

Abstract
The jujubosides are saponin natural products reported to have immunoadjuvant, anticancer, antibacterial, antifungal, and antisweet activities. The triterpene component, jujubogenin contains a unique tricyclic ketal motif comprising the DEF ring system. Herein, we describe our efforts toward the total synthesis of jujubogenin, using a sterically-demanding intermolecular Diels–Alder reaction to assemble the C-ring and a tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reaction to form the DF-ring system. Acid-catalyzed cyclization of the resulting bicyclic enol ether then closes the E-ring to provide the hexacyclic core of jujubogenin.
Keywords: Natural Products, Total Synthesis, Wolff rearrangement, Ketene hetero-Diels–Alder, Tandem reaction
Graphical abstract
1. Introduction
The jujubosides are glycosylated triterpene natural products isolated mainly from Chinese and Indian herbal plants. They exhibit a wide range of bioactivities including immunoadjuvant, anticancer, antifungal, antibacterial, and antisweet activities.1–3 In particular, jujuboside A (2) has been reported to generate higher antibody titers in mouse vaccinations and lower in vitro toxicity compared to QS-21,2–3 another saponin immunoadjuvant that is a component of the Mosquirix (RTS,S/AS01) malaria vaccine4 and Shingrix shingles vaccine5 and has been investigated as the immunoadjuvant of choice in numerous other vaccine clinical trials.6–12 Based on our laboratory’s long-standing interest in saponin immunoadjvuants13–20 we initiated a research program toward the total synthesis of jujuboside A to enable detailed structure–activity relationships studies. We recently disclosed our synthesis of the complex, doubly-branched pentasaccharide portion of jujuboside A.21 Herein, we describe our efforts toward the total synthesis of triperpene portion of jujuboside A, called jujubogenin (1).22
Jujubogenin is a complex triterpene with nine contiguous stereocenters, six of which are quaternary, including four all-carbon quaternary centers (Figure 1). In addition, the tricyclic DEF ring system bearing a C16 ketal is unique to jujubogenin. Despite these striking structural features and numerous reported biological activities of the jujubosides, a chemical synthesis of the jujubogenin triterpene has not been reported. Developing a synthetic strategy that would address the complexity of these structural features and allow structure–activity relationship studies of this triterpene and related natural products presents a significant challenge.
Figure 1.

Structures of jujubogenin (1) and jujuboside A (2).
2. Results and Discussion
2.1. Retrosynthetic analysis of jujubogenin (1)
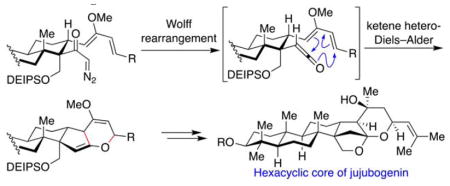
Retrosynthetically, we envisioned late-stage installation of the C21 methyl group of jujubogenin (1) by addition of methyl lithium to the convex α-face of ketone 3 (Scheme 1). Ketone 3 could be assembled from bis(enol ether) 4 through a series of functional group interconversions including an acid-catalyzed tandem C30 alcohol deprotection, ketalization, and methyl enol ether hydrolysis.
Scheme 1.

Retrosynthetic analysis of jujubogenin (1).
The structural complexity of bis(enol ether) 4 could be dramatically reduced by a tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reaction starting from diazoketone 6 via ketene intermediate 5. The highly functionalized C-ring of the jujubogenin would be furnished using a sterically-demanding intermolecular Diels–Alder reaction between diene 8 and dienophile 7. Diene 8 would be prepared in enetioenriched form from the Wieland–Miescher ketone (9).23
2.2. Model study of sterically demanding Diels–Alder reaction
One of the key transformations in our synthetic plan for the synthesis of jujubogenin involved a sterically-demanding Diels–Alder reaction between diene 8 and dienophile 7. Such sterically-demanding cycloadditions are challenging despite many advances in Diels–Alder reaction methodology due to several reasons: 1) the diene 8 bears a C18 methyl group that hinders the adoption of s-cis conformation of the diene required for the Diels-Alder reaction; 2) the dienophile 7 is trisubstituted, thus the desired reaction pathway involves a highly congested transition state with two adjacent all-carbon quaternary centers; 3) the diene 8 lacks a strong electron-donating substiutent to control the regioselectivity of the reaction.
In support of the challenges highlighted above, it has been shown that dienes that are structurally similar to our proposed diene 8 react only with highly activated and sterically accessible dienophiles, such as tetracyanoethylene and acetylene dienes with an electron-withdrawing group, or under forcing condition such as high pressure.24–25 Diactivated dienophiles such as dimethyl fumarate/maleate and maleic anhydride do not react with this type of diene. On the other hand, (carbomethoxy)maleic anhydride 7 is a highly activated dienophile.26 Kinetic studies by Hall and coworkers revealed that it is more reactive than tetracyanoethylene. Interestingly, dienophile 7 reacts even with styrene as a diene to give the corresponding Wagner–Jauregg type27 adduct. Despite this high reactivity, there has been limited use of this dienophile to deliver substituted cyclohexanes.26, 28–33 In fact, only one application to natural product synthesis has appeared in the literature.33
We anticipated that the high reactivity of dienophile 7 would compensate for the poor reactivity of diene 8 in our proposed sterically-demanding Diels–Alder reaction. To test this hypothesis, we first conducted a model study with readily accessible diene 10.34 We were delighted to find that the reaction between diene 10 and dienophile 7 proceeded smoothly at ambient temperature to give tricycle 11 as a single regio- and stereoisomer (Scheme 2). Structural assignment was based on extensive 2D-NMR and nOe studies of the dicarboxylic acid derivative 12, which was recovered after silica gel purification of anhydride 11. Mechanistically, the cycloadduct 11 arises from an endo transition state relative to the anhydride portion of the dienophile.
Scheme 2.

Model study of sterically-demanding Diels–Alder reaction.
Although this achiral model system 10 does not address the facial selectivity of dienophile approach, it validated the feasibility of this approach with respect to reactivity, setting the stage for its evaluation in the context of the more complex jujubogenin system below.
2.3. Model study of tandem Wolff rearrangement–intramolecular ketene hetero-Diels-Alder reaction
Both the C=C and C=O π-systems of ketenes can react with electron-rich dienes to produce [4+2] and [2+2] cycloaddition products and aldol-like reaction products. A literature survey suggests that, out of these possible pathways, [2+2] cycloadditions are the most common (Staudinger ketene cycloaddition).35 In contrast, [4+2] cycloadditions involving either the C=C or C=O bonds of ketenes are rare and usually regarded as exceptions.36–54
In our retrosynthetic analysis, we proposed a [4+2] cycloaddition reaction between an electron-rich diene and the C=O bond of ketene intermediate 5. Examples of such cycloaddition reactions are limited to stabilized ketenes such as dichloro-, diphenyl-, and bis(trifluoromethyl)ketenes and often result in mixtures of cycloadducts. However, in the context of our proposed approach to jujubogenin, use of the ketene intermediate 5 in a conformationally-constrained hetero-[4+2] cycloaddition appeared promising based on the fact that the products arising from alternative undesired pathways ([2+2] and ketene C=C bond [4+2] cycloadditions) were expected to suffer from prohibitively high strain energy.
Considering that hetero-[4+2] cycloadditions of ketenes are rare and that such a cycloaddition of a simple monoalkyl substituted ketene has not been reported, we decided to test the feasibility of the proposed [4+2] cycloaddition in a model system, using diazoketone 13, in which the cyclohexane ring mimics the C-ring of jujubogenin (Scheme 3). Thus, diazoketone 13 was prepared from readily available starting materials in nine steps (see Supporting Information). Gratifyingly, reaction of diazoketone 13 with AgOTf in the presence of triethylamine to generate corresponding ketene intermediate 14, led to formation of tricyclic bis(enol ether) 16, indicating that the tandem Wolff rearrangement–Diels–Alder reaction proceeded via the desired ketene C=O [4+2] cycloaddition pathway. This set the stage for investigation of this synthetic approach in the context of the more complex jujubogenin system below.
Scheme 3.

Model study of tandem Wolff rearrangement–intramolecular hetero-[4+2] cycloaddition across ketene C=O bond.
2.4. Synthesis and sterically-demanding Diel–-Alder reaction of Wieland–Miescher ketone-derived diene 8
Synthesis of diene 8 started from the Wieland–Miescher ketone (9) (Scheme 4), which was converted to ketone 16 through a previously described reaction sequence (see Supporting Information). Addition of vinyl magnesium bromide to ketone 16 gave allylic alcohol 17 as an epimeric mixture of vinyl carbinols (major isomer shown). Dehydration delivered the desired diene 8 in 32% overall yield from the Wieland–Miescher ketone (9). Importantly, the reaction sequence was easy scalable to multigram quantities, facilitating material throughput.
Scheme 4.

Synthesis of diene 8 from the Wieland–Miescher ketone (9).
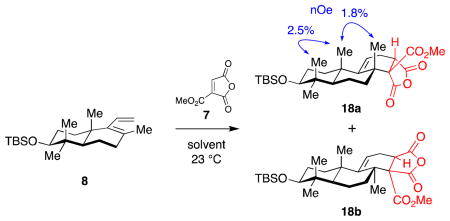
With diene 8 in hand, we began investigations into the envisioned sterically-demanding Diels–Alder reaction. In our initial attempt, diene 8 was reacted with triactivated dieneophile 7 in dichloromethane (Table 1, entry 1). Under these conditions, the reaction went to completion over several days to give a 1.5:1 mixture of the diastereomers 18a and 18b in nearly quantitative yield.
Table 1.
Effect of solvents on diastereoselectivity of sterically-demanding Diels–Alder reaction of 7 and 8.a
| ||
|---|---|---|
| entry | solvent | 18a: 18b |
|
| ||
| 1 | CH2Cl2 | 1.5: 1 |
| 2 | THF | decomposition |
| 3 | Et2O | 2.2: 1 |
| 4 | acetone | decomposition |
| 5 | EtOAc | 2: 1 |
| 6 | PhH | 2.2: 1b |
| 7 | PhMe | 2: 1 |
| 8 | CF3Ph | 1.8: 1 |
| 9 | C6F6 | 1.85: 1 |
| 10 | mesytylene | 2: 1 |
| 11 | pentane | 2: 1 |
All reactions were carried out with 0.2 mmol of diene 8 and 0.22 mmol of dienophile 7 in 1mL of the solvent at ambient temperature. Ratios of products was determined by 1H NMR analysis of the crude reaction mixtures.
54% isolated yield of 18a.
After screening a number of solvents, we found that the best diastereoselectivity was obtained in diethyl ether or benzene (entries 3, 6), although a small amount of dienophile decomposition was observed in diethyl ether. The overall reaction proceeded faster in aromatic solvents (entries 6–10) while nucleophilic solvents resulted in dienophile decomposition (entries 2, 4). The structure of the anhydride 18a was confirmed through extensive 2D-NMR and nOe studies. We were unable to assign the stereochemistry of the second diastereomer unambiguously, but presume it to be 18b, arising from an endo transition state and approach of the dienophile from the β-face of the diene 8.
Lewis acids can dramatically alter the reactivity and stereoselectivity of Diels–Alder reactions.55–56 Thus, we also investigated a number of Lewis-acid catalysts (B(C6F5)3, Yb(OTf)3, Sc(OTf)3, La(OTf)3, Zn(OTf)2, Cu(OTf)2, Fe(OTf)3, In(OTf)3, SnCl4, Et2AlCl, EtAlCl2) in CH2Cl2 or PhMe, at 0 and −78 °C. Unfortunately, no clear trend or improvement in diastereoselectivity (1:1 to 2:1) was observed. Moreover, these reactions all generated significant side products, presumably arising from product and/or dienophile decomposition. Accordingly, the optimized reaction was run in benzene without any additives (entry 6), delivering the desired anhydride 18a in 54% isolated yield.
2.5. Elaboration of the C-ring of jujubogenin
To advance the synthesis from anhydride 18a, several functional group manipulations were required to elaborate the Cring. We started with hydrogenation of the double bond, which we anticipated to be challenging as both the α and β faces of the double bond are hindered by functional groups present in B and C rings. In fact, all attempts to hydrogenate the double bond of anhydride 18a using variety of heterogeneous and homogeneous catalysts, elevated hydrogen pressure (550 psi), and even heating failed. Finally, we found that directed hydrogenation could be achieved using carboxylic acid 19, obtained by methanolysis of anhydride 18a, affording a 90% yield of the reduced product 20 under optimized conditions (30 mol% Crabtree catalyst, 0.1 M substrate concentration) (Scheme 5). Reduction of catalyst loading or increase in olefin concentration resulted in incomplete hydrogenation.
Scheme 5.

Elaboration of jujubogenin C-ring from anhydride 18a to generate α-diazoketone 29.
Successful α-hydrogenation of the double bond in carboxylic acid 19 established all of the stereocenters present in the C-ring of jujubogenin. Subsequently, we turned our attention to functional group transformations of the three carbonyl groups in 20. Thus, carboxylic acid 20 was first methylated to give trimethylester 21, which was then converted to monoaldehyde 22 by reduction of the sterically more accessible C17 methyl ester using DIBAL, followed by Dess–Martin oxidation. Olefination of the aldehyde followed by reduction of the remaining ester groups afforded diol 23. Several oxidants were then screened for selective monooxidation of the diol to equatorial aldehyde 24. While Dess–Martin periodinane gave high selectivity for oxidation of the axially disposed alcohol, other oxidants (e.g., SO3·pyr, TEMPO, TPAP, PCC) provided large amounts of overoxidized dialdehyde product. Ultimately, we found that IBX oxidation with sonication provided a 1.2:1 mixture of monoaldehydes, favoring the desired equatorial aldehyde 24. The undesired axial aldehyde, as well as small amount of dialdehyde, was then reduced to diol 23 and recycled, affording the desired aldehyde 24 in 74% yield based on recovered starting material.
Aldehyde 24 is a versatile intermediate, as a number of protecting groups can be put on the C30 primary alcohol at this stage of the synthesis. We found that diethylisopropylsilyl (DEIPS) was the optimal protecting group for masking the primary C30 alcohol. In later stages of the synthesis, a less-hindered triethylsilyl (TES) group was partially cleaved under strongly basic conditions, while a more-hindered tert-butyldimethylsilyl (TBS) group required harsh deprotection conditions that were not selective and resulted in the cleavage of the C3-TBS protecting group as well. Addition of the methyl lithium to the DEIPS-protected aldehyde derived from 24 followed by oxidation of the resulting secondary alcohol yielded methyl ketone 25, which was then advanced to ketoaldehyde 26 through ozonolysis and reductive workup. Horner–Wadsworth–Emmons olefination of aldehyde 26 with phosphonate 27 gave triene 28 as a 2:1 mixture of Z- and E-enol ethers. Subsequent deformylative diazotransfer then delivered the α-diazoketone 29, setting the stage for assembly of the DEF-ring system.
2.6. Tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reaction of diazoketone 29
Next, we explored the pivotal tandem Wolff rearrangement–ketene hetero-Diels–Alder reaction of α-diazoketone 29. Unfortunately, under the conditions applied in our model studies above (AgOTf, Et3N) (Scheme 3), no reaction was observed. It has been documented that silver-promoted Wolff rearrangement with sterically-hindered diazoketones is often sluggish.57 On the other hand, irradiation of the α-diazoketone 29 at 310 nm in C6D6 delivered the desired bis(enol ether) 31 based on 1H-NMR analysis of the crude reaction mixture (Scheme 6). Product formation was clearly supported by comparison of this spectrum to that of the previously synthesized model bis(enol ether) 15 (Scheme 3).
Scheme 6.

Tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reaction of α-diazoketone 29
Unfortunately, this cycloadduct 31 readily decomposed upon attempted purification on silica gel deactivated with Et3N, or basic or neutral alumina. The major product isolated after purification appeared to be triene 32, presumably arising from fragmentation of the F-ring. However, complete characterization of 32 was difficult as a result of keto–enol tautomerization, which led to various stereoisomers.
The difference in stability between the original model bis(enol ether) 15 and the elaborated bis(enol ether) 31 was striking. Careful analysis of the structural features of 15 and 31 revealed a key difference. We noted that, compared to the model system product 15, the elaborated product 31 bore an additional exocyclic olefin adjacent to the labile C23–O bond (Figure 2). Thus, we hypothesized that donation of electrons from the π-orbital of the additional olefin substituent into the σ*-antibonding orbital of the C23–O bond resulted in weakening of this bond in 31 compared to 15, with this π to σ* donation promoting the observed fragmentation.
Figure 2.

Comparison of tandem Wolff rearrangement–ketene hetero-Diels–Alder products 15 and 31.
Thus, we hypothesized that masking the C24–C25 double bond, or introducing it at a later stage in the synthesis, should improve the stability of the corresponding cycloadduct. Toward this we designed two new α-diazoketones, 33 and 35 (Scheme 7), the synthesis of which involved a similar process to that of previously prepared diazoketone 29 (see Supporting Information for details). To our delight, irradiation of diazoketones 33 and 35 gave the respective bis(enol ethers) 34 and 36 in good yields. In contrast to 31, both products were stable to the purification using basic alumina, thus supporting our hypothesis that the main source of relative instability of 31 compared to the model product 15 was the olefin substituent at C23. Of note, the stereochemical configuration of the C23 substituent in 36 remained unassigned at this stage.
Scheme 7.

Tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reactions of alternative α-diazoketone substrates 33 and 35
2.7. Formation of the E-ring via C16 ketalization
Successful application of the tandem Wolff rearrangement–ketene hetero-Diels–Alder reaction furnished the ABCDF-ring system of jujubogenin (1). We then focused our attention to formation of the C16 ketal which would complete the hexacyclic core of jujubogenin (1). As a model system, we first advanced the C23-unsubstituted cycloadduct 34. Selective removal of the DEIPS protecting group set the stage for acid-catalyzed ketalization (Scheme 8). Treatment of alcohol 37 with Amberlyst-15 in wet CH2Cl2 resulted in formation of the desired keto-ketal 38. Addition of MeLi at −78 °C proceeded with the desired attack from the exo-face to deliver the complete hexacyclic core of jujubogenin 39.
Scheme 8.

Completion of the hexacyclic core of jujubogenin via formation of the C16 ketal.
Encouraged by the successful conversion of the C23-unsubstituted enol ether 34 to hexacycle 39, we turned our attention to the C23-substituted enol ether 36. While TBAF deprotection of the C30-DEIPS group proceeded smoothly, treatment of the resulting alcohol with Amberlyst-15 resulted in formation of lactone 42 and vinylogous ester 43, with only small amount of the desired ketal 41 (Scheme 9).
Scheme 9.

Attempted C16-ketalization of C23-substituted enol ether substrate 36.
Mechanistic understanding of the formation of lactone 42 lies in the innate reactivity of the jujubogenin triterpene under acidic conditions.22 Initial isolation reports documented that treatment of jujubogenin (1) with acid induced ring fragmentation, yielding ebelin lactone (45) (Scheme 10a). We hypothesized that, in case of C23-substituted enol ether 36, after initial cyclization and hydrolysis of the methyl enol ether, the desired ketone 41 is formed, but then undergoes further reaction with acid to generate oxocarbenium 46, which undergoes a similar fragmentation to that of jujubogenin (1), yielding lactone 42 (Scheme 10b). Indeed, when isolated ketone 41 was treated with Amberlyst-15 in CH2Cl2, lactone 42 was produced efficiently. On the other hand, the other byproduct, vinylogous ester 43, can be generated through C23–O bond fragmentation of the oxocarbenium generated by protonation of the C15–C16 enol ether (not shown).
Scheme 10.

(a) Acid-catalyzed fragmentation of jujubogenin (1). (b) Proposed acid-catalyzed fragmentation of ketone 41.
Based on this mechanistic analysis, we posited that the fragmentation leading to vinylogous ester 43 would most likely happen under acidic conditions, regardless of the strength of the acid. However, the stability of the desired product 41 under the reaction conditions could depend on the strength of the acid. Accordingly, we investigated weaker acids and found that dichloroacetic acid was the optimal acid for this transformation, delivering ketone product 41 in 63% isolated yield (Scheme 11).
Scheme 11.

Sucessful C16-ketalization of C23-substituted enol ether substrate 36 with dichloroacetic acid (DCA).
Stereochemical assignment of ketone 41 was then conducted using extensive 2D NMR studies. The previously published X-ray structure of jujubogenin (1)58 revealed that the F-ring is in a chair conformation, putting the bulkier C21 (methyl) and C23-isobutenyl group in equatorial positions (Figure 3). Thus, if ketone 41 had the same stereochemistry, no nOe should be observed between C23-H and C17-H due to their 1,4-diaxial relationship. However, after complete structural assignment, nOe analysis indicated that these two protons reside on the same face of the F-ring. In conjunction with the lack of nOe between C17-H and bridging C15-methylene, or between the C24-methylene and bridging C15-methylene, these data were consistent with a the C23 carbon of ketone 41 having the opposite stereochemical configuration to that in jujubogenin (1). Furthermore, these nOe analyses indicated that the F-ring in ketone 41 adopts a boat conformation (41b) to avoid an unfavorable syn-pentane interaction between the axial C24-methylene and bridging C15-methylene groups in the corresponding chair conformation (41a).
Figure 3.

Stereochemical assignment of ketone 41.
This C23 stereochemical outcome, while undesired, shed light on the mechanism of the ketene hetero-Diels–Alder reaction (Scheme 12). In a concerted, pericyclic reaction mechanism, the stereochemistry observed in ketone 41 would be predicted. However, in a stepwise reaction mechanism, initial addition of the enol ether to the ketene would give zwitterion 49, which could then undergo 6-endo-trig cyclization to either face of the C22–C23 double bond to provide a mixture of C23 epimers. In our retrosynthetic analysis, we assumed that the reaction would proceed through the stepwise pathway, as both the enol ether portion of the diene and carbonyl of the ketene are highly polarized and such Mukaiyama-aldol/oxy-conjugate addition reactions are well known.59 Unfortunately, formation of the bis(enol ether) 36 indicates that the ketene hetero-Diels–Alder reaction most likely proceeds through concerted mechanism.
Scheme 12.

Predicted stereochemical outcomes at C23 depending on mechanism of the ketene hetero-Diels–Alder reaction.
Although the C23 stereochemistry was incorrect, we decided to advance ketone 41 forward, anticipating that it might be possible to correct this stereochemical configuration at a later stage in the synthesis. Thus, addition of methyl lithium followed by removal of the benzyl protecting group delivered diol 51 in good overall yield (Scheme 13). By exploiting the difference in steric hindrance between the C20 and C25 tertiary alcohols, we then selectively effected elimination of the C25 alcohol using a bulky dehydrating agent, Martin sulfurane, which gave a 1:1 mixture of olefins 52 and 53. Olefin 52 has all of the structural features of jujubogenin (in C3-O-TBS protected form) except for the stereochemical configuration at C23.
Scheme 13.

Synthesis of C3-O-TBS protected 23-epi-jujubogenin (52)
2.8. Attempts to correct the C23 stereochemistry
With olefin 52 in hand, we sought to epimerize the C23 stereogenic center to finish the total synthesis of jujubogenin (1). Our plan involved oxidative cleavage of the olefin to form aldehyde 54, which then would undergo epimerization under thermodynamically-controlled conditions (anticipating that the energy difference of ~5.5 kcal/mol60 between chair and boat comformations would be sufficient to drive the equilibrium toward the desired aldehyde) to deliver the epimeric aldehyde 55 (Scheme 14). Unfortunately, ozonolysis of the olefin 52 followed by reductive workup led to hemiacetal 56 instead of the desired aldehyde 54. Attempts to epimerize the hemiacetal 56 under basic conditions were not fruitful, leading to decomposition under forcing conditions.
Scheme 14.

Attemped epimerization of the C23 stereocenter via aldehyde 54.
3. Conclusion
In conclusion, we have developed a synthetic approach toward the total synthesis of jujubogenin (1) featuring two pivotal Diels–Alder reactions. First, we developed a sterically-demanding Diels–Alder reaction using a model system, which was then applied successfully to the synthesis of the C-ring of jujubogenin. The product of this reaction was then advanced to the diazoketones 28, 33, and 35 via several key transformations including stereoselective hydrogenation using Crabtree’s catalyst, chemoselective carboxylic acid reduction, Horner–Wadsworth–Emmons olefination, and deformylative diazotransfer. Subsequently, we developed a novel tandem Wolff rearrangement–intramolecular ketene hetero-Diels–Alder reaction using a model study and successfully applied it to the construction of the DF-ring system. Finally, we developed a ketalization reaction to close the E-ring, completing the hexacyclic ring system of jujubogenin. This required us to avoid several perilous decomposition pathways via judicious choice of acid catalyst and masking of the pendant C22–C23 olefin. Although we were unable to correct the undesired stereochemical configuration at C23, this overall synthetic strategy provided expedient access to the complete hexacyclic triterpene core of jujubogenin (52), and future synthetic efforts may be able to leverage the mechanistic insights derived herein to access jujubogenin (1), by either epimerization at C23 or direct installation of the desired configuration at the stage of the ketene hetero Diels–Alder reaction. This will enable detailed structure–activity relationship studies of the jujuboside family of natural products.
4. Experimental Section
4.1. General Materials and Methods
Reactions were performed in flame-dried sealed tubes or modified Schlenk (Kjeldahl shape) flasks fitted with a glass stopper under a positive pressure of argon, unless otherwise noted. Air- and moisture-sensitive liquids and solutions were transferred via syringe. The appropriate starting materials and reagents were dried via azeotropic removal of water with toluene. Molecular sieves were activated at 350 °C and were crushed immediately prior to use, then flame-dried under vacuum. Organic solutions were concentrated by rotary evaporation below 30 °C. Flash column chromatography was performed employing 230–400 mesh silica gel. Thin-layer chromatography was performed using glass plates pre-coated to a depth of 0.25 mm with 230–400 mesh silica gel impregnated with a fluorescent indicator (254 nm) and visualized under UV light (254 and 360 nm), or stained with Ceric Ammonium Molybdate in conc. H2SO4.
Dichloromethane, tetrahydrofuran, diethyl ether, and toluene were purified by passage through two packed columns of neutral alumina under an argon atmosphere.61 Methanol was distilled from magnesium at 760 Torr. Pyridine and trimethylamine were freshly distilled from CaH2 prior to use. All other chemicals were obtained from commercial vendors and were used without further purification unless otherwise noted.
Automated flash chromatography was performed with an Isco Combiflash medium-pressure liquid chromatography with Redisep silica columns (47–60 μm). Infrared (IR) spectra were obtained using a Perkin Elmer Spectrum BX or a Bruker Tensor 27 spectrophotometer. Data are presented as the frequency of absorption (cm−1). Proton and carbon-13 nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded on a Bruker Avance III instrument; chemical shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and are referenced to residual proton in the NMR solvent (d-chloroform: δ 7.26 for 1H NMR, δ 77.16 for 13C NMR; d6-benzene: δ 7.16 for 1H NMR, δ 128.06 for 13C NMR; d4-methanol: δ 3.31 for 1H NMR, δ 49.00 for 13C NMR; d3-acetonitrile: δ 1.94 for 1H NMR, δ 1.32 for 13C NMR; deuterium oxide: δ 4.79 for 1H NMR). Data are presented as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet and/or multiple resonances), coupling constant in Hertz (Hz), integration, assignment.
4.2. Model bis(enol ether) 15
To a solution of diazoketone 13 (5.8 mg, 0.021 mmol, 1.0 equiv) in THF (250 μl) at 45 °C, was added Et3N (3.4 μl, 0.021 mmol, 1.0 equiv) followed by AgOTf (1.1 mg, 0.0042 mmol, 0.20 equiv). The reaction mixture was stirred for 40 min at 45 °C, then cooled to rt and filtered through a pad of SiO2. The SiO2 layer was washed with Et2O five times (10 mL total). The combined THF and Et2O fractions were concentrated and analyzed by 1H-NMR, which indicated complete conversion and formation of the desired product (50% yield). The analytically pure bis(enol ether) 15 was obtained by flash column chromatography using basic alumina (hexanes:EtOAc 25:1). TLC Rf 0.14 (hexanes:EtOAc 25:1); FTIR (NaCl film) 2959, 1452, 1240, 1034, 835, 716; 1H NMR (600 MHz, C6D6) δ 5.51 (d, J = 2.0 Hz, 1H), 4.42 (dt, J = 13.9, 2.5 Hz, 1H), 4.33 (dt, J = 13.8, 2.9 Hz, 1H), 4.09 (td, J = 3.3, 1.9 Hz, 1H), 3.53 (d, J = 8.4 Hz, 1H), 3.23 (d, J = 8.6 Hz, 2H), 3.11 (s, 3H), 3.09 (s, 3H), 2.23 (ddd, J = 12.8, 4.7, 2.0 Hz, 1H), 2.17 (ddt, J = 12.7, 4.7, 2.5 Hz, 1H), 1.92 (ddd, J = 13.6, 11.3, 2.8 Hz, 1H), 1.79 – 1.70 (m, 1H), 1.62 – 1.32 (m, 5H), 1.28 (td, J = 12.9, 4.6 Hz, 1H); 13C NMR (151 MHz, C6D6) δ 157.69, 157.14, 110.77, 90.07, 74.97, 67.27, 59.31, 54.40, 53.94, 46.92, 43.18, 33.21, 27.04, 24.59, 21.38; HRMS (ESI) m/z Calcd for C15H23O3 [M+H]+ 251.1647, found 251.1650.
4.3. Allylic alcohol 17
The known ketone 16 (5.08 g, 15.0 mmol, 1.0 equiv), prepared as previously described,62 was azeotropically dried with PhMe (15 mL), dissolved in THF (150 mL), and cooled to −78 °C. Vinylmagnesium bromide (1 M in THF, 37.5 mL, 37.5 mmol, 2.5 equiv) was added via syringe. The cooling bath was removed and the solution was stirred for 10 h then quenched with satd aq NH4Cl (200 mL) and H2O (100 mL). The aqueous layer was extracted with EtOAc (3 x 100 mL), and the combined organic extracts were dried over MgSO4, filtered, and concentrated to give crude allylic alcohol 17, which was carried to the next step without further purification.
4.4. Diene 8
Crude allylic alcohol 17 (1 equiv.) was azeotropically dried with PhMe (15 mL), dissolved in pyridine (110 mL) and cooled to −30 °C. SOCl2 (1.32 mL, 18 mmol, 1.2 equiv.) was added via syringe. The solution was stirred for 4 h at this temperature, diluted with pentane (1.2 L) and washed sequentially with H2O (3 x 200) and satd aq CuSO4 (3 x 300 mL). The organic layer was dried over MgSO4, filtered, and concentrated to give crude diene 8. Purification by flash column chromatography (hexanes) afforded diene 8 (3.56 g, 68% over 2 steps) as a white solid. TLC Rf 0.60 (hexanes); [α]D19 +38.5 (c 1.00, C6H6); FTIR (NaCl film) 2934, 2855, 1472, 1361, 1254, 1104, 1070, 1004, 934, 915, 883, 835, 772, 665; 1H NMR (600 MHz, CDCl3) δ 6.07 (dddt, J = 17.7, 11.3, 2.2, 1.2 Hz, 1H), 5.20 (dd, J = 11.2, 2.8 Hz, 1H), 4.87 (dd, J = 17.6, 2.7 Hz, 1H), 3.17 (dd, J = 11.3, 4.5 Hz, 1H), 2.07 – 2.00 (m, 2H), 1.68 – 1.41 (m, 5H), 1.61 (s, 3H), 1.17 (td, J = 13.0, 3.3 Hz, 1H), 1.07 (dd, J = 12.6, 2.0 Hz, 1H), 0.96 (s, 3H), 0.89 (s, 3H), 0.86 (s, 9H), 0.74 (s, 3H), −0.00 (s, 3H), −0.01 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 141.62, 135.10, 127.18, 118.60, 79.50, 77.37, 77.16, 76.95, 50.64, 39.63, 37.44, 36.33, 33.83, 28.59, 28.30, 26.08, 21.11, 20.18, 18.91, 18.28, 16.05, −3.61, −4.79.
4.5. Anhydride 18a
Diene 8 (1.60 g, 10.3 mmol, 1.0 equiv) was azeotropically dried with PhMe (5 mL) and dissolved in PhH. Dienophile 7 (1.90 g, 14.6 mmol, 1.4 equiv) was added as a solid and the resulting bright yellow solution was stirred for 24 h, at which point the bright yellow color of the solution had disappeared. The reaction mixture was directly purified by flash column chromatography as described below. A column (22 cm length, 5.5 cm width of dry silica) was slurry packed with the aid of hexanes containing 2% AcOH. Then all of the solvent was removed using N2 and the column was repacked with hexanes containing 2% AcOH. The reaction mixture was diluted with hexanes, loaded directly onto the column, and eluted with hexanes:PhH:AcOH (50:1:1) to yield anhydride 18a (646 mg, 54%) and its diastereomer 18b (275 mg, 23%) as white solids. 18a: TLC Rf 0.21 (hexanes:PhH:AcOH 50:1:1); [α]D20 +69.7 (c 1.00, C6H6); FTIR (NaCl film) 2936, 2856, 1797, 1728, 1462, 1388, 1249, 1168, 1094, 928, 885, 836, 773, 665; 1H NMR (600 MHz, CDCl3) δ 5.52 (dd, J = 7.1, 2.4 Hz, 1H), 3.80 (s, 3H), 3.70 (dd, J = 11.4, 1.6 Hz, 1H), 3.21 (dd, J = 11.1, 4.9 Hz, 1H), 2.84 (td, J = 13.2, 5.1 Hz, 1H), 2.74 (ddd, J = 18.0, 11.5, 2.4 Hz, 1H), 2.47 (ddd, J = 17.9, 7.1, 1.6 Hz, 1H), 1.86 (dt, J = 13.1, 3.6 Hz, 1H), 1.72 (ddt, J = 13.8, 4.9, 2.2 Hz, 1H), 1.65 – 1.46 (m, 4H), 1.31 – 1.22 (m, 2H), 1.29 (s, 3H), 1.13 (s, 3H), 0.97 (s, 3H), 0.88 (s, 9H), 0.76 (s, 3H), 0.04 (s, 3H), 0.04 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 172.62, 169.12, 167.82, 158.19, 114.91, 78.64, 77.37, 77.16, 76.95, 66.71, 53.69, 48.63, 44.09, 39.93, 39.80, 39.54, 36.71, 35.14, 28.76, 28.14, 26.04, 25.62, 25.37, 22.60, 18.23, 16.37, −3.65, −4.74; HRMS (ESI) m/z Calcd for C28H45O6Si [M+H]+ 505.2985, found 505.2973.
18b: TLC Rf 0.20 (hexanes:PhH:AcOH 50:1:1); [α]D20 −28.1 (c 1.00, C6H6); FTIR (NaCl film) 2935, 1785, 1733, 1462, 1251, 1174, 1098, 1075, 956, 884, 836, 773, 665; 1H NMR (600 MHz, CDCl3) δ 5.59 (dd, J = 7.6, 2.6 Hz, 1H), 3.89 (dd, J = 9.6, 1.6 Hz, 1H), 3.82 (s, 3H), 3.12 (dd, J = 11.4, 4.2 Hz, 1H), 2.88 (dt, J = 14.2, 10.0 Hz, 1H), 2.74 (ddd, J = 17.3, 9.6, 2.6 Hz, 1H), 2.59 (ddd, J = 17.2, 7.6, 1.6 Hz, 1H), 1.82 – 1.62 (m, 5H), 1.59 – 1.52 (m, 1H), 1.33 – 1.22 (m, 2H), 1.20 (s, 3H), 1.03 (s, 3H), 0.88 (s, 9H), 0.88 (s, 3H), 0.84 (s, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 173.01, 167.81, 167.53, 157.57, 115.75, 79.33, 77.37, 77.16, 76.95, 65.99, 53.72, 44.90, 43.30, 41.47, 40.47, 39.07, 38.15, 28.14, 27.88, 26.58, 26.03, 26.01, 25.01, 23.72, 22.03, 18.23, 17.33, 15.32, −3.75, −4.78; HRMS (ESI) m/z Calcd for C28H45O6Si [M+H]+ 505.2985, found 505.2966.
4.6. Monocarboxylic acid 19
A solution of anhydride 18a (4.00 g, 7.93 mmol, 1.0 equiv) was stirred at rt in MeOH/PhMe (250 ml/250 mL). After 24 h, the reaction mixture was concentrated to give monocarboxylic acid 19 (4.25 g, 100%). Residual PhMe was removed azeotropically with MeOH. (Note that it is important to remove all residual solvents at this stage as residual PhMe or MeOH deactivates Crabree’s catalyst in the subsequent hydrogenation step.) TLC Rf 0.41 (hexanes:EtOAc 3:2); [α]D20 +37.0 (c 1.00, C6H6) FTIR (NaCl film) 2951, 1790, 1738, 1436, 1387, 1249, 1109, 1077, 994, 925, 885, 836, 774, 738, 666; 1H NMR (600 MHz, CDCl3) δ 11.70 (bs, 1H), 5.45 (t, J = 3.9 Hz, 1H), 3.79 (s, 3H), 3.72 (s, 3H), 3.63 (dd, J = 10.9, 8.2 Hz, 1H), 3.16 (dd, J = 11.3, 4.7 Hz, 1H), 2.68 (ddd, J = 18.3, 10.9, 3.5 Hz, 1H), 2.52 (ddd, J = 18.3, 8.3, 4.1 Hz, 1H), 2.07 (td, J = 12.9, 4.5 Hz, 1H), 1.80 (dt, J = 12.9, 3.6 Hz, 1H), 1.69 – 1.48 (m, 5H), 1.38 (td, J = 13.4, 3.9 Hz, 1H), 1.25 (d, J = 1.0 Hz, 3H), 1.14 (s, 3H), 1.04 (dd, J = 12.0, 1.9 Hz, 1H), 0.89 (s, 4H), 0.88 (s, 9H), 0.74 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 176.06, 174.55, 169.75, 151.16, 116.91, 78.69, 77.37, 77.16, 76.95, 63.59, 53.17, 52.72, 50.35, 43.13, 41.47, 39.68, 39.61, 37.74, 35.90, 28.45, 28.37, 27.49, 27.03, 26.05, 25.48, 18.46, 18.23, 16.06, −3.67, −4.80; HRMS (ESI) m/z Calcd for C29H49O7Si [M+H]+ 559.3067, found 559.3069.
4.7. Saturated monocarboxylic acid 20
A flask containing alkene monocarboxylic acid 19 (2.700 g, 5.030 mmol, 1.0 equiv) and Crabtree’s catalyst (1.333 g, 1.660 mmol, 0.3 equiv) was evacuated and backfilled with H2 (balloon). CH2Cl2 (50 mL) was added and the reaction mixture was stirred for 6 h at rt, then concentrated. Purification by column chromatography (hexanes: EtOAc 5:1 with 2% AcOH) yielded the saturated carboxylic acid 20 (2.4351 g, 90%). TLC Rf 0.41 (hexanes:EtOAc 3:2); [α]D20 +4.8 (c 1.00, C6H6); FTIR (NaCl film) 2953, 2855, 1728, 1681, 1438, 1391, 1286, 1246, 1223, 1100, 1015, 884, 836, 774, 735, 666; 1H NMR (600 MHz, CDCl3) δ 12.25 (s, 1H), 3.77 (s, 3H), 3.66 (s, 3H), 3.47 (dd, J = 12.9, 5.6 Hz, 1H), 3.14 (dd, J = 11.4, 4.6 Hz, 1H), 2.45 (qd, J = 13.2, 4.6 Hz, 1H), 2.23 – 2.15 (m, 1H), 1.78 (dt, J = 12.9, 3.2 Hz, 1H), 1.72 – 1.63 (m, 2H), 1.63 – 1.51 (m, 4H), 1.51 – 1.45 (m, 1H), 1.36 – 1.26 (m, 2H), 1.01 (td, J = 13.6, 4.0 Hz, 1H), 0.97 (s, 3H), 0.87 (s, 9H), 0.85 (s, 6H), 0.77 (dd, J = 12.1, 1.9 Hz, 1H), 0.70 (s, 3H), 0.02 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 177.34, 175.18, 170.33, 78.92, 77.37, 77.16, 76.95, 64.72, 54.70, 53.51, 52.45, 49.94, 44.62, 42.64, 39.44, 38.64, 37.89, 37.67, 28.25, 27.78, 26.05, 25.68, 20.02, 18.93, 18.40, 18.24, 16.97, 16.07, −3.68, −4.82; HRMS (ESI) m/z Calcd for C29H51O7Si [M+H]+ 539.3404, found 539.3391.
4.8. Triester 21
To the solution of monocarboxylic acid 20 (2.71 g, 5.03 mmol, 1.0 equiv) in DMF (25 mL) was added KHCO3 (1.51 g, 15.1 mmol, 3.0 equiv) and MeI (3.57 g, 25.1 mmol, 5.0 equiv). The reaction mixture was stirred at rt for 16 h then diluted with Et2O (300 mL), and washed with water (50 mL), 1 N HCl (50 mL), and satd aq NaHCO3 (50 mL). The Et2O layer was dried over MgSO4, filtered, and concentrated. Purification by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 80 g column, 1–70% EtOAc in hexanes) yielded triester 21 (2.47 g, 89%) as a white solid. TLC Rf 0.13 (hexanes:EtOAc 10:1); [α]D20 +1.62 (c 1.00, C6H6); FTIR (NaCl film) 2951, 1738, 1434, 1390, 1361, 1257, 1197, 1171, 1101, 1071, 884, 835, 773, 737; 1H NMR (600 MHz, CDCl3) δ 3.76 (s, 3H), 3.72 (s, 3H), 3.67 (s, 3H), 3.19 (dd, J = 11.4, 4.6 Hz, 1H), 3.07 (dd, J = 13.5, 4.4 Hz, 1H), 2.44 (dt, J = 12.9, 3.0 Hz, 1H), 2.09 (dd, J = 12.7, 2.5 Hz, 1H), 2.04 (td, J = 13.3, 4.3 Hz, 1H), 1.95 (dtd, J = 12.9, 4.2, 2.7 Hz, 1H), 1.67 (ddt, J = 12.8, 7.5, 3.8 Hz, 2H), 1.63 – 1.52 (m, 2H), 1.52 – 1.44 (m, 1H), 1.41 – 1.21 (m, 4H), 1.03 (td, J = 13.3, 4.0 Hz, 1H), 0.94 (s, 3H), 0.87 (s, 9H), 0.86 (s, 3H), 0.83 (s, 3H), 0.80 (dd, J = 11.9, 2.2 Hz, 1H), 0.71 (s, 3H), 0.02 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 175.60, 172.22, 170.62, 79.00, 77.37, 77.16, 76.95, 65.96, 54.59, 52.40, 52.33, 52.04, 50.28, 46.44, 42.72, 39.43, 38.90, 37.69, 37.34, 28.24, 27.90, 26.54, 26.06, 19.96, 18.50, 18.25, 17.64, 16.82, 15.98, −3.66, −4.78; HRMS (ESI) m/z Calcd for C30H52O7Si [M+H]+ 553.3561, found 553.3558.
4.9. Monoaldehyde 22
To the solution of triester 21 (2.00 g, 3.62 mmol, 1.0 equiv) in PhMe (50 mL) cooled to −78 °C was added DIBAL (1 M in PhMe, 7.4 mL, 7.4 mmol, 2.1 equiv). The reaction mixture was stirred at −78 °C for 30 min, then at −50 °C for 30 min, and quenched by adding EtOAc (10 mL). Rochelle’s salt solution (20%, 100 mL) was added and the mixture stirred for 20 min. The aqueous layer was extracted with EtOAc (3 x 40 mL). The combined EtOAc extracts were dried over MgSO4, filtered, and concentrated to give the corresponding crude alcohol, which was used in the subsequent oxidation step without further purification. 1H NMR (600 MHz, CDCl3) δ 3.82 – 3.74 (m, 1H), 3.71 (s, 3H), 3.64 (s, 3H), 3.21 (dd, J = 11.5, 4.6 Hz, 1H), 3.14 (dt, J = 11.6, 6.1 Hz, 1H), 2.40 (ddt, J = 13.1, 7.1, 3.5 Hz, 1H), 2.33 (dd, J = 12.6, 2.7 Hz, 1H), 2.04 (dq, J = 13.1, 3.4 Hz, 1H), 1.84 (td, J = 13.1, 4.2 Hz, 1H), 1.73 (dt, J = 13.1, 3.4 Hz, 1H), 1.68 (dt, J = 13.0, 3.6 Hz, 1H), 1.65 – 1.52 (m, 5H), 1.49 (dq, J = 13.6, 3.9 Hz, 1H), 1.36 (qd, J = 13.0, 3.5 Hz, 1H), 1.27 (qd, J = 13.0, 3.7 Hz, 1H), 1.21 – 1.12 (m, 1H), 1.03 (td, J = 13.4, 3.8 Hz, 1H), 0.92 (s, 4H), 0.88 (d, J = 3.5 Hz, 12H), 0.83 (s, 3H), 0.71 (s, 3H), 0.02 (s, 5H), 0.02 (s, 4H); 13C NMR (151 MHz, CDCl3) δ 171.28, 170.46, 79.19, 77.37, 77.16, 76.95, 66.66, 66.51, 54.21, 51.90, 51.63, 50.64, 42.32, 41.70, 39.47, 38.98, 37.70, 36.58, 28.27, 27.94, 27.03, 26.07, 26.05, 20.30, 18.84, 18.42, 18.26, 16.91, 15.95, −3.64, −4.77.
To the solution of the crude alcohol above (1.634 g, 3.114 mmol, 1.0 equiv) in CH2Cl2 (100 mL) at rt was added NaHCO3 (2.62 g, 31.1 mmol, 10 equiv) followed by the Dess–Martin periodinane (1.32 g, 3.11 mmol, 1.0 equiv). The reaction mixture was stirred for 30 min, then quenched by adding a solution of Na2S2O3 (20% w/w, 50 mL) and satd aq NaHCO3 (50 mL) and stirring for 40 min. The aqueous layer was extracted with CH2Cl2 (3 x 80 mL). The combined CH2Cl2 extracts were dried over Na2SO4, filtered, and concentrated. Purification by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 40 g column, 1–60% EtOAc in hexanes) yielded monoaldehyde 22 (1.44 g, 79% over 2 steps) as a white solid. TLC Rf 0.13 (hexanes:EtOAc 10:1); [α]D20 +18.0 (c 1.00, C6H6); FTIR (NaCl film) 2952, 1721, 1433, 1390, 1253, 1100, 1070, 884, 835, 773; 1H NMR (600 MHz, CDCl3) δ 9.65 (s, 1H), 3.77 (s, 3H), 3.63 (s, 3H), 3.21 (dd, J = 11.4, 4.6 Hz, 1H), 3.06 (dd, J = 13.1, 3.7 Hz, 1H), 2.25 (tdd, J = 13.4, 4.8, 1.4 Hz, 1H), 2.15 – 2.08 (m, 1H), 2.00 (dd, J = 12.8, 3.0 Hz, 1H), 1.68 – 1.52 (m, 6H), 1.49 (dq, J = 13.6, 3.8 Hz, 1H), 1.45 – 1.36 (m, 1H), 1.34 – 1.24 (m, 1H), 1.13 (qd, J = 13.5, 4.3 Hz, 1H), 1.02 (d, J = 1.0 Hz, 3H), 1.01 – 0.95 (m, 2H), 0.90 (s, 3H), 0.88 (s, 9H), 0.84 (d, J = 0.9 Hz, 3H), 0.72 (s, 3H), 0.02 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 201.80, 170.10, 169.54, 79.10, 77.37, 77.16, 76.95, 65.84, 54.05, 52.45, 52.06, 50.55, 50.29, 42.12, 39.48, 38.77, 37.60, 35.74, 28.36, 27.84, 26.06, 22.82, 19.54, 18.84, 18.44, 18.25, 16.60, 15.94, −3.65, −4.77; HRMS (ESI) m/z Calcd for C29H51O6Si [M+H]+ 523.3455, found 523.3438.
4.10. Olefin diol 23
To the slurry of Ph3PMeBr (1.972 g, 5.520 mmol, 2.0 equiv) in THF (20 mL) cooled to 0 °C was added KHMDS (0.826 g, 4.41 mmol, 1.5 equiv). The cooling bath was removed and the orange solution was stirred for 30 min at rt. In a separate flask, aldehyde 22 (1.443 g, 2.760 mmol, 1 equiv) was azeotropically dried with toluene (5 mL), then dissolved in THF (20 mL). The phosphorous ylide solution was then added to the aldehyde solution via cannula. The resulting solution was stirred for 15 h at rt, at which time TLC analysis indicated complete conversion. The solution was poured into ice cold 1 N HCl (100 mL) and extracted with Et2O (3 x 80 mL). The combined Et2O extracts were washed with water (100 mL), dried over MgSO4, filtered, and concentrated. Purification by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 40 g column, 1–60% EtOAc in hexanes) yielded the corresponding olefin (1.41 g, 98%) as a white solid. TLC Rf 0.21 (hexanes:EtOAc 10:1); [α]D20 +13.7 (c 0.84, C6H6); FTIR (NaCl film) 2949, 1722, 1461, 1389, 1243, 1099, 1071, 884, 835, 773; 1H NMR (600 MHz, CDCl3) δ 5.84 (ddd, J = 17.0, 10.6, 6.0 Hz, 1H), 4.99 (dt, J = 17.3, 1.7 Hz, 1H), 4.95 (dt, J = 10.7, 1.5 Hz, 1H), 3.71 (s, 3H), 3.59 (s, 3H), 3.21 (dd, J = 11.5, 4.6 Hz, 1H), 3.10 – 3.00 (m, 1H), 2.38 (dd, J = 12.6, 2.6 Hz, 1H), 1.89 (dt, J = 13.1, 3.4 Hz, 1H), 1.80 – 1.65 (m, 3H), 1.65 – 1.53 (m, 4H), 1.49 (dq, J = 13.6, 3.9 Hz, 1H), 1.46 – 1.22 (m, 4H), 1.04 (td, J = 13.3, 3.8 Hz, 1H), 0.94 (d, J = 0.9 Hz, 3H), 0.92 (dd, J = 12.4, 2.1 Hz, 1H), 0.88 (s, 12H), 0.84 (s, 3H), 0.71 (s, 3H), 0.02 (s, 3H), 0.02 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.81, 170.38, 140.78, 114.25, 79.20, 77.37, 77.16, 76.95, 68.68, 54.34, 51.65, 51.01, 50.27, 42.03, 41.07, 39.47, 38.98, 37.67, 37.17, 28.24, 27.96, 27.54, 26.08, 26.06, 20.13, 18.80, 18.35, 18.27, 17.02, 15.96, −3.64, −4.77; HRMS (ESI) m/z Calcd for C30H53O5Si [M+H]+ 521.3656, found 521.3662.
To the solution of olefin above (1.59 g, 3.05 mmol, 1.0 equiv) in Et2O (50 mL) at 0 °C was added LiAlH4 (0.23 g, 6.1 mmol, 2.0 equiv). The reaction mixture was allowed to warm to rt and stirred for 15 h. The reaction was cooled to 0 °C and quenched by sequential, slow addition of water (0.23 mL) then a 15% solution of NaOH (0.23 mL), then water (0.69 mL), and stirred for 2 h. The solids were removed by filtration and washed with Et2O (2x10 mL). The combined Et2O layers were concentrated to give diol 23 (1.36 g, 96%) as a white solid, which was analytically pure and used without further purification. TLC Rf 0.23 (hexanes:EtOAc 5:1); [α]D20 +10.3 (c 1.00, C6H6); FTIR (NaCl film) 3356, 2928, 1462, 1387, 1255, 1046, 907, 883, 835, 772; 1H NMR (600 MHz, CDCl3) δ 6.15 (dt, J = 17.2, 9.7 Hz, 1H), 5.22 (dd, J = 17.0, 2.0 Hz, 1H), 5.08 (dd, J = 10.1, 2.0 Hz, 1H), 4.09 (dd, J = 12.6, 7.7 Hz, 1H), 3.96 (dd, J = 11.8, 8.6 Hz, 1H), 3.87 (d, J = 12.4 Hz, 1H), 3.73 (d, J = 11.7 Hz, 1H), 3.16 (dd, J = 11.4, 4.6 Hz, 1H), 3.03 – 2.92 (m, 1H), 2.36 (ddd, J = 12.9, 9.3, 3.7 Hz, 1H), 2.32 – 2.26 (m, 1H), 1.84 (td, J = 13.2, 3.9 Hz, 1H), 1.73 (dt, J = 13.4, 3.4 Hz, 1H), 1.65 (dt, J = 12.9, 3.6 Hz, 1H), 1.62 – 1.52 (m, 3H), 1.48 (dq, J = 13.7, 3.8 Hz, 1H), 1.43 (dq, J = 13.2, 3.3 Hz, 1H), 1.36 (qd, J = 13.0, 3.2 Hz, 1H), 1.32 – 1.12 (m, 4H), 0.93 (s, 3H), 0.88 (s, 12H), 0.84 (s, 3H), 0.79 (dd, J = 12.3, 2.2 Hz, 1H), 0.72 (s, 3H), 0.03 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 143.78, 116.04, 79.35, 77.37, 77.16, 76.95, 67.28, 62.01, 55.41, 52.45, 49.55, 47.73, 41.34, 39.55, 39.13, 37.50, 35.24, 29.38, 28.42, 27.92, 26.05, 20.81, 18.44, 18.24, 16.89, 16.03, 15.85, −3.63, −4.78; HRMS (ESI) m/z Calcd for C28H53O3Si [M+H]+ 465.3764, found 465.3756.
4.11. Aldehyde 24
To the solution of diol 23 (1.324 g, 2.848 mmol, 1.0 equiv) in acetone/CH3CN (25 ml/25 mL) was added IBX (0.7975 g, 2.848 mmol, 1.0 equiv) as a solid. The reaction mixture was sonicated for 8 h, then concentrated. Et2O (20 mL) was added and the mixture was sonicated for 2 min, then filtered through a pad of celite. The celite layer was washed with additional Et2O (2 x 20 mL) and the Et2O layers were combined and concentrated. The residue was purified by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 40 g column, 1–100% CH2Cl2 in hexanes) to yield aldehyde 24 (422 mg, 32%) as a white solid. TLC Rf 0.17 (hexanes:CH2Cl2); [α]D20 +10.0 (c 1.00, C6H6); FTIR (NaCl film) 3385, 2934, 1709, 1462, 1388, 1253, 1098, 1069, 884, 835, 773; 1H NMR (600 MHz, CDCl3) δ 9.77 (s, 1H), 6.12 (ddd, J = 17.6, 10.6, 6.0 Hz, 1H), 5.08 (dt, J = 17.6, 1.7 Hz, 1H), 5.04 (dt, J = 10.6, 1.5 Hz, 1H), 4.36 (dd, J = 12.3, 8.3 Hz, 1H), 4.07 (dd, J = 12.4, 4.1 Hz, 1H), 3.17 (dd, J = 11.4, 4.6 Hz, 1H), 3.04 (dddd, J = 12.7, 5.8, 3.9, 1.6 Hz, 1H), 2.06 – 1.92 (m, 2H), 1.84 (dtd, J = 13.5, 4.2, 2.1 Hz, 1H), 1.70 – 1.36 (m, 11H), 1.09 (d, J = 1.1 Hz, 3H), 0.98 (dd, J = 12.6, 3.0 Hz, 1H), 0.89 (s, 4H), 0.88 (s, 10H), 0.84 (d, J = 0.9 Hz, 3H), 0.72 (s, 4H), 0.03 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 208.54, 208.53, 141.90, 114.80, 79.19, 77.37, 77.16, 76.95, 61.13, 60.87, 55.26, 51.36, 42.51, 39.55, 38.77, 38.22, 37.69, 36.10, 28.52, 27.77, 26.05, 25.68, 20.35, 18.27, 18.25, 18.22, 16.57, 16.08, −3.62, −4.77; HRMS (ESI) m/z Calcd for C28H51O3Si [M+H]+ 463.3607, found 463.3598.
4.12. Methyl ketone 25
To the solution of aldehyde 24 (500 mg, 1.08 mmol, 1.0 equiv) in CH2Cl2 (20 mL) cooled to 0 °C was added pyridine (435 μl, 5.40 mmol, 5.0 equiv) followed by DEIPSOTf (488 μl, 2.16 mmol, 2.0 equiv). The reaction mixture was stirred at 0 °C for 20 min, then diluted with Et2O (150 mL) and washed with brine (50 mL). The organic layer was dried over MgSO4, filtered, and concentrated to obtain the crude diethylisopropylsilyl ether, which was carried to the next step without further purification. TLC Rf 0.44 (hexanes:CH2Cl2 1:1); [α]D20 +22.5 (c 1.12, C6H6); FTIR (NaCl film) 2951, 1718, 1462, 1388, 1252, 1097, 1008, 913, 882, 835, 773, 726; 1H NMR (600 MHz, CDCl3) δ 9.69 (s, 1H), 5.89 (ddd, J = 17.2, 10.5, 6.6 Hz, 1H), 4.99 (dt, J = 17.3, 1.6 Hz, 1H), 4.93 (dt, J = 10.5, 1.4 Hz, 1H), 4.12 (d, J = 10.8 Hz, 1H), 4.06 (d, J = 10.8 Hz, 1H), 3.16 (dd, J = 11.4, 4.6 Hz, 1H), 2.99 – 2.90 (m, 1H), 1.96 (td, J = 13.1, 12.4, 4.5 Hz, 1H), 1.80 – 1.73 (m, 1H), 1.66 (dt, J = 13.0, 3.6 Hz, 1H), 1.63 – 1.22 (m, 11H), 1.02 (s, 3H), 1.01 – 0.95 (m, 13H), 0.90 (s, 3H), 0.88 (s, 10H), 0.83 (s, 3H), 0.77 (dd, J = 11.9, 2.0 Hz, 1H), 0.72 (s, 3H), 0.68 – 0.57 (m, 4H), 0.03 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 207.38, 141.35, 114.07, 79.37, 77.37, 77.16, 76.95, 61.42, 59.07, 55.39, 51.63, 42.15, 39.72, 39.53, 38.76, 37.71, 36.16, 28.57, 27.82, 27.61, 26.05, 20.34, 18.26, 18.25, 17.93, 17.52, 16.77, 16.01, 12.64, 7.29, 7.28, 3.22, 3.21, −3.63, −4.80; HRMS (ESI) m/z Calcd for C35H67O3Si2 [M+H]+ 591.4629, found 591.4633.
The crude diethylisopropylsilyl ether above was azetropically dried using PhMe (5 mL), dissolved in Et2O, and cooled to −78 °C. MeLi (1 M in Et2O, 2.16 mL, 2.16 mmol, 2.0 equiv) was added and the reaction mixture was stirred for 2 h at −78 °C. The reaction was quenched by adding AcOH (180 μl, 3.0 mmol) then the Et2O was removed using a stream of N2, and the resulting crude secondary alcohol was dried under high vacuum, then used without further purification.
The crude secondary alcohol above was dissolved in CH2Cl2 (25 mL). NaHCO3 (910 mg, 10.8 mmol, 10 equiv) and Dess–Martin periodinane (458 mg, 1.08 mmol, 1.0 equiv) were added promptly. The reaction mixture was stirred for 30 min and quenched by addition of 20% aq Na2S2O3 (25 mL) and saturated NaHCO3 (25 mL). The mixture was stirred for 40 min, then extracted with CH2Cl2 (3 x 40 mL). The combined CH2Cl2 extracts were dried over Na2SO4, filtered, and concentrated. The residue was purified by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 40 g column, 1–60% EtOAc in hexanes) to yield methyl ketone 25 (554 mg, 87%) as a white solid. TLC Rf 0.28 (hexanes:CH2Cl2 1:1); [α]D22 +4.7 (c 1.00, C6H6); FTIR (NaCl film) 2876, 1632, 1430, 1359, 1215, 1069, 974, 835, 813, 726, 716; 1H NMR (600 MHz, CDCl3) δ 5.88 (ddd, J = 17.1, 10.5, 6.4 Hz, 1H), 4.90 (dt, J = 17.4, 1.7 Hz, 1H), 4.80 (dt, J = 10.5, 1.7 Hz, 1H), 4.25 (dd, J = 10.7, 1.2 Hz, 1H), 4.16 (d, J = 10.7 Hz, 1H), 3.18 (dd, J = 11.4, 4.6 Hz, 1H), 3.13 – 3.03 (m, 1H), 2.13 (s, 3H), 1.91 (td, J = 13.0, 4.0 Hz, 1H), 1.84 – 1.77 (m, 1H), 1.67 (dt, J = 13.0, 3.6 Hz, 1H), 1.61 – 1.34 (m, 10H), 1.05 (dd, J = 12.6, 2.9 Hz, 1H), 1.02 – 0.95 (m, 12H), 0.93 (s, 3H), 0.90 (s, 3H), 0.88 (s, 9H), 0.82 (s, 3H), 0.72 (s, 3H), 0.70 – 0.61 (m, 5H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 210.82, 142.68, 112.55, 79.30, 77.37, 77.16, 76.95, 64.08, 61.06, 55.12, 51.58, 41.32, 39.97, 39.51, 38.93, 37.81, 37.33, 31.97, 28.55, 27.88, 27.64, 26.04, 20.46, 18.77, 18.25, 18.12, 17.64, 17.63, 16.59, 16.08, 12.63, 7.41, 7.40, 3.28, −3.61, −4.76; HRMS (ESI) m/z Calcd for C36H69O3Si2 [M+H]+ 605.4785, found 605.4796.
4.13. Keto-aldehyde 26
Keto-alkene 25 (160 mg, 0.282 mmol, 1 equiv) was dissolved in CH2Cl2 (10 mL) and cooled to −78 °C. Ozone was bubbled through the solution until a blue color appeared (~3 min). The reaction mixture was stirred for 15 min, then N2 was bubbled through the solution to remove excess ozone. Solid Ph3P (81.4 mg, 0.310 mmol, 1.1 equiv) was then added and reaction was stirred for 24 h at rt, then concentrated. Purification by automated SiO2 column chromatography (Isco CombiFlash Rf: 10 g solid loading cartridge, 40 g column, 1–60% EtOAc in hexanes) yielded keto-aldehyde 26 (100 mg, 65%) as a white solid. TLC Rf 0.17 (CH2Cl2) [α]D19 +39.1 (c 1.00, C6H6); 1H NMR (600 MHz, CDCl3) δ 9.40 (s, 1H), 4.42 (d, J = 10.2 Hz, 1H), 4.09 (d, J = 10.2 Hz, 1H), 3.20 (dd, J = 11.4, 4.6 Hz, 1H), 3.12 (dd, J = 12.1, 4.1 Hz, 1H), 2.29 (s, 3H), 2.13 (ddd, J = 10.1, 4.3, 2.6 Hz, 1H), 1.67 (dtq, J = 20.2, 7.9, 4.2 Hz, 4H), 1.62 – 1.47 (m, 5H), 1.47 – 1.23 (m, 4H), 1.04 – 0.98 (m, 1H), 0.98 – 0.93 (m, 17H), 0.91 (s, 3H), 0.88 (s, 10H), 0.83 (s, 3H), 0.72 (s, 3H), 0.68 (dd, J = 12.3, 2.0 Hz, 1H), 0.62 (dqd, J = 10.3, 7.9, 2.2 Hz, 4H), 0.03 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 211.31, 203.00, 79.11, 77.37, 77.16, 76.95, 65.58, 60.80, 55.32, 52.55, 48.30, 41.07, 39.54, 39.02, 37.97, 37.77, 30.78, 28.49, 27.82, 26.03, 21.29, 19.89, 18.76, 18.34, 18.25, 17.46, 17.45, 16.81, 16.07, 12.37, 7.24, 7.13, 3.02, 3.00, −3.59, −4.74; HRMS (ESI) m/z Calcd for C35H67O4Si2 [M+H]+ 607.4578, found 607.4562.
4.14. Triene 28
To a solution of phosphonate 27 (150 mg, 0.63 mmol, 2.5 equiv) in THF (8 mL) cooled to −78 °C was added LiHMDS (85 mg, 0.51 mmol, 2.0 equiv). The resulting orange solution was stirred for 35 min at −78 °C. In a separate flask, aldehyde 26 (150 mg, 0.25 mmol, 1.0 equiv.) was azeotropically dried with toluene (5 mL), then dissolved in THF (7 mL) and cooled to −78 °C. The phosphorous ylide solution was added to the aldehyde solution via cannula. The resulting solution was stirred for 4 h at −78 °C, by which time TLC analysis indicated complete conversion. The reaction was quenched by addition of satd aq NaHCO3 (3 mL). The resulting mixture was diluted with Et2O (100 mL), washed with brine (20 mL), dried over MgSO4, filtered, and concentrated. The residue was purified by automated column chromatography (Isco CombiFlash Rf: basic Al2O3, 10 g solid loading cartridge, 40 g column, 1–15% EtOAc in hexanes) to yield triene 28 (220 mg, 63%) as a colorless oil. TLC Rf 0.44 (hexanes:EtOAc 25:1); FTIR (NaCl film) 2951, 1696, 1462, 1388, 1359, 1253, 1100, 1068, 1009, 956, 884, 835, 774, 732; 1H NMR (600 MHz, C6D6) For (Z)-eno lether isomer: δ 6.89 (dd, J = 15.2, 11.2 Hz, 1H), 6.09 (d, J = 15.1 Hz, 1H), 6.01 (d, J = 11.2 Hz, 1H), 5.94 (d, J = 9.7 Hz, 1H), 4.54 (s, 2H), 3.95 (ddd, J = 13.5, 9.8, 4.4 Hz, 1H), 3.80 (s, 3H), 3.12 (dd, J = 11.5, 4.3 Hz, 1H), 2.27 (s, 3H), 2.04 – 1.89 (m, 2H), 1.84 (qd, J = 13.2, 4.9 Hz, 1H), 1.74 – 1.66 (m, 2H), 1.65 (s, 2H), 1.63 (s, 4H), 1.61 (d, J = 3.2 Hz, 1H), 1.56 – 1.35 (m, 6H), 1.27 (ddd, J = 12.4, 8.9, 3.3 Hz, 2H), 1.10 – 1.00 (m, 30H), 0.98 (s, 3H), 0.89 (s, 1H), 0.86 (s, 3H), 0.76 (s, 1H), 0.73 (s, 3H), 0.71 – 0.57 (m, 5H), 0.09 (s, 4H), 0.07 (s, 3H); 13C NMR (151 MHz, C6D6) For (Z) enol ether isomers: δ 209.38, 154.14, 135.43, 128.35, 128.22, 128.14, 128.06, 127.98, 127.90, 126.62, 125.96, 125.10, 122.74, 79.64, 63.01, 62.44, 59.11, 55.71, 52.20, 42.03, 39.69, 39.09, 37.97, 37.96, 37.07, 35.54, 31.67, 30.81, 28.73, 28.27, 26.22, 26.21, 26.15, 21.38, 18.98, 18.63, 18.42, 18.39, 17.78, 17.73, 16.76, 16.37, 13.00, 7.58, 7.56, 7.54, 3.58, 3.52, −3.58, −3.59, −4.67; HRMS (ESI) m/z Calcd for C43H79O4Si2 [M+H]+, 715.5517 found 715.5524.
4.15. Diazoketone 29
Methyl ketone 28 (50 mg, 0.071 mmol, 1.0 equiv) was dissolved in THF (8 mL) in a sealed tube and cooled to 0 °C. NaH (12 mg, 0.20 mmol, 7.0 eqiuv) and ethyl formate (250 μL, 3.10 mmol, 44 equiv.) were added and the reaction vessel was sealed and heated to 45 °C for 24 h, by which point TLC analysis (basic alumina, hexanes:EtOAc 5:1) indicated complete conversion. The volatiles were removed under high vacuum. The residue was dissolved in EtOH/CH3CN (8 mL/6 mL) and AcNHPhSO2N3 (72 mg, 2.840 mmol, 4.0 equiv.) was added at rt. The reaction mixture was stirred for 3 h, diluted with Et2O (100 mL), and washed with brine (2x 20 mL) and water (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. Purification by flash column chromatography (toluene then toluene:EtOAc 10:1) afforded diazoketone 29 (37 mg, 71%) as a yellow oil. TLC Rf 0.43 (hexanes:EtOAc 25:1); FTIR (NaCl film) 2952, 2096, 1625, 1462, 1359, 1251, 1100, 1070, 884, 835, 773, 725; 1H NMR (600 MHz, C6D6) δ 6.88 (dd, J = 15.1, 11.2 Hz, 1H), 6.16 (s, 1H), 6.07 (d, J = 15.1 Hz, 1H), 5.97 (t, J = 11.3 Hz, 2H), 4.62 (d, J = 10.6 Hz, 1H), 4.58 (d, J = 10.5 Hz, 1H), 3.61 (s, 3H), 3.47 (ddd, J = 12.4, 10.5, 4.3 Hz, 1H), 3.17 (dd, J = 11.4, 4.8 Hz, 1H), 2.13 – 1.73 (m, 5H), 1.67 – 1.64 (m, 3H), 1.63 (s, 3H), 1.61 – 1.40 (m, 3H), 1.38 (s, 3H), 1.15 (s, 3H), 1.12 – 1.05 (m, 15H), 1.03 (s, 10H), 0.99 (s, 3H), 0.88 (s, 3H), 0.85 – 0.62 (m, 10H), 0.11 (s, 3H), 0.09 (s, 3H); 13C NMR (151 MHz, C6D6) δ 196.58, 153.22, 136.15, 128.35, 128.22, 128.14, 128.06, 127.98, 127.90, 126.00, 125.73, 125.42, 123.05, 79.75, 62.99, 61.58, 59.74, 55.76, 55.53, 52.39, 42.40, 39.72, 39.16, 38.06, 36.76, 35.05, 31.73, 30.49, 28.71, 28.35, 26.27, 26.22, 26.20, 26.14, 21.42, 18.98, 18.81, 18.43, 18.38, 17.80, 17.77, 17.69, 16.98, 16.38, 13.09, 13.08, 7.58, 7.52, 7.47, 7.45, 3.64, 3.57, 3.47, −3.58, −4.65, −4.66; HRMS (ESI) m/z Calcd for C43H77O4Si2 [M+H]+ 763.5241 found 763.5240.
4.16. Bis(enol ether) 31
A solution of diazoketone 29 (3.5 mg, 0.0048 mmol, 1.0 equiv) in C6D6 (0.6 mL) was irradiated in a photobox using UV-B lights (centered at 310 nm) for 20 min, by which time 1H-NMR analysis of the crude reaction mixture indicated complete conversion. The resulting bis(enol ether) 31 was not stable to purification and was not fully characterized.
4.17. Bis(enol ether) 34
A solution of diazoketone 33 (11.5 mg, 0.0167 mmol, 1.0 equiv) (see Supporting Information) in C6D6 (1.5 mL) was irradiated in a photobox using UV-B lights (centered at 310 nm) for 20 min, by which time 1H-NMR analysis of the crude reaction mixture indicated complete conversion. The reaction mixture was concentrated and purified by basic Al2O3 column chromatography (hexanes:EtOAc 100:1) to give bis(enol ether) 34 (7.4 mg, 67 %) as a colorless oil. TLC Rf 0.24 (hexanes:EtOAc 100:1); FTIR (NaCl film) 2935, 1641, 1462, 1386, 1221, 1064, 883, 835, 772, 728; 1H NMR (600 MHz, C6D6) δ 5.23 (d, J = 1.8 Hz, 1H), 4.47 (dt, J = 13.7, 2.5 Hz, 1H), 4.37 (ddd, J = 13.8, 3.5, 2.2 Hz, 1H), 4.18 (d, J = 10.3 Hz, 1H), 4.13 (q, J = 2.9 Hz, 1H), 3.97 (d, J = 10.3 Hz, 1H), 3.40 (dt, J = 10.9, 2.1 Hz, 1H), 3.15 (s, 3H), 3.15 (m, 1H), 2.36 (td, J = 13.2, 12.5, 7.6 Hz, 2H), 1.94 (td, J = 13.0, 3.8 Hz, 1H), 1.74 – 1.54 (m, 6H), 1.54 – 1.15 (m, 9H), 1.13 – 1.04 (m, 18H), 1.03 (s, 9H), 1.00 (s, 3H), 0.98 – 0.91 (m, 1H), 0.90 (s, 3H), 0.89 – 0.79 (m, 5H), 0.76 (s, 3H), 0.72 – 0.60 (m, 4H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (151 MHz, C6D6) δ 157.54, 156.89, 128.44, 128.36, 128.32, 128.20, 128.07, 127.99, 127.96, 127.91, 127.86, 127.83, 127.75, 127.58, 127.46, 106.19, 89.98, 79.68, 67.17, 63.75, 56.53, 56.31, 53.88, 52.14, 47.81, 43.17, 40.31, 39.74, 38.85, 37.66, 36.18, 31.84, 28.66, 28.24, 26.08, 25.61, 22.93, 22.59, 18.93, 18.86, 18.28, 17.64, 17.60, 16.28, 16.14, 14.24, 13.10, 7.42, 3.52, 3.47, −3.70, −4.80; HRMS (ESI) m/z Calcd for C39H71O4Si2 [M+H]+ 659.4891 found 659.4883.
4.18. Bis(enol ether) 36
A solution of diazoketone 35 (10.6 mg, 0.0125 mmol, 1.0 equiv) (see Supporting Information) in C6D6 (1.5 mL) was irradiated in a photobox using UV-B lights (centered at 310 nm) for 20 min, by which time 1H-NMR analysis of the crude reaction mixture indicated complete conversoion. The reaction mixture was concentrated and purified by basic Al2O3 column chromatography (hexanes:EtOAc 100:1) to give bis(enol ether) 36 (7.2 mg, 70 %) as a colorless oil. TLC Rf 0.11 (hexanes:EtOAc 100:1); [α]D20 +39.7 (c 0.95, C6H6); FTIR (NaCl film) 2950, 1651, 1463, 1386, 1250, 1218, 1187, 1146, 1103, 1062, 883, 835, 773, 730; 1H NMR (600 MHz, C6D6) δ 7.41 – 7.37 (m, 2H), 7.21 (t, J = 7.7 Hz, 2H), 7.13 – 7.10 (m, 1H), 5.20 (d, J = 2.1 Hz, 1H), 5.14 – 5.08 (m, 1H), 4.46 (dd, J = 3.2, 1.9 Hz, 1H), 4.39 (d, J = 11.4 Hz, 1H), 4.33 (d, J = 11.3 Hz, 1H), 4.19 (d, J = 10.4 Hz, 1H), 3.97 (d, J = 10.3 Hz, 1H), 3.40 – 3.33 (m, 1H), 3.17 (s, 3H), 3.17 (m, 1H), 2.39 – 2.30 (m, 2H), 2.10 (dd, J = 14.7, 7.9 Hz, 1H), 1.97 (td, J = 13.1, 3.8 Hz, 1H), 1.89 (dd, J = 14.6, 3.6 Hz, 1H), 1.74 – 1.43 (m, 11H), 1.41 (s, 4H), 1.25 (s, 3H), 1.17 – 1.04 (m, 18H), 1.02 (s, 10H), 1.00 (s, 3H), 0.98 – 0.92 (m, 1H), 0.90 (s, 3H), 0.89 – 0.80 (m, 3H), 0.77 (s, 3H), 0.72 – 0.58 (m, 4H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (151 MHz, C6D6) δ 158.07, 155.92, 140.50, 128.47, 128.35, 128.22, 128.14, 128.10, 128.06, 127.98, 127.90, 127.59, 127.28, 107.00, 96.27, 79.83, 74.59, 73.86, 63.88, 56.89, 56.69, 54.05, 52.32, 48.08, 48.04, 43.36, 40.41, 39.89, 39.00, 37.83, 36.40, 28.81, 28.38, 27.07, 26.23, 25.72, 25.34, 22.74, 19.10, 19.01, 18.43, 17.80, 17.78, 16.43, 16.29, 13.26, 7.59, 3.70, 3.64, −3.55, −4.65; HRMS (ESI) m/z Calcd for C50H85O5Si2 [M+H]+ 821.5936 found 821.5959.
4.19. Keto-ketal 38
To the solution of bis(enol ether) 34 (7 mg, 0.0106 mmol) in THF (1 mL) was added TBAF (1 M in THF, 16 μl, 0.016 mmol, 1.5 equiv). The reaction mixture was stirred at rt for 16 h, then diluted with Et2O (100 mL), washed with satd aq NaHCO3 (5 x 10 mL). The Et2O layer was dried over MgSO4, filtered, and concentrated to give the crude alcohol 37 (6.9 mg, 100%), which was used without further purification.
To the solution of the crude alcohol 37 above (3.6 mg, 0.0068 mmol, 1.0 equiv) in wet CH2Cl2 (1 mL) was added Amberlyst-15 (1 mg). The reaction mixture was stirred for 20 min at rt then quenched by addition of Et3N (10 μl). The mixture was diluted with Et2O (100 mL), washed with satd aq NaHCO3 (5x 10 mL), dried over MgSO4, filtered, and concentrated. The residue was purified by SiO2 column chromatography (hexanes:EtOAc 5:1) to afford keto-ketal 38 (1.8 mg, 50%) as a white solid. TLC Rf 0.26 (hexanes:EtOAc 10:1); FTIR (NaCl film) 2949, 1721, 1463, 1387, 1253, 1108, 1003, 883, 836, 773, 737; 1H NMR (600 MHz, C6D6) δ 4.04 (dd, J = 7.8, 2.1 Hz, 1H), 4.00 (d, J = 7.7 Hz, 1H), 3.99 – 3.94 (m, 1H), 3.48 (dt, J = 12.0, 5.0 Hz, 1H), 3.12 (dd, J = 11.5, 4.5 Hz, 1H), 2.68 (dddd, J = 12.3, 7.0, 4.6, 2.1 Hz, 1H), 2.18 (dd, J = 7.3, 1.6 Hz, 1H), 2.09 (ddd, J = 17.1, 8.9, 5.4 Hz, 1H), 1.91 (dtd, J = 11.9, 4.6, 2.1 Hz, 1H), 1.82 (dt, J = 17.0, 4.5 Hz, 1H), 1.64 – 1.55 (m, 2H), 1.52 – 1.08 (m, 17H), 1.04 (s, 9H), 0.96 (s, 3H), 0.86 (s, 3H), 0.82 (s, 3H), 0.65 (s, 3H), 0.58 (ddd, J = 22.6, 13.1, 3.5 Hz, 2H), 0.45 (dt, J = 9.4, 2.3 Hz, 1H), 0.13 (s, 3H), 0.12 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 206.47, 112.13, 79.59, 66.46, 62.89, 59.25, 55.87, 53.18, 52.90, 40.43, 39.76, 39.33, 38.58, 38.01, 37.31, 37.11, 35.80, 31.99, 28.73, 28.19, 27.07, 26.21, 23.08, 21.29, 18.60, 18.53, 18.43, 16.35, 16.30, 14.39, −3.50, −4.61.
4.20. Jujubogenin hexacyclic core (39)
To the solution of ketone 38 (0.5 mg, 0.001 mmol, 1.0 equiv) in Et2O (0.3 mL) cooled to −78 C was added MeLi (1M, 5 μl, 0.005 mmol, 5.0 equiv). The reaction mixture was stirred for 30 min then diluted with Et2O (100 mL) and washed with satd aq NaHCO3 (30 mL). The Et2O layer was dried over MgSO4, filtered, and concentrated. The residue was purified by SiO2 column chromatography to afford tertiary alcohol 39 (0.4 mg, 73%) as a white solid. TLC Rf 0.25 (hexanes:EtOAc 5:1); 1H NMR (600 MHz, C6D6) δ 4.09 (d, J = 1.7 Hz, 2H), 3.92 – 3.87 (m, 2H), 3.79 (ddd, J = 11.4, 4.7, 3.2 Hz, 1H), 3.13 (dd, J = 11.5, 4.4 Hz, 1H), 2.40 – 2.31 (m, 1H), 2.05 (d, J = 8.5 Hz, 1H), 1.54 – 1.15 (m, 24H), 1.05 (s, 9H), 1.02 (s, 3H), 0.96 (s, 3H), 0.88 (s, 4H), 0.86 (s, 3H), 0.84 (s, 3H), 0.72 (s, 3H), 0.72 – 0.69 (m, 1H), 0.67 – 0.59 (m, 1H), 0.50 (dd, J = 11.5, 2.2 Hz, 1H), 0.14 (s, 3H), 0.13 (s, 3H); HRMS (ESI) m/z Calcd for C32H56O4SiNa [M+Na]+ 555.3846, found 555.3852.
4.21. Keto-ketal 41
To a solution of bis(enol ether) 36 (12 mg, 0.015 mmol, 1 equiv) in THF (1 mL) was added TBAF (1M, 29 μl, 0.029 mmol, 2.0 equiv). The reaction mixture was stirred at rt for 16 h, then diluted with Et2O (100 mL), and washed with satd aq NaHCO3 (5 x 10 mL). The Et2O layer was dried over MgSO4, filtered, and concentrated to give the crude alcohol 40, which was used without further purification.
To a solution of the crude alcohol 40 above (2.6 mg, 0.0038 mmol, 1.0 equiv) in wet CH2Cl2 (1 mL) was added dichloroacetic acid (0.1 mg, 0.0004 mmol, 0.1 equiv). The reaction mixture was stirred for 20 min at rt, then quenched by addition of Et3N (10 μl). The mixture was diluted with Et2O (100 mL), washed with satd aq NaHCO3 (5x 10 mL), dried over MgSO4, filtered, and concentrated. The residue was purified by SiO2 column chromatography (hexanes:EtOAc 5:1) to afford keto-ketal 41 (1.6 mg, 63%) as a white solid. TLC Rf 0.26 (hexanes:EtOAc 7:1); [α]D18 +17.0 (c 0.18, C6H6) FTIR (NaCl film) 2929, 2855, 1775, 1720, 1462, 1387, 1297, 1254, 1185, 1106, 930, 884, 836, 773, 735, 697 1H NMR (600 MHz, C6D6) δ 7.42 – 7.36 (m, 2H), 7.23 (t, J = 7.7 Hz, 2H), 7.12 (t, J = 7.5 Hz, 1H), 4.86 (ddt, J = 11.1, 7.8, 3.2 Hz, 1H), 4.34 (d, J = 11.5 Hz, 1H), 4.24 (d, J = 11.4 Hz, 1H), 4.11 (dd, J = 7.8, 2.2 Hz, 1H), 4.03 (d, J = 7.7 Hz, 1H), 3.09 (dd, J = 11.5, 4.5 Hz, 1H), 2.76 – 2.64 (m, 1H), 2.31 (dd, J = 7.4, 1.5 Hz, 1H), 2.18 (dd, J = 18.3, 11.1 Hz, 1H), 2.09 (dd, J = 18.3, 3.7 Hz, 1H), 1.89 (ddt, J = 11.1, 5.3, 2.6 Hz, 1H), 1.81 (dd, J = 9.3, 1.5 Hz, 1H), 1.76 (dd, J = 14.7, 7.8 Hz, 1H), 1.63 – 1.52 (m, 2H), 1.49 – 1.30 (m, 9H), 1.30 – 1.18 (m, 3H), 1.16 (s, 4H), 1.04 (s, 9H), 0.93 (s, 3H), 0.85 (s, 3H), 0.82 (s, 3H), 0.65 (s, 3H), 0.63 – 0.53 (m, 2H), 0.44 (dd, J = 11.7, 2.5 Hz, 1H), 0.41 (s, 4H), 0.12 (d, J = 1.4 Hz, 6H); 13C NMR (151 MHz, C6D6) δ 207.01, 140.45, 128.50, 128.47, 128.35, 128.22, 128.14, 128.06, 127.98, 127.90, 127.73, 127.70, 127.61, 127.57, 127.46, 127.37, 111.50, 79.54, 74.46, 70.39, 66.53, 63.99, 58.57, 55.84, 53.18, 52.89, 47.31, 46.49, 40.82, 39.74, 38.59, 37.75, 37.30, 37.14, 35.81, 28.68, 28.19, 27.03, 26.79, 26.21, 25.06, 21.30, 18.57, 18.53, 18.43, 16.36, 16.33, −3.51, −4.60; HRMS (ESI) m/z Calcd for C42H66O5SiNa [M+Na]+ 701.4577, found 701.4543.
4.22. Diol 51
To the solution of ketone 41 (1.5 mg, 0.0022 mmol, 1.0 equiv) in Et2O (0.3 mL) cooled to −78 C was added MeLi (1.2 M, 9.2 μl, 0.011 mmol, 5.0 equiv). The reaction mixture was stirred for 30 min then diluted with Et2O (100 mL) and washed with satd aq NaHCO3 (30 mL). The Et2O layer was dried over MgSO4, filtered, and concentrated. The residue was purified by SiO2 column chromatography to afford the corresponding tertiary alcohol (1.1 mg, 73%) as a white solid. TLC Rf 0.21 (hexanes:EtOAc 5:1); [α]D18 +28.0 (c 0.12, C6H6) FTIR (NaCl film) 3427, 2927, 2855, 1670, 1458, 1387, 1255, 1095, 932, 884, 836, 773, 735, 969, 668; 1H NMR (600 MHz, C6D6) δ 7.42 (d, J = 7.5 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 4.49 – 4.41 (m, 1H), 4.39 (d, J = 11.5 Hz, 1H), 4.31 (d, J = 11.5 Hz, 1H), 4.15 (dd, J = 7.6, 2.0 Hz, 1H), 4.03 (d, J = 7.7 Hz, 1H), 3.10 (dd, J = 11.5, 4.4 Hz, 1H), 2.33 (dtd, J = 12.4, 4.9, 2.5 Hz, 1H), 1.91 – 1.82 (m, 2H), 1.78 – 1.66 (m, 3H), 1.66 – 1.49 (m, 4H), 1.49 – 1.19 (m, 17H), 1.03 (d, J = 8.1 Hz, 13H), 0.96 (s, 3H), 0.94 (s, 3H), 0.87 (s, 3H), 0.70 (s, 3H), 0.63 (dd, J = 12.6, 2.9 Hz, 2H), 0.51 – 0.47 (m, 1H), 0.13 (s, 6H); 13C NMR (151 MHz, C6D6) δ 140.70, 128.47, 128.44, 128.35, 128.22, 128.14, 128.11, 128.06, 127.98, 127.90, 127.73, 127.61, 127.57, 127.23, 109.92, 79.55, 74.86, 69.99, 68.42, 65.94, 63.92, 55.83, 53.32, 53.14, 53.11, 47.98, 47.63, 40.28, 39.76, 38.66, 38.44, 37.34, 37.32, 36.01, 28.69, 28.34, 28.23, 27.23, 26.96, 26.22, 25.12, 21.52, 18.70, 18.62, 18.43, 16.43, 16.40, −3.52, −4.59; HRMS (ESI) m/z Calcd for C43H71O5Si [M+H]+ 695.5071, found 695.5056.
To a solution of the tertiary alcohol above (1.1 mg, 0.0016 mmol, 1 equiv) in THF/EtOAc (0.5 mL/0.5 mL) was added Pd/C (10%, 2 mg, 0.0019 mmol, 1.2 equiv). The reaction was stirred under H2 (balloon) atmosphere for 12 h, then filtered through a pad of celite and concentrated to give diol 51 (0.7 mg, 75%). TLC Rf 0.11 (hexanes:EtOAc 5:1); FTIR (NaCl film); 3412, 2949, 2853, 1433, 1218, 1109, 1087, 867 1H NMR (600 MHz, Benzene-d6) δ 4.33 (dddd, J = 11.5, 9.4, 6.9, 2.3 Hz, 1H), 4.07 (dd, J = 7.8, 2.0 Hz, 1H), 3.98 (d, J = 7.7 Hz, 1H), 3.38 (s, 1H), 3.11 (dd, J = 11.5, 4.4 Hz, 1H), 2.28 (dddd, J = 9.4, 7.0, 4.9, 2.3 Hz, 1H), 1.81 – 1.74 (m, 2H), 1.68 – 1.52 (m, 4H), 1.49 – 1.15 (m, 18H), 1.04 (s, 9H), 1.03 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.88 (s, 3H), 0.70 (s, 3H), 0.66 – 0.57 (m, 2H), 0.50 (dd, J = 11.8, 2.3 Hz, 1H), 0.13 (s, 6H); 13C NMR (151 MHz, C6D6) δ 109.56, 79.53, 70.13, 69.98, 69.60, 66.06, 55.84, 53.35, 53.10, 53.08, 48.27, 46.84, 39.95, 39.77, 38.66, 38.42, 37.31, 37.30, 36.03, 31.15, 28.97, 28.70, 28.34, 28.23, 27.35, 26.22, 21.50, 18.63, 18.62, 18.43, 16.43, 16.40, −3.52, −4.59; HRMS (ESI) m/z Calcd for C36H65O5Si [M+H]+ 605.4601, found 605.4611.
4.23. 3-O-TBS-23-epi-jujubogenin (52)
To a solution of diol 51 (0.7 mg, 0.0012 mmol, 1 equiv) in CH2Cl2 (0.3 mL) cooled to 0 °C, was added Martin sulfurane (1.1 mg, 0.0014 mmol, 1.2 equiv). The reaction was stirred for 30 min then quenched with satd aq NaHCO3 (3 drops) and 20% aq Na2S2O3 (3 drops). The mixture was diluted with CH2Cl2 (20 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by SiO2 column chromatography (CH2Cl2) to afford 3-O-TBS-23-epi-jujubogenin 52 (0.3 mg, 43%). TLC Rf 0.18 (CH2Cl2); FTIR (NaCl film) 3422, 2953, 1653, 1447, 1226, 1128 1015, 9, 821 1H NMR (600 MHz, C6D6) δ 5.32 (d, J = 8.0 Hz, 1H), 4.90 (q, J = 8.1 Hz, 1H), 4.16 (dd, J = 7.8, 2.1 Hz, 1H), 4.06 (d, J = 7.7 Hz, 1H), 3.13 (dd, J = 11.6, 4.3 Hz, 1H), 2.37 (dt, J = 12.2, 5.7 Hz, 1H), 1.86 (d, J = 8.8 Hz, 1H), 1.80 (d, J = 13.3 Hz, 1H), 1.71 (d, J = 8.7 Hz, 1H), 1.68 (s, 0H), 1.62 (s, 4H), 1.59 (s, 3H), 1.55 – 1.13 (m, 58H), 1.09 (s, 3H), 1.05 (s, 10H), 0.97 (s, 4H), 0.95 (s, 3H), 0.88 (s, 3H), 0.71 (s, 3H), 0.64 (dtd, J = 12.7, 7.5, 6.6, 3.8 Hz, 2H), 0.50 (d, J = 11.5 Hz, 1H), 0.13 (d, J = 2.7 Hz, 6H); 13C NMR (151 MHz, C6D6) δ 109.73, 79.62, 70.01, 68.47, 66.07, 55.88, 53.69, 53.13, 53.08, 46.81, 40.34, 39.78, 38.68, 38.56, 37.32, 37.31, 35.95, 32.38, 30.25, 28.74, 28.45, 28.24, 27.15, 26.23, 25.66, 21.53, 18.68, 18.60, 18.51, 18.44, 16.43, 16.38, 14.43, −3.50, −4.59; HRMS (ESI) m/z Calcd for C36H63O4Si [M+H]+ 587.4496, found 587.4501.
Supplementary Material
Acknowledgments
We thank Prof. Samuel Danishefsky and Dr. William Walkowicz for helpful discussions and Dr. G. Sukenick, Dr. H. Liu, H. Fang, and Dr. S. Rusli (MSKCC) for expert mass spectral analyses. Financial support from the NIH (R01 GM058833 to D.S.T. and D.Y.G. and P30 CA008748 to C. B. Thompson) is gratefully acknowledged.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at https\\:. These data include experimental details of synthesis of ketone 16 from Wieland Miescher ketone 9 and synthesis of the diazoketones 13, 33 and 35.
Footnotes
Dedicated to Professor Seth B. Herzon in recognition of his receipt of the 2018 Tetrahedron Young Investigator Award
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dinda B, Debnath S, Mohanta BC, Harigaya Y. Chem Biodivers. 2010;7(10):2327–2580. doi: 10.1002/cbdv.200800070. [DOI] [PubMed] [Google Scholar]
- 2.Oda K, Matsuda H, Murakami T, Katayama S, Ohgitani T, Yoshikawa M. Biol Chem. 2000;381(1):67–74. doi: 10.1515/BC.2000.009. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda H, Murakami T, Ikebata A, Yamahara J, Yoshikawa M. Chem Pharm Bull. 1999;47(12):1744–1748. doi: 10.1248/cpb.47.1744. [DOI] [PubMed] [Google Scholar]
- 4.Rts SCTP. PLoS Med. 2014;11(7):e1001685. doi: 10.1371/journal.pmed.1001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Med Lett Drugs Ther. 2017;59(1535):195–196. [PubMed] [Google Scholar]
- 6.Kennedy JS, Co M, Green S, Longtine K, Longtine J, O’Neill MA, Adams JP, Rothman AL, Yu Q, Johnson-Leva R, Pal R, Wang S, Lu S, Markham P. Vaccine. 2008;26(35):4420–4424. doi: 10.1016/j.vaccine.2008.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandepapeliere P, Horsmans Y, Moris P, Van Mechelen M, Janssens M, Koutsoukos M, Van Belle P, Clement F, Hanon E, Wettendorff M, Garcon N, Leroux-Roels G. Vaccine. 2008;26(10):1375–1386. doi: 10.1016/j.vaccine.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 8.Von Eschen K, Morrison R, Braun M, Ofori-Anyinam O, De Kock E, Pavithran P, Koutsoukos M, Moris P, Cain D, Dubois MC, Cohen J, Ballou WR. Hum Vaccin. 2009;5(7):475–482. doi: 10.4161/hv.8570. [DOI] [PubMed] [Google Scholar]
- 9.Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M, Team ANS. Curr Alzheimer Res. 2009;6(2):144–151. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragupathi G, Gardner JR, Livingston PO, Gin DY. Expert Rev Vaccines. 2011;10(4):463–470. doi: 10.1586/erv.11.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Braeckel E, Bourguignon P, Koutsoukos M, Clement F, Janssens M, Carletti I, Collard A, Demoitie MA, Voss G, Leroux-Roels G, McNally L. Clin Infect Dis. 2011;52(4):522–531. doi: 10.1093/cid/ciq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, Methogo BG, Doucka Y, Flamen A, Mordmuller B, et al. N Engl J Med. 2011;365(20):1863–1875. doi: 10.1056/NEJMoa1102287. [DOI] [PubMed] [Google Scholar]
- 13.Fernández-Tejada A, Tan DS, Gin DY. Acc Chem Res. 2016;49:1741–1756. doi: 10.1021/acs.accounts.6b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang P, Kim YJ, Navarro-Villalobos M, Rohde BD, Gin DY. J Am Chem Soc. 2005;127(10):3256–3257. doi: 10.1021/ja0422007. [DOI] [PubMed] [Google Scholar]
- 15.Kim YJ, Wang PF, Navarro-Villalobos M, Rohde BD, Derryberry J, Gin DY. J Am Chem Soc. 2006;128(36):11906–11915. doi: 10.1021/ja062364i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng K, Adams MM, Gin DY. J Am Chem Soc. 2008;130(18):5860–5861. doi: 10.1021/ja801008m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng K, Adams MM, Damani P, Livingston PO, Ragupathi G, Gin DY. Angew Chem Int Ed. 2008;47(34):6395–6398. doi: 10.1002/anie.200801885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams MM, Damani P, Perl NR, Won A, Hong F, Livingston PO, Ragupathi G, Gin DY. J Am Chem Soc. 2010;132(6):1939–1945. doi: 10.1021/ja9082842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chea EK, Fernández-Tejada A, Damani P, Adams MM, Gardner JR, Livingston PO, Ragupathi G, Gin DY. J Am Chem Soc. 2012;134(32):13448–13457. doi: 10.1021/ja305121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernández-Tejada A, Chea EK, George C, Pillarsetty N, Gardner JR, Livingston PO, Ragupathi G, Lewis JS, Tan DS, Gin DY. Nat Chem. 2014;6(7):635–643. doi: 10.1038/nchem.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karimov RR, Tan DS, Gin DY. Chem Commun. 2017;53(43):5838–5841. doi: 10.1039/c7cc01783a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawai KI, Akiyama T, Ogihara Y, Shibata S. Phytochemistry. 1974;13(12):2829–2832. [Google Scholar]
- 23.Wieland P, Miescher K. Helv Chim Acta. 1950;33(7):2215–2228. [Google Scholar]
- 24.Engler TA, Naganathan S, Takusagawa F, Yohannes D. Tetrahedron Lett. 1987;28(44):5267–5270. [Google Scholar]
- 25.Engler TA, Sampath U, Naganathan S, Velde DV, Takusagawa F, Yohannes D. J Org Chem. 1989;54(24):5712–5727. [Google Scholar]
- 26.Hall HK, Nogues P, Rhoades JW, Sentman RC, Detar M. J Org Chem. 1982;47(8):1451–1455. [Google Scholar]
- 27.Wagner-Jauregg T. Synthesis. 1980;(10):769–798. [Google Scholar]
- 28.Evans SB, Abdelkader M, Padias AB, Hall HK. J Org Chem. 1989;54(12):2848–2852. [Google Scholar]
- 29.Padias AB, Tien TP, Hall HK. J Org Chem. 1991;56(19):5540–5544. [Google Scholar]
- 30.Goldschmidt Z, Antebi S, Gottlieb HE, Cohen D, Shmueli U, Stein Z. J Organomet Chem. 1985;282(3):369–381. [Google Scholar]
- 31.Goldschmidt Z, Genizi E, Gottlieb HE, Hezronilangermann D, Berke H, Bosch HW, Takats J. J Organomet Chem. 1991;420(3):419–429. [Google Scholar]
- 32.Goldschmidt Z, Genizi E, Gottlieb HE. J Organomet Chem. 1999;587(1):81–87. [Google Scholar]
- 33.Rydberg DB, Meinwald J. Tetrahedron Lett. 1996;37(8):1129–1132. [Google Scholar]
- 34.Zhang J, Li LQ, Wang YX, Wang WJ, Xue JJ, Li Y. Org Lett. 2012;14(17):4528–4530. doi: 10.1021/ol3020013. [DOI] [PubMed] [Google Scholar]
- 35.Tidwell TT. Eur J Org Chem. 2006;(3):563–576. [Google Scholar]
- 36.Martin JC, Gott PG, Goodlett VW, Hasek RH. J Org Chem. 1965;30(12):4175–4180. [Google Scholar]
- 37.Cheburkov YA, Mukhamadaliyev N, Knunyants IL. Tetrahedron. 1968;24(3):1341–1356. [Google Scholar]
- 38.England DC, Krespan CG. J Org Chem. 1970;35(10):3300–3307. [Google Scholar]
- 39.Gouesnar JP. Tetrahedron. 1974;30(17):3113–3117. [Google Scholar]
- 40.Brady WT, Agho MO. Synthesis. 1982;(6):500–502. [Google Scholar]
- 41.Trahanovsky WS, Surber BW, Wilkes MC, Preckel MM. J Am Chem Soc. 1982;104(24):6779–6781. [Google Scholar]
- 42.Brady WT, Agho MO. J Heterocycl Chem. 1983;20(3):501–506. [Google Scholar]
- 43.Falshaw CP, Lakoues A, Taylor GA. J Chem Res, Synop. 1985;(4):106–106. [Google Scholar]
- 44.Mayr H, Heigl UW. J Chem Soc, Chem Comm. 1987;(23):1804–1805. [Google Scholar]
- 45.Clennan EL, Lewis KK. J Am Chem Soc. 1987;109(8):2475–2478. [Google Scholar]
- 46.Downing W, Latouche R, Pittol CA, Pryce RJ, Roberts SM, Ryback G, Williams JO. J Chem Soc, Perkin Trans 1. 1990;(9):2613–2615. [Google Scholar]
- 47.Maurya R, Pittol CA, Pryce RJ, Roberts SM, Thomas RJ, Williams JO. J Chem Soc, Perkin Trans 1. 1992;(13):1617–1621. [Google Scholar]
- 48.Ito T, Aoyama T, Shioiri T. Tetrahedron Lett. 1993;34(41):6583–6586. [Google Scholar]
- 49.Pittol CA, Roberts SM, Sutton PW, Williams JO. J Chem Soc, Chem Comm. 1994;(7):803–804. [Google Scholar]
- 50.Merino I, Hegedus LS. Organometallics. 1995;14(5):2522–2531. [Google Scholar]
- 51.Yamabe S, Dai TS, Minato T, Machiguchi T, Hasegawa T. J Am Chem Soc. 1996;118(27):6518–6519. [Google Scholar]
- 52.Machiguchi T, Hasegawa T, Ishiwata A, Terashima S, Yamabe S, Minato T. J Am Chem Soc. 1999;121(20):4771–4786. [Google Scholar]
- 53.Machiguchi T, Okamoto J, Takachi J, Hasegawa T, Yamabe S, Minato T. J Am Chem Soc. 2003;125(47):14446–14448. doi: 10.1021/ja030191g. [DOI] [PubMed] [Google Scholar]
- 54.Ussing BR, Hang C, Singleton DA. J Am Chem Soc. 2006;128(23):7594–7607. doi: 10.1021/ja0606024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yates P, Eaton P. J Am Chem Soc. 1960;82(16):4436–4437. [Google Scholar]
- 56.Kagan HB, Riant O. Chem Rev. 1992;92(5):1007–1019. [Google Scholar]
- 57.Kirmse W. Eur J Org Chem. 2002;(14):2193–2256. [Google Scholar]
- 58.Kawai KI, Iitaka Y, Shibata S. Acta Crystallogr, Sect B: Struct Sci. 1974;30:2886–2888. [Google Scholar]
- 59.Ishihara K, Sakakura A. 5.10 Hetero-Diels–Alder Reactions. In: Knochel P, Molander GA, editors. Comprehensive Organic Synthesis II. 2. Elsevier; Amsterdam, Netherlands: 2014. pp. 409–465. [Google Scholar]
- 60.Johnson WS, Margrave JL, Bauer VJ, Frisch MA, Dreger LH, Hubbard WN. J Am Chem Soc. 1960;82(5):1255–1256. [Google Scholar]
- 61.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15(5):1518–1520. [Google Scholar]
- 62.Jung ME, Duclos BA. Tetrahedron. 2006;62(40):9321–9334. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.