

Abstract
Sorsby fundus dystrophy (SFD), an autosomal dominant, fully penetrant, degenerative disease of the macula, is manifested by symptoms of night blindness or sudden loss of visual acuity, usually in the third to fourth decades of life due to choroidal neovascularization (CNV). SFD is caused by specific mutations in the Tissue Inhibitor of Metalloproteinase-3, (TIMP3) gene. The predominant histo-pathological feature in the eyes of patients with SFD are confluent 20–30 m thick, amorphous deposits found between the basement membrane of the retinal pigment epithelium (RPE) and the inner collagenous layer of Bruch’s membrane. SFD is a rare disease but it has generated significant interest because it closely resembles the exudative or “wet” form of the more common age-related macular degeneration (AMD). In addition, in both SFD and AMD donor eyes, sub-retinal deposits have been shown to accumulate TIMP3 protein. Understanding the molecular functions of wild-type and mutant TIMP3 will provide significant insights into the patho-physiology of SFD and perhaps AMD. This review summarizes the current knowledge on TIMP3 and how mutations in TIMP3 cause SFD to provide insights into how we can study this disease going forward. Findings from these studies could have potential therapeutic implications for both SFD and AMD.
Clinical Disease:
Sorsby Fundus Dystrophy (SFD) is a rare late-onset autosomal dominant inherited retinal dystrophy originally described in 1949 by Arnold Sorsby and Mary E. Joll Mason. They noted striking retinal and choroidal findings of a distinct pattern common among members of five affected families (Sorsby and Joll Mason, 1949). The initial presentation involved central visual blurring, usually around the age of 40 years, with physical exam findings of macular edema and hemorrhage. This was followed, within months or a few years, by similar symptoms and clinical findings in the contralateral eye. Over the course of the next several years, central macular scarring with pigmentation would extend from the macula into the periphery. At the late stage of the disease, approximately 35 years after presentation, extensive peripheral choroidal atrophy and vessel sclerosis was noted with severe central and peripheral vision loss. Causative mutations have since been identified in the tissue inhibitor of metalloproteinases-3 (TIMP3) gene.
Clinical Presentation.
Almost 70 years after the initial description of the disease, our understanding of the clinical symptoms of SFD have remained largely unchanged, with a few exceptions. While central visual changes usually affect individuals in the 4th to 6th decades of life, visual symptoms have been reported to manifest as early as the second decade (Eriksson et al., 1990; Felbor et al., 1997; Gliem et al., 2015b). Clinical symptoms at presentation typically include metamorphopsia, central scotomas, reduced color vision, and sudden loss of central vision (Capon et al., 1988; Hamilton et al., 1989). Abrupt changes in central vision usually result from complications of choroidal neovascular membranes (CNVM), while more slowly progressive vision loss results from expanding areas of geographic atrophy. The latter can occur more rapidly than is usually seen in age-related macular degeneration (AMD). While the original Sorsby report specifically noted that affected individuals did not experience nyctalopia (night blindness), impaired night vision, delayed dark adaptation, and progressive visual field defects have been reported at multiple stages of the disease (Burn, 1950; Capon et al., 1988; Gliem et al., 2015b; Hamilton et al., 1989).
Typical clinical exam findings include the presence of drusen in the posterior pole and along the vascular arcades. Drusen are extracellular deposits located between the basal lamina of the retinal pigmented epithelium (RPE) and the inner collagenous layer of Bruch’s membrane (BM). Reticular drusen (or reticular pseudodrusen) have also been reported in individuals with SFD (Gliem et al., 2015a). Unlike drusen, reticular drusen are subretinal deposits located between the photoreceptor outer segments (POS) and the apical surface of the RPE. They are best appreciated clinically on fundus autofluorescence (FAF) imaging where they can be seen distributed in a distinct reticular pattern (Spaide and Curcio, 2010). Reticular pseudodrusen are often associated with worse visual function and higher likelihood for progression to CNVM and geographic atrophy in AMD patients (Finger et al., 2016; Zhou et al., 2016). In contrast, SFD individuals can develop CNVM at any time, concurrently with drusen deposits or even in their absence. Similar to AMD, active CNVM in SFD individuals can lead to intraretinal edema, subretinal fluid, and subretinal hemorrhages. These findings are detectable by fundoscopic exam, spectral domain optical coherence tomography (SD-OCT), fluorescein angiography (FA), or OCT angiography (OCTA). Geographic atrophy can be observed in the central macula and result in progressive vision loss over time. Late in the disease, there can be widespread central and peripheral chorioretinal atrophy with multilobulated borders best appreciated on FAF (Sivaprasad et al., 2008; Sorsby and Joll Mason, 1949). Of note, there can be wide variations in expressivity between SFD individuals with different TIMP3 mutations as well as among family members with the same genetic mutation. Reasons for the disparity in age of onset and disease severity remain unclear, although interactions with additional genes or environmental factors may contribute to these differences.
The significant central and peripheral chorioretinal atrophy appreciated throughout the course of the disease implicate choroidal vascular changes in either the pathogenesis or as an early marker of disease progression. Gliem et al report that marked choroidal thinning was noted in SFD individuals with significant chorioretinal atrophy as measured by horizontal enhanced depth imaging (EDI) OCT. Separately, the same study reported that reduced late-phase central macular fluorescence on indocyanine green angiography (ICG-A) could be observed in otherwise asymptomatic individuals with normal funduscopic findings, fundus autofluorescence (FAF), SD-OCT, fluorescein angiography, and NIR reflective imaging, implying that ICG-A may be useful for detection of early signs of disease, which may include changes in Bruch’s membrane and RPE (Gliem et al., 2015b). Choroidal perfusion may also be clinically relevant in SFD individuals with reticular pseudodrusen in light of recent findings demonstrating that AMD patients with reticular pseudodrusen and poor vision are associated with significantly larger areas of choriocapillaris nonperfusion as determined by OCTA (Chatziralli et al., 2017; Nesper et al., 2017). Future studies utilizing OCTA imaging may be useful for detecting early disease and for predicting disease progression and vision loss.
Differential Diagnosis.
Retinal diseases with similar presentation to SFD include AMD, Malattia Leventinese (familial dominant drusen or Doyne honeycomb retinal dystrophy; EFEMP1), pattern dystrophy (PRPH2), or other dominantly inherited macular dystrophies, including late stage Best disease (BEST1). A recent report of a young patient with SFD presenting with sub-retinal fluid and no drusen was initially mis-diagnosed as central serous retinopathy, and this entity can be included in the differential diagnosis as well (Menassa et al., 2017). While the clinical features of SFD, including drusen, CNVM development, and central geographic atrophy, are similar to AMD, the earlier onset, strong inheritance pattern, and late involvement of peripheral chorioretinal atrophy are important distinguishing characteristics. Genetic testing is often crucial to confirm or rule out clinical diagnosis. Furthermore, early SFD diagnosis is particularly important for treatment to preserve central vision, monitoring of the disease, and for appropriate counseling. Fortunately, genetic testing is available in most instances for a definitive diagnosis.
Treatment.
Early attempts at treating SFD involved ameliorating the night blindness symptoms experienced by some patients (Jacobson et al., 1995b). The authors of this study posited that SFD night blindness was due to a chronic deprivation of vitamin A of the photoreceptors due to the thickened Bruch’s membrane separating the RPE from the choriocapillaris. After administration of oral Vitamin A at 50,000 IU/d, there was a short-term reversal of night blindness in patients at early stages of disease(Jacobson et al., 1995b). While these initial results were promising, it has not become a widely used treatment due to the potential toxicity of long term high dose Vitamin A and reports of lack of efficacy at lower doses (15,000 IU/day) in advanced disease (Gliem et al., 2015b).
Current treatment for SFD largely revolves around management of the complications resulting from choroidal neovascularization in the macula. Prior to widespread intravitreal anti- VEGF use, multiple treatment modalities including photodynamic therapy (PDT) for subfoveal CNVM and argon laser photocoagulation for extrafoveal CNVM were attempted with variable success (Keller et al., 2015). An early study reported resolution of active CNVM in an SFD patient in response to a series of systemic infusions of bevacizumab (Prager et al., 2007). Intravitreal injections of bevacizumab and ranibizumab have been reported to be successful in the treatment of active CNVM in SFD patients (Balaskas et al., 2013; Fung et al., 2013; Gemenetzi et al., 2011; Gliem et al., 2015b; Gray et al., 2012; Kapoor and Bakri, 2013; Menassa et al., 2017). Early CNVM detection and treatment with anti-VEGF agents result in improved visual prognosis and reduced subretinal scarring and is now considered the mainstay of SFD treatment (Keller et al., 2015; Menassa et al., 2017). The frequency of CNVM recurrences is variable, and no particular treatment protocol has been studied. However, protocols similar to those used for neovascular AMD, with an initial 3 monthly injections, then pro re nata regimen with monthly monitoring have been reported (Gliem et al., 2015b; Menassa et al., 2017). Because of the importance of early detection and treatment in preserving central vision, genetic screening of asymptomatic family members should be considered.
Pathology:
The predominant histopathological feature of SFD appears to be thick, widespread, confluent, lipid-enriched amorphous deposits located between the inner collagenous layer of Bruch’s membrane and the basement membrane of the RPE (Capon et al., 1989). This is different from the discrete drusen seen in AMD. One of the limitations in reporting of histopathology in SFD stems from the dearth of post-mortem ocular tissue due to the rarity of the disease. A immunohistochemical study of the eyes of a 77 year old woman carrying the p.(Ser204Cys) (originally S181C) mutation showed advanced pathology with almost complete loss of the photoreceptors and advanced gliosis of the remnant retina. A markedly thickened Bruch’s membrane contained extracellular periodic acid Schiff (PAS) positive deposits localized between the elastin layer of Bruch’s membrane and the RPE. The deposits contained TIMP3 as determined by immunohistochemistry but were not a consequence of increased mRNA expression of TIMP3 {Chong, 2000 #5106;Chong, 2003 #5443}. Patchy areas of RPE thinning were observed and interestingly localized loss of TIMP3 was noted in the Bruch’s membrane underlying these regions (Fariss et al., 1998). The accumulation of TIMP3 in subretinal deposits in SFD and in drusen in AMD (Crabb et al., 2002) as well as the frequent finding of reticular pseudodrusen in patients with SFD and AMD (Gliem et al., 2015a) suggests a common role of Bruch’s membrane and RPE in the pathogenesis of both these diseases.
Mutations in TIMP3:
SFD is caused by mutations in the TIMP3 gene that are inherited in an autosomal dominant fashion. TIMP3 encodes the tissue inhibitor of metalloproteinases-3, a multifunctional protein that targets enzymes degrading matrix components or catalysing the shedding of ectodomains from cell surface proteins. Furthermore, TIMP3 possesses pro-apoptotic and anti-angiogenic activities. TIMP3 is expressed and secreted by RPE and choroidal endothelial cells. To this date, sixteen different mutations have been associated with the SFD phenotype (Figure 1). Protein sequence variants were described based on the protein reference sequence which includes the 23-amino-acid signal peptide the according to HGVS recommendations:2016 update. With the exception of stop mutation p.(Glu162*) (Langton et al., 2000) and c.439–2dupA at the intron 4/exon 5 boundary (Tabata et al., 1998), fourteen mutations constitute missense mutations that introduce amino acid substitutions: p.(Ser38Cys) (Schoenberger and Agarwal, 2013), p.(Glu162Lys) (Saihan et al., 2009), p.(Tyr151Cys) (Gliem et al., 2015b), p.(Asp167Asn) (Riera et al., 2017), p.(Tyr177Cys) (Gliem et al., 2015b), p.(Ser179Cys) (Felbor et al., 1995), p.(His181Arg) (Lin et al., 2006), p.(Tyr182Cys) (Gliem et al., 2015b), p.(Gly189Cys) (Felbor et al., 1997), p.(Gly190Cys) (Jacobson et al., 1995a). p.(Tyr191Cys) (Weber et al., 1994), p.(Ser193Cys) (Barbazetto et al., 2005), p.(Tyr195Cys) (Jacobson et al., 2002), and p.(Ser204Cys) (Weber et al., 1994). The analysis of ECM/cell lysates from gingival fibroblasts from a patient carrying the p.(Glu162*) mutation revealed the expression of a truncated TIMP3 molecule (Arris et al., 2003), the effect of the c.439–2dupA splice mutation on the protein level has not been investigated. Most of the SFD-associated mutations are located in the last exon of the TIMP3 gene and/or affect the number of cysteine residues in the respective mutant protein. Recently, two unrelated families with a syndromic association of SFD and pulmonary disease have been reported, thus broadening the phenotypic spectrum of TIMP3-associated disorders (Meunier et al., 2016).
Figure 1:
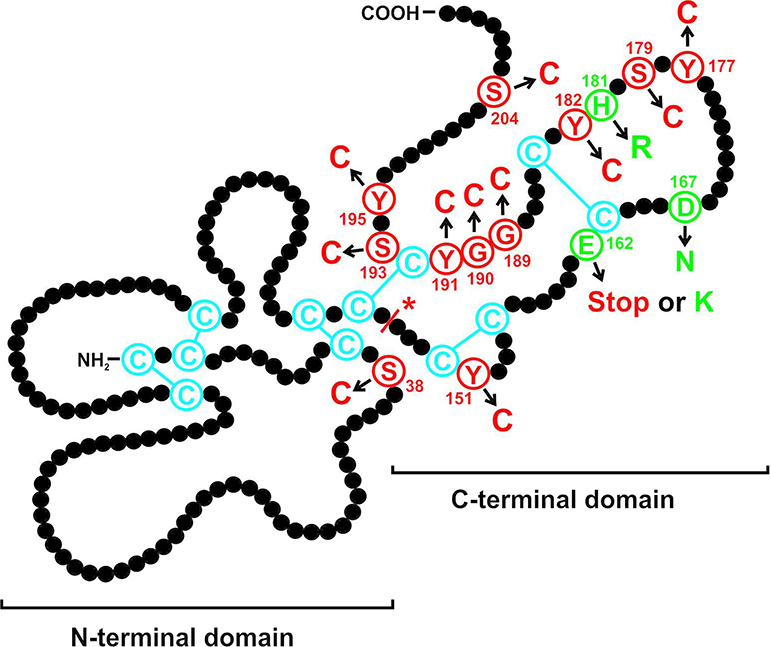
Schematic illustration of the TIMP3 protein displaying mutations that cause autosomal dominant Sorsby fundus dystrophy. Each amino acid residue of the mature protein is shown by a black filled circle. The twelve cysteine residues predicted to form six disulphide bonds are indicated in blue. Mutations leading to an unpaired cysteine are shown in red, the three missense mutations leading to amino acid exchanges other than cysteines are depicted in green. An asterisk indicates the splice mutation. The figure has been modified from Figure 10 in Gliem et al., 2015, copyright holder: Association for Research in Vision and Ophthalmology
A genome-wide association scan for age-related macular degeneration (AMD), a complex and common late-onset disease with striking clinical similarities to SFD, in 2010 was the first to identify a susceptibility locus within intron 5 of the synapsin III (SYN3) gene that also harbors the entire TIMP3 gene (Chen et al., 2010). The association of this non-coding region with AMD was later confirmed by independent studies (Cascella et al., 2017; Fritsche et al., 2013; Fritsche et al., 2016; Yu et al., 2011). The molecular mechanism by which certain common single nucleotide polymorphisms in the genomic region approximately 100 kb upstream of exon 1 of TIMP3 affects the risk to develop macular degeneration is currently unknown. Interestingly, targeted genotyping of known TIMP3 sequence variants and another 44 point mutations predicted to generate an additional cysteine residue in a hypothetical mutant TIMP3 protein revealed a >30-fold excess of mutations among subjects with macular degeneration in a large set of more than 30,000 subjects (29 in 16,144 cases versus 1 in 17,832 controls) (Fritsche et al., 2016). This observation has led to the hypothesis that rare TIMP3 protein-altering variants together with alleles at other AMD risk loci contribute to AMD development(Fritsche et al., 2016). A retrospective, observational case series has recently shown that patients initially diagnosed with neovascular AMD carry the SFD-associated p.(Ser38Cys)-TIMP3 mutation(Warwick and Lotery, 2017). SFD phenotypic expressivity varies considerably including large intra- and interfamilial variability in the age of onset of visual symptoms(Gliem et al., 2015b). Thus, TIMP3 missense mutations identified in patients from AMD cohorts may mimic AMD but actually represent late-onset SFD.
Linking SFD and CADASIL:
The mutation spectrum of TIMP3 causing SFD is very similar to that of NOTCH3 causing CADASIL, a cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. NOTCH3 encodes a transmembrane protein belonging to the evolutionarily conserved NOTCH receptor family and is involved in signalling pathways in vascular smooth-muscle cells(Wang et al., 2008). The large majority of pathogenic mutations in CADASIL are associated with changes in the number of cysteines causing an odd number of cysteines in the mutant NOTCH3 (Joutel et al., 1997). This facilitates the crosslinking of SH-groups and leads to misfolding of the NOTCH3 receptor, enhanced spontaneous formation of oligomers and aggregation of the extracellular domain (NOTCH3-ECD)(Duering et al., 2011; Opherk et al., 2009). Moreover, both, SFD and CADASIL are characterized by an abnormal accumulation of extracellular material harboring the respective causative proteins. In CADASIL, the typical extracellular deposits are located close to the surface of vascular smooth-muscle cells, the so-called granular osmiophilic material (GOM), and consist of excess levels of NOTCH3-ECD(Ishiko et al., 2006). It has recently been shown that mutant NOTCH3-ECD interacts with TIMP3 (and other ECM proteins) promoting the accumulation of TIMP3 in those deposits(Monet-Lepretre et al., 2013). There is evidence that the increased amounts of TIMP3 retaining its MMP inhibitory activity contribute to CADASIL pathophysiology since haploinsufficiency of Timp3 in mice rescues reduced cerebrovascular reactivity, one of CADASIL-related phenotypic features(Capone et al., 2016). Thus, elevated levels of extracellular TIMP3 also seem to be crucial for small vessel disease. Similar to mutant NOTCH3-ECD, mutant TIMP3 may abnormally recruit and sequester other proteins in BM/drusen, thereby eliciting pathological effects leading to SFD development. Proteome analysis of drusen-like deposits isolated from SFD patient-derived cell culture systems will facilitate the identification of candidate proteins.
Molecular mechanisms of SFD - in vitro studies.
The functional consequences of different SFD-associated TIMP3 mutations have been investigated intensively in previous years and consisted of in vitro and ex vivo analyses of effects on dimerization, the inhibition of different matrix metalloproteases, the interaction with the extracellular matrix, glycosylation and the binding to the VEGFR2 receptor. These studies have been performed using various TIMP3 mutants, cell lines and assays and have so far not given a clear answer about the disease pathomechanism (details of these studies are reviewed in Stohr and Anand-Apte, 2012 (Stohr and Anand-Apte, 2012), and more recently in Christensen et al., 2017 (Christensen et al., 2017). The analysis of several normal non-diseased donor eyes has shown that TIMP3 protein accumulates in BM with age (Fariss et al., 1998; Kamei and Hollyfield, 1999; Sohn et al., 2014) and it has been suggested that this deposition may be accelerated in SFD due to an increased tendency of mutant TIMP3 to form turnover-resistant multimeric molecules (Langton et al., 2005). Excess of TIMP3 in BM possibly as a consequence of inadequate clearance or other abnormal mechanisms could potentially disturb ECM homeostasis, lead to BM thickening over time and cause downstream pathogenic processes like atrophy or CNV.
Apart from histopathologic studies on human cadaver eyes obtained from SFD patients and TIMP3 knock-in (TIMP3+/S156C, TIMP3S156C/S156C) and TIMP3 knock-out (TIMP3−/−) mouse models, the cell culture platform has been extensively utilized to study the effect of disease-causing mutations on TIMP3 gene/protein expression and function.
Physiological function(s) of TIMP3.
The molecular/pathologic effect of overexpression and knockdown of wild-type (WT) TIMP3 has been assessed in cell culture studies. Recombinant TIMP3 in overexpression heterologous expression systems is largely N-glycosylated and retains the ability to inhibit the known TIMP3-target enzymes matrix metalloproteinases (MMPs), MMP1, MMP2, MMP3 and MMP9 (Apte et al., 1995). Furthermore, TIMP3 overexpression has been shown to cause dose-related cell death in several cell lines, including corneal stromal cells, HELA, HT1080, MCF-7 cells, vascular smooth muscle cells, cultured adult human RPE cells and monkey choroidal endothelial cells (Baker et al., 1999; Baker et al., 1998; Majid et al., 2002a, b; Matthews et al., 2007; Qi and Anand-Apte, 2015). Interestingly, like WT TIMP3 overexpression, TIMP3 knockdown has also been shown to cause increased cell death (Fata et al., 2001). Furthermore, knockdown studies of WT TIMP3, have shown a role of TIMP3 in angiogenesis and inflammation, mostly by impacting VEGF-VEGFR2 interaction and/or TNF signaling (Lee et al., 2002; Qi et al., 2003a; Qi et al., 2003b; Smookler et al., 2006).
Loss of function or gain of function in SFD?
To interrogate the molecular mechanisms of SFD, cell culture studies comparing WT and mutant proteins have incorporated several different disease causing TIMP3 mutations in overexpression studies utilizing ARPE19 cells, endothelial cells (porcine arotic endothelial cells) or a more generic mammalian cell line (e.g. COS7, BHK, MCF-7). Furthermore, fibroblasts obtained from human SFD patients and TIMP3 mutant mouse model have also been utilized to study the impact of TIMP3 mutation on TIMP3 expression and/or function (Arris et al., 2003; Soboleva et al., 2003). Specific TIMP3 expression and functional attributes have been evaluated in studies comparing WT vs. mutant TIMP3, including the level of TIMP3 glycosylation, secretion and aggregation/dimerization of TIMP3 in the extracellular matrix (ECM), TIMP3’s competence to bind ECM proteins like LAM and COL1, and TIMP3’s ability to inhibit specific matrix metalloproteinases or MMPs (e.g. MMP2, MMP9). In addition, the ability of WT vs. mutant TIMP3 to modulate cellular apoptosis and VEGF-VEGFR2 interaction has also been evaluated {Qi, 2015 #7928}. Interestingly, the impact of TIMP3 mutation(s) on TIMP3 expression and/or function has been cell-type and/or mutation dependent. For instance, mutant TIMP3 [(p.(Ser204Cys)] has been shown to retain normal glycosylation pattern in SFD patient-derived fibroblasts and BHK cells (Arris et al., 2003; Soboleva et al., 2003; Yeow et al., 2001). However, [(p.(Ser179Cys)] mutant TIMP3 in endothelial cells leads to increased TIMP3 glycosylation (Qi et al., 2009). Likewise, TIMP3 turnover is reduced in ARPE19 cells overexpressing p.(Ser179Cys), p.(Glu162*) and [(p.(Ser204Cys)] TIMP3 mutants, but remains unaffected in mouse fibroblasts expressing the p.(Ser179Cys) TIMP3 mutant. Similarly, several studies on distinct TIMP3 variants in several cell lines (BHK, COS7, ARPE-19, patient-derived fibroblasts) have demonstrated that mutant TIMP3 maintains its MMP2/MMP9 inhibitory function (Arris et al., 2003; Langton et al., 2000; Langton et al., 2005; Saihan et al., 2009; Yeow et al., 2001). In contrast, overexpression of (p.(Ser179Cys) and p.(Glu162*) TIMP3 mutants in COS7, BHK, endothelial cell, mouse fibroblast and ARPE19 cells has yielded contradictory results with respect to the retention of TIMP3’s MMP inhibitory activity (Arris et al., 2003; Langton et al., 2000; Langton et al., 2005; Qi et al., 2002; Soboleva et al., 2003; Yeow et al., 2001). Another characteristic of TIMP3 that has been thoroughly interrogated in cell culture, is the propensity of mutant TIMP3 to form dimers and protein aggregates. These studies have also yielded confounding results. Specifically, p.(Ser204Cys), p.(Ser179Cys), p.(Glu162Lys), p.Gly189Cys), p.(Gly190Cys), p.(Tyr191Cys) and p.(Glu162*) TIMP3 mutations lead to increased TIMP3 dimerization/aggregation in several distinct cell types, including ARPE19, COS7, BHK, and patient-derived fibroblast (Arris et al., 2003; Langton et al., 1998; Langton et al., 2000; Langton et al., 2005; Saihan et al., 2009; Weber et al., 2002; Yeow et al., 2001). In contrast the propensity of p.(Ser179Cys) TIMP3 mutant to dimerize in ARPE-19 cells has yielded conflicting results (Langton et al., 2005; Qi et al., 2002). The localization of TIMP3 mutant to the ECM is also controversial with studies showing 1) both proper localization in the ECM of specific TIMP3 variants (p.(Ser204Cys), p.(Ser179Cys), p.Gly189Cys p.(Glu162*) in COS7 cells (Langton et al., 1998; Langton et al., 2000) and 2) mislocalization of mutant TIMP3 (p.(Ser179Cys), p.(Glu162*) in the soluble conditioned media fraction opposed to the ECM in endothelial cell and primary gingival fibroblast cultures from SFD patients (Arris et al., 2003; Qi et al., 2009). Although the effect of multiple mutations and cell type(s) for specific TIMP3 associated functional attributes (apoptosis, VEGF-VEGF-R2 binding) has not been investigated, specific TIMP3 mutations seem to affect apoptosis and VEGF binding to VEGFR2. Specifically, p.(Ser179Cys), TIMP3 expression in endothelial cells positively influences VEGF binding to VEGFR2 (Qi et al., 2009). However these results are not consistent with the effect of p.(Ser179Cys), TIMP3 mutation to block VEGF-VEGFR2 binding in mouse-derived tissue cultures from TIMP3+/S156C mice(Fogarasi et al., 2008). Similarly p.(Ser179Cys), p.(Gly190Cys), p.(Tyr191Cys) and p.(Ser204Cys) TIMP3 mutant overexpression compared to WT TIMP3 overexpression in adult human RPE cells and MCF-7 cells led to increased cellular apoptosis (Majid et al., 2002b).
Overall, studying the effect of TIMP3 mutation on TIMP3 function in an heterologous expression system has provided evidence for both a plausible gain of function (e.g. excess TIMP3, reduced TIMP3 turnover) and loss of function (e.g. reduced MMP inhibitory activity) in the disease. The impact of TIMP3 mutation on TIMP3 function in different cell type(s) (e.g. fibroblast vs. COS7 vs. ARPE 19) yielded contradictory results even for the same “disease-causing TIMP3 variant”. One possible solution to overcome the underlying discrepancies due to TIMP3’s cell-type specific behavior and to confirm the effect of specific TIMP3 mutation on in the human eye would be to employ a cell culture RPE/CC mimetic. Ideally this would 1) display key physical and functional attributes of its in vivo counterpart and 2) be sufficient to develop key pathological manifestations (e.g. drusen, neo-vascularization) as a consequence of TIMP3 dysfunction and thus help interrogate the underlying disease mechanism of an “in vitro disease in a dish”.
Molecular mechanisms of SFD - Potential hiPSC-approach.
Human induced pluripotent stem cell (hiPSC) provide a unique approach to model and study human diseases in patient’s own cells. This is especially relevant to retinal diseases, like SFD, where lack of a suitable animal or cell culture model capable of recapitulating important pathological aspects of the human disease, has hampered the pursuit of the underlying disease mechanism. With regard to SFD and other similar maculopathies that affect multiple distinct cell layers in the eye (e.g. RPE and choriocapillaris), patient-derived hiPSCs also provide a suitable strategy to interrogate the contribution of a singular cell type (RPE vs. choroidal endothelial cells (CECs)) to the disease pathophysiology. Importantly, differentiation protocols to generate both RPE and CE Cs, the cell type(s) relevant to SFD pathology, from hiPSCs have already been developed (Buchholz et al., 2009; Buchholz et al., 2013; Kokkinaki et al., 2011; Singh et al., 2013a; Songstad et al., 2015). Furthermore, several studies have successfully utilized patient-derived hiPSC-derived RPE cells to investigate the molecular mechanisms underlying several retinal degenerative diseases, including both monogenic maculopathies (e.g. Best vitelliform macular dystrophy or BVMD, MFRP-associated retinitis pigmentosa(RP)) and AMD (Golestaneh et al., 2016; Li et al., 2014; Li et al., 2017; Saini et al., 2017; Singh et al., 2013b; Yang et al., 2014). Using hiPSC-RPE from patients with disease causing mutations in BEST1 gene, Li et al, recently confirmed a role of BEST1 in mediating Ca2+-dependent Cl- current in RPE cells (Li et al., 2017). In earlier studies, using hiPSC-RPE based disease modeling, impaired processing of POS by RPE cells was linked for the first time to BVMD development (_S1_Reference72Singh et al., 2013b). Similarly, the comparison of hiPSC-RPE from control individual and Membrane Frizzled-related Protein (MFRP)-associated -RP patients highlighted abnormalities in actin cytoskeleton organization in the disease that could be rescued by AAV-mediated delivery of wild-type MFRP (Li et al., 2014). Similar to inherited maculopathies, patient-derived hiPSC-RPE studies utilizing AMD patient samples have confirmed the involvement of specific cellular pathways, including oxidative stress and complement system in the disease development and progression (Golestaneh et al., 2016; Saini et al., 2017; Yang et al., 2014). It is noteworthy that patient-derived hiPSC-RPE cells as an “in vitro disease model” have been shown to manifest phenotypic hallmarks defining specific human maculopathies. For example, accumulation of autofluorescent material was observed in hiPSC-RPE generated from BVMD patients when cultured in the presence of a physiologically-relevant stressor, POS. This possibly recapitulates the lipofuscin accumulation in the RPE of BVMD patient eyes (Singh et al., 2013b). Similarly, the formation of confluent-drusen like deposits in hiPSC-RPE monocultures derived from patients with SFD and another inherited macular dystrophies, Doyne honeycomb retinal dystrophy (DHRD) has been reported (Galloway et al., 2017). It is plausible that the ability of hiPSCs to differentiate into RPE cells that display several physical and functional characteristics akin to the human adult RPE in vivo contributes to the development of disease-relevant pathology in culture (Buchholz et al., 2009; Buchholz et al., 2013; Kokkinaki et al., 2011; Singh et al., 2013a). For instance, hiPSC-RPE cells have been shown to phagocytose and degrade POS better than human fetal RPE cells, the current gold standard of cultured human RPE cells in culture (Singh et al., 2013a; Westenskow et al., 2012). Similarly, hiPSC-RPE cells have been shown to deposit a basement membrane containing basal infoldings and synthesize and secrete several known RPE-basement membrane constituents (e.g. TIMP3, LAM, COL4) in vivo (Galloway et al., 2017; Kamao et al., 2014).
However, there are also legitimate concerns with the use of hiPSC-derived target cells for disease modeling. For instance, generation of hiPSC resets the developmental clock and hence the RPE and other cell type(s) derived from hiPSC are relatively young and thus may not be able to recapitulate key pathological and functional defects of adult-onset diseases, like SFD and AMD. To address and alleviate these issues, we and others have utilized numerous approaches. Specifically, pharmacological and physiological stressors have been utilized to mimic metabolic stress and “age” RPE cells in culture(Singh et al., 2013b; Yang et al., 2014). It has also been shown that hiPSC-derived RPE cells (unlike previous cell culture models) can be cultured for an extended period of time (up to 3 months), providing a sufficient timeframe to mimic and model specific aspects of disease-associated chronic pathology in a dish (Singh et al., 2013b; Yang et al., 2014). In fact, the relatively long culture life of hiPSC-RPE to stress the cells assisted the development of drusen in patient-derived SFD, DHRD and ADRD hiPSC-RPE cells (Galloway et al., 2017). It is also important to highlight that unlike RPE cells, a homogenous cell population that can be consistently derived from hiPSCs, other cell type(s) in the eye, including CEC’s still need to be evaluated in disease modeling studies.
Given that TIMP3 in the eye is primarily synthesized by RPE cells (Della et al., 1996), hiPSC-derived RPE cells plausibly provide a suitable platform to complement the existing cell culture and animal model platforms for investigating the SFD disease mechanism(s). Furthermore, the fact that a) hiPSC-RPE in culture lay a basement membrane containing TIMP3, and secrete TIMP3 into the ECM (Galloway et al., 2017) (Fig. 2) with banding pattern on Western blotting consistent with known monomer (~24 kDa), glycosylated-monomer (~27 KDa) and dimer (~55 kDa) forms of TIMP3 and b) patient-derived SFD hiPSC-RPE develop confluent drusen-like deposits containing known drusen-resident proteins (e.g. APOE, TIMP3)(Galloway et al., 2017) (Fig. 2) highlights their utility for investigating the role of TIMP3 mutations in the development of specific disease-relevant phenotypes of SFD (e.g. drusen). It is plausible that future efforts involving hiPSCs might also incorporate CECs or even a more complex hiPSC-derived model, like comprehensive RPE-choriocapillaris mimetic that displays both drusn formation and SFD-associated neo-vascularization phenotype, to investigate the singular role of an individual cell type (RPE vs. CEC), RPE-choriocapillaris interaction in SFD disease pathophysiology.
Figure 2:
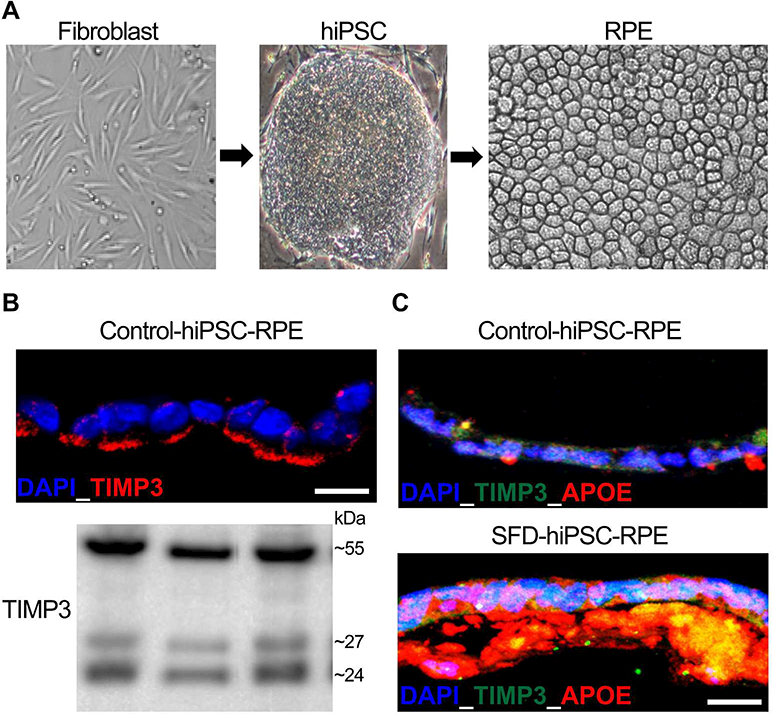
hiPSC-RPE as a tool to study SFD pathophysiology. A) Representative images showing patient-derived fibroblasts p.(Ser204Cys), hiPSCs and consequent differentiation of hiPSCs to obtain hiPSC-RPE monolayer. (B) Immunocytochemical and Western blotting analyses showing robust expression of TIMP3 in the basement membrane/ECM underlying control hiPSC-RPE cultures. Of note, the bottom panel shows the presence of TIMP3 bands consistent with unglycosylated, glycosylated and dimer forms of TIMP3 in ECM extracted from hiPSC-RPE cultures derived from three distinct control hiPSC lines. (C) Representative Immunoflurosecence labeling showing presence of APOE and TIMP3 positive drusen-like deposits underlying control vs. SFD hiPSC-RPE monolayer.
Significance Statement.
The clinical and histopathological similarities between Age-related macular Degeneration (AMD) and Sorsby Fundus Dystrophy (SFD) and the identification of variants in the TIMP3 and matrix metalloproteinase pathway genes in AMD suggest that similar downstream effectors might be in play in both conditions. Understanding the functions of TIMP3 and the effect of potential loss of those functions on retinal pathology will provide insight into the pathophysiology in SFD and likely the more commonly seen AMD.
Funding Acknowledgements:
BA-A -NIH Grants RO1EY026181, RO1EY027083, T32EY024236, P30EY025585, Foundation Fighting Blindness Center Grant, Cleveland Clinic Foundation and an Unrestricted Grant from Research To Prevent Blindness.
VRC (NIH Grants RO1EY026030, P30EY001730, BrightFocus Foundation MDR Grant, and Unrestricted Grant from Research to Prevent Blindness.
RS NIH Grant RO1EY028167, BrightFocus Foundation MDR Grant, Research to Prevent Blindness-Unrestricted Grant and Career Development Award and Foundation Fighting Blindness Individual Investigator Award
References
- Apte SS, Olsen B, Murphy G, 1995. The gene structure of tissue inhibitor of metalloproteinases (TIMP)-3 and its inhibitory activities define the distinct TIMP gene family. J. Biol. Chem 270, 14313–14318. [DOI] [PubMed] [Google Scholar]
- Arris CE, Bevitt DJ, Mohamed J, Li Z, Langton KP, Barker MD, Clarke MP, McKie N, 2003. Expression of mutant and wild-type TIMP3 in primary gingival fibroblasts from Sorsby’s fundus dystrophy patients. Biochim Biophys Acta 1638, 20–28. [DOI] [PubMed] [Google Scholar]
- Baker A, George S, Zaltsman A, Murphy G, Newby A, 1999. Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br J Cancer 79, p1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AH, Zaltsman AB, George SJ, Newby AC, 1998. Divergent effects of tissue inhibitor of metalloproteinase-1, −2, or −3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest 101, 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaskas K, Hovan M, Mahmood S, Bishop P, 2013. Ranibizumab for the management of Sorsby fundus dystrophy. Eye (Lond) 27, 101–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazetto IA, Hayashi M, Klais CM, Yannuzzi LA, Allikmets R, 2005. A novel TIMP3 mutation associated with Sorsby fundus dystrophy. Arch Ophthalmol 123, 542–543. [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Hikita ST, Rowland TJ, Friedrich AM, Hinman CR, Johnson LV, Clegg DO, 2009. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells 27, 2427–2434. [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Pennington BO, Croze RH, Hinman CR, Coffey PJ, Clegg DO, 2013. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem cells translational medicine 2, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn RA, 1950. Further cases of a fundus dystrophy with unusual features. Br J Ophthalmol 34, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capon MR, Marshall J, Krafft JI, Alexander RA, Hiscott PS, Bird AC, 1989. Sorsby’s fundus dystrophy. A light and electron microscopic study. Ophthalmology 96, 1769–1777. [DOI] [PubMed] [Google Scholar]
- Capon MR, Polkinghorne PJ, Fitzke FW, Bird AC, 1988. Sorsby’s pseudoinflammatory macula dystrophy--Sorsby’s fundus dystrophies. Eye 2, 114–122. [DOI] [PubMed] [Google Scholar]
- Capone C, Dabertrand F, Baron-Menguy C, Chalaris A, Ghezali L, Domenga-Denier V, Schmidt S, Huneau C, Rose-John S, Nelson MT, Joutel A, 2016. Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, Potenza S, Mauriello S, Novelli G, Ricci F, Cusumano A, Giardina E, 2017. Towards the application of precision medicine in Age-Related Macular Degeneration. Prog Retin Eye Res. [DOI] [PubMed] [Google Scholar]
- Chatziralli I, Theodossiadis G, Panagiotidis D, Pousoulidi P, Theodossiadis P, 2017. Choriocapillaris’ alterations in the presence of reticular pseudodrusen compared to drusen: study based on OCTA findings. Int Ophthalmol. [DOI] [PubMed] [Google Scholar]
- Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J, Tosakulwong N, Pericak-Vance MA, Campochiaro PA, Klein ML, Tan PL, Conley YP, Kanda A, Kopplin L, Li Y, Augustaitis KJ, Karoukis AJ, Scott WK, Agarwal A, Kovach JL, Schwartz SG, Postel EA, Brooks M, Baratz KH, Brown WL, Brucker AJ, Orlin A, Brown G, Ho A, Regillo C, Donoso L, Tian L, Kaderli B, Hadley D, Hagstrom SA, Peachey NS, Klein R, Klein BE, Gotoh N, Yamashiro K, Ferris Iii F, Fagerness JA, Reynolds R, Farrer LA, Kim IK, Miller JW, Corton M, Carracedo A, Sanchez-Salorio M, Pugh EW, Doheny KF, Brion M, Deangelis MM, Weeks DE, Zack DJ, Chew EY, Heckenlively JR, Yoshimura N, Iyengar SK, Francis PJ, Katsanis N, Seddon JM, Haines JL, Gorin MB, Abecasis GR, Swaroop A, 2010. Genetic variants near TIMP3 and high-density lipoprotein- associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A 107, 7401–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DRG, Brown FE, Cree AJ, Ratnayaka JA, Lotery AJ, 2017. Sorsby fundus dystrophy - A review of pathology and disease mechanisms. Exp Eye Res 165, 35–46. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG, 2002. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 99, 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della NG, Campochiaro PA, Zack DJ, 1996. Localization of TIMP-3 mRNA expression to the retinal pigment epithelium. Invest Ophthalmol Vis Sci 37, 1921–1924. [PubMed] [Google Scholar]
- Duering M, Karpinska A, Rosner S, Hopfner F, Zechmeister M, Peters N, Kremmer E, Haffner C, Giese A, Dichgans M, Opherk C, 2011. Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum Mol Genet 20, 3256–3265. [DOI] [PubMed] [Google Scholar]
- Eriksson AW, Suvanto EA, Frants RR, Forsius HR, 1990. Pseudoinflammatory fundus dystrophy: a follow-up study. Clinical genetics 38, 21–32. [DOI] [PubMed] [Google Scholar]
- Fariss RN, Apte SS, Luthert PJ, Bird AC, Milam AH, 1998. Accumulation of tissue inhibitor of metalloproteinases-3 in human eyes with Sorsby’s fundus dystrophy or retinitis pigmentosa. Br J Ophthalmol 82, 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata JE, Leco KJ, Voura EB, Yu HY, Waterhouse P, Murphy G, Moorehead RA, Khokha R, 2001. Accelerated apoptosis in the Timp-3-deficient mammary gland. J Clin Invest 108, 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felbor U, Stohr H, Amann T, Schonherr U, Weber BH, 1995. A novel Ser156Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsby’s fundus dystrophy with unusual clinical features. Hum Mol Genet 4, 2415–2416. [DOI] [PubMed] [Google Scholar]
- Felbor U, Suvanto EA, Forsius HR, Eriksson AW, Weber BH, 1997. Autosomal recessive Sorsby fundus dystrophy revisited: molecular evidence for dominant inheritance. Am J Hum Genet 60, 57–62. [PMC free article] [PubMed] [Google Scholar]
- Finger RP, Chong E, McGuinness MB, Robman LD, Aung KZ, Giles G, Baird PN, Guymer RH, 2016. Reticular Pseudodrusen and Their Association with Age-Related Macular Degeneration: The Melbourne Collaborative Cohort Study. Ophthalmology 123, 599–608. [DOI] [PubMed] [Google Scholar]
- Fogarasi M, Janssen A, Weber BH, Stohr H, 2008. Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol 27, 381–392. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP Jr., Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, o I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, e GR, 2013. Seven new loci associated with age-related macular degeneration. Nat Genet 45, 433–439, 439e431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YT, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GH, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Said S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanche H, Deleuze JF, Igo RP Jr., Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA Jr., Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NT, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CC, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JR Allikmets R, Wang JJ, Schaumberg DA, Klein BE, Hagstrom SA, Chowers I, Lotery AJ, Leveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BH, Abecasis GR, Heid IM, 2016. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung AT, Stohr H, Weber BH, Holz FG, Yannuzzi LA, 2013. Atypical sorsby fundus dystrophy with a novel tyr159cys timp-3 mutation. Retin Cases Brief Rep 7, 71–74. [DOI] [PubMed] [Google Scholar]
- Galloway CA, Dalvi S, Hung SSC, MacDonald LA, Latchney LR, Wong RCB, Guymer RH, Mackey DA, Williams DS, Chung MM, Gamm DM, Pebay A, Hewitt AW, Singh R, 2017. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc Natl Acad Sci U S A 114, E8214–e8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemenetzi MK, Luff AJ, Lotery AJ, 2011. Successful treatment of choroidal neovascularization secondary to sorsby fundus dystrophy with intravitreal bevacizumab. Retin Cases Brief Rep 5, 132–135. [DOI] [PubMed] [Google Scholar]
- Gliem M, Muller PL, Mangold E, Bolz HJ, Stohr H, Weber BH, Holz FG, Charbel Issa P, 2015a. Reticular Pseudodrusen in Sorsby Fundus Dystrophy. Ophthalmology 122, 1555–1562. [DOI] [PubMed] [Google Scholar]
- Gliem M, Muller PL, Mangold E, Holz FG, Bolz HJ, Stohr H, Weber BH, Charbel Issa P, 2015b. Sorsby Fundus Dystrophy: Novel Mutations, Novel Phenotypic Characteristics, and Treatment Outcomes. Invest Ophthalmol Vis Sci 56, 2664–2676. [DOI] [PubMed] [Google Scholar]
- Golestaneh N, Chu Y, Cheng SK, Cao H, Poliakov E, Berinstein DM, 2016. Repressed SIRT1/PGC-1alpha pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J Transl Med 14, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TL, Wong HC, Raymond GL, 2012. Choroidal neovascularization secondary to sorsby fundus dystrophy treated with intravitreal bevacizumab. Retin Cases Brief Rep 6, 193–196. [DOI] [PubMed] [Google Scholar]
- Hamilton WK, Ewing CC, Ives EJ, Carruthers JD, 1989. Sorsby’s fundus dystrophy. Ophthalmology 96, 1755–1762. [DOI] [PubMed] [Google Scholar]
- Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N, 2006. Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol 112, 333–339. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Bennett J, Kingsley RM, Sheffield VC, Stone EM, 2002. Novel mutation in the TIMP3 gene causes Sorsby fundus dystrophy. Arch Ophthalmol 120, 376–379. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Regunath G, Rodriguez FJ, Vandenburgh K, Sheffield VC, Stone EM, 1995a. Night blindness in Sorsby’s fundus dystrophy reversed by vitamin A. Nat Genet 11, 27–32. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Regunath G, Rodriguez FJ, Vandenburgh K, Sheffield VV, 1995b. Night blindness in Sorsby’s fundus dystrophy is reversed by vitamin A. Nature Genetics 11, 27–32. [DOI] [PubMed] [Google Scholar]
- Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C, Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF, Tournier-Lasserve E, 1997. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 350, 1511–1515. [DOI] [PubMed] [Google Scholar]
- Kamao H, Mandai M, Okamoto S, Sakai N, Suga A, Sugita S, Kiryu J, Takahashi M, 2014. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem cell reports 2, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei M, Hollyfield J, 1999. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci 40, p2367–2375. [PubMed] [Google Scholar]
- Kapoor KG, Bakri SJ, 2013. Intravitreal anti-vascular endothelial growth factor therapy for choroidal neovascularization due to Sorsby macular dystrophy. J Ocul Pharmacol Ther 29, 444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller J, Giralt J, Alforja S, Casaroli-Marano RP, 2015. Altering the clinical course of Sorsby fundus dystrophy with the use of anti-vascular endothelial growth factor intraocular therapy. Retin Cases Brief Rep 9, 104–105. [DOI] [PubMed] [Google Scholar]
- Kokkinaki M, Sahibzada N, Golestaneh N, 2011. Human induced pluripotent stem-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native RPE. Stem Cells 29, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langton KP, Barker MD, McKie N, 1998. Localization of the functional domains of human tissue inhibitor of metalloproteinases-3 and the effects of a Sorsby’s fundus dystrophy mutation. J Biol Chem 273, 16778–16781. [DOI] [PubMed] [Google Scholar]
- Langton KP, McKie N, Curtis A, Goodship JA, Bond PM, Barker MD, Clarke M, 2000. A novel tissue inhibitor of metalloproteinases-3 mutation reveals a common molecular phenotype in Sorsby’s fundus dystrophy. J Biol Chem 275, 27027–27031. [DOI] [PubMed] [Google Scholar]
- Langton KP, McKie N, Smith BM, Brown NJ, Barker MD, 2005. Sorsby’s fundus dystrophy mutations impair turnover of TIMP-3 by retinal pigment epithelial cells. Hum Mol Genet 14, 3579–3586. [DOI] [PubMed] [Google Scholar]
- Lee MH, Verma V, Maskos K, Nath D, Knauper V, Dodds P, Amour A, Murphy G, 2002. Engineering N-terminal domain of tissue inhibitor of metalloproteinase (TIMP)-3 to be a better inhibitor against tumour necrosis factor-alpha-converting enzyme. Biochem J 364, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu WH, Hsu CW, Nguyen HV, Tsai YT, Chan L, Nagasaki T, Maumenee IH, Yannuzzi LA, Hoang QV, Hua H, Egli D, Tsang SH, 2014. Gene therapy in patient-specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled-related protein defects. Mol Ther 22, 1688–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang Y, Xu Y, Kittredge A, Ward N, Chen S, Tsang SH, Yang T, 2017. Patient-specific mutations impair BESTROPHIN1’s essential role in mediating Ca(2+)-dependent Cl(−) currents in human RPE. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Blumenkranz MS, Binkley J, Wu K, Vollrath D, 2006. A novel His158Arg mutation in TIMP3 causes a late-onset form of Sorsby fundus dystrophy. Am J Ophthalmol 142, 839–848. [DOI] [PubMed] [Google Scholar]
- Majid MA, Smith VA, Easty DL, Baker AH, Newby AC, 2002a. Adenovirus mediated gene delivery of tissue inhibitor of metalloproteinases-3 induces death in retinal pigment epithelial cells. Br J Ophthalmol 86, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majid MA, Smith VA, Easty DL, Baker AH, Newby AC, 2002b. Sorsby’s fundus dystrophy mutant tissue inhibitors of metalloproteinase-3 induce apoptosis of retinal pigment epithelial and MCF-7 cells. FEBS Lett 529, 281–285. [DOI] [PubMed] [Google Scholar]
- Matthews FJ, Cook SD, Majid MA, Dick AD, Smith VA, 2007. Changes in the balance of the tissue inhibitor of matrix metalloproteinases (TIMPs)-1 and −3 may promote keratocyte apoptosis in keratoconus. Exp Eye Res 84, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Menassa N, Burgula S, Empeslidis T, Tsaousis KT, 2017. Bilateral choroidal neovascular membrane in a young patient with Sorsby fundus dystrophy: the value of prompt treatment. BMJ case reports 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier I, Bocquet B, Labesse G, Zeitz C, Defoort-Dhellemmes S, Lacroux A, Mauget- Faysse M, Drumare I, Gamez AS, Mathieu C, Marquette V, Sagot L, Dhaenens CM, Arndt C, Carroll P, Remy-Jardin M, Cohen SY, Sahel JA, Puech B, Audo I, Mrejen S, Hamel CP, 2016. A new autosomal dominant eye and lung syndrome linked to mutations in TIMP3 gene. Sci Rep 6, 32544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monet-Lepretre M, Haddad I, Baron-Menguy C, Fouillot-Panchal M, Riani M, Domenga- Denier V, Dussaule C, Cognat E, Vinh J, Joutel A, 2013. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain : a journal of neurology 136, 1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesper PL, Soetikno BT, Fawzi AA, 2017. Choriocapillaris Nonperfusion is Associated With Poor Visual Acuity in Eyes With Reticular Pseudodrusen. Am J Ophthalmol 174, 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opherk C, Duering M, Peters N, Karpinska A, Rosner S, Schneider E, Bader B, Giese A, Dichgans M, 2009. CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum Mol Genet 18, 2761–2767. [DOI] [PubMed] [Google Scholar]
- Prager F, Michels S, Geitzenauer W, Schmidt-Erfurth U, 2007. Choroidal neovascularization secondary to Sorsby fundus dystrophy treated with systemic bevacizumab (Avastin((R))). Acta Ophthalmol Scand. [DOI] [PubMed] [Google Scholar]
- Qi JH, Anand-Apte B, 2015. Tissue inhibitor of metalloproteinase-3 (TIMP3) promotes endothelial apoptosis via a caspase-independent mechanism. Apoptosis 20, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JH, Dai G, Luthert P, Chaurasia S, Hollyfield J, Weber BH, Stohr H, Anand-Apte B, 2009. S156C mutation in tissue inhibitor of metalloproteinases-3 induces increased angiogenesis. J Biol Chem 284, 19927–19936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Anand-Apte B, 2003a. Tissue inhibitor of metalloproteinases-3 and Sorsby fundus dystrophy. Adv Exp Med Biol 533, 97–105. [DOI] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand- Apte B, 2003b. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 9, 407–415. [DOI] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Yeow K, Edwards DR, Fox PL, Anand-Apte B, 2002. Expression of Sorsby’s fundus dystrophy mutations in human retinal pigment Epithelial cells reduces matrix metalloproteinase inhibition and may promote angiogenesis. J Biol Chem 30, 30. [DOI] [PubMed] [Google Scholar]
- Riera M, Navarro R, Ruiz-Nogales S, Mendez P, Bures-Jelstrup A, Corcostegui B, Pomares E, 2017. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci Rep 7, 42078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saihan Z, Li Z, Rice J, Rana NA, Ramsden S, Schlottmann PG, Jenkins SA, Blyth C, Black GC, McKie N, Webster AR, 2009. Clinical and biochemical effects of the E139K missense mutation in the TIMP3 gene, associated with Sorsby fundus dystrophy. Mol Vis 15, 1218–1230. [PMC free article] [PubMed] [Google Scholar]
- Saini JS, Corneo B, Miller JD, Kiehl TR, Wang Q, Boles NC, Blenkinsop TA, Stern JH, Temple S, 2017. Nicotinamide Ameliorates Disease Phenotypes in a Human iPSC Model of Age- Related Macular Degeneration. Cell Stem Cell 20, 635–647.e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberger SD, Agarwal A, 2013. A novel mutation at the N-terminal domain of the TIMP3 gene in Sorsby fundus dystrophy. Retina 33, 429–435. [DOI] [PubMed] [Google Scholar]
- Singh R, Phillips MJ, Kuai D, Meyer J, Martin JM, Smith MA, Perez ET, Shen W, Wallace KA, Capowski EE, Wright LS, Gamm DM, 2013a. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Invest Ophthalmol Vis Sci 54, 6767–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, Verhoeven AD, Capowski EE, Zhang X, Yin Y, Halbach PJ, Fishman GA, Wright LS, Pattnaik BR, Gamm DM, 2013b. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet 22, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaprasad S, Webster AR, Egan CA, Bird AC, Tufail A, 2008. Clinical course and treatment outcomes of Sorsby fundus dystrophy. Am J Ophthalmol 146, 228–234. [DOI] [PubMed] [Google Scholar]
- Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R, 2006. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 176, 721–725. [DOI] [PubMed] [Google Scholar]
- Soboleva G, Geis B, Schrewe H, Weber BH, 2003. Sorsby fundus dystrophy mutation Timp3(S156C) affects the morphological and biochemical phenotype but not metalloproteinase homeostasis. J Cell Physiol 197, 149–156. [DOI] [PubMed] [Google Scholar]
- Sohn EH, Khanna A, Tucker BA, Abramoff MD, Stone EM, Mullins RF, 2014. Structural and biochemical analyses of choroidal thickness in human donor eyes. Invest Ophthalmol Vis Sci 55, 1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songstad AE, Wiley LA, Duong K, Kaalberg E, Flamme-Wiese MJ, Cranston CM, Riker MJ, Levasseur D, Stone EM, Mullins RF, Tucker BA, 2015. Generating iPSC-Derived Choroidal Endothelial Cells to Study Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 56, 8258–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorsby A, Joll Mason ME, 1949. A fundus dystrophy with unusual features. British journal of Ophthalmology, 67–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaide RF, Curcio CA, 2010. Drusen characterization with multimodal imaging. Retina 30, 1441–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr H, Anand-Apte B, 2012. A review and update on the molecular basis of pathogenesis of Sorsby fundus dystrophy. Adv Exp Med Biol 723, 261–267. [DOI] [PubMed] [Google Scholar]
- Tabata Y, Isashiki Y, Kamimura K, Nakao K, Ohba N, 1998. A novel splice site mutation in the tissue inhibitor of the metalloproteinases-3 gene in Sorsby’s fundus dystrophy with unusual clinical features. Hum Genet 103, 179–182. [DOI] [PubMed] [Google Scholar]
- Wang T, Baron M, Trump D, 2008. An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol 96, 499–509. [DOI] [PubMed] [Google Scholar]
- Warwick A, Lotery A, 2017. Genetics and genetic testing for age-related macular degeneration. Eye (Lond). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber BH, Lin B, White K, Kohler K, Soboleva G, Herterich S, Seeliger MW, Jaissle GB, Grimm C, Reme C, Wenzel A, Asan E, Schrewe H, 2002. A mouse model for Sorsby fundus dystrophy. Invest Ophthalmol Vis Sci 43, 2732–2740. [PubMed] [Google Scholar]
- Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U, 1994. Mutations in the tissue inhibitor of metalloproteinase-3 (TIMP-3) in patients with Sorsby’s fundus dystrophy. Nature Genetics 8, 352–356. [DOI] [PubMed] [Google Scholar]
- Westenskow PD, Moreno SK, Krohne TU, Kurihara T, Zhu S, Zhang ZN, Zhao T, Xu Y, Ding S, Friedlander M, 2012. Using flow cytometry to compare the dynamics of photoreceptor outer segment phagocytosis in iPS-derived RPE cells. Invest Ophthalmol Vis Sci 53, 6282–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Li Y, Chan L, Tsai YT, Wu WH, Nguyen HV, Hsu CW, Li X, Brown LM, Egli D, Sparrow JR, Tsang SH, 2014. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum Mol Genet 23, 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeow KM, Kishnani NS, Hutton M, Hawkes SP, Murphy G, Edwards DR, 2001. Sorsby’s fundus dystrophy Tissue Inhibitor of Metalloproteinases-3 (TIMP-3) mutants have unimpaired matrix metalloproteinase inhibitory activities but affect cell adhesion to the extracellular matrix. Matrix Biology In Press. [DOI] [PubMed] [Google Scholar]
- Yu Y, Bhangale TR, Fagerness J, Ripke S, Thorleifsson G, Tan PL, Souied EH, Richardson AJ, Merriam JE, Buitendijk GH, Reynolds R, Raychaudhuri S, Chin KA, Sobrin L, Evangelou E, Lee PH, Lee AY, Leveziel N, Zack DJ, Campochiaro B, Campochiaro P, Smith RT, Barile GR, Guymer RH, Hogg R, Chakravarthy U, Robman LD, Gustafsson O, Sigurdsson H, Ortmann W, Behrens TW, Stefansson K, Uitterlinden AG, van Duijn CM, Vingerling JR, Klaver CC, Allikmets R, Brantley MA Jr., Baird PN, Katsanis N, Thorsteinsdottir U, Ioannidis JP, Daly MJ, Graham RR, Seddon JM, 2011. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum Mol Genet 20, 3699–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Daniel E, Maguire MG, Grunwald JE, Martin ER, Martin DF, Ying GS, 2016. Pseudodrusen and Incidence of Late Age-Related Macular Degeneration in Fellow Eyes in the Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 123, 1530–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]