

Abstract
Objectives
To demonstrate the equivalent efficacy and compare the safety and immunogenicity of an etanercept biosimilar, GP2015, with reference etanercept (ETN) in patients with moderate-to-severe, active rheumatoid arthritis (RA), characterised by an inadequate response to synthetic or biologic disease-modifying antirheumatic drugs (DMARDs).
Methods
In the EQUIRA study, eligible patients (n=376) were randomised 1: 1 to 50 mg GP2015 or ETN subcutaneously, once weekly, for 24 weeks (treatment period 1). Patients from both groups, with at least moderate European League Against Rheumatism response at week 24, received GP2015 up to week 48 (treatment period 2). All patients continued to receive concomitant methotrexate at a stable dose (10–25 mg/week) until end of the study. The 24-week results are presented here.
Results
Equivalent efficacy between GP2015 and ETN was demonstrated if the 95% CI for the difference in disease activity score 28-joint count C reactive protein (DAS28-CRP) change from baseline to week 24 between treatment arms was contained within the prespecified equivalence margin range of −0.6 to 0.6. The least squares mean difference (GP2015–ETN) in change from baseline in DAS28-CRP up to week 24 was −0.07 (95% CI −0.26 to 0.12 [primary endpoint)]. The incidence of treatment-emergent adverse events was comparable between GP2015 (43.5%) and ETN (49.5%). None of the GP2015-treated patients developed neutralising anti-drug antibodies (NAbs) whereas 1.6% and 0.6% of patients in ETN group were NAb positive at weeks 4 and 12, respectively.
Conclusion
In patients with RA who had an inadequate response to DMARDs, GP2015 demonstrated a similar efficacy and a comparable safety and immunogenicity profile with ETN.
Trial registration
Keywords: bioequivalence, biosimilar, etanercept, GP2015, rheumatoid arthritis
Key messages.
What is already known about this subject?
GP2015 is an etanercept biosimilar.
Analytical similarity and bioequivalence between GP2015 and reference etanercept (ETN) has been demonstrated.
What does this study add?
GP2015 demonstrated an equivalent efficacy to ETN in patients with rheumatoid arthritis with moderate to severe disease with inadequate responses to DMARDs, including methotrexate and biologics. No new safety findings were reported.
How might this impact on clinical practice?
The results from this study providing evidence of biosimilarity between GP2015 and ETN may aid clinical decision-making around the use of biologics/biosimilars in the treatment of rheumatoid arthritis.
Introduction
Etanercept is a recombinant dimeric fusion protein that binds to and neutralises the biological activity of tumour necrosis factor (TNF), a naturally occurring cytokine. TNF plays a key role in the pathogenesis of a range of immune-mediated inflammatory diseases.1 Etanercept is approved for the treatment of multiple immune-mediated inflammatory diseases including moderate-to-severe rheumatoid arthritis (RA)2 3 and has been used successfully in clinical practice over many years.
A biosimilar is a biologic agent that contains a similar version of the active substance of an already authorised reference biologic.4 Regulatory decisions for approval of biosimilars require confirmation and evaluation of biosimilarity between a proposed biosimilar and the approved reference biologic in terms of physicochemical characteristics, biological activity, efficacy and safety.5 6
GP2015 (Erelzi®; Sandoz Inc., Princeton, New Jersey, USA) is an etanercept biosimilar. Analytical similarity and bioequivalence between GP2015 and reference etanercept (ETN; Enbrel® (European Union authorised)) has been demonstrated previously.7 The phase III EGALITY study demonstrated equivalent clinical efficacy of GP2015 and ETN in plaque-type psoriasis.8 GP2015 is currently approved in the EU9 and the USA.10
The objective of the present comparative, phase III, EQUIRA study was to demonstrate equivalent efficacy and to compare the safety and immunogenicity of GP2015 with ETN in patients with moderate-to-severe, active RA, who had an inadequate response to either conventional synthetic and/or biologic disease modifying antirheumatic drugs (DMARDs). Here, the results from the first 24-week treatment period of the study are reported.
Methods
Study endpoints
The primary endpoint was the change from baseline in disease activity score 28-joint count C reactive protein (DAS28-CRP) up to week 24. Secondary endpoints were other efficacy endpoints assessed up to week 24 including (1) change from baseline in DAS28-CRP scores,11 (2) proportion of patients achieving good and moderate European League Against Rheumatism (EULAR) response12 based on DAS28-erythrocyte sedimentation rate (ESR), (3) proportion of patients achieving a 20/50/70% improvement in the American College of Rheumatology (ACR) core set of measurements (ACR20/50/70), (4) physical function assessed by the Health Assessment Questionnaire Disability Index (HAQ-DI) score13 14 and (5) impact of fatigue on patients assessed by the Functional Assessment of Chronic Illness Therapy (FACIT) fatigue scale.15 16
Safety endpoints included adverse event monitoring and evaluation of local tolerability at the injection sites of both medications as assessed by the investigator during the study. The proportion of patients with anti-drug antibodies (ADAs) was assessed at baseline and at weeks 2, 4, 12 and 24 by a three-step procedure comprising a validated screening, confirmatory and titre determination electrochemiluminescence bridging assay.
Immunogenicity assessment
The applied method for the detection of ADAs is based on a bridging electrochemiluminescence assay format17 including acid dissociation steps. Study samples were first analysed in a screening assay based on a 5% false-positive rate. Assay sensitivity (<100 ng/mL using a polyclonal ADA-positive control) and drug tolerance (50 µg/mL for GP2015 and ETN) was determined during method validation. Samples that were positive in the screening assay were subsequently analysed in a confirmatory assay based on a 1% false-positive rate. The titre of confirmed positive ADA samples was determined and the neutralising capacity of ADAs was assessed in a competitive ligand binding assay.
Study design
EQUIRA was a 48-week, phase III, parallel-group, randomised, double-blind study conducted at 113 sites across 16 countries from November 2015 to June 2017. Eligible patients were randomised 1:1 to self-administer 50 mg GP2015 or ETN subcutaneously, once weekly, for 24 weeks (treatment period 1 [TP1]; see online supplementary appendix 1 for details on the randomisation schedule). At week 24, patients from both treatment groups achieving at least a moderate treatment response according to EULAR response criteria (responders) received GP2015 up to week 48 (TP2; online supplementary figure S1).
rmdopen-2018-000757supp001.docx (3.8MB, docx)
This study was conducted in accordance with the ethical principles derived from the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practices and in compliance with local regulatory requirements.
Study population
Patients of either sex, aged ≥18 years, were included if they met the following criteria: (1) had RA classified according to the ACR 1987 or ACR/EULAR 2010 criteria18 for ≥6 months before baseline; (2) had active disease defined as DAS28-CRP ≥3.2; (3) had CRP >5 mg/L or ESR ≥28 mm/h; (4) had an inadequate clinical response to methotrexate (MTX) at a dose of 10–25 mg/week following dose escalation according to local standards (those who had failed a DMARD other than MTX, and any other DMARD used in combination with MTX prior to baseline, were allowed after an appropriate wash-out period of 4 weeks); (5) were on MTX therapy for ≥3 months and on a stable dose for ≥28 days prior to baseline; (6) were on stable dose of folic acid (≥5 mg per week) for ≥28 days before baseline.
The key exclusion criteria included (1) any previous exposure to ETN; (2) treatment with any other biologic therapy for RA, including TNF inhibitors, anti-CD20, immune-modulator drug(s), other investigational drug(s) and/or device(s) within 3 months or five half-lives at the time of enrolment, whichever was longer; (3) previous use of >2 biologics (allowed only if the therapy was efficient and not failing and was withdrawn because of other reasons that were not due to efficacy failure or safety issues); (4) investigator-determined primary-line and secondary-line biologic therapy failures; (5) patients taking high-potency opioid analgesics, and/or any intramuscular corticosteroid injection, and/or any therapy by intra-articular injection required for treatment of acute RA flare within 4 weeks before baseline; (6) functional status class IV according to the ACR 1991 revised criteria19; (7) systemic manifestations of RA, with the exception of Sjögren’s syndrome; (8) any other active inflammatory or autoimmune diseases other than RA; and (9) history of tuberculosis and latent tuberculosis detected by imaging and/or by the QuantiFERON®-TB Gold test at screening (see online supplementary appendix 1 for detailed exclusion criteria).
Concomitant and prohibited medications
All patients continued to receive concomitant MTX at a stable dose (10–25 mg/week) and folic acid (≥5 mg/week) until the end of the study. Patients on a stable dose of oral corticosteroids (prednisone ≤7.5 mg or equivalent) or non-steroidal anti-inflammatory drugs/cyclooxygenase-2 inhibitors and paracetamol/low-strength opioids for at least 4 weeks before randomisation were allowed to continue the treatment during the study, if necessary. Use of rescue medication was not allowed.
During the study, the following medications were prohibited: (1) any biologic or conventional synthetic DMARDs and/or immunosuppressants other than MTX; (2) any intra-articular injection (ie, corticosteroid, hyaluronan); (3) any intramuscular corticosteroid injection; (4) any intra-articular or any systemic corticosteroid treatment; (5) administration of live vaccines.
Statistical analysis
Assuming no difference between GP2015 and ETN, a sample size of 183 patients per treatment group was calculated (to obtain 155 evaluable patients per treatment group, with an assumed drop-out and major protocol deviation rate of 15%), which was estimated to provide 90% power for the primary endpoint analysis.
DAS28-CRP was chosen as the primary endpoint of this study as it is a continuous measure and is more sensitive compared with categorical measures, such as ACR20. In addition, DAS28-CRP is a clinically relevant and widely accepted endpoint across RA studies and has been extensively validated for its use in clinical trials in combination with EULAR response criteria.20
Therapeutic equivalence, in terms of change from baseline in DAS28-CRP, could be established if the 95% CI for the difference in mean changes was contained within the interval (−0.6 to 0.6). A mixed-model repeated measures analysis was performed for the primary variable to calculate the 95% CI at week 24 including treatment as a fixed effect. The model adjusted for the following stratification factors: weight (<90 kg), prior DMARD strata time (visits), the interaction between time (visits) and treatment, all as categorical variables, and baseline DAS28-CRP as a continuous variable. No imputation was performed for the missing components of the DAS28-CRP score.
All efficacy analyses were performed on the TP1 per-protocol set (PPS) that consisted of all patients who completed the study until week 24 without major protocol deviations. The analyses were repeated on the TP1 full analysis set (TP1 FAS) that included all randomised patients to whom study treatment had been assigned. Safety and immunogenicity results were summarised descriptively for the TP1 safety set (SAF) that included all patients who received at least one dose of study treatment during the treatment period.
Results
Patient disposition
Of the 376 patients randomised, 353 completed TP1 (figure 1) while 23 patients were discontinued in both treatment groups; the main reason for discontinuation was withdrawal of consent. The TP1 FAS and SAF included all 376 randomised patients, and the TP1 PPS included 323 patients. The main reasons for excluding patients from the PPS included (1) deviation from key (diagnostic) and efficacy-related eligibility criteria, (2) prohibited concomitant medication or procedure, and (3) failure to perform key and safety procedures.
Figure 1.
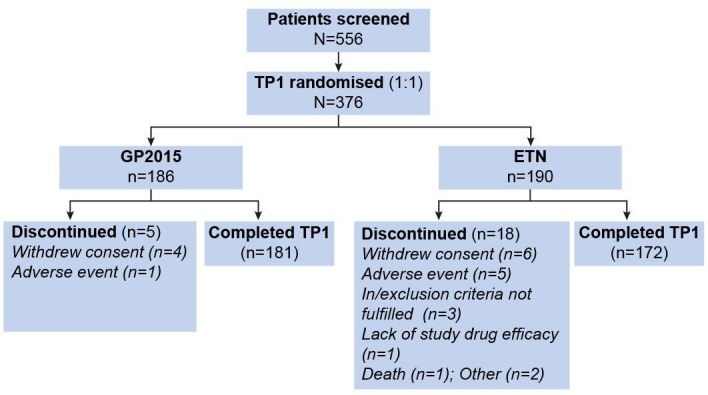
Patient disposition (full analysis set). Patients completing week 24 visit are considered completing TP1. ETN, reference etanercept; TP1, treatment period 1.
Baseline characteristics
Patients had a mean age of 54.1 years (SD 12.02), and the majority were females (n=308; 81.9%) and Caucasian (n=365; 97.1%). The majority (68.6%) of patients had received prior treatment with one or two DMARDs. The baseline demographics and disease characteristics of patients were comparable across treatment groups (table 1). Prior anti-TNF and other biologics use was well balanced between treatment arms.
Table 1*.
Baseline characteristics and disease history (TP1 full analysis set)
| Characteristics | GP2015 (n=186) |
ETN (n=190) |
| Age (years) | 55.2 (11.22) | 53.1 (12.70) |
| Male, n (%) | 28 (15.1) | 40 (21.1) |
| Race, n (%) | ||
| Caucasian | 180 (96.8) | 185 (97.4) |
| African American | 5 (2.7) | 1 (0.5) |
| Asian | 0 | 3 (1.6) |
| American Indian or Alaska Native | 1 (0.5) | 1 (0.5) |
| DAS28-CRP | 5.43 (0.92) | 5.55 (0.78) |
| Tender 28-joint count | 14.2 (6.18) | 14.8 (5.78) |
| Swollen 28-joint count | 10.5 (5.28) | 11.1 (5.39) |
| C reactive protein (mg/L) | 11.7 (21.09) | 11.0 (15.83) |
| HAQ-DI score | 1.45 (0.55) | 1.44 (0.58) |
| FACIT fatigue score | 26.95 (9.60) | 25.28 (10.09) |
| Duration of rheumatoid arthritis (years) | 8.79 (8.25) | 8.18 (6.92) |
| Rheumatoid factor, positive, n (%) | 137 (73.70) | 140 (73.70) |
| Anti-CCP, positive, n (%) | 144 (77.40) | 140 (73.70) |
| Prior therapy*, n (%) | ||
| MTX only | 56 (30.1) | 58 (30.5) |
| MTX+any DMARDs | 73 (39.2) | 72 (37.9) |
| MTX+any anti-TNF | 31 (16.7) | 33 (17.4) |
| MTX+any other biologic | 26 (14.0) | 27 (14.2) |
| Previous DMARDs used, n (%) | ||
| 1 | 56 (30.1) | 58 (30.5) |
| 2 | 75 (40.3) | 69 (36.3) |
| 3 | 36 (19.4) | 39 (20.5) |
| 4 or more | 19 (10.2) | 24 (12.6) |
| MTX dose (mg/week) | 16.0 (4.9) | 17.1 (4.6) |
| Prior duration of MTX (months) | 55.6 (49.7) | 59.5 (51.7) |
Prior therapy strata is arranged according to the hierarchy; values are mean (SD) unless otherwise stated; rheumatoid factor ≤10 IU/mL and anti-CCP <17 U/mL are considered negative.
CCP, cyclic citrullinated peptide; DAS28-CRP, disease activity score 28-joint count C reactive protein; DMARD, disease-modifying antirheumatic drug; ETN, reference etanercept; FACIT, Functional Assessment of Chronic Illness Therapy; HAQ-DI, Health Assessment Questionnaire Disability Index; MTX, methotrexate; TNF, tumour necrosis factor; TP1, treatment period 1.
Primary outcome
In the TP1 PPS, the least squares mean difference for change in DAS28-CRP from baseline to week 24 between GP2015 and ETN (GP2015–ETN) was −0.07 (95% CI −0.26 to 0.12; figure 2). The 95% CI was well contained within the prespecified equivalence margin of −0.6 to 0.6 demonstrating therapeutic equivalence between GP2015 and ETN. Equivalence was further confirmed with the analysis on the FAS (least squares mean difference: −0.06 (95% CI −0.25 to 0.12)).
Figure 2.
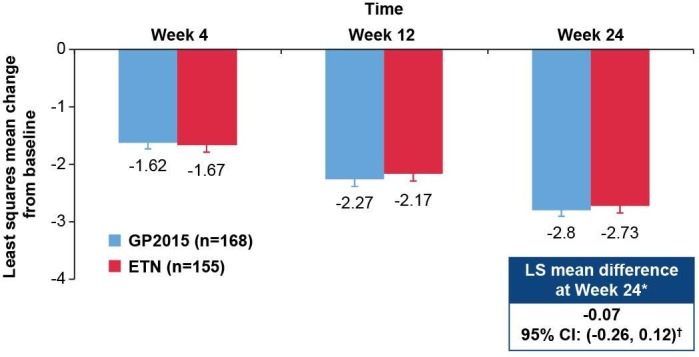
DAS28-CRP change from baseline over 24 weeks (TP1 per-protocol set) at baseline, the mean DAS28-CRP was 5.42 and 5.53 for GP2015 and ETN groups, respectively. The per-protocol set consists of all patients completing the study until week 24 without major protocol deviations. Error bars represent the SE. *Primary endpoint; †95% CI was contained within the prespecified equivalence margin of −0.6 to 0.6; a mixed-model repeated measures analysis was performed for DAS28-CRP change from baseline using the TP1 per-protocol set. Equivalence was further confirmed with the analysis on the FAS (least squares mean difference: −0.06 (95% CI −0.25 to 0.12)). Per-protocol set: GP2015 (n=168), ETN (n=155). DAS28-CRP, disease activity score 28-joint count C reactive protein; ETN, reference etanercept; LS, least squares; TP1, treatment period 1.
Key secondary outcomes
Efficacy
The mean (SD) DAS28-CRP change from baseline to week 24 was similar between the GP2015 (−2.78 [1.1]) and ETN groups (−2.78 [1.0]; figure 3A).
Figure 3.
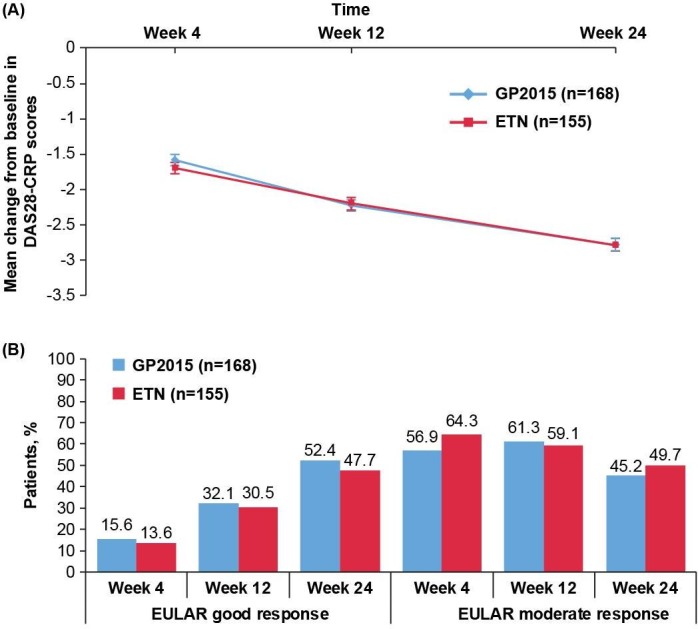
(A) Mean change from baseline in DAS28-CRP scores over 24 weeks (TP1 per-protocol set). The per-protocol set consists of all patients completing the study until week 24 without major protocol deviations. Error bars represent the SE. Per-protocol set: GP2015 (n=168), ETN (n=155). (B) EULAR response rates over 24 weeks (TP1 per-protocol set). The per-protocol set consists of all patients completing the study until week 24 without major protocol deviations. EULAR good response is defined as DAS28 ≤3.2 at week 24 and improvement from baseline >1.2; EULAR moderate response is defined as DAS28 >3.2 and ≤5.1 at week 24 and improvement from baseline >0.6 or DAS28 >5.1 at week 24 and improvement from baseline >1.2. Per-protocol set: GP2015 (n=168), ETN (n=155). DAS28-CRP, disease activity score 28-joint count C reactive protein; ETN, reference etanercept; EULAR, European League Against Rheumatism; TP1, treatment period 1.
At 24 weeks, EULAR moderate and good response rates improved and were again comparable between the GP2015 and ETN (figure 3B) groups.
The ACR20, ACR50 and ACR70 response rates were comparable between the GP2015 and ETN groups over 24 weeks (figure 4). At week 24, ACR20, ACR50 and ACR70 response rates were 88%, 64.1% and 33.5%, respectively, in the GP2015 group and 92.9%, 71.0% and 42.6%, in the ETN group, respectively.
Figure 4.
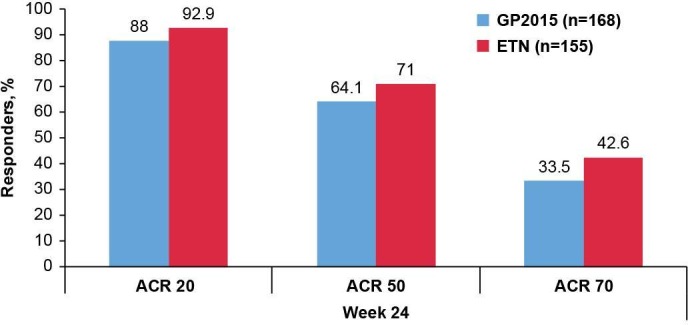
ACR20/50/70 response rates at week 24 (TP1 per-protocol set). The per-protocol set consists of all patients completing the study until week 24 without major protocol deviations. Per-protocol set: GP2015 (n=168), ETN (n=155). ACR20/50/70, American College of Rheumatology 20%/50%/70% response criteria; ETN, reference etanercept; TP1, treatment period 1.
Improvements from baseline in HAQ-DI and FACIT fatigue scores were comparable between the GP2015 and ETN groups over 24 weeks. At week 24, the mean change from baseline in HAQ-DI score was −0.57 in the GP2015 group and −0.67 in the ETN group (online supplementary figure S2). The mean change from baseline in FACIT fatigue score at week 24 was 9.45 in the GP2015 group and 11.82 in the ETN group, respectively (online supplementary figure S3). The DAS28-CRP change from baseline at week 24 for the different subgroups by prior treatment is provided in online supplementary table S1.
Safety
The median duration of exposure was similar between the GP2015 and ETN groups (162 days). The number of patients with at least one treatment-emergent adverse event (TEAE) up to 24 weeks was comparable between the GP2015 (n=81 [43.5%]) and ETN (n=94 [49.5%]) group (table 2). At least one TEAE and the most commonly (≥2% incidence) reported TEAEs up to 24 weeks occurred in a similar proportion of patients in the GP2015 and ETN groups (table 2). Injection-site reactions were reported as TEAEs in fewer patients in the GP2015 group (13 [7.0%]) compared with the ETN group (35 [18.4%]); all were mild or moderate in intensity and none were considered as a serious adverse event.
Table 2.
TEAEs and SAEs during the 24-week treatment period (TP1 safety set)
| Preferred term | GP2015 (n=186) n (%) |
ETN (n=190) n (%) |
| ≥1 TEAE | 81 (43.5) | 94 (49.5) |
| ≥1 SAE | 1 (0.5) | 5 (2.6) |
| ≥1 Treatment-related TEAE | 39 (21.0) | 46 (24.2) |
| ≥1 Treatment-related SAE | 0 | 0 |
| Discontinuations due to TEAE | 2 (1.1) | 7 (3.7) |
| ≥1 AE of special interest | 12 (6.5) | 9 (4.7) |
| Deaths | 0 | 1 (0.5) |
| TEAEs with a ≥2% incidence in any of the treatment groups | ||
| Injection-site reaction | 13 (7.0) | 35 (18.4) |
| Nasopharyngitis | 9 (4.8) | 4 (2.1) |
| Alanine aminotransferase increased | 8 (4.3) | 4 (2.1) |
| Urinary tract infection | 8 (4.3) | 8 (4.2) |
| Upper respiratory tract infection | 6 (3.2) | 7 (3.7) |
| Back pain | 5 (2.7) | 1 (0.5) |
| Diarrhoea | 3 (1.6) | 4 (2.1) |
| Bronchitis | 2 (1.1) | 4 (2.1) |
| Cystitis | 2 (1.1) | 4 (2.1) |
A patient with multiple occurrences of event within the same system organ class or preferred term under one treatment is counted only once. TEAEs are events started after the first dose of study treatment and before study discontinuation or 30 days after the last dose, whichever occurs later. Adverse event terms are coded using MedDRA V.19.1.
ETN, reference etanercept; MedDRA, Medical Dictionary for Regulatory Activities; SAE, serious adverse event; TEAE, treatment-emergent adverse event; TP1, treatment period 1.
In both groups during TP1, injection-site reactions, mostly mild, were the treatment-related TEAE with higher incidence in the ETN (18.4%) group compared with the GP2015 group (6.5%; online supplementary table S2). Serious AEs were reported in one (0.5%) patient in the GP2015 group (spinal fracture caused by injury) and five (2.6%) patients in the ETN group (atrial fibrillation, cholelithiasis, hepatic enzyme increase, worsening RA, lung neoplasm [n=1 each]). In the ETN group, one patient died during the study due to malignant lung neoplasm. As assessed by the investigator, the serious adverse event (SAE) lung cancer was severe with no suspected causal relationship to study drug. The patient was a smoker (40 cigarettes per day) with a family history of cancer. Two (1.1%) patients in the GP2015 group and seven (3.7%) in the ETN group discontinued due to TEAEs during TP1, with one patient each with pruritic rash and decreased platelet count in the GP2015 group. In the ETN group, discontinuations were due to urticaria, RA flare, thrombocytopenia/injection-site reaction, lung neoplasm in one case each and injection site reactions in another three cases. TEAEs of special interest were infrequently reported in either the GP2015 or ETN group (6.5% vs 4.7%, respectively; online supplementary table S3).
Immunogenicity
In both groups, ADAs were detected between weeks 2 and 12. While ADA incidence in general was low, ADA positivity peaked in a singular point at week 4 (GP2015: 1.6%; ETN: 22.7%). The titre of all measured ADAs was low and near the detection limit of a highly sensitive method. None of the GP2015-treated patients developed neutralising antibodies (NAbs) whereas 1.6% and 0.6% of patients in ETN group were NAb positive at weeks 4 and 12, respectively. Of note, all ADA/NAb responses were transient and none of the patients had detectable ADA or NAb levels at week 24 (online supplementary figure S4).
Discussion
The EQUIRA study met the primary endpoint showing that GP2015 has equivalent efficacy to ETN in patients with RA, who had an inadequate response to synthetic or biologic DMARDs. This finding was further supported by the evidence that secondary efficacy endpoints were clearly comparable between GP2015 and ETN. The data were also strengthened by the evidence that other items such as patient-reported outcomes were also comparable between the two treatments.
The study design attributes, as well as the key features of the EQUIRA study, such as patient population, inclusion criteria, prior therapy including stable dose of MTX and efficacy evaluations, were aligned with the key phase III trials for RA conducted with ETN. Although the dose of ETN used in the pivotal trials was 25 mg twice weekly, similar clinical outcomes, safety and pharmacokinetic profiles have previously been demonstrated for the 50 mg dose administered once weekly in patients with RA.21 The population was predominantly Caucasian in this study.
In the EQUIRA study, the use of DAS28-CRP as the primary endpoint allowed for a direct comparison with relevant literature data and a comprehensive interpretation of the study results. The therapeutic equivalence for the DAS28-CRP change from baseline to week 24 was achieved. In addition, considering that previous ETN studies have assessed both DAS28 and ACR response,22–24 ACR20 and other ACR responses were assessed as secondary endpoints in order to better compare the results of the current study with historical data and to ensure complete evaluation of clinical efficacy.
The ACR20 response rates in the GP2015 and ETN groups at week 24 (88% vs 92.9%, respectively) were comparable although slightly higher than those reported in previous ETN studies.25 26 The biosimilar study design uses active treatment arms and lacks a placebo comparator, and it has been reported previously that higher ACR20 response rates are observed in biosimilar studies compared with placebo-controlled studies.27–29 The higher ACR20 response rates at week 24 in both treatment groups in this study may also be due to high numbers of (1) biologic-naïve patients and (2) patients with low or no prior exposure to DMARDs in combination with MTX.
It is important to remark that over 24 weeks, the incidence of TEAEs was comparable between the two treatment groups. No new safety findings or concerns were reported. Overall, GP2015 was well tolerated with a similar safety profile to the well-established safety profile of ETN.3 The incidence of injection site reactions was lower in the GP2015 (7.0%) than in the ETN group (18.4%), similar to those observed in recent biosimilar ETN studies.28 30 31 Similar differences between GP2015 and ETN were also observed in the EGALITY study in patients with plaque-type psoriasis.8 This could be due to differences in the formulations between the two drugs, as reported for other ETN biosimilars.28 30
In this study, immunogenicity assessment was performed using a validated state-of-the-art technique comprising a high sensitivity and drug tolerance which enables the detection of low titre and transient ADAs even during the drug treatment phase. As recently recommended,32 a false-positive rate for confirmatory assay of 1% was applied instead of the commonly used 0.1% rate.33 Applying both a sensitive and drug-tolerant assay as well as a contemporary biostatistical approach may lead to a higher reported ADA incidence compared with historical data.17 While ADA incidence was consistently low in the GP2015 treatment group, a singular peak of ADA positivity was observed at week 4 for ETN-treated patients also including NAbs. Beside the evaluation of ADA/NAb incidence, a correlation of immunogenicity outcome to efficacy or patients’ safety was not observed, indicating that detected ADAs were not clinically relevant. This may be explained by the transient nature of the detected ADAs (including NAbs) as well as the low titre results.
Conclusions
The EQUIRA study demonstrated an equivalent efficacy of GP2015 and ETN in patients with RA with moderate to severe disease with inadequate responses to DMARDs, including MTX and biologics. The safety profiles of GP2015 and ETN were comparable and no new safety findings or concerns were reported. These results obtained in patients with RA are a further complement to prior evidence indicating equivalent efficacy of GP2015 and ETN in plaque-type psoriasis.
Acknowledgments
The authors thank all investigators (ClinicalTrials.gov: NCT02638259) and participating patients who contributed to the successful conduct of this study, and Divya Chandrasekhar (Product Lifecycle Services—NBS, Novartis Healthcare Pvt. Ltd., Hyderabad, India) for medical writing and editorial assistance.
Footnotes
Contributors: MM-C contributed to the conception of the study and interpretation of the results. YA contributed to the conception of the study and interpretation of the results. AK contributed to the conception of the study and interpretation of the results. MHB contributed to the conception of the study and interpretation of the results. HS-K contributed to the conception of the study and interpretation of the results. EJK contributed to the conception of the study and interpretation of the results. HW contributed to the statistical planning of the study and the interpretation of the results. GB contributed to the study design, the study conduct and the interpretation of the results. JP generated immunogenicity data and contributed to interpretation of results. AS contributed to the study design, the study conduct and the interpretation of the results. AD was involved in the study protocol design and administration. All authors agree to be accountable for all aspects of the work and in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding: This study was funded by Hexal AG, a Sandoz company.
Competing interests: MM-C reports no conflicts of interest. YA reports personal fees from Sandoz, grants and personal fees from Pfizer, grants and personal fees from Roche, during the conduct of the study. AK reports personal fees from Sandoz, outside the submitted work. MHB reports personal fees from Sandoz during the conduct of the study. HS-K reports personal fees from Sandoz/Hexal during the conduct of the study, personal fees from Sandoz/Hexal, grants and personal fees from Novartis, outside the submitted work. EJK reports no conflict of interest. HW, GB, JP and AS are employees of Hexal AG. AD is an employee of Sandoz Inc.
Patient consent: Obtained.
Ethics approval: The study protocol was approved by the Independent Ethics Committee or Institutional Review Board for each centre.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All data generated or analysed are included in this article and the supplementary information files.
References
- 1. Goffe B, Cather JC. Etanercept: an overview. J Am Acad Dermatol 2003;49:105–11. 10.1016/mjd.2003.554 [DOI] [PubMed] [Google Scholar]
- 2. Amgen , 1998. Enbrel US prescribing information. Available from: http://pi.amgen.com/united_states/enbrel/derm/enbrel_pi.pdf [Accessed 16 Oct 2017].
- 3. European Medicines Agency , 2017. Enbrel, summary of product characteristics. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000262/WC500027361.pdf [Accessed 16 Oct 2017].
- 4. World Health Organization , Guidelines on evaluation of similar biotherapeutic products (SBPs). 2009. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf [accessed 6 Jan 2018].
- 5. Weise M, Bielsky MC, De Smet K, et al. Biosimilars—why terminology matters. Nat Biotechnol 2011;29:690–3. 10.1038/nbt.1936 [DOI] [PubMed] [Google Scholar]
- 6. European Medicines Agency , Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf [Accessed 18 Dec 2017].
- 7. von Richter O, Skerjanec A, Afonso M, et al. GP2015, a proposed etanercept biosimilar: pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br J Clin Pharmacol 2017;83:732–41. 10.1111/bcp.13170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffiths CEM, Thaçi D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol 2017;176:928–38. 10.1111/bjd.15152 [DOI] [PubMed] [Google Scholar]
- 9. European Medicines Agency , 2018. Assessment report Erelzi. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004192/WC500230144.pdf [accessed on 11 Sep].
- 10. Accessdata , 2018. Erelzi Prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761042lbl.pdf [Accessed 11 Sep].
- 11. Prevoo ML, van 't Hof MA, Kuper HH, et al. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. 10.1002/art.1780380107 [DOI] [PubMed] [Google Scholar]
- 12. Van Gestel AM, Prevoo MLL, Van 't Hof MA. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Arthritis Rheum 1996;39:34–40. [DOI] [PubMed] [Google Scholar]
- 13. Fries JF, Spitz P, Kraines RG, et al. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. 10.1002/art.1780230202 [DOI] [PubMed] [Google Scholar]
- 14. Bruce B, Fries JF. The Health Assessment Questionnaire (HAQ). Clin Exp Rheumatol 2005;23(5 Suppl 39):S14–18. [PubMed] [Google Scholar]
- 15. Cella DF, Tulsky DS, Gray G, et al. The functional assessment of cancer therapy scale: development and validation of the general measure. J Clin Oncol 1993;11:570–9. 10.1200/JCO.1993.11.3.570 [DOI] [PubMed] [Google Scholar]
- 16. Yellen SB, Cella DF, Webster K, et al. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage 1997;13:63–74. 10.1016/S0885-3924(96)00274-6 [DOI] [PubMed] [Google Scholar]
- 17. Poetzl J, Arlt I, von Richter O. State-of-the-art immunogenicity evaluation in phase 3 confirmatory study (EGALITY) with etanercept biosimilar GP. J Eur Acad Dermatol Venereol 2015. [DOI] [PubMed] [Google Scholar]
- 18. Kay J, Upchurch KS. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 2012;51(suppl 6):vi5–vi9. 10.1093/rheumatology/kes279 [DOI] [PubMed] [Google Scholar]
- 19. Hochberg MC, Chang RW, Dwosh I, et al. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum 1992;35:498–502. 10.1002/art.1780350502 [DOI] [PubMed] [Google Scholar]
- 20. Wells G, Becker JC, Teng J, et al. Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis 2009;68:954–60. 10.1136/ard.2007.084459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keystone EC, Schiff MH, Kremer JM, et al. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2004;50:353–63. 10.1002/art.20019 [DOI] [PubMed] [Google Scholar]
- 22. Bathon JM, Martin RW, Fleischmann RM, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med 2000;343:1586–93. 10.1056/NEJM200011303432201 [DOI] [PubMed] [Google Scholar]
- 23. Genovese MC, Bathon JM, Fleischmann RM, et al. Longterm safety, efficacy, and radiographic outcome with etanercept treatment in patients with early rheumatoid arthritis. J Rheumatol 2005;32:1232–42. [PubMed] [Google Scholar]
- 24. Genovese MC, Bathon JM, Martin RW, et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: two-year radiographic and clinical outcomes. Arthritis Rheum 2002;46:1443–50. 10.1002/art.10308 [DOI] [PubMed] [Google Scholar]
- 25. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med 1999;340:253–9. 10.1056/NEJM199901283400401 [DOI] [PubMed] [Google Scholar]
- 26. Combe B, Codreanu C, Fiocco U, et al. Etanercept and sulfasalazine, alone and combined, in patients with active rheumatoid arthritis despite receiving sulfasalazine: a double-blind comparison. Ann Rheum Dis 2006;65:1357–62. 10.1136/ard.2005.049650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bae SC, Kim J, Choe JY, et al. A phase III, multicentre, randomised, double-blind, active-controlled, parallel-group trial comparing safety and efficacy of HD203, with innovator etanercept, in combination with methotrexate, in patients with rheumatoid arthritis: the HERA study. Ann Rheum Dis 2017;76:65–71. 10.1136/annrheumdis-2015-207613 [DOI] [PubMed] [Google Scholar]
- 28. Emery P, Vencovský J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 2017;76:51–7. 10.1136/annrheumdis-2015-207588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013;72:1613–20. 10.1136/annrheumdis-2012-203090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsuno H, Tomomitsu M, Hagino A, et al. Phase III, multicentre, double-blind, randomised, parallel-group study to evaluate the similarities between LBEC0101 and etanercept reference product in terms of efficacy and safety in patients with active rheumatoid arthritis inadequately responding to methotrexate. Ann Rheum Dis 2018;77:488–94. 10.1136/annrheumdis-2017-212172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O’Dell J, Takeuchi T, Tanaka Y. Randomized, double-blind study comparing CHS-0214 with etanercept in patients with active rheumatoid arthritis (RA) despite methotrexate (MTX) therapy [abstract]. Ann Rheum Dis 2016;75(Suppl 2):P143. [Google Scholar]
- 32. FDA , 2018. Assay development and validation for immunogenicity testing of therapeutic protein products (draft guidance, 2016). Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM192750.pdf [Accessed 31 Jan 2018].
- 33. Shankar G, Devanarayan V, Amaravadi L, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal 2008;48:1267–81. 10.1016/j.jpba.2008.09.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2018-000757supp001.docx (3.8MB, docx)