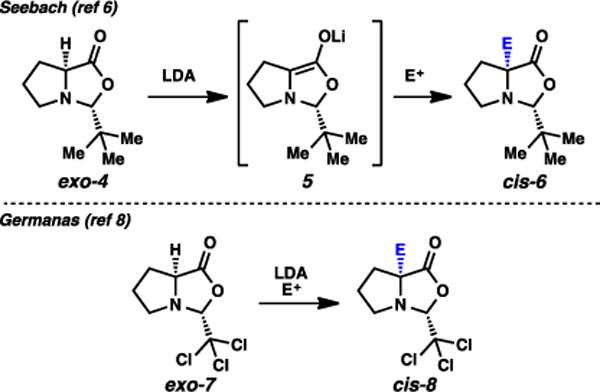
Seebach and Germanas SRS approaches to α-quaternary proline derivatives.
A report from Hughes and Trauner on the syntheses of amathaspiramide F described an intriguing reversal of selectivity using structurally similar proline-based imidazolidinones (Fig. 4).10 In this particular case, the key transformation involved the pregeneration of a silyl enol ether and a subsequent conjugate addition to a nitroolefin. Here, stereoselectivity is hypothesized to be governed by the staggered orientations of the t-Bu group, the amide methyl substituent, and the bulky TBS groups in the enol ether. Both large groups reside on the convex face of the bicycle, and thus the electrophile approaches from the less hindered, concave face. This stereochemical hypothesis was supported by the alkylation of lithium enolate 5, which demonstrated opposite facial selectivity in the conjugate addition, more aligned with Seebach’s and others’ original cases.6–9
Fig. 4.
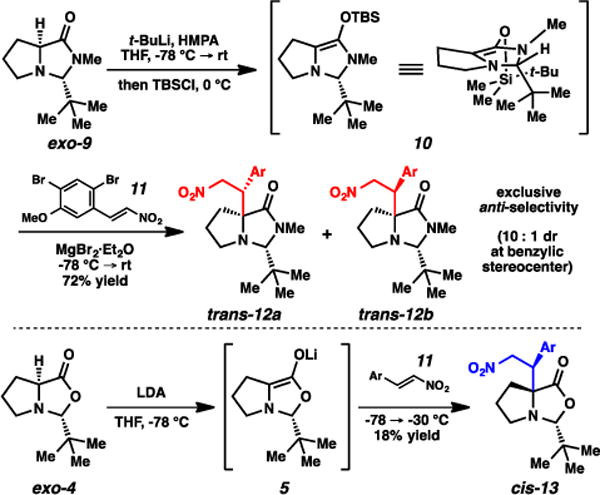
Trauner observation of trans-selectivity in silyl enol ether alkylations.
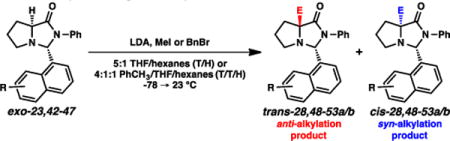
Results and Discussion
With this backdrop, we set out to establish the stereogenicity that would arise from alkylation processes of the proline-based imidazolidinones using SRS chemistry. Taking into account Trauner’s findings specifically,10 it was important to confirm the stereochemical outcome of these alkylations. The reversal he had observed could have potentially arisen via two contributing factors: (1) potentially differential alkylations of oxazolindinones vs. imidazolidinones, and (2) potentially differential alkylations of lithium enolates vs. silyl enol ethers. We opted to investigate lithium enolates as our nucleophilic species, motivated by our desire to install a range of alkyl and other non-carbonyl electrophiles.
Imidazolidinone exo-15 was synthesized via combination of the phenyl amide of proline with isobutyraldehyde under acidic conditions (Scheme 1).11 The imidazolidinone was formed exclusively as the exo diastereomer, confirmed by NOE analysis. This compound was then alkylated using LDA and MeI, and product cis-16a was formed as a single diastereomer. The (S) stereochemistry was confirmed via subsequent acidic cleavage of the imidazolidinone, where the optical rotation of resulting amino amide matched the rotation of the amino amide independently synthesized via the Germanas/Wang protocol.8,12 This stereochemical outcome is therefore consistent with the facial selectivity expected based on Seebach’s original observations,6,7 in that the electrophile adds syn to the isopropyl group. The outcome also further corroborates Trauner’s hypothesis for his observed stereochemical reversal,10 in that the silyl group of enol ether 10 (Fig. 4) was impactful, likely blocking the convex face from reactivity.
Scheme 1.
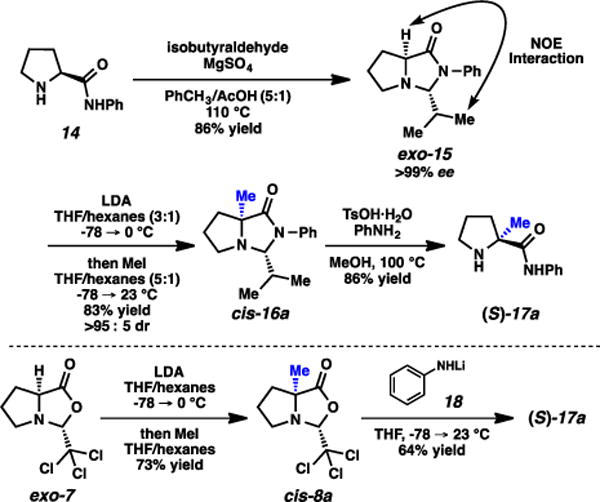
Imidazolidinone formation, alkylation, and stereochemical confirmation.
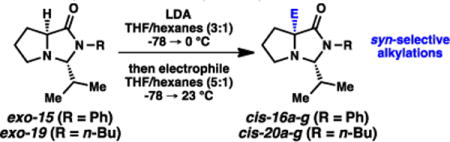
The highly selective alkylations established a convenient method for accessing the α-quaternary amino amide motif with high levels of enantioenrichment. Illustrated in Table 1, we evaluated both imidazolidinone exo-15 and the N-n-Bu variant (exo-19) with a variety of electrophiles under these alkylative conditions (LDA, THF/hexanes, −78 → 23 °C). Good yields were generally observed, with all transformations proceeding with excellent diastereoselecivity (>95:5 favoring syn alkylation). Included in these electrophiles is 2-fluoropyridine, representing a straightforward strategy for synthesizing our aforementioned targeted molecular scaffolds.2 Lithium chloride as an additive was notably helpful for this specific electrophile. The benefits of LiCl as an additive in anionic additions to glycine methyl ester have been noted;13 a similar Lewis acid coordination of the pyridyl electrophile may be operative here.
Table 1.
Syn-selective alkylations of isobutyraldehyde-based imidazolidinones.
| |||||
|---|---|---|---|---|---|
| Entry | R | Electrophile | Product | Alkylation dr (syn/anti)a | Isol. yield of cis diast. (%) |
| 1 | Ph | Mel | cis-16a | >95: 5 | 83 |
| 2 | n-Bu | cls-20a | >95: 5 | 73 | |
|
| |||||
| 3 | Ph | BnBr | cis-16b | >95: 5 | 91 |
| 4 | n-Bu | cis-20b | >95: 5 | 85 | |
|
| |||||
| 5 | Ph | n-BuBr | cls-16c | >95: 5 | 77 |
| 6 | n-Bu | cls-20c | >95: 5 | 83 | |
|
| |||||
| 7 | Ph |
|
cls-16d | >95: 5 | 82 |
| 8 | n-Bu | cls-20d | >95: 5 | 74 | |
|
| |||||
| 9 | Ph |
|
cls-16e | >95: 5 | 95 |
| 10 | n-Bu | cls-20e | >95: 5 | 62 | |
|
| |||||
| 11 | Ph |
|
cis-16f | >95: 5 | 76 |
| 12 | n-Bu | cls-20f | >95: 5 | 31 | |
|
| |||||
| 13b | Ph |
|
cis-16g | >95: 5 | 92 |
| 14b | n-Bu | cls-20g | >95: 5 | 64 | |
Diastereomeric ratio measured by 1H NMR.
LiCI (1.1 equiv) added.
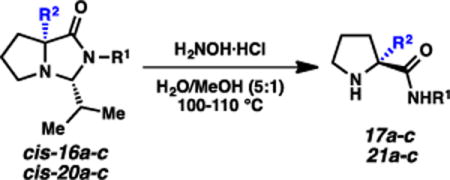
Cleavage of the imidazolidinones would result in a convenient route to enantioenriched α-quaternary amino amides. As mentioned above, acid-mediated aminolysis (PhNH2, TsOH•H2O, MeOH, 100 °C) was effective for the formation of amide 17a (Scheme 1), but reactivity was not uniform. Sterically hindered systems in general were much less reactive. We hypothesized that hydroxylamine would lead to enhanced imidazolidinone cleavage, due to its increased nucleophilicity14 and/or the resulting stability of the oxime byproduct.15,16 Indeed, we found that this proved to be true, and several α-quaternary amino amides were afforded via this aminolysis (Table 2).
Table 2.
Aminolysis of alkylated isobutyraldehyde-based imidazolidinones.
| |||||
|---|---|---|---|---|---|
| Entry | Imidazolidinone | R1 | R2 | Amide | Isolated yield (%) |
| 1 | cis-16a | Ph | Me | 17a | >99 |
| 2 | cis-16b | Ph | Bn | 17b | 80 |
| 3 | cis-16C | Ph | n-Bu | 17c | >99 |
| 4 | cis-20a | n-Bu | Me | 21a | >99 |
| 5a | cis-20b | n-Bu | Bn | 21b | 70 |
| 6a | cis-20c | n-Bu | n-Bu | 21c | 88 |
H2O/1,4-Dioxane (3:2) solvent.
During the course of evaluating this overall process, we questioned if we may be able to access the opposite enantiomeric series via a comparable SRS strategy. Although the simplest approach would be to apply the same sequence to D-proline, the unnatural enantiomer is appreciably more expensive and we considered this process unattractive. Two other strategies, both starting from L-proline, were envisaged (Fig. 5). Strategy A would entail a unique endo-selective condensation17 followed by a syn-selective alkylation, while Strategy B would involve an exo-selective condensation followed by a novel anti-selective alkylation.18 We anticipated either or both strategies could potentially be realized by investigating the nature of the condensing aldehyde; the nature of the “temporary” group, if different from a bulky aliphatic moiety, may influence the diastereoselectivity of the condensation and/or the alkylation.
Fig. 5.
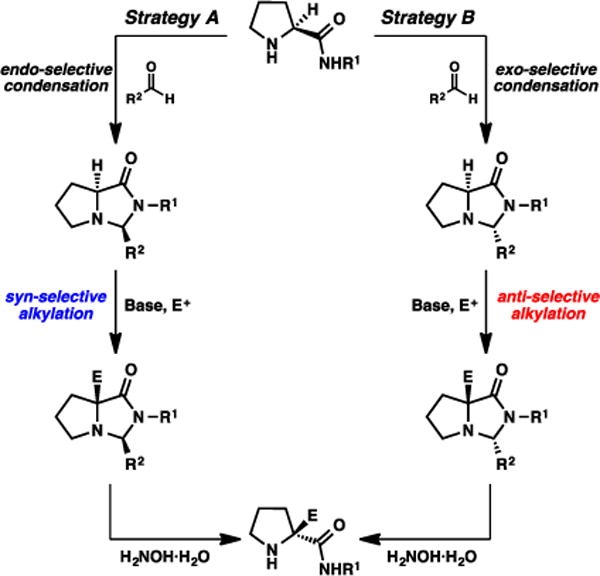
Two potential strategies for accessing enantiomeric series of α-quaternary amino amides via SRS.
We started by investigating benzaldehyde and 1-naphthaldehyde as our condensing units, positing that their flat nature and electronic tunability could induce differential reactivity from those featuring the isopropyl group. Initial condensation of the aromatic aldehydes, however, yielded surprising results. For example, under acidic conditions using amino amide 14 and 1-naphthaldehyde, the exo diastereomer was observed, but the enantioenrichment was substantially eroded (Scheme 2). Presumably, an intermediate iminium, which may be more long-lived when conjugated to an arene (e.g., 22) acidifies the α-proton and leads to racemization. This issue was circumvented using basic condensation conditions (K2CO3, alcohol solvent, heat), and we could access the exo imidazolidinone in good yield and with complete conservation of chirality. The relative stereochemistry of this imidazolidinone (exo-23) was confirmed by X-ray.5
Scheme 2.
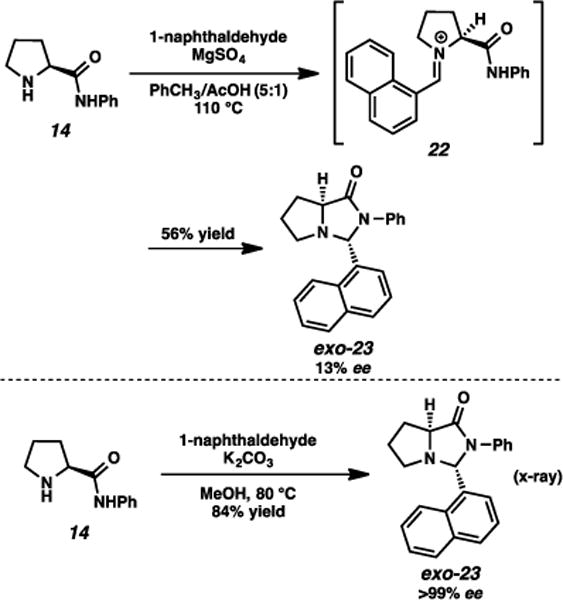
Dependency of conditions for imidazolidinone formation on enantioenrichment.
Interestingly, this method for condensation was capricious for attempts at endo-diastereoselection, affording inconsistent endo/exo ratios of these imidazolidinone diastereomers when varying substrate. An example is illustrated in Scheme 3. When the condensation was performed for a relatively short period of time (5.5 h), using amino amide 24, a 78:22 mixture of endo and exo diastereomers of the imidazolidinone was observed. A longer reaction time (7 h) produced a 56:44 endo/exo mixture. When the condensation was performed for 24 h in ethylene glycol, the exo diastereomer was the lone observed imidazolidinone. These and other experiments suggested that the initial condensation was somewhat endo-selective, but gradually proceeded toward the thermodynamically-favored exo product. Further experimentation toward endo-selectivity did not prove fruitful, as no method for general endo enrichment could be achieved. Ultimately our lack of confidence in developing a reliable endo-selective condensation compelled us to focus on pursuing Strategy B, relying on an exo-selective condensation and subsequent anti-selective alkylation.
Scheme 3.
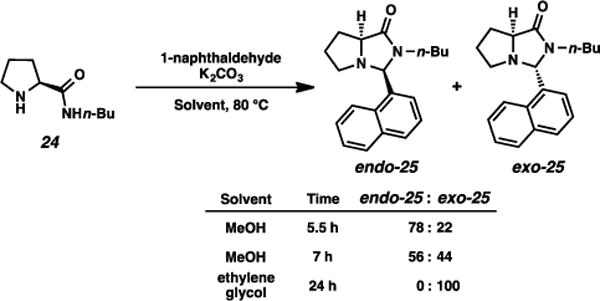
Attempted endo-selective condensations.
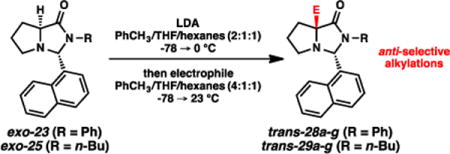
Promising results for the anti-selective alkylation were obtained with the aromatic aldehyde-derived imidazolidinones (Scheme 4). When imidazolidinone exo-26 was treated with LDA and BnBr, the alkylation facial selectivity was very low, yielding a 60:40 mixture of cis and trans isomers, respectively. The 1-naphthaldehyde-derived imidazolidinone (exo-23), however, afforded remarkable selectivity for the trans isomer (92:8 dr). This outcome was highly surprising to us, representing a substantial departure from the generally observed selectivities for these bicyclic enolates. More notably, this example represents the lone case to our knowledge of complementary stereoselectivities in the family of SRS reactions based solely on the “temporary” substituent, where the i-Pr and 1-naphthyl groups induce markedly contrasting stereochemical outcomes.19
Scheme 4.
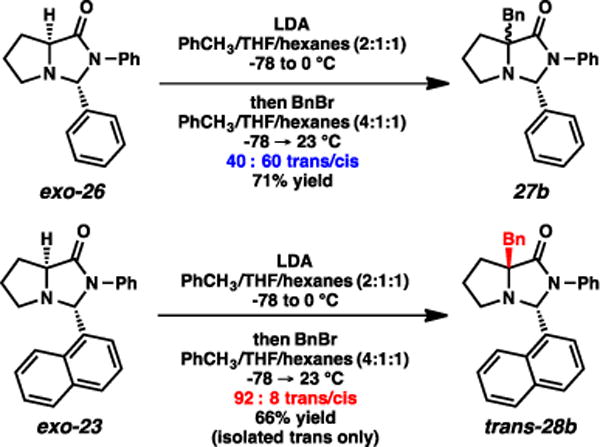
Alkylations of aromatic aldehyde-based imidazolidinones.
We explored the scope of this anti-selective reactivity and in general obtained high levels of efficiency for formation of the trans diastereomer (Table 3). Although THF/hexanes (5:1) could be used as the solvent system (entries 2 and 9), we found that a toluene/THF/hexanes (4:1:1) was more effective for anti-selectivity. The smallest electrophile evaluated (MeI) gave the lowest diastereoselectivity, albeit still significantly favoring an anti process. A spectrum of electrophiles generally afforded the trans products with good to excellent selectivity; moreover, the diastereomers were chromatographically separable, thereby enabling ready access to the pure trans diastereomers.
Table 3.
Anti-selective alkylations of 1-naphthaldehyde-based imidazolidinones.
| |||||
|---|---|---|---|---|---|
| Entry | R | Electrophile | Product | Alkylation dr (antl/syn)a | Isol. yield of trans diast. (%) |
| 1 | Ph | Mel | trans-28a | 68:32 | 53 |
| 2b | Ph | trans-28a | 54:46 | 44 | |
| 3 | n-Bu | trans-29a | 91:9 | 91 | |
|
| |||||
| 4 | Ph | BnBr | trans-28b | 92:8 | 66 |
| 5 | n-Bu | trans-29b | >95:5 | 90 | |
|
| |||||
| 6 | Ph | n-BuBr | trans-28c | 78:22 | 60 |
| 7 | n-Bu | trans-29c | >95:5 | 99 | |
|
| |||||
| 8 | Ph |
|
trans-28d | 84:16 | 84 |
| 9b | Ph | trans-28d | 70:30 | 63c | |
| 10 | n-Bu | trans-29d | 94:6 | 63 | |
|
| |||||
| 11 | Ph |
|
trans-28e | 74:26 | 64 |
| 12 | n-Bu | trans-29e | >95:5 | 93 | |
|
| |||||
| 13 | Ph |
|
trans-28f | 91:9 | 77 |
| 14 | n-Bu | trans-29f | 92:8 | 69 | |
|
| |||||
| 15d | Ph |
|
trans-28g | 86:14 | 77 |
| 16d | n-Bu | trans-29g | >95:5 | 96 | |
Diastereomeric ratio measured by 1H NMR.
Alkylation performed in THF/hexanes (5:1).
Isolated yield of both diastereomers.
LiCI (1.1 equiv) added.
As mentioned above, the alkylations using methyl iodide as the electrophile afforded the lowest diastereoselectivities for imidazolidinones exo-23 and exo-25. These cases are likely reflective of the size of the electrophile, where the two faces of the enolate can be accessed more competitively with smaller species. Consistent with this hypothesis, using THF as the solvent a simple protonation of the enolate by quenching with H2O generated a 34:66 mixture of endo and exo imidazolinones (Scheme 5, endo- and exo-23), respectively. Curiously, attempts to subsequently enolize this isolated endo imidazolidinone led primarily to decomposition.20
Scheme 5.
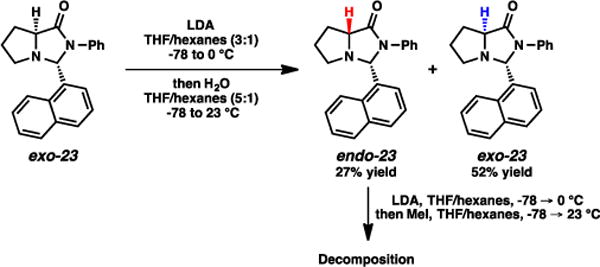
Deprotonation/reprotonation of imidazolidinone exo-23.
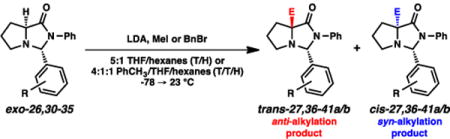
We attempted to interrogate these anti-selective alkylations by evaluating other imidazolidinones based on a range of aromatic aldehydes. The imidazolidinones were tested using either MeI or BnBr as the electrophile, and using either 5:1 THF/hexanes (T/H) or 4:1:1 toluene/THF/hexanes (T/T/H) as the reaction medium. Select examples are presented in Tables 4 and 5. As can be seen, several of the imidazolidinones bearing benzene derivatives predominantly afforded syn-selective alkylations instead of anti-selective ones. Notable are the p-trifluoromethylphenyl and p-cyanophenyl examples (Table 4, entries 4-9); compared to the parent phenyl case, these systems generally showed a greater preference for syn alkylations. Ortho-substituted phenyl systems also showed a significant trend for syn-selectivity in most cases.
Table 4.
Further analyses of benzaldehyde-based imidazolidinones.
| ||||
|---|---|---|---|---|
| Entry | Phenyl derivative | Electrophile | Solvent | Alkylation dr (anti/syn)a |
| 1 |
|
Mel | T/H | 45:55 |
| 2 | Mel | T/T/H | 46:54 | |
| 3 | BnBr | T/T/H | 40:60 | |
|
| ||||
| 4 |
|
Mel | T/H | 35:65 |
| 5 | Mel | T/T/H | 50:50 | |
| 6 | BnBr | T/H | 24:76 | |
| 7 | BnBr | T/T/H | 20:80 | |
|
| ||||
| 8 |
|
BnBr | T/H | 26:74 |
| 9 | BnBr | T/T/H | 29:71 | |
|
| ||||
| 10 |
|
Mel | T/T/H | 55:45 |
| 11 | BnBr | T/T/H | 36:64 | |
|
| ||||
| 12 |
|
Mel | T/H | 14:86 |
| 13 | Mel | T/T/H | 10:90 | |
| 14 | BnBr | T/H | 16:84 | |
| 15 | BnBr | T/T/H | 19:81 | |
|
| ||||
| 16 |
|
Mel | T/H | 39:61 |
| 17 | Mel | T/T/H | 67:33 | |
|
| ||||
| 18 |
|
Mel | T/H | 9:91 |
Diastereomeric ratio measured by 1H NMR.
Table 5.
Further analyses of naphthaldehyde-based imidazolidinones.
| ||||
|---|---|---|---|---|
| Entry | Naphthyl derivative | Electrophile | Solvent | Alkylation dr (anti/syn)a |
| 1 |
|
Mel | T/H | 54:46 |
| 2 | Mel | T/T/H | 68:32 | |
| 3 | BnBr | T/H | 91:9 | |
| 4 | BnBr | T/T/H | 92:8 | |
|
| ||||
| 5 |
|
Mel | T/H | 64:36 |
| 6 | Mel | T/T/H | 68:32 | |
| 7 | BnBr | T/H | 91:9 | |
| 8 | BnBr | T/T/H | 94:6 | |
|
| ||||
| 9 |
|
Mel | T/H | 53:47 |
| 10 | Mel | T/T/H | 50:50 | |
| 11 | BnBr | T/H | 27:73 | |
|
| ||||
| 12 |
|
Mel | T/H | >95:5 |
| 13 | Mel | T/T/H | >95:5 | |
| 14 | BnBr | T/H | <5:95 | |
| 15 | BnBr | T/T/H | <5:95 | |
|
| ||||
| 16 |
|
Mel | T/H | 7:93 |
| 17 | Mel | T/T/H | –b | |
| 18 | BnBr | T/H | –b | |
|
| ||||
| 19 |
|
Mel | T/H | –c |
|
| ||||
| 20 |
|
Mel | T/H | –c |
Diastereomeric ratio measured by 1H NMR.
Only starting material and slight decomposition were observed.
Complete decomposition was observed.
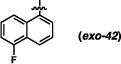
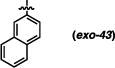
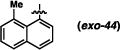
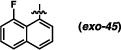
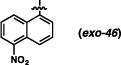
Derivatives of the 1-naphthyl-based imidazolidinones were unfortunately not much more informative (Table 5). A 5-fluoro substituent afforded similar selectivities to the parent system (entry 1-4 vs. 5-8). The β-naphthyl variant (exo-43) was less selective overall (entries 9-11). An 8-methyl substituent appeared to boost selectivity for the methyl iodide electrophile, but BnBr was exclusively syn-selective instead.21 Fluorine at the 8-position yielded a near complete reversal of selectivity (93:7 favoring the cis diastereomer) under one set of conditions; other tests of this imidazolidinone gave slight decomposition and nothing more. The 5-nitro-1-naphthyl and 9-anthracenyl imidazolidinones (exo-46 and exo-47) were not effective reactants, decomposing under these reaction conditions. It seems, based on this series of results, that there are not readily discernible trends of reactivity that can explain the variances in anti-selectivity.
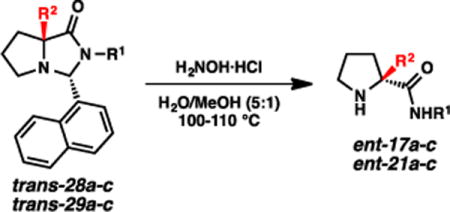
Analogous to the alkylations affording the cis diastereomers, the imidazolidinone products of the successful anti-selective alkylations (i.e., Table 3) could be readily aminolyzed to afford the enantiomeric set of the α-quaternary amino amides. Identical conditions (H2NOH•HCl, H2O/MeOH) were employed, and the products were obtained in excellent yields (Table 6). As a whole, this series of processes constitutes an SRS approach to both enantiomeric series of α-quaternary amino amides and acids22 from the same inexpensive and readily available natural enantiomer of proline.
Table 6.
Aminolysis of alkylated 1-naphthaldehyde-based imidazolidinones.
| |||||
|---|---|---|---|---|---|
| Entry | Imidazolidinone | R1 | R2 | Amide | Isolated yield (%) |
| 1 | trans-28a | Ph | Me | ent-17a | >99 |
| 2 | trans-28b | Ph | Bn | ent-17b | 92 |
| 3 | trans-28c | Ph | n-Bu | ent-17c | >99 |
| 4 | trans-29a | n-Bu | Me | ent-21a | 98 |
| 5 | trans-29b | n-Bu | Bn | ent-21b | 90 |
| 6 | trans-29c | n-Bu | n-Bu | ent-21c | 99 |
The origins of the stereochemical bifurcation of this transformation intrigued us. A “memory effect” was likely not operational here,23 although the ability of imidazolidinone enolate carbons to planarize as opposed to maintaining pyramidalization was not fully established. We tested this by the evaluation of enantioenriched imidazolidinone 54, featuring no substituent at the aminal position. Alkylation under the standard conditions with MeI as the electrophile afforded imidazolidinone 55 as a racemate. Although it does not wholly eliminate a “memory effect” rationalization for the substituted imidazolidinones, this observation highly implicates that enolization proceeds via planarization of the α-carbon prior to addition to the electrophile.
If planarization of the enolate carbon indeed occurs, the stereochemistry of the addition to the electrophile should be dictated by the nature of the group that has been installed. We believe the isopropyl-based alkylation are aligned with the prior alkylative transformations using proline-derived oxazolidinones.6–9 Diastereoselectivity arises primarily from the steric influence of the bulky, aliphatic isopropyl group. In enolate 56, the all-staggered orientation of the substituents across the N–C–N bonds positions the pyrrolidine ring above the enolate plane (Fig. 6).24 As the alkylating agent approaches the bicyclic system, the enolate carbon pyramidalizes toward forming the cis-fused 5,5-ring framework. In this reaction trajectory, the i-Pr group moves toward a position on the convex face of the forming bicycle, avoiding steric congestion on the concave side.
Fig. 6.
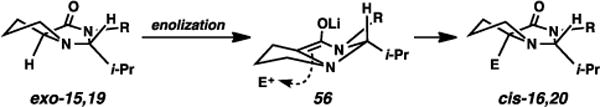
Rationalization for syn-selective alkylations.
The anti-selective processes are much more difficult to rationalize. With the 1-naphthyl imidazolidinone derivatives (i.e., Table 3), the magnitude and direction of stereoselectivity were relatively consistent for the N-phenyl and N-n-butyl amides, suggesting that potential arene-arene interactions25 for the N-phenyl system are not primarily contributing to stereoselection. We had originally hypothesized that the anti-selectivity arises from lone pair delocalization from the central nitrogen atom into the C–C σ* orbital (Fig. 7). This delocalization would necessitate that the 1-naphthyl group is situated in a pseudoaxial position, but perhaps the arene planarity would render the group sufficiently “small” to adopt this position. The alkylation of the phenyl-based imidazolidinone (Scheme 4, exo-26 → 27b) appeared to support this idea, as the phenyl group should be less electron-withdrawing than a 1-naphthyl group.
Fig. 7.
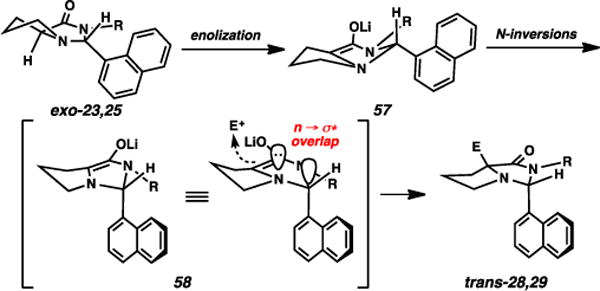
Original hypothesis of anti-selectivity; inconsistent with complete electronic analysis.
The substituted phenyl-based imidazolidinone systems that were examined (Table 4) appear to refute this hypothesis, however. Specifically, the electron-withdrawing trifluoromethyl and cyano groups should inductively increase the delocalization capacity by lowering the C–C σ* orbital in question, thereby increasing overall anti-selectivity relative to the parent phenyl case. Instead, these two moieties appear to induce higher syn-selectivity. Since steric attributes are unlikely to factor into this specific comparison, these observations appear to negate a purely inductive rationalization for anti-selectivity.
We had also considered an electrostatic interaction as a potential influence on reactivity. Specifically, if the naphthyl ring of enolate 57 were to rotate to reside underneath the bicycle, then the (modestly acidic) naphthyl C-8 hydrogen could engage in a favorable interaction with the electron-rich enolate (60, Fig. 9).26,27 The increased anti-selectivity using toluene over THF as the primary solvent (Table 3, entries 1/8 vs. 2/9) agreed with this hypothesis, as electrostatic interactions should be accentuated in nonpolar media. Although a reversal of selectivity was observed with the 8-fluorinated derivative (Table 5, entry 16), this was a lone case, and the unusual results with the 8-methyl derivative (Table 5, entries 12-15) render this hypothesis speculative at best. Certainly the comparisons between phenyl and naphthyl systems have several differences that could be quite influential beyond electronic factors (e.g., rotational barriers); the data we have obtained thus far unfortunately does not lend itself to immediate direct and obvious interpretation, and additional experimental and computational evidence will ideally yield a clearer picture of this intriguing differential reactivity.
Fig. 9.
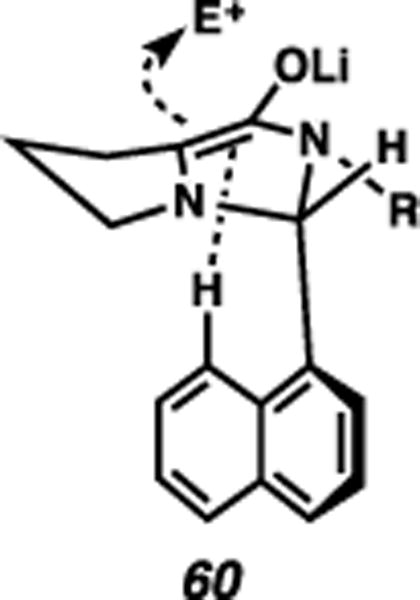
Potential arene C-H/enolate electrostatic interaction.
It should also be noted that attempts to compare the unique effects of the 1-naphthyl group in proline-based oxazolidinone alkylations were unsuccessful. We anticipated that the alkylations of oxazolidinone 63 (Scheme 7) could be compared to the Seebach and Germanas examples6–8 to provide analogous relationships, and thus ideally shed insight into the unique behavior of the 1-naphthyl species. Unfortnuately, we were unable to isolate the oxazolidinone in question via condensation of proline with 1-naphthaldehyde; Blackmond and coworkers had noted a similar difficulty in isolating oxazolidinones from aromatic aldehydes.28
Scheme 7.
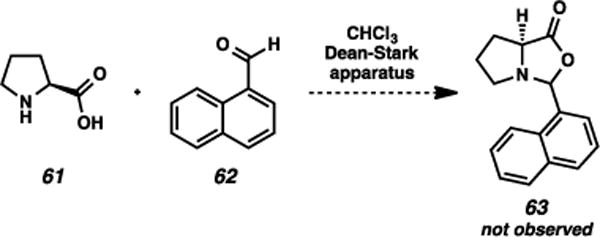
Failed oxazolidinone formation.
Conclusion
To summarize, we have described a highly unique complementarity in stereoselective alkylations in the SRS reaction class. Proline-based imidazolidinones can be readily synthesized and subsequently alkylated with excellent diastereoselectivities. These species can then be aminolyzed to produce enantiomeric series of α-quaternary amino amides from the same natural enantiomer of proline. The different characteristics of the isopropyl and 1-naphthyl imidazolidinone substituents dictate this stereodifferentiation, although the exact origins of these outcomes remain unclear. To our knowledge, this system is the only example of contrasting stereoselectivities in the family of SRS reactions where complementarity is dictated solely by the “temporary” substituent. Importantly, we anticipate that this method will serve as a general and convenient approach toward accessing both enantiomers of α-quaternary amino acids and amides in highly enantioenriched form.
Experimental Section
Materials and Methods
Reactions were performed under an argon atmosphere unless otherwise noted. Tetrahydrofuran, dichloromethane, and toluene were purified by passing through activated alumina columns. Diisopropylamine was distilled over CaH2. 2-Fluoropyridine was freshly distilled before use. LiCl was dried at 150 °C for 12 h under high vacuum (<0.1 torr) before use. All other reagents were used as received unless otherwise noted. Commercially available chemicals were purchased from Alfa Aesar (Ward Hill, Ma), Sigma-Aldrich (St. Louis, MO), Oakwood Products (West Columbia, SC), Strem (Newport, MA), TCI America (Portland, OR), and Bachem (Torrance, CA). Qualitative TLC was performed on 250 mm thick, 60 Å, glass backed, F254 silica (Silicycle, Quebec City, Canada). Visualization was accomplished with UV light and exposure to ninhydrin, p-anisaldehyde, or KMnO4 stain solutions followed by heating. Qualitative TLC of amino amide products required pretreatment of TLC plates with 9:1 hexanes/triethylamine followed by evaporation under reduced pressure (basic plates). Visualization of amino amide products required pretreatment of the plate with 19:1 1.0 N HCl(aq)/isobutyraldehyde and heating to dryness before using ninhydrin stain solution. Flash column chromatography was performed using Silicylce silica gel (230-400 mesh).
General procedure for the formation of isobutyraldehyde-derived proline imidazolidinones
A suspension of amino amide, isobutyraldehyde (1.3 equiv), and MgSO4 (1.5 equiv) in 5:1 PhCH3/glacial AcOH (0.08 M) was heated to reflux and stirred overnight. Upon cooling, to the solution was added sat. aq. NaHCO3 and the mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated by rotary evaporation. The crude mixture was purified by flash column chromatography to afford pure imidazolidinone.
General procedure for the alkylation of isobutyraldehyde-derived proline imidazolidinones
In a flame-dried flask, to a suspension of isobutyraldehyde-derived proline imidazolidinone in THF (1.0 M) cooled to −78 °C was added a cooled solution of lithium diisopropylamide (1.1 equiv, 0.9 to 1.1 M in ~1:1 hexanes/THF, −4 °C). The solution was then allowed to warm to 0 °C and stirred for 20 min, generally turning yellow. The solution was recooled to −78 °C, and to the solution was added an equal volume of THF (bringing the reaction concentration to ~0.3 M; the additional THF improves stirring upon addition of the electrophile). The electrophile (1.1 equiv) was added at −78 °C, and the reaction mixture stirred at −78 °C for 1.5 h before the ice bath was removed and the suspension was allowed to warm gradually to ambient temperature. Upon completion, the reaction was quenched with water, and the mixture was extracted into EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was then removed by rotary evaporation, and the crude imidazolidinone was purified by flash column chromatography.
General procedure for the formation of aromatic aldehyde-derived proline imidazolidinones
A solution of amino amide, aromatic aldehyde (1.3 equiv), K2CO3 (2.0 equiv) in either methanol or ethylene glycol (0.33 M) was heated to 80 °C and stirred for >2 h. Upon cooling, to the solution was added water, and the mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude mixture was purified by flash column chromatography to afford pure imidazolidinone.
General procedure for the alkylation of aromatic aldehyde-derived proline imidazolidinones (THF/hexanes conditions)
In a flame-dried flask, to a suspension of aromatic aldehyde-derived proline imidazolidinone in THF (1.0 M) cooled to −78 °C was added a cooled solution of lithium diisopropylamide (1.1 equiv, 0.9 to 1.1 M in ~1:1 hexane/THF, −4 °C). The solution was then allowed to warm to 0 °C and stirred for 70 min. The solution was recooled to −78 °C; and to the solution was added an equal volume of PhCH3 (bringing the reaction to ~0.3 M; the additional THF improves stirring upon addition of the electrophile). The electrophile (1.1 equiv) was added at −78 °C, and the reaction mixture stirred at −78 °C for 1.5 h before the cooling bath was removed, and the suspension was allowed to gradually warm to ambient temperature. Upon completion, the reaction was quenched with water, and the mixture was extracted into EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was then removed by rotary evaporation, and the crude imidazolidinone was purified by flash column chromatography.
General procedure for the alkylation of aromatic aldehyde-derived proline imidazolidinones (Toluene/THF/hexanes conditions)
In a flame-dried flask, to a suspension of aromatic aldehyde-derived proline imidazolidinone in PhCH3 (1.0 M) cooled to −78 °C was added a cooled solution of lithium diisopropylamide (1.1 equiv, 0.9 to 1.1 M in ~1:1 hexane/THF, −4 °C). The solution was then allowed to warm to 0 °C and stirred for 70 min. The solution was recooled to −78 °C; and to the solution was added an equal volume of PhCH3 (bringing the reaction to ~0.3 M; the additional PhCH3 improves stirring upon addition of the electrophile). The electrophile (1.1 equiv) was added at −78 °C, and the reaction mixture stirred at −78 °C for 1.5 h before the cooling bath was removed, and the suspension was allowed to gradually warm to ambient temperature. Upon completion, the reaction was quenched with water, and the mixture was extracted into EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was then removed by rotary evaporation, and the crude imidazolidinone was purified by flash column chromatography.
General procedure for the imidazolidinone aminolysis for formation of α-alkylated proline amides
In a sealed vial, a suspension of imidazolidinone and H2NOH•HCl (3 equiv) in 5.2:1 H2O/MeOH (0.16 M) or 1.5:1 H2O/dioxane (0.20 M) was heated at 100-110 °C and stirred for >3 h. The imidazolidinones slowly dissolved over the course of the reaction. Upon cooling, the reaction was added to 1 M KHSO4 and was washed with EtOAc. To the aqueous layer was added sat. Na2CO3 until it reached pH ~12. The aqueous solution was extracted with EtOAc (the products are very water soluble), and the combined organic layers were dried over Na2SO4 and concentrated by rotary evaporation. The crude film was dissolved in CHCl3, and the solution was transferred to another container to remove any insoluble materials. This solution was concentrated by rotary evaporation to afford analytically pure amino amides.
Characterization Data
Characterization data has been previously reported for the following compounds: cis-8a,8 14,2 exo-15,2 cis-16a,c-f,5 cis-16b,g,2 17a-c,5 exo-19,5 cis-20a-g,5 21a-c,5 endo-23,5 exo-23,5 24,5 endo-25,5 exo-25,5 exo-26,2 27b,5 trans-28a-g,5 trans-29a-g,5 54,5 and 55.5
(3R,7aS)-2-phenyl-3-(4-(trifluoromethyl)phenyl)hexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-30)
89% yield. Beige solid (Rf = 0.21 in 39:1 CHCl3/Et2O). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.8 Hz, 2H), 7.31 (dd, J = 8.0, 7.2 Hz, 2H), 7.13 (t, J = 7.2 Hz, 1H), 5.74 (s, 1H), 4.00 (t, J = 7.0 Hz, 1H), 3.45 (dt, J = 10.0, 4.5 Hz, 1H), 2.90 (q, J = 4.5 Hz, 1H), 2.22 (q, J = 7.0 Hz, 2H), 1.95-1.85 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 174.9, 143.6, 137.4, 130.9 (q, J = 32.4 Hz), 129.3, 126.7, 126.2 (q, J = 13.9 Hz), 125.5, 124.0 (q, J = 270.6 Hz), 121.2, 83.0, 64.5, 56.3, 27.7, 25.0; 19F NMR (377 MHz, CDCl3) δ −63.9; IR (film) 1697, 1496, 1379, 1329, 1111 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C19H17F3N2O + H]+: 347.1366, found 347.1366.
4-((3R,7aS)-1-oxo-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-3-yl)benzonitrile (exo-31)
36% yield. Beige solid (Rf = 0.30 in 7:3 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 4H), 7.31 (t, J = 8.0 Hz, 2H), 7.14 (t, J = 8.0 Hz, 1H), 5.72 (s, 1H), 3.97 (t, J = 6.4 Hz, 1H), 3.43 (dt, J = 10.0, 4.8 Hz, 1H), 2.90 (q, J = 4.8 Hz, 1H), 2.25-2.17 (comp. m, 2H), 1.96-1.86 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 174.7, 144.8, 137.2, 132.9, 129.5, 127.2, 125.7, 121.2, 118.5, 112.6, 82.9, 64.5, 56.3, 27.8, 25.0; IR (film) 2232, 1700, 1494, 1379, 730 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C19H17N3O + H]+: 304.1444, found 304.1445.
(3R,7aS)-3-(4-methoxyphenyl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-32)
72% yield. White solid (Rf = 0.18 in 3:2 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.0 Hz, 2H), 7.28 (dd, J = 8.0, 7.6 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 7.10 (t, J = 7.6 Hz, 1H), 6.86 (d, J = 8.4 Hz, 2H), 5.63 (s, 1H), 4.03 (t, J = 7.2 Hz, 1H), 3.76 (s, 3H), 3.41 (dt, J = 9.4, 5.2 Hz, 1H), 2.85 (dt, J = 9.4, 8.4 Hz, 1H), 2.20 (q, J = 7.2 Hz, 2H), 1.93-1.84 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.0, 159.8, 137.8, 131.7, 129.1, 127.4, 125.2, 121.3, 114.5, 83.4, 64.5, 56.0, 55.4, 27.6, 24.9; IR (film) 1700, 1379, 1252, 910, 731 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C19H20N2O2 + H]+: 309.1598, found 309.1599.
(3R,7aS)-3-(2,6-difluorophenyl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-33)
73% yield. White solid (Rf = 0.21 in 7:3 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.0 Hz, 2H), 7.28 (t, J = 8.0 Hz, 2H), 7.23-7.14 (m, 1H), 7.10 (t, J = 7.2 Hz, 1H), 6.82 (dd, J = 8.0, 7.2 Hz, 2H), 6.14 (s, 1H), 4.24 (dd, J = 8.8, 5.2 Hz, 1H), 3.41 (dt, J = 10.0, 6.4 Hz, 1H), 2.97 (dt, J = 10.0, 6.4 Hz, 1H), 2.31-2.11 (comp. m, 2H), 1.87 (quint, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 174.5, 160.9 (dd, J = 248.1, 7.5 Hz), 136.7, 130.5 (t, J = 11.0 Hz), 129.2, 125.8, 121.9, 116.1 (t, J = 14.7 Hz), 112.2 (dd, J = 22.3, 3.6 Hz), 76.3 (t, J = 3.3 Hz), 65.9, 57.7, 28.9, 25.3; 19F NMR (377 MHz, CDCl3) δ −118.2; IR (film) 1700, 1468, 1379, 1120, 1005 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C18H16F2N2O + H]+: 315.1303, found 315.1303.
(3R,7aS)-2-phenyl-3-(o-tolyl)hexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-34)
82% yield. White solid (Rf = 0.33 in 7:3 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 2H), 7.32-7.25 (comp. m, 3H), 7.22 (t, J = 7.6 Hz, 1H), 7.15-7.04 (comp. m, 3H), 5.87 (s, 3H), 3.92 (dd, J = 8.8, 4.0 Hz, 1H), 3.49-3.42 (m, 1H), 2.86 (dt, J = 9.2, 7.2 Hz, 1H), 2.54 (s, 3H), 2.31-2.13 (comp. m, 2H), 1.97-1.84 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.9, 138.2, 136.5, 135.8, 131.6, 129.1, 128.5, 126.6, 124.8, 124.3, 120.1, 80.3, 64.1, 55.8, 27.1, 25.0, 19.3; IR (film) 1700, 1594, 1500, 1379, 757 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C19H20N2O + H]+: 293.1648, found 293.1649.
(3R,7aS)-3-(2-methoxyphenyl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-35)
95% yield. White solid (Rf = 0.17 in 3:2 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.0 Hz, 2H), 7.30-7.23 (comp. m, 3H), 7.10-7.04 (comp. m, 2H), 6.94 (d, J = 8.4 Hz, 1H), 6.85 (t, J = 7.6 Hz, 1H), 6.11 (s, 1H), 4.03 (t, J = 6.8 Hz, 1H), 3.93 (s, 3H), 3.47 (dt, J = 9.0, 5.2 Hz, 1H), 2.87 (dt, J = 9.0, 8.4 Hz, 1H), 2.21 (q, J = 6.8 Hz, 2H), 1.93-1.84 (comp. m, 2 H); 13C NMR (100 MHz, CDCl3) δ 175.7, 156.9, 137.9, 129.7, 129.0, 127.0, 125.8, 124.8, 120.8, 120.5, 111.3, 78.5, 64.5, 56.3, 55.7, 27.6, 25.0; IR (film) 1700, 1594, 1500, 1379, 757 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C19H20N2O2 + H]+: 309.1598, found 309.1598.
(3R,7aS)-3-(5-fluoronaphthalen-1-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-42)
74% yield. Beige solid (Rf = 0.61 in 7:3 hexane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.60 (dt, J = 8.0, 5.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 2H), 7.43 (app. t, J = 7.8 Hz, 1H), 7.33 (app. d, J = 7.1 Hz, 1H), 7.29-7.22 (comp m, 3H), 7.09 (t, J = 7.6 Hz, 1H), 6.44 (s, 1H), 3.94 (dd, J = 9.2, 3.6 Hz, 1H), 3.68-3.61 (m, 1H), 3.00 (td, J = 9.4, 7.0 Hz, 1H), 2.36-2.25 (m, 1H), 2.25-2.14 (m, 1H), 2.03-1.89 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 176.0, 159.6 (d, J = 251.3 Hz), 138.4, 133.4 (d, J = 2.4 Hz), 132.4 (d, J = 4.4 Hz), 129.2, 127.0 (d, J = 8.5 Hz), 125.8 (d, J = 1.8 Hz), 125.0 (d, J = 16.2 Hz), 124.8, 123.6, 121.8 (d, J = 6.1 Hz), 119.8, 118.7 (d, J = 4.0 Hz), 110.0 (d, J = 20.0 Hz), 80.0, 64.5, 55.4, 27.1, 25.1; 19F NMR (272 MHz, CDCl3) δ −120.8; IR (film) 1701, 1599, 1499, 1388, 1229 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C22H19FN2O + H]+: 347.1554, found 347.1548.
(3R,7aS)-3-(naphthalen-2-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-43)
64% yield. Beige solid (Rf = 0.11 in 7:3 hexane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.4 Hz, 1H), 7.85-7.75 (comp m, 2H), 7.70 (s, 1H), 7.53 (d, J = 7.6 Hz, 2H), 7.51-7.44 (comp. m, 3H), 7.27 (t, J = 7.6 Hz, 2H), 7.09 (t, J = 7.6 Hz, 1H), 5.83 (s, 1H), 4.10 (dd, J = 7.6, 6.0 Hz, 1H), 3.51-3.44 (m, 1H), 3.94 (q, J = 4.4 Hz, 1H), 2.28-2.20 (comp. m, 2H), 1.98-1.88 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.2, 137.8, 136.9, 133.4, 133.3, 129.4, 129.1, 128.3, 127.8, 126.6, 126.5, 125.2, 125.1, 124.0, 121.2, 83.9, 64.6, 56.1, 27.7, 25.0; IR (film) 1684, 1594, 1494, 1405, 820 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C22H20N2O + H]+: 329.1648, found 329.1649.
(3R,7aS)-3-(8-methylnaphthalen-1-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-44)
67% yield. White solid (Rf = 0.28 in 7:3 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.80 (dd, J = 7.6, 1.6 Hz, 1H), 7.75 (dd, J = 7.2, 2.4 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.42-7.21 (comp. m, 6H), 7.05 (t, J = 7.6 Hz, 1H), 6.90 (s, 1H), 3.94 (dd, J = 9.2, 2.8 Hz, 1H), 3.60 (ddd, 9.6, 6.8, 3.2 Hz, 1H), 3.14 (s, 3H), 2.84 (dt, J = 9.6, 2.0 Hz, 1H), 2.36-2.27 (m, 1H), 2.15-2.04 (m, 1H), 1.98-1.82 (comp. m, 2H); 13C NMR (100 MHz, CDCl3) δ 176.5, 138.6, 136.7, 134.2, 133.4, 131.4, 131.30, 131.25, 129.2, 128.8, 125.7, 125.0, 124.6, 124.5, 119.9, 81.2, 63.9, 54.3, 26.5, 26.1, 24.5; IR (film) 1699, 1598, 1498, 1390, 1296 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C23H22N2O + H]+: 343.1805, found 343.1812.
(3R,7aS)-3-(8-fluoronaphthalen-1-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-45)
74% yield. Colorless oil (Rf = 0.61 in 7:3 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 7.47 (dt, J = 7.6, 4.8 Hz, 1H), 7.40-7.25 (comp. m, 5H), 7.09 (t, J = 7.2 Hz, 1H), 6.82 (s, 1H), 3.89 (dd, J = 9.2, 3.2 Hz, 1H), 3.66-3.64 (m, 1H), 2.97-2.87 (m, 1H), 2.33-2.23 (m, 1H), 2.21-2.10 (m, 1H), 1.98-1.81 (comp. m, 2 H); 13C NMR (100 MHz, CDCl3) δ 176.3, 159.5 (d, J = 249.8 Hz), 138.4, 137.2 (d, J = 5.9 Hz), 132.7 (d, J = 7.3 Hz), 129.2 (d, J = 2.7 Hz), 129.1, 126.3 (d, J = 4.8 Hz), 126.2 (d, J = 3.0 Hz), 125.5 (d, J = 3.7 Hz), 124.6, 123.5, 121.1 (d, J = 13.1 Hz), 119.8, 112.6 (d, J = 24.1 Hz), 80.8 (d, J = 14.3 Hz), 63.7, 55.3, 26.8, 24.8; 19F NMR (272 MHz, CDCl3) δ −107.9; IR (film) 1699, 1597, 1498, 1388, 1308 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C22H19FN2O + H]+: 347.1554, found 347.1557.
(3R,7aS)-3-(5-nitronaphthalen-1-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-46)
58% yield. Red foam (Rf = 0.38 in 3:2 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 8.47 (d, J = 8.4 Hz, 1H), 8.41, (d, J = 8.8 Hz, 1H), 8.19 (dd, J = 8.8, 7.2 Hz, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.57 (dd, J = 8.4, 7.6 Hz, 1H), 7.51 (d, J = 7.6 Hz, 2H), 7.40 (d, J = 7.2 Hz, 1H), 7.26 (dd, J = 8.0, 7.6 Hz, 2H), 7.09 (t, J = 7.6 Hz, 1H), 6.45 (s, 1H), 3.89 (dd, J = 9.2, 3.6 Hz, 1H), 3.67-3.60 (m, 1H), 3.00 (dt, J = 7.2, 2.4 Hz, 1H), 2.34-2.23 (m, 1H), 2.23-2.12 (m, 1H), 2.05-1.86 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.7, 148.1, 138.1, 134.1, 132.0, 129.3, 128.9, 128.7, 126.3, 125.3, 125.1, 124.4, 124.1, 123.5, 119.8, 79.9, 64.4, 55.3, 27.0, 25.0; IR (film) 1701, 1522, 1498, 1388, 1341 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C22H19N3O3 + H]+: 374.1499, found 374.1498.
(3R,7aS)-3-(anthracen-9-yl)-2-phenylhexahydro-1H-pyrrolo[1,2-c]imidazol-1-one (exo-47)
79% yield. Orange powder (Rf = 0.33 in 1:1 hexanes/EtOAc). 1H NMR (400 MHz, CDCl3) δ 8.57 (br. s, 2H), 8.39 (s, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.57 (t, J = 7.6 Hz, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.18 (d, J = 7.6 Hz, 2H), 7.11 (s, 1H), 6.97 (t, J = 7.6 Hz, 2H), 6.85 (t, J = 7.6 Hz, 1H), 4.50 (t, J = 7.2 Hz, 1H), 3.26-3.05 (comp. m, 2H), 2.42-2.32 (comp. m, 2H), 2.21-2.07 (m, 1H), 2.06-1.95 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 173.4, 139.5, 130.4, 129.9, 128.6, 127.1, 125.9, 124.8, 123.3, 79.2, 66.7, 55.2, 30.0, 24.9; IR (film) 1693, 1598, 1499, 1395, 1312 cm−1; HRMS (ESI+) m/z calc’d for (M + H)+ [C26H22N2O + H]+: 379.1805, found 379.1813.
Supplementary Material
Fig. 8.
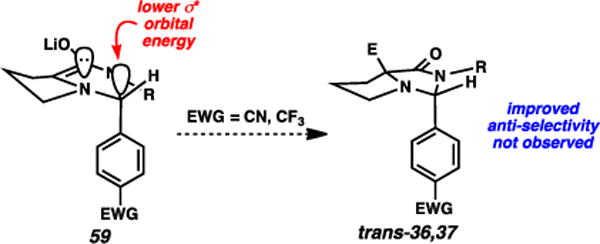
Electron-withdrawing groups do not induce improved anti-alkylation selectivity.
Scheme 6.
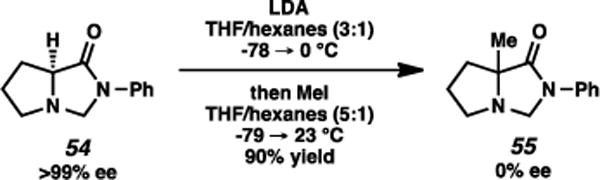
Absence of “memory effect” in imidazolidinone alkylation.
Acknowledgments
The Donors of the American Chemical Society Petroleum Research Fund are acknowledged for support of this research. Curtis Seizert, Tyler Miller, and Angella Greenawalt are acknowledged for experimental contributions.
References and notes
- 1.For a review on SRS, see:; Seebach D, Sting AR, Hoffmann M. Angew Chem, Int Ed Engl. 1996;35:2708–2748. [Google Scholar]
- 2.Stache EE, Seizert CA, Ferreira EM. Chem Sci. 2012;3:1623–1628. [Google Scholar]
- 3.For a review on α-quaternary proline derivatives, see:; Calaza MI, Cativiela C. Eur J Org Chem. 2008:3427–3448. doi: 10.1002/ejoc.200800225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For select examples of syntheses and applications of α-quaternary proline-based amino acids and derivatives, see:; (a) Frebault F, Simpkins NS, Fenwick A. J Am Chem Soc. 2009;131:4214–4215. doi: 10.1021/ja900688y. [DOI] [PubMed] [Google Scholar]; (b) Crick PJ, Simpkins NS, Highton A. Org Lett. 2011;13:6472–6475. doi: 10.1021/ol202769f. [DOI] [PubMed] [Google Scholar]; (c) Su B, Cai C, Wang Q. J Org Chem. 2012;77:7981–7987. doi: 10.1021/jo3012122. [DOI] [PubMed] [Google Scholar]; (d) Tong ST, Harris PWR, Barker D, Brimble MA. Eur J Org Chem. 2008:164–170. [Google Scholar]; (e) Bittermann H, Böckler F, Einsiedel J, Gmeiner P. Chem Eur J. 2006;12:6315–6322. doi: 10.1002/chem.200600432. [DOI] [PubMed] [Google Scholar]; (f) Vartak AP, Johnson RL. Org Lett. 2006;8:983–986. doi: 10.1021/ol0600335. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hoffmann T, Lanig H, Waibel R, Gmeiner P. Angew Chem, Int Ed. 2001;40:3361–3364. doi: 10.1002/1521-3773(20010917)40:18<3361::aid-anie3361>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (h) Genin MJ, Johnson RL. J Am Chem Soc. 1992;114:8778–8783. [Google Scholar]
- 5.Knight BJ, Stache EE, Ferreira EM. Org Lett. 2014;16:432–435. doi: 10.1021/ol403320d. [DOI] [PubMed] [Google Scholar]
- 6.Seebach D, Boes M, Naef R, Schweizer WB. J Am Chem Soc. 1983;105:5390–5398. [Google Scholar]
- 7.Beck AK, Blank S, Job K, Seebach D, Sommerfeld T. Org Synth. 1995;72:62–73. [Google Scholar]
- 8.Wang H, Germanas JP. Synlett. 1999:33–36. [Google Scholar]
- 9.For a method using the more conveniently accessed 2,2,2-trichloro-1-ethoxyethanol, see:; Artman GD, III, Rafferty RJ, Williams RM. Org Synth. 2009;86:262–274. [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes CC, Trauner D. Angew Chem, Int Ed. 2002;41:4556–4559. doi: 10.1002/1521-3773(20021202)41:23<4556::AID-ANIE4556>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 11.We chose to use isobutyraldehyde over the traditional pivalaldehyde because of cost and its similar behavior in SRS with amino amides. For a recent example, see; Wang X, Xu Y, Zhang L, Krishnamurthy D, Wirth T, Nicola T, Senanayake CH. Org Lett. 2010;12:4412–4415. doi: 10.1021/ol101960x. [DOI] [PubMed] [Google Scholar]
- 12.Although it stands to reason that an alternative approach to the target α-amino amides would be to use this Wang/Germanas protocol followed by amide bond formation, we found that this latter bond formation was generally difficult and low yielding with more sterically hindered α-substituted oxazolidinones.
- 13.Myers AG, Yoon T, Gleason JL. Tetrahedron Lett. 1995;36:4555–4558. [Google Scholar]
- 14.For references discussing the α-effect, see:; (a) Fina NJ, Edwards JO. Int J Chem Kinet. 1973;5:1–26. [Google Scholar]; (b) Buncel E, Um IK. Tetrahedron. 2004;60:7801–7825. [Google Scholar]
- 15.(a) Kalia J, Raines RT. Angew Chem, Int Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wiberg KB, Glaser R. J Am Chem Soc. 1992;114:841–850. [Google Scholar]
- 16.For a related example of imidazolidinone aminolysis with H2NOH·HCl, see; Feenstra RW, Stokkingreef EHM, Reichwein AM, Lousberg WBH, Ottenheihm HCJ, Kamphuis J, Boesten WHJ, Schoemaker HE, Meijer EM. Tetrahedron. 1990;46:1745–1756. [Google Scholar]
- 17.Although the formation of the exo diastereomer is generally preferred, formation of the endo diastereomer has been reported for imidazolidinones derived from pyrrolidine-based amino amide compounds. For examples and discussions, see:; (a) Shibatomi K, Uozumi Y. Tetrahedron: Asymmetry. 2002;13:1769–1772. [Google Scholar]; (b) Plaskon AS, Ryabukhin SV, Volochnyuk DM, Shivanyuk AN, Tolmachev AA. Tetrahedron. 2008;64:5933–5943. [Google Scholar]; (c) Fuentes de Arriba ÁL, Simón L, Raposo C, Alcázar V, Sanz F, Muñiz FM, Morán JR. Org Biomol Chem. 2010;8:2979–2985. doi: 10.1039/b926284a. [DOI] [PubMed] [Google Scholar]; (d) Trachsel A, Buchs B, Godin G, Crochet A, Fromm KM, Herrmann A. Eur J Org Chem. 2012:2837–2854. [Google Scholar]; (e) Tin S, Fuentes JA, Lebl T, Clarke ML. Eur J Org Chem. 2013:141–147. [Google Scholar]; (f) Khan FA, Ahmad S. Tetrahedron Lett. 2013;54:2996–2998. [Google Scholar]
- 18.The stereochemical course of alkylations can be subject to notably minor structural modifications in the enolate precursor. See ref 1 for a thorough discussion.
- 19.There is a lone report of a syn-selective alkylation based on mandelic acid derivatives:; Liu YQ, Liu H, Zhong B, Deng YL, Liu K. Synth Commun. 2005;35:1403–1412. [Google Scholar]; In this case, the benzaldehyde acetal is illustrated to provide the opposite stereochemical outcome than what has been established anti-selectivity with pivalaldehyde-based acetals. The configuration of this acetal stereocenter may be illustrated incorrectly, however, making this process an anti-selective alkylation. This notion is corroborated by another report from the same authors, where anti-selective alkylations of the benzaldehyde-derived mandelic acid derivative are performed:; Han XY, Liu H, Liu CH, Wu B, Zhong BH, Liu KL. J Chem Res. 2004:816–817. [Google Scholar]
- 20.It is unclear why the endo diastereomer was so difficult to enolize effectively, but we note that this observation further validated our decision to abandon Strategy A and focus on Strategy B.
- 21.The yields for these specific alkylations (entries 14 and 15) were significantly lower (~15-20%) than the others that were evaluated (generally >40%).
- 22.Related imidazolidinone hydrolysis to amino acid:; Seebach D, Dziadulewicz E, Behrendt L, Cantoreggi S, Fitzi R. Liebigs Ann Chem. 1989:1215–1232. [Google Scholar]
- 23.For seminal examples of memory effects in asymmetric alkylations of α-amino acid derivatives, see:; (a) Kawabata T, Wirth T, Yahiro K, Suzuki H, Fuji K. J Am Chem Soc. 1994;116:10809–10810. [Google Scholar]; (b) Kawabata T, Suzuki H, Nagae Y, Fuji K. Angew Chem, Int Ed. 2000;39:2155–2157. doi: 10.1002/1521-3773(20000616)39:12<2155::aid-anie2155>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 24.Pyramidalization of the nitrogen atoms of the imidazolidinone should occur upon enolization. This orientation is consistent with a proposed transition state in the alkylations of imidazolidinones based on glycine derivatives. See:; Seebach D, Juaristi E, Miller DD, Schickli C, Weber T. Helv Chim Acta. 1987;70:237–261. [Google Scholar]
- 25.Meyer EA, Castellano RK, Diederich F. Angew Chem, Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 26.For a summary of computational and spectroscopic studies of anion/arene C–H interactions, see:; Hay BP, Bryantsev VS. Chem Commun. 2008:2417–2428. doi: 10.1039/b800055g. [DOI] [PubMed] [Google Scholar]; For select examples, see:; (a) French MA, Ikuta S, Kebarle P. Can J Chem. 1982;60:1907–1918. [Google Scholar]; (b) Bryantsev VS, Hay BP. J Am Chem Soc. 2005;127:8282–8283. doi: 10.1021/ja0518272. [DOI] [PubMed] [Google Scholar]; (c) Schneider H, Vogelhuber KM, Schinle F, Weber JM. J Am Chem Soc. 2007;129:13022–13026. doi: 10.1021/ja073028k. [DOI] [PubMed] [Google Scholar]
- 27.For recent examples of the naphthyl α-C–H bond being invoked as H-bond donors, see:; (a) Laursen JS, Engel-Andreasen H, Fristrup P, Harris P, Olsen CA. J Am Chem Soc. 2013;135:2835–2844. doi: 10.1021/ja312532x. [DOI] [PubMed] [Google Scholar]; (b) Huang Z, Lim LH, Chen Z, Li Y, Zhou F, Su H, Zhou J. Angew Chem, Int Ed. 2013;52:4906–4911. doi: 10.1002/anie.201300621. [DOI] [PubMed] [Google Scholar]; (c) Huang Z, Chen Z, Lim LH, Quang GCP, Hirao H, Zhou J. Angew Chem, Int Ed. 2013;52:5807–5812. doi: 10.1002/anie.201300481. [DOI] [PubMed] [Google Scholar]
- 28.Zotova N, Franzke A, Armstrong A, Blackmond DG. J Am Chem Soc. 2007;129:15100–15101. doi: 10.1021/ja0738881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.