

Abstract
Insulin is an important therapeutic protein for the treatment of diabetes, but it is unstable and aggregates upon exposure to environmental stressors encountered during storage and transport. To prevent degradation of the protein in this manner and retain as much in vivo bioactivity as possible, a well-defined insulin-trehalose glycopolymer conjugate was synthesized. To accomplish this, a strategy was employed to site-specifically modify insulin with a polymerization initiator at a particular conjugation site; this also facilitated purification and characterization. Lysine of the B chain was preferentially modified by conducting the reaction at high pH, taking advantage of its higher nucleophilicity than the N-terminal amines. Trehalose monomer was polymerized directly from this macroinitiator to form a well-defined conjugate. Bioactivity of the site-specific conjugate was shown to be higher compared to the non-specific conjugate and the same as the analogous site-specific polyethylene glycol (PEG) conjugate as confirmed by the insulin tolerance test (ITT) in mice. The conjugated trehalose glycopolymer also stabilized insulin to heat as measured by high-performance liquid chromatography (HPLC).
Graphical Abstract
INTRODUCTION
Diabetes is a growing worldwide problem – affecting over 30 million people1 in the United States alone – with its prevalence continuing to rise.1–2 Insulin is an important therapeutic protein for the treatment of diabetes; type I diabetes requires several injections or infusions of insulin daily.3 However, exposure to environmental stressors such as heat and mechanical agitation encountered during storage and transportation can lead to protein degradation and aggregation.4–6 Inadequate dosing resulting from degradation increases risk to the patient. For example, diabetic ketoacidosis can result from degradation of insulin from exposure to heat and extended hyperglycemia can increase risk for complications such as retinal damage and kidney disease.4
Our group has synthesized polymers with trehalose side chains that stabilize a variety of proteins to stressors such as lyophilization, heat, and mechanical agitation.7–11 We have shown that conjugating insulin with a trehalose glycopolymer stabilized the protein to heat and mechanical agitation and also improved its pharmacokinetics.9 However, the bioactivity was significantly lower than the native protein, requiring 5-fold dosage to achieve the same decrease in blood glucose as the native protein. The conjugate was synthesized by reductive amination at pH 8.0 using a benzaldehyde-functionalized trehalose polymer resulting in modification at both GlyA1 and LysB29; it has been shown that under similar pH conditions, the relative reactivity of the three amines in insulin is GlyA1>LysB29≫PheB1.12 We hypothesized that this non-specific “grafting to” conjugation strategy contributed to the observed decrease in activity. Modification of GlyA1 decreases insulin affinity for its receptor and even conjugation of a 2000 Da poly(ethylene glycol) (PEG) can decrease bioactivity.12 Modification at either PheB1 or LysB29 does not impact bioactivity to the same extent.12–13 Furthermore, PheB1 has been shown to react more slowly than the other two amines of insulin and modification at lysine can be favored by increasing the pH of the reaction above pH 9.5, taking advantage of its higher nucleophilicity than the N-terminal amines.12 Therefore, we decided to target modification with the trehalose glycopolymer at LysB29 by conducting the conjugation at higher pH and purifying the macroinitiator to contain only the LysB29 modification.
Polymerizing directly from a protein, or “grafting from” a protein, is a strategy that facilitates characterization of conjugation site and purification of the conjugate. Our group was the first to report this strategy by modifying biotin with an initiator, forming a biotin/streptavidin macroinitiator, and polymerizing N-isopropylacrylamide (NIPAAm) from the protein.14 Subsequently, our group grafted NIPAAm directly from bovine serum albumin (BSA) and lysozyme macroinitiators – with the latter remaining active.15 In addition, Matyjaszewski and Russell prepared polymers by first modifying amines and polymerizing poly(ethylene glycol) methacrylate (PEGMA).16 Many examples of grafting from proteins have followed. Matyjaszewski and coworkers used activators generated by electron transfer atom transfer radical polymerization (AGET ATRP) to polymerize oligo(ethylene glycol) methacrylate (OEGMA) from BSA in phosphate buffer.17 Bulmus and Davis were the first to demonstrate reversible addition–fragmentation chain transfer (RAFT) polymerization from a protein by conjugating the Z-group of a thiol-reactive chain transfer agent (CTA) to BSA and polymerizing acrylate and acrylamide monomers from the protein.18–19 Sumerlin and coworkers subsequently employed RAFT to polymerize NIPAAm from BSA by conjugating the R-group of the CTA to the protein.20 Additionally, Haddleton and coworkers polymerized acrylate and acrylamide monomers from a variety of proteins under mild, aqueous conditions.21
We devised a strategy to synthesize the insulin-trehalose glycopolymer conjugate involving targeted modification of LysB29 with an initiator. We envisioned that modification with a small molecule would also facilitate characterization of the conjugation site because the trehalose glycopolymer is not directly amenable to mass spectral analysis, and also purification of the LysB29 only species. It was expected that polymerizing from this insulin macroinitiator with trehalose monomer in mild, aqueous conditions would result in a conjugate with improved bioactivity compared to our previous approach, while retaining stabilization properties (Figure 1). The results are described herein.
Figure 1.

Scheme of experimental design
RESULTS AND DISCUSSION
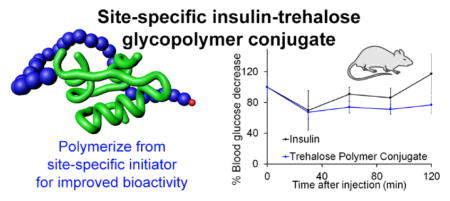
The designed approach required polymerization of a trehalose monomer under mild, aqueous conditions. AGET ATRP is a technique that has been performed in aqueous buffer at room temperature,17 and our group used this technique to polymerize OEGMA monomers from siRNA.22 A methacrylate trehalose monomer (Figure 2A) was selected to facilitate controlled polymerization at room temperature in place of the styrenyl acetal trehalose monomer used in previous work.8–9 First the polymerization was explored under the conditions that would be suitable for the protein. Polymerization in DPBS with the small molecule initiator 2-hydroxyethyl-2-bromoisobutyrate (HEBIB), tris(2-pyridylmethyl)amine (TPMA) as the ligand, and ascorbic acid (AA) as the reducing agent was accomplished at 23 °C. Complete conversion was observed within 4 hours and dispersity was low (Figure 2B). This polymer exhibited excipient stabilization of insulin to heating with 93 ± 3 % intact insulin (Figure S8) compared to 68 ± 2 % intact insulin remaining with the styrenyl trehalose glycopolymer used in our previous work,9 demonstrating that the polymethacrylate backbone polymer also prevented heat-induced aggregation of insulin.
Figure 2.
Aqueous polymerization of methacrylate trehalose. (A) Reaction scheme to polymerize trehalose monomer by AGET ATRP ([HEBIB]/[M]/[CuBr2]/[TPMA]/[AA] = 1/23/1/1/0.6) in DPBS pH 7.4 at 23 °C 3.5 h and (B) SEC trace of trehalose glycopolymer.
With the success of the trehalose monomer polymerization and resulting polymer stabilization of insulin, the protein was then modified with an initiator. Specifically, the insulin macroinitiator was synthesized with a nitrophenyl carbonate-activated initiator (NPC initiator) in borate buffer at pH 11 (Figure 3A). Even at this high pH, modification of multiple amines in addition to the desired product and unmodified insulin was observed after 1 hour (Figure S9 and S10). No increase in the 40% conversion to the desired product was observed for longer reaction times and this yield is consistent with other site-selective modifications for insulin.23–24 The singly modified product was separated and recovered after semi-preparative HPLC. The purified macroinitiator exhibited a single peak by analytical HPLC and matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) (Figure 4A and B). Further, the macroinitiator was reduced with dithiothreitol (DTT) for analysis by MALDI MS and modification was observed only on the B chain (Figure S11). To confirm the modification site, electrospray ionization (ESI) tandem MS was performed on the insulin macroinitiator, which showed fragments consistent with modification on LysB29 and not PheB1 (Table S1). Although only one species was observed for the insulin macroinitiator by HPLC, MALDI MS, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE), two bands were observed for the macroinitiator after purification by native PAGE (Figure 4D and S13). To determine if insulin was degrading during synthesis or purification of the macroinitiator, the insulin macroinitiator was run on a highly cross-linked Tris-Tricine gel under reducing and non-reducing conditions (Figure S13). Insulin and the insulin macroinitiator both exhibited one band near 6 kDa under non-reducing conditions and shifted bands near 3 kDa indicating reduction of the interchain disulfide bonds. These results indicate that insulin does not degrade by disulfide reduction once modified. Insulin readily forms dimers near neutral pH at which the native gel was run. LysB29 is involved in facilitating dimerization and monomeric rapid-acting insulin analogues have been prepared by mutating the amino acid at this position.25–26 These two bands may be due to dissociation of insulin dimers into monomers with modification at LysB29 in the macroinitiator interfering with this interaction.
Figure 3.

Synthesis of insulin-trehalose glycopolymer conjugate. (A) Preparation of the insulin macroinitiator and (B) grafting from the macroinitiator with AGET ATRP ([Resin]/[M]/[CuBr2]/[TPMA]/[AA] = 1/30/1/10/0.6).
Figure 4.

Characterization of insulin macroinitiator and insulin-trehalose glycopolymer conjugate. (A) Analytical HPLC and (B) MALDI MS after purification of the insulin macroinitiator, (C) Coomassie stained SDS PAGE (Lane 1: Ladder, Lane 2: Insulin, Lane 3: Purified insulin macroinitiator, Lane 4: Trehalose glycopolymer, Lane 5: Crude insulin-trehalose glycopolymer conjugate, Lane 6: Purified insulin-trehalose glycopolymer conjugate), (D) native PAGE with Western blot analysis (Lane 1: Insulin, Lane 2: Purified insulin macroinitiator, Lane 3: Trehalose glycopolymer, Lane 4: Crude insulin-trehalose glycopolymer conjugate, Lane 5: Purified insulin-trehalose glycopolymer conjugate Lane 6: Insulin-trehalose glycopolymer conjugate after digestion with Proteinase K), and (E) SEC trace of trehalose glycopolymer after digestion of insulin with Proteinase K.
Attempts to polymerize from the macroinitiator were initially unsuccessful under a range of AGET ATRP conditions, likely due to the low concentration of initiating sites from the small amount of insulin macroinitiator. To facilitate polymerization from the insulin macroinitiator, a sacrificial resin was prepared as described previously.22 This resin was modified with an initiator and increased the concentration of initiating sites. With the addition of the resin, the trehalose monomer was then successfully polymerized using AGET ATRP in DPBS pH 7.4 at 23 °C (Figure 3B) although it took 14 hours to polymerize indicating that the reaction was slower for the macroinitiator than the small molecule initiator. Also, a large molecular weight peak was observed in addition to the main peak by the right-angle light scattering detector equipped on the SEC that was not observed during polymerization with the small molecule initiator. This peak had also been detected during preparation of the monomer if the temperature was high or pressure too low when removing water and observed when insulin was added at 1 mg/mL to the polymerization mixture for the small molecule initiator HEBIB (Figure S14). We expected that this peak resulted from uncontrolled polymerization of the monomer. Increasing the amount of TPMA from 1 to 10 molar equivalents with respect to copper eliminated this peak (Figure S15). We hypothesize that adding excess ligand displaces copper from interactions with insulin that result in uncontrolled polymerization.
The polymerization product exhibited a shift by native and SDS PAGE and this band contained insulin by Western blot analysis (Figure 4C, 4D, and S12). To determine if this band formed without the macroinitiator, several control polymerizations were performed. Unmodified insulin was substituted for the insulin macroinitiator, initiator was excluded with unmodified insulin included, or no insulin or initiator was included under the standard AGET ATRP conditions. No new bands were observed when the polymerization mixtures were analyzed by native PAGE (Figure S16). Polymer formed (팶 >1.6) for conditions without insulin or initiator, which was likely due to autopolymerization. It is interesting to note that the trehalose glycopolymer did not stain by Coomassie except when these large molecular weight species were present (Figure 4C and S12).
When the crude conjugate mixture was analyzed by analytical HPLC, peaks corresponding to free polymer likely hydrolyzed from the resin, insulin-trehalose glycopolymer conjugate, and residual insulin macroinitiator were observed (Figure S17). The insulin-trehalose glycopolymer conjugate could be separated using the same HPLC method as was used to purify the macroinitiator, yielding the final purified product. To characterize the polymer formed when grafting from the insulin macroinitiator, insulin was digested using Proteinase K as described in literature.27 Polymer alone in the presence of Proteinase K did not change dispersity or molecular weight (Figure S18) showing that Proteinase K does not degrade the polymer or overlap with the polymer signal. By SEC analysis, the polymer was 8.7 kDa and exhibited similar dispersity as with the small molecule initiator (Figure 4E). The analogous insulin-PEG conjugate was also prepared with a 10 kDa methoxyPEG nitrophenyl carbonate in borate buffer pH 11. The singly modified species was separated by semi-preparative HPLC. The insulin-PEG conjugate exhibited single a band at approximately 16 kDa by SDS PAGE, which is consistent with one site modification (Figure S19). Additionally, analytical HPLC of the purified insulin-PEG conjugate also exhibited a single peak (Figures S21). This was corroborated by MALDI MS (Figure S20A) and modification was confirmed on the B chain following reduction of the disulfides with DTT (Figure S20B). The modification is most likely at LysB29 because of the similarity of conditions to the macroinitiator and the reportedly higher reactivity of LysB29 over PheB1 under similar conditions.12
To evaluate the bioactivity of the conjugates, insulin tolerance test (ITT) was performed in mice (Figure 5). Animals were injected with insulin, insulin-trehalose glycopolymer conjugate, and the analogous insulin-PEG conjugate. Insulin remained active after exposure to HPLC purification conditions, exhibiting a decrease in blood glucose 30 min after injection. Both the insulin-trehalose glycopolymer conjugate and insulin-PEG conjugate required only a 3-fold dosage relative to insulin alone (48 μg/kg versus 16 μg/kg) to achieve the same change in blood glucose, which is a significant improvement over the 5-fold dosage (48 μg/kg versus 80 μg/kg) required previously with a non-selective conjugate. Insulin conjugated to 20 kDa PEG through LysB29 had significantly lower binding affinity to the receptor than insulin alone.28 Additionally, 2000 Da PEG-insulin conjugates showed decreased in vivo bioactivity, while 600 Da PEG-insulin conjugates retained bioactivity.12 Thus, tuning the length of the polymer may further improve the retention of bioactivity.10 A balance between stabilization properties and bioactivity may be necessary, yet the site-specific conjugation strategy described herein clearly significantly improved retention of bioactivity.
Figure 5.
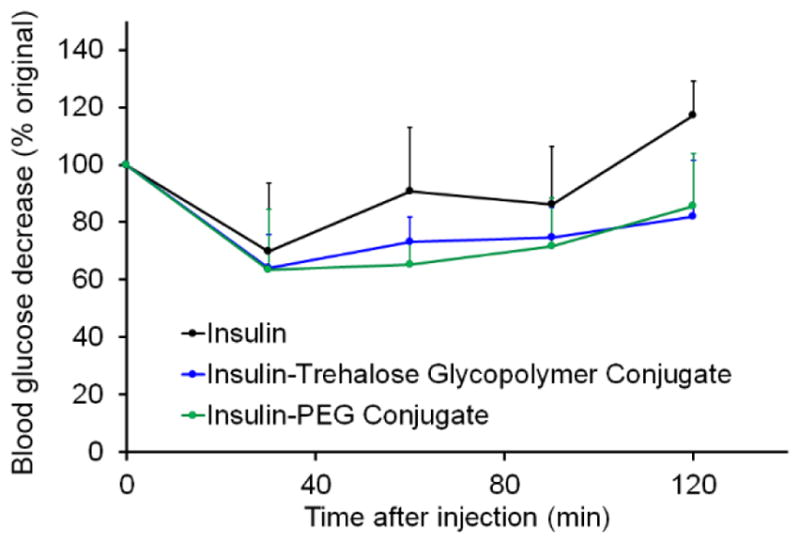
ITT in mice with insulin (16 μg/kg), insulin-PEG conjugate (48 μg/kg), and insulin-trehalose glycopolymer conjugate (48 μg/kg) (n=4, p > 0.05 at all points between insulin and conjugates).
Next, the stability of the insulin-trehalose glycopolymer was evaluated using an accelerated heating assay. Insulin and the insulin-trehalose glycopolymer conjugate were heated for 90 °C for 30 minutes and the amount of intact insulin was determined by measuring the area under the curve (AUC) of the insulin peak. The amount of intact insulin remaining for the conjugate after heating was found to be significantly greater than for insulin without the trehalose glycopolymer (Figure 6). This demonstrates that one polymer chain may be conjugated in a site-specific manner and yet still stabilize the protein to heat-induced aggregation.
Figure 6.
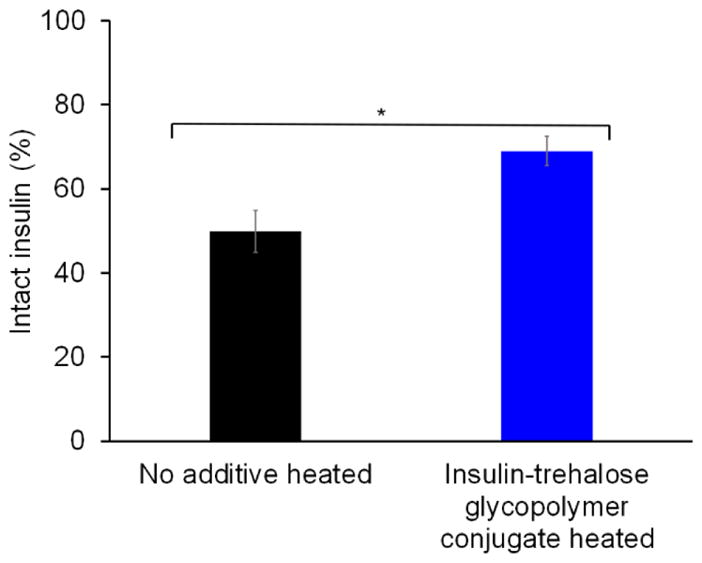
Biochemical stability assay of insulin and insulin-trehalose glycopolymer conjugate after heating to 90 °C for 30 minutes by HPLC AUC (n=3, * p = 0.0056).
CONCLUSION
In conclusion, we synthesized a site-specific trehalose glycopolymer conjugate with improved bioactivity retention compared to non-specific conjugation. An insulin macroinitiator was prepared at high pH to favor modification at LysB29. Following separation by HPLC, modification at the desired position was confirmed with tandem MS. Trehalose glycopolymer was polymerized directly from this macroinitiator with AGET ATRP under mild, aqueous conditions. The insulin-trehalose glycopolymer conjugate had greater retention of bioactivity than the non-specific conjugate, requiring just over half the dosage used to achieve the same decrease in blood glucose. Moreover, the insulin-trehalose glycopolymer conjugate retained its stabilization properties by HPLC monitoring of intact insulin after heating.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health (NIBIB R01EB020676). K.M.M. thanks the NIH for the Biotechnology Training Fellowship (T32 GM067555). The authors thank Marco S. Messina, Neil Forsythe, and Mary Waddington for their assistance with LC-MS.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Methods and materials, analytical techniques, experimental details, insulin conjugate syntheses and characterization, insulin macroinitiator characterization by MS and HPLC, evaluation of conjugates by PAGE, and SEC traces of trehalose glycopolymer and insulin-trehalose glycopolymer conjugates (PDF)
References
- 1.Centers for Disease Control and Prevention. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services; 2014. [Google Scholar]
- 2.Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.American Diabetes Association. Standards of medical care in diabetes—2017 abridged for primary care providers. Clinical Diabetes. 2017;35:5–26. doi: 10.2337/cd16-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pryce R. Diabetic ketoacidosis caused by exposure of insulin pump to heat and sunlight. Br Med J. 2009;338 doi: 10.1136/bmj.a2218. [DOI] [PubMed] [Google Scholar]
- 5.Sluzky V, Tamada JA, Klibanov AM, Langer R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc Natl Acad Sci U S A. 1991;88:9377–9381. doi: 10.1073/pnas.88.21.9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliva A, Fariña J, Llabrés M. Influence of temperature and shaking on stability of insulin preparations: degradation kinetics. Int J Pharm. 1996;143:163–170. [Google Scholar]
- 7.Mancini RJ, Lee J, Maynard HD. Trehalose Glycopolymers for Stabilization of Protein Conjugates to Environmental Stressors. J Am Chem Soc. 2012;134:8474–8479. doi: 10.1021/ja2120234. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Lin EW, Lau UY, Hedrick JL, Bat E, Maynard HD. Trehalose Glycopolymers as Excipients for Protein Stabilization. Biomacromolecules. 2013;14:2561–2569. doi: 10.1021/bm4003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Lee J, Mansfield KM, Ko JH, Sallam S, Wesdemiotis C, Maynard HD. Trehalose Glycopolymer Enhances Both Solution Stability and Pharmacokinetics of a Therapeutic Protein. Bioconjugate Chem. 2017;28:836–845. doi: 10.1021/acs.bioconjchem.6b00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelegri-O’Day EM, Paluck SJ, Maynard HD. Substituted Polyesters by Thiol–Ene Modification: Rapid Diversification for Therapeutic Protein Stabilization. J Am Chem Soc. 2017;139:1145–1154. doi: 10.1021/jacs.6b10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Messina MS, Ko JH, Yang Z, Strouse MJ, Houk KN, Maynard HD. Effect of trehalose polymer regioisomers on protein stabilization. Polym Chem. 2017;8:4781–4788. [Google Scholar]
- 12.Uchio T, Baudyš M, Liu F, Song SC, Kim SW. Site-specific insulin conjugates with enhanced stability and extended action profile. Adv Drug Delivery Rev. 1999;35:289–306. doi: 10.1016/s0169-409x(98)00078-7. [DOI] [PubMed] [Google Scholar]
- 13.Gliemann J, Gammeltoft S. The biological activity and the binding affinity of modified insulins determined on isolated rat fat cells. Diabetologia. 1974;10:105–113. doi: 10.1007/BF01219665. [DOI] [PubMed] [Google Scholar]
- 14.Bontempo D, Maynard HD. Streptavidin as a Macroinitiator for Polymerization: In Situ Protein–Polymer Conjugate Formation. J Am Chem Soc. 2005;127:6508–6509. doi: 10.1021/ja042230+. [DOI] [PubMed] [Google Scholar]
- 15.Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. In Situ Preparation of Protein–“Smart” Polymer Conjugates with Retention of Bioactivity. J Am Chem Soc. 2005;127:16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
- 16.Lele BS, Murata H, Matyjaszewski K, Russell AJ. Synthesis of Uniform Protein–Polymer Conjugates. Biomacromolecules. 2005;6:3380–3387. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- 17.Averick S, Simakova A, Park S, Konkolewicz D, Magenau AJD, Mehl RA, Matyjaszewski K. ATRP under Biologically Relevant Conditions: Grafting from a Protein. ACS Macro Lett. 2012;1:6–10. doi: 10.1021/mz200020c. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Bulmus V, Herlambang DL, Barner-Kowollik C, Stenzel MH, Davis TP. In situ formation of protein–polymer conjugates through reversible addition fragmentation chain transfer polymerization. Angew Chem, Int Ed. 2007;46:3099–3103. doi: 10.1002/anie.200604922. [DOI] [PubMed] [Google Scholar]
- 19.Boyer C, Bulmus V, Liu J, Davis TP, Stenzel MH, Barner-Kowollik C. Well-Defined Protein–Polymer Conjugates via in Situ RAFT Polymerization. J Am Chem Soc. 2007;129:7145–7154. doi: 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]
- 20.De P, Li M, Gondi SR, Sumerlin BS. Temperature-Regulated Activity of Responsive Polymer–Protein Conjugates Prepared by Grafting-from via RAFT Polymerization. J Am Chem Soc. 2008;130:11288–11289. doi: 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Li M, Zhu C, Nurumbetov G, Li Z, Wilson P, Kempe K, Haddleton DM. Well-Defined Protein/Peptide–Polymer Conjugates by Aqueous Cu-LRP: Synthesis and Controlled Self-Assembly. J Am Chem Soc. 2015;137:9344–9353. doi: 10.1021/jacs.5b04139. [DOI] [PubMed] [Google Scholar]
- 22.Lin EW, Maynard HD. Grafting from Small Interfering Ribonucleic Acid (siRNA) as an Alternative Synthesis Route to siRNA–Polymer Conjugates. Macromolecules. 2015;48:5640–5647. [Google Scholar]
- 23.Baudyš M, Uchio T, Mix D, Kim SW, Wilson D. Physical stabilization of insulin by glycosylation. J Pharm Sci. 1995;84:28–33. doi: 10.1002/jps.2600840108. [DOI] [PubMed] [Google Scholar]
- 24.Liu F, Kim SW, Baudys M. Synthesis of insulin derivatives. 6323311 B1. US. 2001
- 25.Hirsch IB. Insulin analogues. N Engl J Med. 2005;352:174–183. doi: 10.1056/NEJMra040832. [DOI] [PubMed] [Google Scholar]
- 26.Evans M, Schumm-Draeger P, Vora J, King A. A review of modern insulin analogue pharmacokinetic and pharmacodynamic profiles in type 2 diabetes: improvements and limitations. Diabetes, Obes Metab. 2011;13:677–684. doi: 10.1111/j.1463-1326.2011.01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Zhao W, Liu X, Wang G, Wang Y, Li D, Xie L, Gao Y, Deng H, Gao W. Site-selective in situ growth of fluorescent polymer–antibody conjugates with enhanced antigen detection by signal amplification. Biomaterials. 2015;64:2–9. doi: 10.1016/j.biomaterials.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 28.Beals JM, Cutler GB, Doyle BL, Hansen RJ, Li S, Shirani S, Zhang L. Pegylated Insulin Lispro Compounds. 20110105392 A1. US. 2011
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.