

Summary
Prokaryotic CRISPR-Cas systems provide adaptive immunity by integrating portions of foreign nucleic acids (spacers) into genomic CRISPR arrays. Cas6 proteins then process CRISPR array transcripts into spacer-derived RNAs (crRNAs) that target Cas nucleases to matching invaders. We find that a Marinomonas mediterranea fusion protein combines three enzymatic domains (Cas6, reverse transcriptase (RT), and Cas1), which function in both crRNA biogenesis and spacer acquisition from RNA and DNA. We report a crystal structure of this divergent Cas6, identify amino acids required for Cas6 activity, show the Cas6 domain is required for RT activity and RNA spacer acquisition, and demonstrate CRISPR-repeat binding to Cas6 regulates RT activity. Coevolution of putative interacting surfaces suggests a specific structural interaction between the Cas6 and RT domains, and phylogenetic analysis reveals repeated, stable association of free-standing Cas6s with CRISPR RTs in multiple microbial lineages, indicating a functional interaction between these proteins preceded evolution of the fusion.
Keywords: Adaptive immunity, Cas1, CRISPR adaptation, CRISPR-Cas, crRNA processing, crystal structure, evolution, fusion protein, reverse transcriptase
eTOC
CRISPR RNAs target the CRISPR-Cas immune machinery to invasive nucleic acids. Mohr et al. describe a single Cas enzyme with three biochemical activities (CRISPR RNA processing, reverse transcriptase, and spacer integration) that unite the separate functions of adapting to RNA and DNA invaders and creating molecular guides for combating future infections.
Graphical Abstract
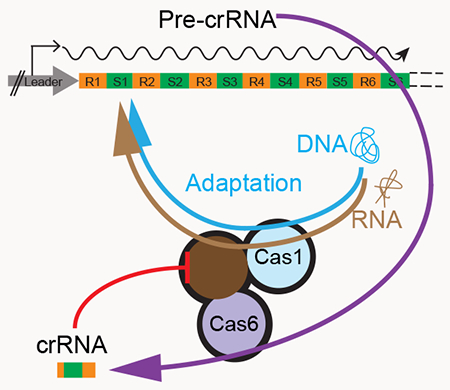
INTRODUCTION
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated proteins) systems confer adaptive immunity by integrating short sequences (“spacers”) derived from invasive nucleic acids into arrays of direct repeats (CRISPR arrays) in bacterial and archaeal genomes (Jackson et al., 2017; Mohanraju et al., 2016). The CRISPR array is typically transcribed into a single, long precursor CRISPR RNA (pre-crRNA), which contains spacers specific for multiple invaders interspersed between the repeats. Before these spacers can be employed to direct CRISPR-Cas interference against pathogens, the pre-crRNA must be processed to generate mature crRNA guides, each containing a single spacer and repeat-derived flanking sequences (Brouns et al., 2008; Carte et al., 2008).
CRISPR-Cas systems have been grouped into six types, which differ in their pre-crRNA processing and interference mechanisms (Koonin et al., 2017; Makarova et al., 2015). Type I and III systems use dedicated ribonucleases, mostly of the Cas6 family, for pre-crRNA processing; type II systems rely on RNase III, a host enzyme not associated with CRISPR-Cas loci, and a trans-activating RNA encoded within the locus; and in some type V and VI systems, the pre-crRNA is processed by a multi-functional effector protein that also functions in degrading targeted nucleic acids (Charpentier et al., 2015; East-Seletsky et al., 2016; Fonfara et al., 2016; Hochstrasser and Doudna, 2015). Many type III systems lack a cas6 gene and are thought to employ a Cas6 RNase encoded by other (usually type I) CRISPR-Cas loci present in the same genome for pre-crRNA processing (Hochstrasser and Doudna, 2015; Mohanraju et al., 2016).
Cas6 ribonucleases show limited sequence conservation, but exhibit common structural features, including two RNA recognition motif (RRM) domains, each with a βαββαβ secondary structure (Haurwitz et al., 2010; Hochstrasser and Doudna, 2015). The active site of the nuclease is typically located in the N-terminal RRM, but the catalytic amino acids can vary among Cas6 family members (Haurwitz et al., 2010; Hochstrasser and Doudna, 2015). The majority of pre-crRNA interactions occur on the face of the protein containing the α-helices of the two RRMs (Gesner et al., 2011; Sashital et al., 2011; Shao and Li, 2013; Wang et al., 2011), with three conserved structural features in the C-terminal RRM performing critical functions. The groove-binding element (GBE) located between β1’ and α1’ (prime indicating the second RRM) probes the major groove of the pre-crRNA stem, often providing sequence specificity (Haurwitz et al., 2010); the glycine-rich loop (G-loop) located between α2’ and β4’ stabilizes the pre-crRNA interaction with Cas6 (Carte et al., 2008); and the β hairpin formed by β2’ and β3’ helps position the scissile phosphate of the pre-crRNA in the active site (Shao and Li, 2013). Processing occurs via acid-base catalysis with no requirement for a metal-ion cofactor and produces crRNAs with 5’ OH and 2’3’-cyclic phosphate termini (Carte et al., 2008). In many type I and III systems, cleavage occurs within the repeat, 8-nt upstream of the 5’ end of the spacer sequence, generating an 8-nt repeat-derived 5’ tag on each mature crRNA, which contributes to self versus non-self discrimination (Brouns et al., 2008; Marraffini and Sontheimer, 2008).
A subset of type III CRISPR-Cas loci encode a reverse transcriptase (RT) that is closely related to and thought to have evolved from the RTs encoded by mobile group II introns (Kojima and Kanehisa, 2008; Makarova et al., 2006). In some RT-encoding CRISPR systems, the RT is fused to Cas1, the endonuclease involved in spacer acquisition, including in the type III-B system of Marinomonas mediterranea MMB-1 for which RT-mediated spacer acquisition from RNA has been demonstrated experimentally (Silas et al., 2016). Two subgroups of RT-Cas1 fusion proteins also contain an N-terminal domain of unknown function, which has a G-loop sequence similar to that of known Cas6 proteins (Makarova et al., 2015; Silas et al., 2016; Toro et al., 2017). The CRISPR loci that encode these fusions lack a free-standing cas6 gene, suggesting that this putative Cas6 domain might be functional (Silas et al., 2017b; Toro et al., 2017). Bioinformatic analyses identified a number of other fusion proteins in which either Cas1 or group II intron-like RTs are linked to different proteins acting on nucleic acids or in cellular regulation (Makarova et al., 2013; Zimmerly and Wu, 2015). Thus far, however, little is known about how the different domains of these proteins function in concert or what selective advantage such fusions might provide.
Here we show that the N-terminal domain of the M. mediterranea RT-Cas1 is a fully functional Cas6 and report its crystal structure, which differs notably from those of other Cas6 proteins. Further, we find that this Cas6 domain not only functions in pre-crRNA processing, but also plays a role in the RT-mediated acquisition of spacers from RNA and the regulation of RT activity. Our results reveal a previously unsuspected link between crRNA biogenesis and adaptation, potentially relevant to many CRISPR systems.
RESULTS
Association of Cas6 with CRISPR RTs and RT-Cas1 Proteins
To elucidate the extent and the origin(s) of Cas6 association with CRISPR RTs and RT-Cas1 proteins, we obtained 4,040 representative Cas6 proteins and domains from complete and partial bacterial and archaeal genomes (Methods and Table S1). We then constructed a phylogenetic tree for all Cas6 sequences by combining an approximate maximum likelihood method and UPGMA clustering. The resulting sequence similarity dendrogram shows that these Cas6 homologs fall into 3 major groups: Cas6e (type I-E CRISPR-Cas systems), Cas6f (type I-F CRISPR-Cas systems), and “Cas6 main”, which contain 15 well-supported major branches (branches 3-17) and include the majority of the Cas6 family proteins (Figure 1 and Data S1)(Makarova et al., 2011).
Figure 1. Phylogeny of a Representative Set of Cas6 proteins.
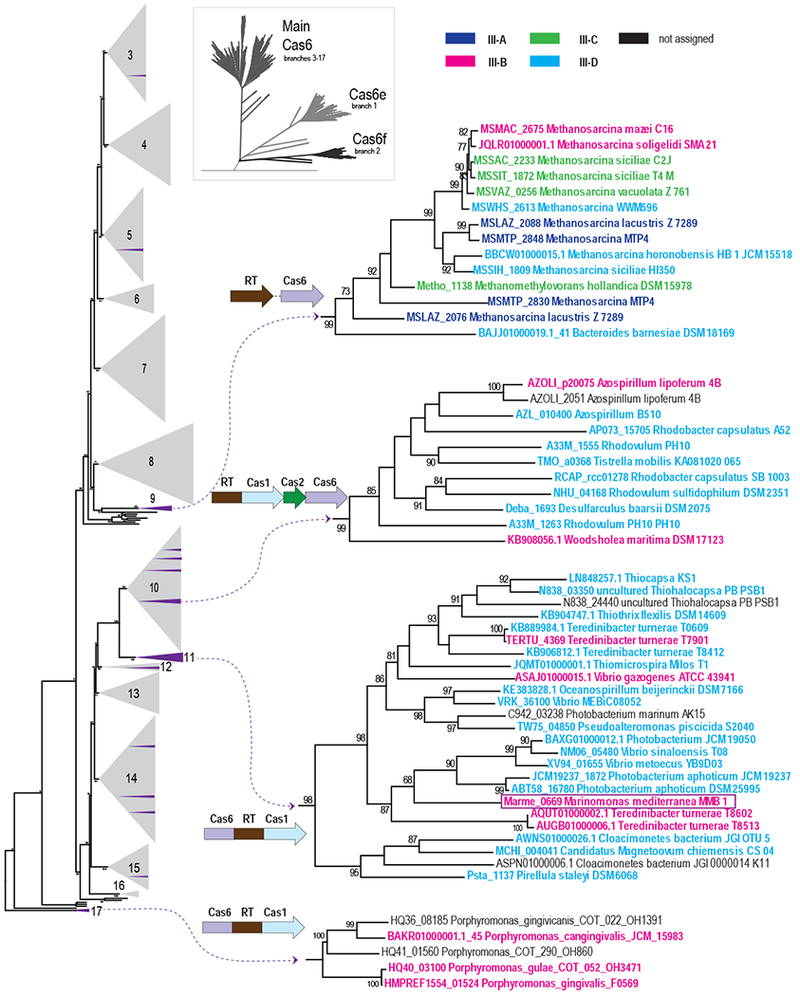
The complete tree was constructed from an alignment of 4,048 Cas6 protein sequences and is depicted schematically in the inset and provided as Data S1. The Cas6 main clade with branches 3 to 17 is shown to the left with collapsed branches shown as numbered gray triangles. Distinct branches that are associated with RT-containing CRISPR-Cas loci are shown in purple with the gray triangles, and four large clades are displayed on the right. Group II intron RTs, RT fragments, and a branch 2 RT associated with Tn7 transposition machinery were excluded from this analysis (see also Figure S1). Each sequence within these clades is identified by a protein locus tag or contig accession number (when a locus tag is unavailable), and a species name; see also Table S1. Branch support values >70% (calculated using FastTree) are indicated. The domain architecture or the most common gene order in the gene neighborhoods of RT and cas6 are shown for each subtree. Additional representative gene orders are shown in Figure S1. The dashed lines connecting the RT and cas6 genes for subtrees from branches 9 indicate that the exact gene arrangement differs among the respective genomes. The CRISPR-Cas subtypes are color-coded as shown at the top right.
The RT genes present in the respective CRISPR-cas loci were mapped onto the tree, (Figure 1; purple branches within gray triangles; Figure S1, Table S1, and Data S1). In agreement with previous observations (Vestergaard et al., 2014), the Cas6 phylogeny did not follow the classification of CRISPR-Cas systems into types and subtypes (Figure 1 and Table S1), supporting the hypothesis that cas6 is a distinct functional module that evolves largely independently of other CRISPR-Cas modules (Makarova et al., 2015). Indeed, some Cas6 proteins show functional promiscuity by cleaving pre-crRNAs from other systems (Carte et al., 2014; Reimann et al., 2017), and the cas6 gene has been identified as a genomic hot spot for recombination between different effector modules (Puigbo et al., 2017). Thus, it is unsurprising that Cas6 proteins from RT-containing CRISPR-cas subtypes are polyphyletic (i.e., scattered across the phylogenetic tree; Figure 1).
Branch 11 contains the great majority of the Cas6-RT-Cas1 fusion proteins, including that found in M. mediterranea MMB-1. As for Cas6, these fusion proteins show no specific association with any type III CRISPR-Cas subtype, nor with any bacterial or archaeal phyla (Figure 1). Clade 11 also includes several Teredinibacter genomes (e.g., T. turnerae T8602) with two III-B loci, one containing Cas6-RT-Cas1 and the other Cas6-RT without a Cas1 domain. Sequence alignments show that the latter contain a conserved Cas6 linked to a truncated RT domain, which lacks the conserved YXDD motif required for RT activity, indicating that these proteins are not functional as RTs (Figure S2A).
Branch 17 contains Cas6-RT-Cas1 fusion proteins found only in several Porphyromonas genomes (Makarova et al., 2015; Toro et al., 2017; 2018). The RT domain of these proteins is highly diverged and forms a distinct long branch in the RT phylogeny (Toro et al., 2018; 2017), and we find the Cas6 domain of these proteins is also highly diverged, resulting in a distinct long branch in the Cas6 tree (Figure 1). Sequence alignments show that the Cas6 domain of the Porphyromonas fusion proteins lacks one of the four conserved glycine residues in the G-loop, while the RT domain is intact, but with a much longer linker between the RT and Cas1 domains than in branch 11 (Figure S2B). Because the branch 17 Cas6-RT-Cas1 fusions form a distinct long branch in the RT phylogeny, Toro and colleagues concluded that Cas6-RT-Cas1 fusions evolved at least twice independently. However, the narrow taxonomic span of this group, the absence of closely related stand-alone Cas6s, and the extreme divergence of both the Cas6 and RT domains leave open the possibility that the Cas6-RT-Cas1 fusion in branch 17 is a “runaway” variant of that in branch 11, which is erroneously placed outside of all major Cas6 clades due to a long branch artifact.
Notably, a number of other Cas6 clades that lack Cas6-RT-Cas1 fusions include CRISPR loci in which a free-standing cas6 gene is located adjacent to or near a gene encoding RT or RT-Cas1 and transcribed in the same direction, with such juxtapositions being most prevalent in branches 9 and 10 (Figure 1 and Figure S1). Branch 9 is specific for Methanomicrobia, mostly Methanosarcina sp., and is associated with a variety of type III CRISPR-Cas systems most of which lack a Cas1-Cas2 adaptation module and acquired an RT independently of other clades (Silas et al., 2017b). In this branch, Cas6 is typically encoded next to but downstream of the RT (Figure S1). Branch 10 corresponds to a group of α-proteobacteria in which the cas6 gene is typically located near an RT-Cas1 gene, sometimes adjacent but often separated by cas2 (Figure 1, Figure S1 and Data S1). The branch 10 adaptation (RT-Cas1) and pre-crRNA processing modules (Cas6) also appear to coevolve and spread horizontally among different type III subtypes. Additional examples of free-standing cas6 genes adjacent to or neighboring and transcribed in the same direction as a CRISPR RT gene are found in branches 3, 12, 14, and 15 (Figure S1). This frequent juxtaposition of genes encoding stand alone Cas6 to those encoding RT or RT-Cas1 in multiple Cas6 branches implies a functional link between Cas6 and RT-containing CRISPR adaptation modules (Figure 1 and Figure S1). Further, the congruence of Cas6 subtrees with the previously published RT and Cas1 trees (Silas et al., 2017b), even in branches where these domains are not fused (e.g., branch 10; Figure S3), suggests that the functional link between Cas6, RT, and Cas1 emerged independently and before the triple fusion. In bacteria, such conserved gene juxtaposition and co-transcription are typically indicative of a functional link and often of formation of a protein complex (Dandekar et al., 1998; Sneppen et al., 2010).
The N-terminal Domain of MMB-1 RT-Cas1 Processes pre-crRNAs
To test whether the N-terminal domain of the M. mediterranea MMB-1 RT-Cas1 protein has pre-crRNA processing activity, we incubated in vitro transcripts of the type III-B CRISPR03 array with purified wild-type (WT) or mutant versions of the protein (Figure 2). The proteins were expressed with a maltose-binding protein (MBP) tag fused to their N-terminus via a non-cleavable rigid linker (denoted MRF) in order to facilitate purification and maintain solubility of the proteins in the absence of bound nucleic acids (Mohr et al., 2013; Silas et al., 2016). Initial assays used either a full-length transcript (1,039 nt), which encompasses the entire CRISPR03 array, or a shorter version with only the first 271 nt of the full-length transcript (up to the first 8 nt of R3; Figure 2A).
Figure 2. The N-terminal Domain of an RT-Cas1 Fusion Protein is a Functional Cas6 Nuclease.
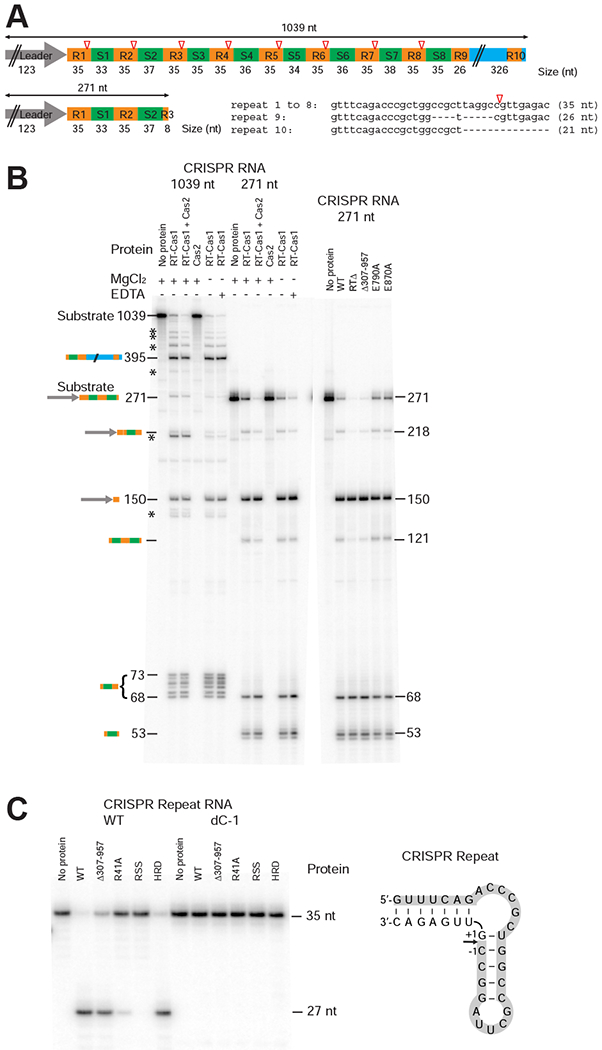
(A) Schematic of the MMB-1 CRISPR03 array transcripts used as substrates for assaying Cas6 activity. The CRISPR array contains a 123-bp leader (gray arrow), 8 identical 35-bp repeats (R1-8; orange), and 8 spacers (S1-8; 33 to 38 bp, green), followed by a partial repeat (R9), and a 326-bp downstream sequence (blue) that includes a truncated repeat (R10). Open red triangles indicate Cas6 cleavage sites 8 nt from the 3’ end of each repeat. (B) Cas6 assays. Reactions were performed using purified WT and mutant proteins (200 nM) and 32P-labeled pre-crRNA in vitro transcripts encompassing the entire 1,039 nt CRISPR03 array (2.75 nM; left) or the first 271-nt of the array from the leader through the first 8 bp of R3 (10 nM; right). The products were analyzed on a denaturing 6% polyacrylamide gel; the identities and sizes (nt) of processed pre-crRNA products are indicated to the left and right of the gels. * indicates partially processed pre-crRNAs. (C) Cas6 activity of WT and mutant Cas6-RT-Cas1 proteins assayed using CRISPR repeat-containing RNA oligonucleotide substrates. 5’-labeled RNA oligonucleotides (35-nt; 100 nM) were incubated with the indicated proteins (250 nM), and the products were analyzed on a denaturing 10% polyacrylamide gel. The sequence and predicted secondary structure of the 35-nt WT RNA oligonucleotide is shown to the right, with shading indicating the labeled 27-nt fragment resulting from Cas6 cleavage. The dC-1 RNA oligonucleotide has a deoxy C residue at position −1 from the cleavage site. The R41A, RSS (R41A, S46A, S51A), and HRD (H196A, R197A, D200A) mutants are described later in the text.
Both pre-crRNA transcripts were processed efficiently into discrete fragments by the WT protein, whereas no cleavage was observed in the absence of the protein (Figure 2B). Three fragments were shared between the cleavage products of the full-length and short transcripts: 218- and 150-nt bands corresponding to the leader and portions of the leader proximal spacer (S1) and repeats (R1 and R2), and a 68-nt band corresponding to S1 (33 nt) with processed repeat-derived flanks (8-nt 5’ and 27-nt 3’ flanks). The short (271-nt) pre-crRNA transcript also yielded a 53-nt product corresponding to the second spacer (37 nt) with an 8-nt 5’ tag derived from the second repeat, and 8 nt of the artificially truncated third repeat. Processing of the full-length (1,039 nt) transcript additionally produced five RNAs in the 69-73 nt range, representing crRNAs containing the other spacers (34-38 nt) with repeat-derived flanks. The identity of all these products was confirmed by RNA-seq, using a strand-specific thermostable group II intron RT (TGIRT) sequencing method that enables the precise mapping of both 5’ and 3’ RNA ends (Figure S4).
The pre-crRNA processing activity of the Cas6-RT-Cas1 protein was only minimally affected by adding Cas2, which functions in complex with RT-Cas1 for CRISPR adaptation (Silas et al., 2016), and purified Cas2 alone did not cleave the pre-crRNA (Figure 2B, left). The Cas2 protein used in these assays was verified to be active in Cas1/Cas2-mediated spacer acquisition. The fusion protein produced the same cleavage patterns in the presence or absence of Mg2+ or EDTA (Figure 2B), indicating that the pre-crRNA cleavage does not require a metal ion cofactor, consistent with acid-base catalysis, as shown for other Cas6 proteins (Carte et al., 2008). The Cas6 cleavage site within the repeat was confirmed by additional assays with WT and mutant fusion proteins using a synthetic 35-nt RNA corresponding to the repeat sequence (Figure 2C, left lanes). Further, replacement of the C residue immediately upstream of the cleavage site in this RNA with a deoxy C-residue (dC-1) abolished cleavage, as expected for acid-base catalysis (Figure 2C, right lanes).
Additional assays showed that mutant Cas6-RT-Cas1 proteins with the RT-domain deleted (RTΔ) or the Cas1 endonuclease inactivated (E790A, E870A) cleaved pre-crRNA with efficiencies similar to WT protein (Figure 2B, right), indicating that the RT and Cas1 activities of the protein are not required for pre-crRNA processing. Confirming this conclusion, a truncated protein consisting of only the N-terminal Cas6 domain (Δ307-957) processed the pre-crRNA to yield the same products as the full-length protein (Figure 2B, right and 2C). Together, these results indicate that the N-terminal domain of the MMB-1 fusion protein is an active Cas6 nuclease that can function independently of the remainder of the protein to process pre-crRNAs.
Crystal Structure of the Cas6 Domain
We obtained a 2.85-Å crystal structure of an N-terminal MBP fusion of the Cas6 domain, comprised of the 306 N-terminal residues shown above to have Cas6 activity in vitro (Table 1 and Figure 3A). The asymmetric unit contains two MBP-Cas6 proteins, which differ slightly in the angle between MBP and Cas6 (Figure S5A). Although the two Cas6 monomers share a symmetric dimer interface (burying 1,150 Å2), it is composed primarily of weak van der Waals interactions and differs from the interfaces in other Cas6 protein dimers (Hochstrasser and Doudna, 2015; Reeks et al., 2013), suggesting a crystallographic dimer that is not physiologically relevant.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Data Collection and Refinement Statistics | MMB-1 Cas6 |
|---|---|
| Data Collection | |
| PDB ID | 6DD5 |
| Wavelength (Å) | 0.9765 |
| Resolution range (Å) | 48.0−2.85 (2.95−2.85) |
| Space group | P212121 |
| Unit cell: a, b, c (Å) | 92.3, 110.8, 192.9 |
| Total reflections | 695,870 (69,476) |
| Unique reflections | 46,940 (4,611) |
| Multiplicity | 14.8 (15.1) |
| Completeness (%) | 100 (100) |
| Mean I/sigma (I) | 17.7 (2.4) |
| Wilson B-factor | 54.1 |
| R-merge | 0.194 (1.43) |
| R-meas | 0.201 (1.48) |
| R-pim | 0.052 (0.380) |
| CC1/2 | 0.997 (0.777) |
| CC* | 1 (0.935) |
| Refinement | |
| Reflections used in refinement | 46,929 (4,610) |
| Reflections used for R-free | 2,277 (202) |
| R-work | 0.187 |
| R-free | 0.225 |
| CC (work) | 0.954 |
| CC (free) | 0.924 |
| Number of non-hydrogen atoms | 10,268 |
| macromolecules | 10,125 |
| ligands | 143 |
| Protein residues | 1,297 |
| RMS (bonds) | 0.006 |
| RMS (angles) | 0.78 |
| Ramachandran favored (%) | 98.0 |
| Ramachandran allowed (%) | 2.0 |
| Ramachandran outliers (%) | 0.0 |
| Rotamer outliers (%) | 0.67 |
| Clashscore | 1.2 |
| Average B-factor | 55.2 |
| macromolecules | 54.8 |
| ligands | 78.8 |
| Number of TLS groups | 6 |
Statistics for the highest resolution shell are shown in parentheses. TLS, translation-libration-screw.
Figure 3. Crystal Structure of the MMB-1 Cas6 Domain.
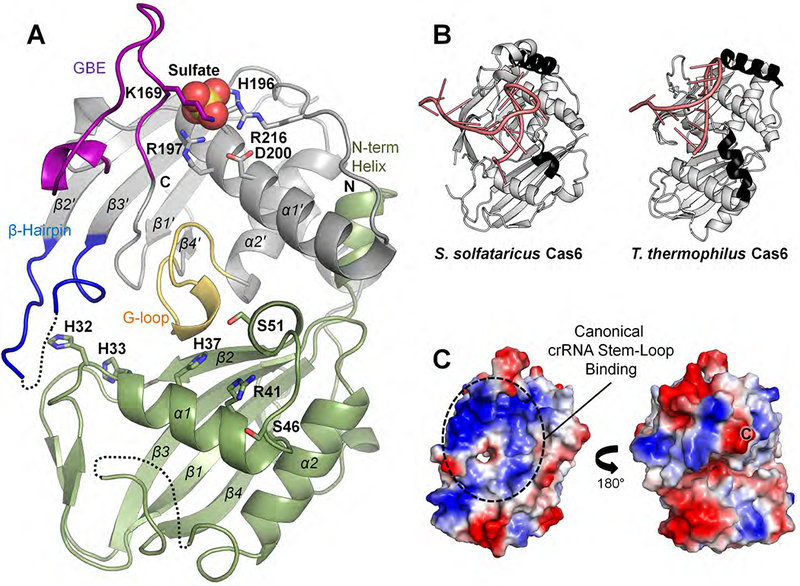
(A) Structure of the MMB-1 Cas6 monomer, with canonical RRM-fold α-helices and β-sheets labeled (N-terminal RRM domain, olive; C-terminal RRM domain, grey; conserved G-loop domain, yellow; groove-binding element (GBE), purple; β-hairpin motif, blue; sulfate ion, spheres). Black dotted lines represent disordered loops. Side chains for key residues discussed in the text are depicted in stick form. (B) Structures of S. solfataricus Cas6 (SsoCas6-1A, PDB: 4ILL) and T. thermophilus Cas6 (TthCas6B, PDB: 4C9D), which are the closest available structural homologs of MMB-1 Cas6. Additional surface helices present on the crRNA (pink) stem-loop binding surface are in black. (C) Two views at 180° rotation of the electrostatic surface potential of the MMB1 Cas6 monomer (red, negative; blue, positive; “C”, C-terminal residue). The left panel shows the same orientation as panel (A).
The MMB-1 Cas6 adopts the typical two-domain ferredoxin/RRM fold that is found in other Cas6 proteins and more broadly in the RAMP (Repeat Associated Mysterious Proteins) superfamily of Cas proteins (Hochstrasser and Doudna, 2015; Makarova et al., 2015)(Figure 3A). Both RRM domains share the β1α1β2β3α2β4 secondary structure arrangement, with the conserved G-loop motif residing at the interface between the two RRM domains. Disordered loops are found between α2 and β4 in the N-terminal RRM, and between β2’ and β3’ at the tip of the β-hairpin loop in the C-terminal RRM, which is often involved in crRNA stem-loop binding (Hochstrasser and Doudna, 2015; Shao and Li, 2013). The GBE between β1’ and α1’, which is occasionally disordered in structures lacking crRNA (Wang et al., 2011), is fully visible in both monomers of the asymmetric unit, likely due to crystallographic interactions with the neighboring MBP moieties. The final 13 residues in our construct are disordered in both monomers.
A comparison to protein structures available at the Protein Data Bank using the DALI software (Holm and Rosenström, 2010) identified free-standing Cas6 proteins from Sulfolobus solfataricus (4ILL and 3ZFV, RMSD = 3.3Å) and Thermus thermophilus (4C9D, RMSD =3.4Å) (branches 13 and 14, respectively; Figure 1) as having the highest structural similarity to MMB-1 Cas6 (Figure 3B). Architecturally, MMB-1 Cas6 differs from these and other free-standing Cas6 structures primarily in the arrangement of helices within the two RRM domains. In many previously solved Cas6 structures, the surface of the protein that binds the crRNA stem-loop contains additional helices (black in Figure 3B), which could be adaptations to accommodate specific crRNA shapes (Hochstrasser and Doudna, 2015; Shao and Li, 2013). By contrast, the MMB-1 Cas6 lacks such additional surface helices, resulting in a deep trench along the putative crRNA stem-loop binding surface (Figure 3A and Figure S5B). Further, the N-terminus of the MMB-1 Cas6 forms an additional short helix, which is not seen in previous Cas6 structures and packs beneath α1’. As a result, the α1’-helix curves upward toward the crRNA loop binding region and forms a pincer-like structure with the GBE at one end of the trench (Figure 3A). The putative crRNA stem-loop binding face is positively charged, similar to most Cas6 family members (Figures 3C and S5B). The opposite face, where single-stranded crRNA binds in some homologues (Figure 3C, right), is less positively charged than in other Cas6 proteins (Hochstrasser and Doudna, 2015). This region might interact with the RT domain in the fusion given that the C-terminus of the Cas6 domain emerges from this face.
In the N-terminal RRM domain of MMB-1 Cas6, the α1-helix contains several amino acid residues that might function in catalyzing the RNA-cleavage reaction. Active-site residues are not strictly conserved in the Cas6 family, but many Cas6 proteins contain an essential His, Arg, or Ser in the active site (Hochstrasser and Doudna, 2015). Combining the evidence from sequence conservation (Figure S1) and structural comparisons, we identified H32, H33, H37, R41 and two nearby serines (S46 and S51), all located in and around the α1-helix, as potential active-site residues (Figure 3A). The H32 and H33 residues are toward the N-terminus of α1, whereas H37 and R41 are near the C-terminus of the same helix, closer to the canonical location of active-site residues in other Cas6 proteins (Hochstrasser and Doudna, 2015).
Finally, at the base of the GBE in the C-terminal RRM domain, we observed a prominent sulfate ion that is sandwiched between the backbone amide and side chain of K169 on one side and R197 and R216 on the other side (Figure 3A), along with a trail of other less well-defined sulfates nearby in the canonical crRNA stem-loop binding region (Figure S5C). The position of the prominent sulfate ion might correspond to that of a pre-crRNA backbone phosphate during substrate binding.
Identification of Cas6 Domain Residues Required for Cas6 Catalytic Activity
To identify residues required for Cas6 activity, we constructed mutants with single, double, or triple alanine substitutions of potential active-site residues located in and around the α1-helix: H32A, H33A, H37A, R41A, S46A, S51A, SS (S46A, S51A double mutant), and RSS (R41A, S46A, S51A triple mutant). Mutant versions of the Cas6-RT-Cas1 protein were expressed with N-terminal MBP and C-terminal 8xHis tags, and the purified proteins were assayed for three biochemical activities: (i) pre-crRNA cleavage (Cas6) activity using the short 271-nt CRISPR RNA or 35-nt repeat RNA oligonucleotide as substrate (Figure 4A, 4B and Figure S6A); (ii) RT activity using the artificial template-primer substrate poly(rA)/oligo(dT)24 (Figure 4C); and (iii) Cas1-Cas2-mediated integration of 29-nt DNA or 35-nt RNA oligonucleotides (protospacers) into a CRISPR03 DNA substrate (Figure 4D).
Figure 4. Biochemical Assays with WT and Mutant MMB1 Cas6-RT-Cas1 Proteins.
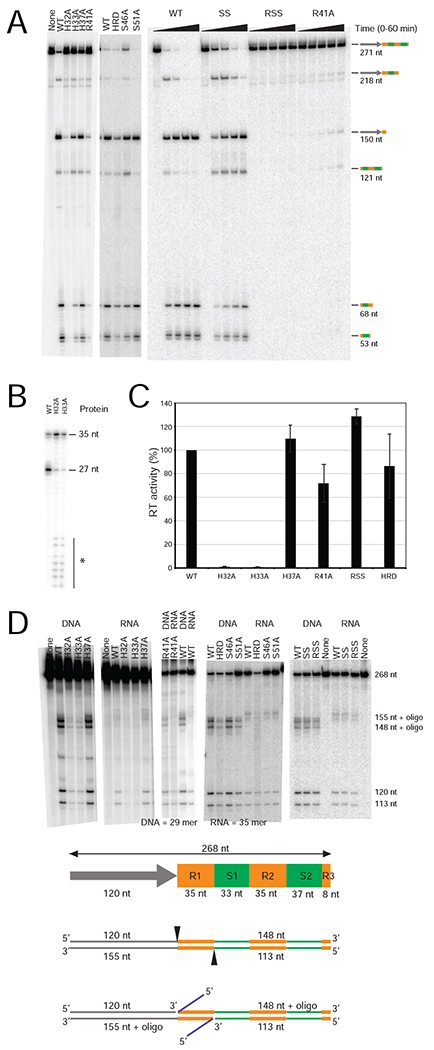
(A) Cas6 assays. Reactions were performed with WT and mutant Cas6-RT-Cas1 proteins (320 nM; left panel; 200 nM; middle and right panels) and 32P-labeled 271-nt CRISPR03 array transcript (10 nM) for 30 min (left gels), or over a time course of 0 to 1 h (right gel). The products were analyzed, as described in Figure 2. (B) Cas6 activity of WT and mutant Cas6-RT-Cas1 proteins assayed using a CRISPR repeat RNA oligonucleotide substrate. 5’-labeled RNA oligonucleotide (35-nt; 250 nM) was incubated with the indicated proteins (250 nM), and the products were analyzed on a denaturing 10% polyacrylamide gel. * indicates small RNA fragments due to nuclease contamination. (C) RT assays. Polymerization of 32P-dTTP was measured using poly(rA)/oligo(dT)24 substrate, and RT activities were normalized to a parallel WT control. The bar graphs show the mean for at least three independent experiments with the error bars indicating the standard deviation. (D) In vitro spacer acquisition assays. WT or mutant Cas6-RT-Cas1 and Cas2 were incubated with an internally 32P-labeled CRISPR03 DNA (268 bp), in the presence or absence of 29-nt ssDNA or 35-nt ssRNA oligonucleotide protospacers, and the products were analyzed on a denaturing 6% polyacrylamide gel. The schematic below the gel shows the 268-bp CRISPR03 DNA consisting of 120-bp leader (gray arrow), two full repeats (35 bp, yellow), two full spacers (33 and 37 bp, green), and a partial repeat (8 bp). Cas6-RT-Cas1 + Cas2 predominantly cleaves at the 5’ ends of the first repeat (black triangles), resulting in 5’ DNA fragments of 113 and 120 nt and ligation of oligonucleotides (blue lines) to the 148 and 155 nt 3’ DNA fragments. “None” indicates no protein added.
The activity of the Cas6 domain in crRNA cleavage was tested first at high protein concentrations relative to the RNA substrate (Figure 4A and4B). Under these conditions, the H32A and H33A mutants showed weak, variable Cas6 activity and weak Cas1 integrase activity with a DNA protospacer, but no detectable RT activity and little Cas1 integrase activity with an RNA protospacer (Figure 4A-D). Both mutant proteins were difficult to purify, suggesting structural defects in vitro. The H37A mutant, which appeared to be the prime candidate for an active-site residue based on its similar location to conserved histidines in the active sites of other Cas6 proteins (Hochstrasser and Doudna, 2015), had no effect on Cas6, RT, or Cas1 activities (Figure 4A-D). The S46A, S51A and SS double mutant (S46A, S51A) showed little (S46A, SS) or no (S51A) decrease in pre-crRNA cleavage and no decrease in the other tested activities (Figure 4A-D).
By contrast, mutant proteins with R41A substitutions (R41A and the RSS triple mutant R41A, S46A, S51A) showed strongly decreased pre-crRNA cleavage activity (<5% WT activity; Figure 4A), whereas its RT and Cas1 integrase activities were only weakly affected (<two-fold decrease relative to WT; Figure 4C and 4D). Likewise, in assays under RNA excess conditions (1 μM RNA/40 nM protein), these R41 mutants showed little or no cleavage of the 35-nt repeat RNA, whereas the WT protein performed multiple turnover catalysis as indicated by the large molar excess of cleaved repeat RNA over input protein (Figure S6A). Additionally, both the R41A and RSS mutants showed strongly decreased binding affinity for the non-cleavable CRISPR-repeat RNA containing dC-1 (Kds = 110 and 182 nM for R41A and RSS respectively, compared to 3.2 nM for WT protein; Figure S6B). In all of the above assays, the RSS mutant cleaved and bound pre-crRNA to a lesser degree than the R41A mutant (Figure 4A and Figure S6A-C), likely due to the S46A mutation, which by itself weakly inhibited Cas6 activity (Figure 4A). Together, these findings identify R41 as a critical residue of the Cas6 active site, possibly with some contribution from S46.
Identification of Cas6 Domain Residues Involved in pre-crRNA binding
We also constructed a triple mutant (HRD; H196A, R197A, D200A) with alanine substitutions in three residues at the sulfate-binding site proximal to the GBE. The HRD mutant showed little decrease in pre-crRNA processing activity under protein excess conditions (Figure 4A), but a pronounced decrease under RNA excess conditions (~30% WT product after 1 hr; Figure S6A). It also exhibited a >30-fold decreased binding affinity for pre-crRNA (Figures S6B and S6C; Kd = 127 nM; koff = 1.5 × 10−2 sec−1 compared to <1.2 ×10−3 sec−1 for the WT protein). By contrast, the HRD mutant retained high RT and Cas1-Cas2 integrase activities with either RNA or DNA oligonucleotides in vitro (Figure 4). These findings suggest that one or more of the residues in the sulfate-binding site in the crystal structure contribute to pre-crRNA binding, as further supported by in vivo assays below.
The Cas6 Activity of the Cas6-RT-Cas1 Protein is Required for Pre-crRNA Processing In Vivo
To assess the requirements for pre-crRNA processing in the type III-B system in vivo, we deleted the entire type III-B CRISPR locus from the M. mediterranea MMB-1 chromosome (III-B OperonΔ) and expressed the pre-crRNA processing and CRISPR adaptation factors (Cas6-RT-Cas1, Cas2, Marme_0670, and the CRISPR03 array) from a plasmid (Figure 5A). Pre-crRNA processing was assayed by RNA-seq, using a protocol that preserves strand information and faithfully reports the 3’ end of RNAs (Silas et al., 2018). The presence of a distinct 3’-end sequence in the population of CRISPR repeat containing RNAs can be used as a readout of pre-crRNA processing at the respective site in the CRISPR repeat.
Figure 5. Pre-crRNA Processing and CRISPR Adaptation by WT and Mutant Cas6-RT-Cas1 Proteins In Vivo.
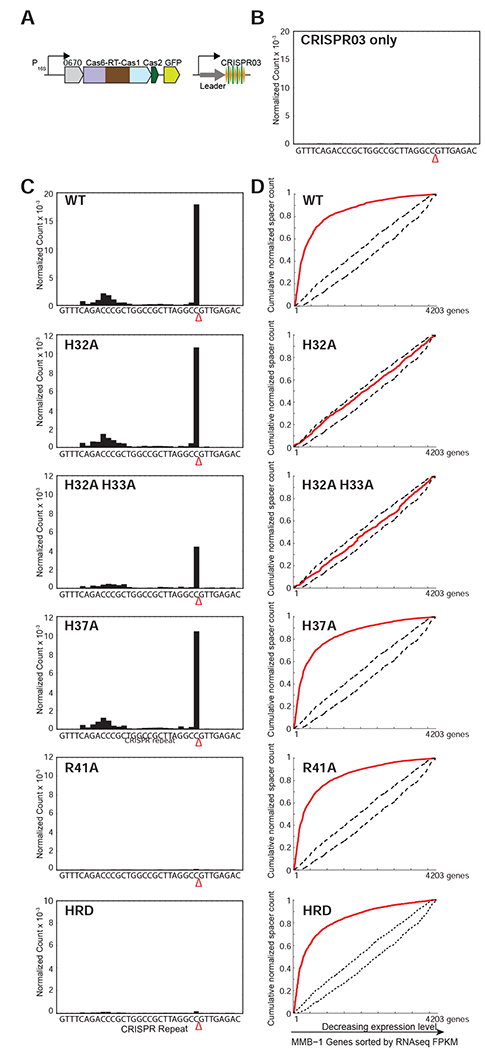
(A) Schematic of type III-B adaptation components (Cas6-RT-Cas1, Cas2, Marme_0670, along with GFP, and the CRISPR03 array) supplied on plasmid pKT230. Experiments were performed in the M. mediterranea MMB-1 III-B OperonΔ strain from which the endogenous type III-B CRISPR-Cas system was deleted, and assays were replicated with two independent transconjugant strains for each mutant. (B) Pre-crRNA processing in an empty vector control in which CRISPR array is supplied on a plasmid without type III Cas proteins. No processed crRNAs were detected by high-throughput small RNA sequencing. (C) Pre-crRNA processing by WT and mutant Cas6-RT-Cas1 proteins. WT and mutant Cas6-RT-Cas1 proteins and the CRISPR03 array were supplied on plasmid pKT230 (see above). The graphs show processed crRNA levels assayed by high-throughput small RNA sequencing. Counts are normalized to isoleucine tRNA levels (consistently the most abundant species encountered) and each experiment includes data from two independent transconjugants. The presence of a distinct 3’ end sequence in the population of CRISPR-repeat containing RNAs indicates site-specific cleavage and processing of pre-crRNA. (D) RNA or DNA spacer acquisition by WT and mutant Cas6-RT-Cas1 proteins in vivo. Assays were performed with the same cultures used in (C). Newly integrated CRISPR spacers were mapped to the MMB-1 genome. Red lines in graphs show cumulative distributions of newly acquired spacer pools (N = 500-10,000 spacers in each pool), plotted against the MMB-1 genes they were derived from sorted by expression level. WT, H37A, R41A, and HRD variants of Cas6-RT-Cas1 show preferred acquisition from highly expressed genes, suggesting spacer acquisition from RNA. Dotted black lines show expectation from random assortment (Monte-Carlo bounds: no transcription-related bias).
We observed robust processing of the type III-B CRISPR03 pre-crRNA when the CRISPR03 array and WT Cas6-RT-Cas1 and Cas2 were supplied, but no discernible signal in the control with only the CRISPR03 array (Figures 5B and 5C). Consistent with reports on other CRISPR systems (Carte et al., 2008) and our in vitro assays (Figure S4), the pre-crRNA cleavage site was found to be within the repeat 8-nt upstream of the spacer sequence (Figure S7).
Next, we tested the effect of mutations in the Cas6 domain in vivo. The H32A, H32A-H33A double substitution, and H37A mutants all retained pre-crRNA processing activity in vivo (Figure 5C), indicating that none of these residues is required for pre-crRNA processing. By contrast, the R41A mutation abolished pre-crRNA processing in vivo, as did the HRD triple mutation of residues identified above as being involved in CRISPR repeat binding (Figure 5C). These results show that the Cas6 domain of the Cas6-RT-Cas1 fusion protein is required in vivo for pre-crRNA processing in the III-B OperonΔ background. The MMB-1 genome also contains a type I-F CRISPR system, which was left intact in our experiments (Silas et al., 2017a). The processing defects in the CRISPR03-only control vector, the R41A active-site mutant, and the HRD mutant indicate that neither the type I-F CRISPR system nor other enzyme present in the host are capable of processing the CRISPR03 transcript.
The Cas6 Domain of Cas6-RT-Cas1 is Required for RNA but not DNA Spacer Acquisition In Vivo
The fusion of the RNA spacer acquisition and pre-crRNA processing machineries in the MMB-1 Cas6-RT-Cas1 protein raises the possibility of a functional relationship between these two processes. Because the RT and Cas1 domains do not contribute to pre-crRNA processing in vitro (see Figure 2), we speculated that this functional relationship could instead involve a requirement for the Cas6 domain for RNA spacer acquisition.
Thus, we tested the WT and various Cas6 mutant proteins in an in vivo spacer acquisition assay (Silas et al., 2016). We expressed WT and mutant Cas6-RT-Cas1 proteins and the CRISPR03 array along with other adaptation factors from the same plasmids that were used for Cas6 assays (Figure 5A) and assayed for integration of new spacers into the plasmid copy of CRISPR03 (Silas et al., 2016). To determine whether new spacers were being acquired from RNA or DNA, we plotted the cumulative spacer count (normalized by the length of the source genes) against MMB-1 genes ordered by decreasing expression level (Figure 5D). A positive correlation between the frequency of spacer acquisition and gene expression level (convex curve) indicates spacer acquisition from RNA, whereas a lack of correlation (linear plot) indicates spacer acquisition from DNA. The WT Cas6-RT-Cas1 protein supported RNA spacer acquisition in vivo, as indicated by the strong preference for spacer capture from highly expressed genes (Figure 5D).
We next tested whether the pre-crRNA processing activity of the Cas6 domain was required for RNA spacer acquisition. The R41A mutation in the Cas6 active site and the HRD mutations in the Cas6 pre-crRNA binding cleft, both of which abrogated pre-crRNA processing in the same strains (Figure 5C), did not affect spacer acquisition from RNA in vivo (Figure 5D). Thus, the catalytic activity of Cas6 is not required for RNA spacer acquisition, and the pre-crRNA processing activity of Cas6-RT-Cas1 can be selectively abrogated while retaining the function of the RT and Cas1 domains.
The H32A and H33A mutants in the Cas6 domain were of particular interest because they retained Cas6 activity in vivo (Figure 5C), but had no detectable RT activity in vitro (Figure 4C). Consistent with the in vitro RT defects, both the H32A and H32A-H33A mutants exhibited a complete loss of correlation between spacer capture and host RNA transcript levels in vivo, indicating that they retained the ability to acquire spacers from DNA, but lost the ability to acquire spacers from RNA (Figure 5D). By contrast, the nearby H37A mutation in the Cas6 domain, which had no effect on RT activity in vitro (Figure 4C), still supported robust spacer acquisition from RNA in vivo (Figure 5D).
We wondered whether the RT defects observed with substitutions of H32 and H33 might indicate a broader requirement of the Cas6 domain for RT activity. We therefore assayed deletions of either the entire Cas6 domain (Δ5-289) or the region containing the signature G-loop of Cas6 alone (Δ256-289) (Figure 6A). Both mutants supported robust spacer acquisition from DNA but not RNA (Figures 6A and 6B). These findings indicate that the Cas6 domain is required for spacer acquisition from RNA. Additionally, the finding that H32A and H33A mutations inhibit RT activity (Figure 4C), while retaining Cas6 and Cas1 activities in vivo suggests that these residues may play a role in mediating a specific interaction between the Cas6 and RT domains (see Discussion).
Figure 6. Interaction Between the Cas6 and RT Domains of the Cas6-RT-Cas1 Fusion Protein.
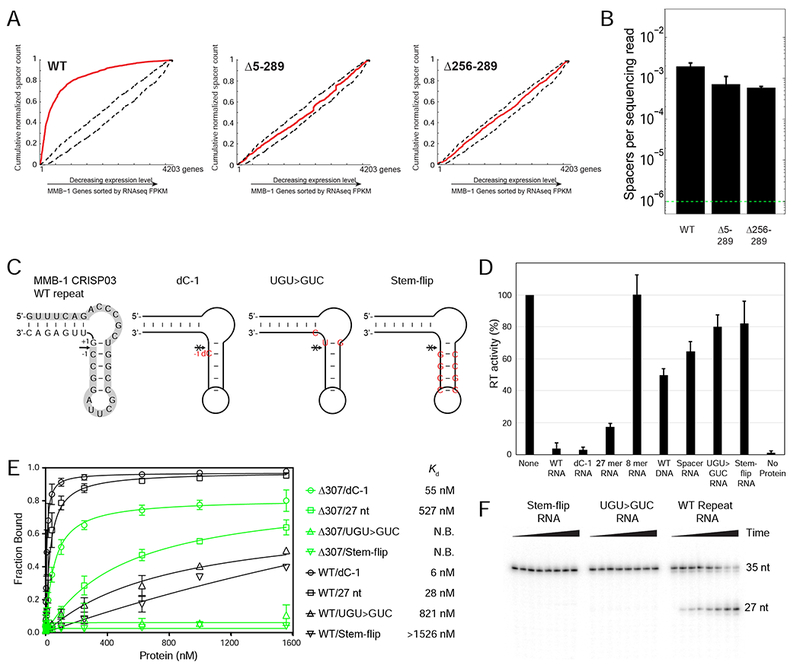
(A) In vivo CRISPR adaptation assays with WT and mutant Cas6-RT-Cas1 proteins from which the Cas6 domain (Δ5-289) or the Cas6 G-loop region (Δ256-289) were deleted. Experiments were performed as in Figure 5D, but in WT MMB-1 instead of the III-B OperonΔ genetic background. (B) Spacer detection frequency upon expression of plasmid-supplied CRISPR adaptation components as in (A). Range bars depict spacer detection frequencies for two biological replicates. The dotted green line indicates the detection limit of the assay. (C) Sequence and predicted structure of the WT and mutant MMB-1 CRISPR repeat RNA oligonucleotides with the arrow indicating the cleavage site and the 5’ 27-nt cleavage fragment highlighted in gray. The WT repeat is on the left, followed by the non-cleavable dC-1 variant, which has a single deoxy C (dC) substitution immediately upstream of the cleavage site (position −1). The UGU>GUC mutant has 3 nucleotide changes (red) that flip the G-U base-pair at the cleavage site and introduce a U to C mutation at position +2 from the cleavage site. The stem-flip variant of the CRISPR repeat has 4-bp in the stem changed to their complements (red). (D) RT activity measured by polymerization of 32P-dTTP using poly(rA)/oligo(dT)24 substrate in the presence or absence of 45 μM of WT or mutant CRISPR repeat RNA oligonucleotides. Activities are expressed as percent of a parallel no-oligonucleotide control (“None”). The bar graphs show the mean for three independent experiments with the error bars indicating the standard deviation. (E) Nitrocellulose filter binding assays for binding of WT Cas6-RT-Cas1 (black) or the isolated Cas6 domain (Δ307-957; green) to WT or mutant CRISPR repeat RNA oligonucleotides. The data were fit to a one-site binding model, and the Kd was obtained from the fit. N.B. denotes no binding. The experiment was repeated three times, with the range bars indicating the standard deviation. (F) Cas6 cleavage assays with WT and mutant CRISPR-repeat RNAs. 5’-labeled RNA oligonucleotides (35-nt; 1 μΜ) were incubated with 40 nM WT Cas6-RT-Cas1 protein. Samples were taken at intervals up to 1 hr, and products were analyzed on a denaturing 12% polyacrylamide gel to detect the appearance of the 5’-labeled 27-nt fragment.
Binding of the crRNA Repeat to the Cas6 Domain Inhibits RT Activity of the Cas6-RT-Cas1 Protein
To further explore the nature of the interaction between the Cas6 and RT domains of the MMB-1 Cas6-RT-Cas1 protein, we performed in vitro RT assays with poly(rA)/oligo(dT)24 in the presence or absence of a roughly equimolar concentration of RNA oligonucleotides corresponding to WT or mutant CRISPR repeat sequences (Figures 6C and 6D). Addition of either a cleavable CRISPR repeat RNA oligonucleotide (WT), a non-cleavable oligonucleotide containing a deoxy C substituted at the cleavage site (dC-1; Figure 6C), or a 27-nt RNA corresponding to the 5’ fragment of cleaved CRISPR repeat strongly inhibited the RT activity of the fusion protein (4%, 3% and 17% of the control reaction in the absence of added oligonucleotide, respectively; Figure 6D). By contrast, the 8-nt cleaved 3’ part of the CRISPR repeat, a 35-nt DNA oligonucleotide with the WT repeat sequence, or an RNA oligonucleotide corresponding to a spacer sequence acquired in one of our in vivo experiments (see Star Methods) had less or no effect on RT activity (100%, 45% and 60% of the control reaction, respectively).
To exclude the possibility that the inhibition of RT activity by the CRISPR repeat RNA was due to non-specific binding of the oligonucleotide to the RT domain, we tested mutant CRISPR repeats in which the 3 nucleotides around the cleavage site were replaced without changing the predicted secondary structure (UGU>CUG mutant), or the 4 GC base pairs forming the central stem of the repeat were flipped (stem-flip mutant; Figure 6C). Both mutations strongly decreased the binding and cleavage of the repeat RNA by the Cas6 domain (Figure 6E, F), and concomitantly, both mutant RNAs lost the ability to substantially inhibit RT activity (Figure 6D). Together, these findings show that specific binding of the crRNA repeat or its 5’-cleavage product by the Cas6 domain inhibits the RT activity of the fusion protein. The results further indicate a structural or functional interaction between the Cas6 and RT domains and suggest that binding of pre-cRNA by the Cas6 domain may regulate RNA spacer acquisition.
DISCUSSION
We characterized a multidomain Cas6-RT-Cas1 fusion protein that is a component of the CRISPR adaptation module in different type III CRISPR-Cas systems in a variety of bacteria. We showed previously that this fusion protein from M. mediterranea MMB-1 uses its RT and Cas1 domains in concert to promote spacer acquisition from RNA (Silas et al., 2016). Here, we show by phylogenetic analysis that the N-terminal domain of the MMB-1 RT-Cas1 protein belongs to a distinct branch of the Cas6 family and demonstrate experimentally that this Cas6 domain is responsible for pre-crRNA processing. This processing activity depends on specific binding of the CRISPR repeat RNA to the Cas6 domain, is multi-turnover, and has the characteristics expected of acid-base catalysis (Figures 2 and S6A). We also identified residues required for Cas6 activity through structural and mutational analyses and assessed the relationship between Cas6 and the fused CRISPR adaptation machinery. Importantly, our phylogenetic analysis indicates that a stable association of Cas6 with RT or RT-Cas1 proteins is a general phenomenon that occurred multiple times in different lineages, whereas Cas6-RT-Cas1 fusion is a relatively rare event that occurred only once or twice, after the functional link between these proteins was already established. Moreover, the repeatedly observed proximity and likely co-transcription of the free-standing cas6 and RT or RT-cas1 genes in different lineages suggests that these proteins might function in a complex (Dandekar et al. 1998; Sneppen et al., 2010), a possibility supported by the functional interaction between the Cas6 and RT domains in the fusion protein.
CRISPR-Cas adaptive immunity includes three stages: (i) adaptation (spacer acquisition); (ii) crRNA biogenesis via pre-crRNA processing; and (iii) target interference. Each stage is mediated by a distinct suite of Cas proteins that comprise functional modules of the CRISPR-Cas system. The partial independence of these modules is supported by the observations of frequent module shuffling between CRISPR-Cas loci in bacteria and archaea (Makarova et al., 2015). Such shuffling is particularly common among type III CRISPR-Cas systems, and our previous analyses showed that the Cas6-RT-Cas1 fusion protein can combine with a broad variety of type III effector modules (Silas et al., 2017b). Nevertheless, the separation between the stages of the CRISPR immune response is far from complete. For example, in subtype V-A and subtype VI-A systems, the same multi-domain protein is responsible for both pre-crRNA processing and interference (East-Seletsky et al., 2016; Swarts et al., 2017), whereas in subtype II-A, the Cas9 effector protein is essential for adaptation although its nuclease activity is not required (Wei et al., 2015).
Our finding that the MMB1 RT-Cas1 fusion protein contains an active Cas6 domain establishes a link between pre-crRNA processing and CRISPR adaptation in certain type III CRISPR-Cas systems. These findings are compatible with the modular evolution of CRISPR-Cas. Indeed, our previous and present analyses of the evolution of RT-containing type III systems ((Silas et al., 2017b) and Figure 1, respectively) strongly suggest pronounced horizontal mobility of the gene that encodes the Cas6-RT-Cas1 fusion protein. The three domains thus comprise a distinct, mobile CRISPR-Cas module.
Our mutational analyses of the three domains (Cas6, RT, Cas1) indicate that the pre-crRNA processing and RNA spacer acquisition functions of the fusion protein can be partially separated. Thus, it is possible to remove the RT domain or mutationally inactivate the Cas1 domain in the fusion protein, while still retaining Cas6 pre-crRNA processing activity, and the isolated Cas6 domain is by itself active in processing pre-crRNAs in vitro. Conversely, the R41A active-site mutation abolishes Cas6 activity in vitro and in vivo, but has little if any effect on RT activity in vitro or the ability to acquire spacers from RNA in vivo.
Despite this partial independence, the deleterious effect of certain mutations in the Cas6 domain on RT activity implies that structural or functional interactions between the Cas6 and RT domains are required for spacer acquisition from RNA. Deletion of all or part of the Cas6 domain as well as mutations in the conserved H32 and adjacent H33 all led to a failure of spacer acquisition from RNA (but not DNA) in vivo, a phenotype that mimics that of deletion of the RT domain (Silas et al., 2016). The H32A substitution is particularly cogent, as it is a Cas6 domain mutation that retains both Cas6 and Cas1 activities in vivo (Figures 5C and 5D), but abolishes RT activity (Figure 4C), accounting for the defect in RNA spacer acquisition (Figure 5D). A functional interaction between the Cas6 and RT domains is further indicated by our finding that specific binding of CRISPR-repeat RNAs to the Cas6 domain inhibits RT activity, a potential mechanism for coordinating the crRNA biogenesis and RNA spacer acquisition functions of the protein (Figure 6).
Exploring further the nature of the interaction between the Cas6 and RT domains, comparison of the sequences of Cas6 domains fused with RT-Cas1 in branch 11 with those of Cas6 and RT-Cas1 proteins encoded by separate genes in branch 10 showed fusion-specific conservation of distinct sequence features in both the Cas6 and RT domains of the fusion (Figure 7). In the Cas6 domain, the fusion-specific amino acid motifs are located in three regions (I, II, and III, Figure 7A and 7B), which are separated in the protein sequence but are clustered on the same surface of the protein around the highly conserved H32 residue in the crystal structure of MMB-1 Cas6 (Figure 7C). In the RT domain, which is closely related to that of a group II intron-encoded RT, whose structure was solved recently (Stamos et al., 2017), the fusion-specific conserved sequences are located near the template-binding pocket region (RT0) and adjacent RT3a loop.
Figure 7. Model of the Cas6-RT Domain Interaction in MMB-1 Cas6-RT-Cas1.
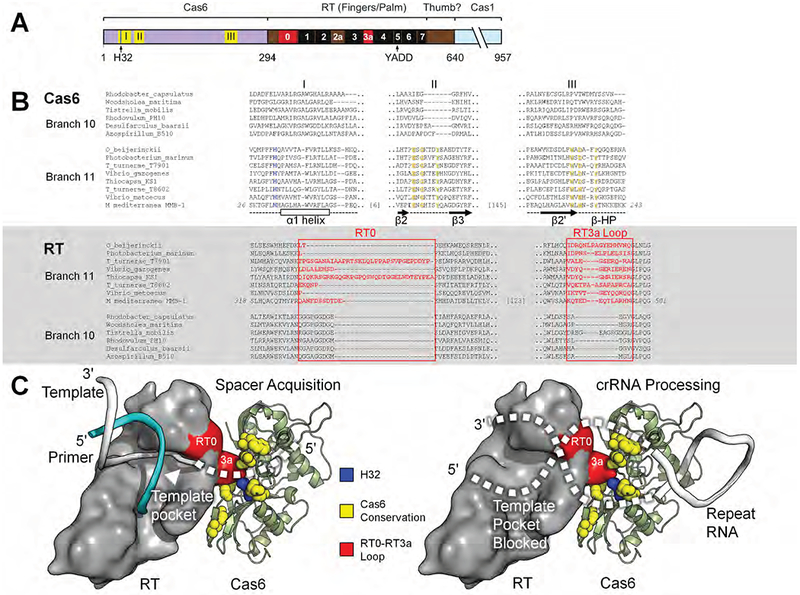
(A) Schematic of the Cas6-RT-Cas1 fusion protein. Yellow (I, II, III) and red (RT0, RT3a) show the location of sequences aligned in panel B; black boxes indicate conserved RT sequence blocks. The location of the conserved H32 residue in motif I is indicated by a blue line. (B) Branch-specific sequence features of Cas6 (top) and RT (bottom) shown in representative alignments from branches 10 and 11. In the Cas6 alignment, H32 is in blue, and additional conserved residues characteristic of Branch 11 are in yellow. Secondary structure elements are indicated below the Cas6 alignment. In the RT alignment, RT0 and RT3a are in red. (C) Model of the potential Cas6-RT domain interaction. The branch-specific RT0-RT3a loop surface (red) is positioned near the branch-specific Cas6 conserved motifs (yellow), including highly conserved H32 (blue). During spacer acquisition, left, the template strand is unobstructed from the template pocket, whereas during crRNA processing, right, the bound CRISPR repeat RNA may sterically block or enter the template pocket unproductively. The RT domain surface is loosely based on a group II intron RT structure in complex with template-primer (PDB: 6AR1; template, white; primer, cyan) and manually docked against MMB-1 Cas6. Features and amino acid residues are colored as in (B).
The apparently correlated sequence conservation on specific surfaces of the Cas6 and RT domains suggests that these two surfaces might form a structural interface in Cas6-RT-Cas1 fusion proteins. A model of the interaction, consistent with the positions of the C-terminus of Cas6 and the N-terminus of RT (Figure 7C, left), positions Cas6 near the RT template-binding pocket during reverse transcription of RNA protospacers. Loss of a key protein contact or local structural disruption in this region as caused by the H32A mutation could lead to a collapse or block of the template-binding pocket in the RT domain, accounting for the loss of RT activity in the H32A mutant (Figure 4). The finding that the H32A and H32A-H33A mutants retain Cas6 activity and can acquire spacers from DNA (but not RNA) in vivo indicates that these Cas6 domain mutations specifically affect RT activity and do not grossly disrupt the structure of either the Cas6 or Cas1 domains. During crRNA processing, the crRNA 5’ or 3’ region could block or enter the RT template-primer binding cleft in an unproductive orientation (Figure 7C, right), which would explain the loss of the RT activity in the presence of crRNA (Figure 6). The model is also consistent with the possibility that the Cas6 domain contributes to RNA binding during spacer acquisition. Protospacer RNAs could bind across the Cas6-RT interface to be fed into the RT active site for reverse transcription to yield nascent spacers in the CRISPR array. An analogous interaction between free-standing Cas6 and RT or RT-Cas1 proteins with similar functional consequences could account for their stable association preceding the origin of the fusion.
STAR*METHODS
CONTACTS FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Alan M. Lambowitz (lambowitz@austin.utexas.edu). Marinomonas mediterranea strains used in this study can be obtained from Andrew Z. Fire (afire@stanford.edu), Sukrit Silas (sukrit@alumni.stanford.edu), or Antonio Sanchez-Amat (antonio@um.es).
METHOD DETAILS
Comparative Genomic, Sequence, and Phylogenetic Analysis
Archaeal and bacterial complete and draft genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/) in March 2016. Protein-coding genes for the draft genomes were annotated as previously described (Shmakov et al., 2017). Altogether, the database includes 4,961 completely sequenced and assembled genomes and 43,599 partially sequenced genomes. Twenty two Cas6 profiles (or multiple sequence alignments) built previously (Makarova et al., 2015) were obtained from ftp://ftp.ncbi.nlm.nih.gov/pub/wolf/suppl/CRISPR2015/. These profiles were used as queries for the PSI-BLAST program (Altschul et al., 1997) to identify Cas6 homologs in the above database. Additionally, we searched the database using the amino acid sequence of the Cas6 domain (1 to 300 aa) from two previously identified (Makarova et al., 2015; Toro et al., 2017) Cas6-RT-Cas1 fusions, MMB-1 (Marme_0669) and PGN_1927 from Porphyromonas gingivalis ATCC 33277, as a query, using the PSI-BLAST program with E-value 0.01, and added the retrieved sequences to the Cas6 protein set in case they were missed by the previous approach. Proteins smaller than 150 amino acids and a few other fragments or false positive assignments were discarded. The final set included 16,404 Cas6-like proteins or domains. We used UCLUST (Edgar, 2010) with identity threshold 0.95 to obtain a non-redundant set of 4,040 sequences, which were then used for phylogenetic and neighborhood analysis. Genomic loci that contained 10 genes upstream and downstream of the respective cas6 genes were retrieved and all proteins encoded in these loci were annotated using PSI-BLAST searches (E-value of 0.01) with 217 profiles of cas genes (Makarova et al., 2015) and with 33,895 profiles (derived from COG, pfam, cd databases) from the NCBI CDD database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (Marchler-Bauer et al., 2015). The CRISPR-Cas system (sub)type identification for all loci was performed using previously described procedures (Makarova et al., 2015).
Multiple alignments and phylogenies of Cas6 protein sequences were constructed as described previously (Peters et al., 2017). Briefly, the sequences were clustered by similarity and a multiple alignment was built for each cluster using the MUSCLE program (Edgar, 2004). Alignments were combined into larger aligned clusters by the HHalign program (Yu et al., 2015) if the resulting relative score between any two alignments was higher than a specified threshold (0.1); otherwise the scores were recorded in a similarity matrix. This matrix was used to reconstruct an UPGMA tree. For each cluster, the respective alignment was filtered as follows: alignment positions with gap character fraction values of <0.5 and homogeneity values of <0.1 were removed. The remaining positions were used for tree reconstruction using the FastTree program (Price et al., 2010) with the WAG evolutionary model, and the discrete gamma model with 20 rate categories. The same program was used to compute the SH (Shimodaira-Hasegawa)-like node support values.
Strains and Culture Conditions
The type IIIB operon-deleted M. mediterranea MMB-1 strain was constructed in a spontaneous rifampicin-resistant genetic background by allelic exchange mutagenesis using sacB/sucrose counter-selection with the pEX18Gm suicide vector backbone and the E. coli S17-1λpir donor strain as described previously (Campillo-Brocal et al., 2013). All bacterial strains were stored at −80°C in 20% glycerol. Expression plasmids encoding MMB-1 type III-B operon components were mobilized into wild-type (WT) or mutant M. mediterranea MMB-1 strains from a donor E. coli strain carrying the pRL443 conjugal plasmid (a gift from M. Davison, Carnegie Institution), as described previously (Solano et al., 2000). Transconjugants were selected on 2216 marine agar (Difco) with 50 μg/mL kanamycin and 50 μg/mL rifampicin at 25°C. Two clones (independent transconjugants) from each conjugation were tested for spacer acquisition and in vivo pre-crRNA processing. For nucleic acid extractions, transconjugant strains were inoculated into 2 mL Km-broth (2216 marine medium (Difco) with 50 μg/mL kanamycin) and shaken at 23-25°C for 4–8 hr. Cultures were immediately expanded in 15-mL Km-broth and grown for an additional 4–8 hr before nucleic acid extraction. The same cultures were used for both spacer acquisition and in vivo pre-crRNA processing assays.
Recombinant proteins were expressed in E. coli Rosetta2(DE3) (Novagen) grown in LB medium at 37°C with shaking at 200 rpm and antibiotics (100 mg/L ampicillin; 25 mg/L chloramphenicol) added as needed. The conditions used to induce protein expression are described further below.
DNA and RNA Constructs
Plasmids for the expression of WT M. mediterranea type III-B adaptation components (Marme_0670, Cas6-RT-Cas1, Cas2, and CRISPR03) and empty-vector controls supplying only CRISPR03 for in vivo assays were described previously (Silas et al., 2017a; 2016). Additional Cas6-RT-Cas1 mutants used in in vivo assays were constructed from the WT expression plasmid using Gibson Assembly Master Mix (New England Biolabs) and verified by sequencing.
The plasmids used for expression of WT Cas6-RT-Cas1 protein for biochemical assays was pMalRF-RTCas1 (Silas et al., 2016). Plasmids used for the expression of mutant proteins were derived from pMalRF-RTCas1 by replacing the N-terminal domain of the protein with PCR fragments generated using primers that introduced the desired mutations. All of the protein constructs had a maltose-binding protein (MBP) tag fused to their N-terminus via a non-cleavable rigid linker (denoted MRF) and carried a C-terminal 8x His tag for purification. All expression constructs have amino acid 2 of the Cas6 domain mutated to Valine.
CRISPR03 array transcripts used as substrates for Cas6 assays were prepared by in vitro transcription with a T7 Megascript Kit (Ambion) from PCR-amplified DNA templates derived from the CRISPR03 plasmid (Silas et al., 2016). The 5’ PCR primer contained a T7 promoter sequence fused to MMB-1 sequence 120-nt upstream of the first repeat of the CRISPR03 locus (MMB1crisp5b+T75’; 5’-ATGAATTCGTAATACGACTCACTATAGGGCACTCGACCGGAATTATCGACGAA), and the downstream primers were either MMB1crisp3full (5’ CTAGCTCTCGAGAGGCCTTCGTCA) or MMB1crisp3 (5’ TCTGAAACTCTGAATACTAACGAAAAATAG) producing 1,065-bp and 297-bp DNAs, respectively. Transcription reactions contained 0.25 or 0.65 μg DNA for the long and short CRISPR03 templates, respectively, and 30 μCi [α-32P] UTP (3,000 Ci/mmol; Perkin-Elmer). After digestion with with TurboDNase (Ambion), RNA was extracted with phenol-chloroform-isoamyl alcohol (phenol-CIA; 25:24:1) followed by CHCl3. Isopropanol was added and RNA was pelleted by centrifugation. The RNA pellet was washed with 70% ethanol, and resuspended in RNase-free water. The purified RNA was then column-purified using P30 columns (BioRad).
Oligonucleotides were 5’ end-labeled in 50 μL total volume containing 1 nmol oligonucleotide, 600 μCi [γ-32P] ATP (6,000 Ci/mmol, Perkin-Elmer), 1 nmol ATP, 40 units T4 Polynucleotide Kinase (New England Biolabs) for 45 min at 37°C in the manufacturer’s buffer. Labeled oligonucleotides were purified on a denaturing 10% polyacrylamide gel; the gel area containing the oligonucleotide was cut out, crushed and incubated with 1 mL of TE (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) per ~1.5 cm gel slice at 4°C overnight on a rotator. The TE was then removed, and the gel was washed with an equal volume of TE. The combined TE fractions were extracted repeatedly with butanol until the volume was about 500 μL, and then cleaned up using an Oligo Clean and Concentrator kit (Zymo). The oligonucleotide was eluted in ~60 μL H20 and quantitated using a LS 6500 scintillation counter (Beckman-Coulter).
Protein Purification
Protein expression plasmids were transformed into E. coli Rosetta2 (EMD Milipore) and single transformed colonies were inoculated in LB medium supplemented with appropriate antibiotics and incubated overnight at 37°C with shaking. Six 1-liter LB broth flasks were each inoculated with 10 mL of the overnight culture, and grown at 37°C with shaking to log phase (O.D.600 = ~0.8). IPTG was then added to a final concentration of 1 mM, and the cultures were incubated at 19°C for 20 to 24 hr. Cells were harvested by centrifugation, and the pellet was dissolved in A1 buffer (25 mM Tris-HCl, pH 7.5; 500 mM NaCl; 10% glycerol; 10 mM ß-mercaptoethanol (BME); 10 mL/g cells) on ice. The cells were incubated with lysozyme (1 mg/mL, 0.5 hr, 4°C) and sonicated (Branson Sonifier 450; 3 bursts of 15 sec each with 15 sec between each burst). The lysate was cleared by centrifugation (29,400 × g, 25 min, 4°C), and polyethyleneimine (PEI) was added to the supernatant in six steps on ice with stirring to a final concentration of 0.4%. After 10 min, precipitated nucleic acids were removed by centrifugation (Beckman-Coulter JA-14 rotor; 29,400 × g, 25 min, 4°C), and proteins were precipitated from the supernatant by adding ammonium sulfate to 60% saturation and incubating on ice for 30 min. Proteins were collected by centrifugation (Beckman-Coulter JA-14 rotor; 29,400 × g, 25 min, 4°C), dissolved in 20 mL A1 buffer, and filtered through a 0.45-mm polyethersulfone membrane (Whatman Puradisc).
Protein purifications were done by using an AKTA start system (GE Healthcare). RT-Cas1 proteins were purified by loading the filtered crude protein onto an amylose column (30 ml; Amylose High Flow resin; New England Biolabs), washing with 50 ml of A1 buffer, followed by 30 ml A1 plus 1.5 M NaCl and 30 ml of A1 buffer. Bound proteins were eluted with 50 ml of 10 mM maltose in A1 buffer. Fractions containing RT-Cas1 were identified by SDS-PAGE, pooled, and diluted to 250 mM NaCl. The protein was then loaded onto a 5-ml heparin-Sepharose column (HiTrap Heparin HP column; GE Healthcare) and eluted with a 0.1 to 1 M NaCl gradient. Peak fractions (~700 mM NaCl) were identified by SDS-PAGE, pooled, and dialyzed into A1 buffer. The dialyzed protein was concentrated to >10 μM using an Amicon Ultra Centrifugal Filter (Ultracel-50K).
The initial steps in the Cas2 purification were similar, except that the cell paste was resuspended in N1 buffer (25 mM Tris-HCl, pH 7.5; 500 mM KCl; 10 mM imidazole; 10% glycerol; 10 mM DTT), and the ammonium sulfate precipitation step was omitted. Instead, the Cas2 supernatant after PEI precipitation was loaded directly onto a 5-ml nickel column (HiTrap Nickel HP column; GE Healthcare) and eluted with an imidazole gradient (60 ml 10 to 500 mM in N1 buffer). Peak fractions containing Cas2 were identified by SDS-PAGE and pooled. After adjusting the KCl concentration to 200 mM, the pooled fractions were loaded onto two 5-ml heparin-Sepharose columns arranged in tandem. The protein was eluted with a linear KCl gradient (50 ml, 100 mM to 1 M), and Cas2 peak fractions (~800 mM KCl) were identified by SDS-PAGE and stored on ice in elution buffer. Protein concentrations were measured using the Qubit Protein assay kit (Life Technologies) according to the manufacturer’s protocol. Proteins were >80% pure as assayed by SDS-PAGE.
For crystallography, MRF-Cas6-RT-Cas1Δ307-957 was purified by loading the filtered crude protein onto an amylose column (30 mL; Amylose High Flow resin; New England Biolabs), washing with 50 ml A1 buffer, followed by 30 mL A1 + 1.5 M NaCl and 30 ml A1 buffer. Bound proteins were eluted with 50 mL of 10 mM maltose in A1 buffer. Fractions containing MRF-Cas6-RT-Cas1D307-957 were identified by SDS PAGE, pooled, and diluted to 250 mM NaCl. The protein was then loaded onto a 5 mL heparin-Sepharose column (HiTrap Heparin HP column; GE Healthcare) and eluted with a 500 mM to 1.5 M NaCl gradient. Peak fractions (~700 mM NaCl) were identified by SDS-PAGE, pooled, and dialyzed into A1 buffer. The dialyzed protein was concentrated to >20 μM using an Ultra Centrifugal Filter (Ultracel-30K; Amicon). The concentrated protein was stored at −80°C. For crystallization, the protein was removed from storage and concentrated further to >23 μM using an Ultra Centrifugal Filter (Ultracel-30K; Amicon) and desalted using Zeba Spin Desalting Column 7K MWCO (Thermo) to exchange into Crystallography Buffer (25 mM Tris-HCl, pH 7, 500 mM NaCl, 10% glycerol, 10 mM BME).
Protein Crystallization, Data Collection, and Structure Refinement
Crystals were grown by sitting drop vapor diffusion at 22°C by combining 0.5 μL of protein stock with 0.5 μL of well solution containing 0.1 M sodium acetate pH 5.2, and 2.5 M ammonium sulfate. Crystals appeared in 1-2 days and grew as long rods over the course of 7 days to final dimensions of ~0.075 mm × 0.075 mm × 0.3 mm.
Crystals were harvested, immersed briefly in well solution containing 25% glycerol, and then flash-frozen in liquid nitrogen. Data were collected on Beamline 5.0.3 at the Advanced Light Source (Berkeley, CA). Crystals diffracted to a resolution of 2.85 Å in space group P212121, a=92.3Å, b=110.8Å, c=192.9Å. Data were processed with XDS (Kabsch, 2010) and Aimless (Evans and Murshudov, 2013). A starting model for the MalE protein was derived from PDB ID: 5B3W and used for a molecular replacement search in Phaser (Adams et al., 2011). Alternating rounds of structure building and refinement were carried out in COOT (Emsley et al., 2010) and Phenix (Adams et al., 2011) to a final Rwork/Rfree of 0.187/0.225.
In Vitro Cas6 Pre-crRNA Cleavage Assays
Pre-crRNA processing assays with 32P-labeled pre-cRNA in vitro transcript substrates (10 nM of short (271 nt) or 2.75 nM of long (1,039 nt) pre-crRNA) were performed in 20 μL reaction medium containing 50 mM KCl, 7.5 mM MgCl2, and 20 mM Tris-HCl, pH 7.5, with 25 mM EDTA added where indicated. Purified wild-type and mutant Cas6-RT-Cas1 proteins were added last at concentrations indicated in the figure legends. The reactions were pre-incubated on ice for 5 min, incubated at 37°C for up to 1 hr, and terminated by placing on ice followed by extraction with phenol-CIA. The aqueous phase was then mixed at a 2:1 ratio with loading dye (90% formamide, 50 mM EDTA, and 0.25 mg/ml bromophenol blue and xylene cyanol), and reaction products were analyzed by electrophoresis on a denaturing 6% polyacrylamide gel, which was dried and scanned with a phosphorimager (Typhoon FLA 9500, GE Healthcare). Pre-crRNA processing assays with 32P-labeled RNA oligonucleotides were done as above but analyzed on a denaturing 10% polyacrylamide gel.
Pre-crRNA processing reactions with excess RNA (to assess protein turnover) were performed with 1 μM 5’-labeled 35-nt repeat RNA oligonucleotide and 40 nM protein in TKN reaction medium (100 mM KCl and 50 mM NaCl (from protein input) and 20 mM Tris-HCl, pH 7.5). After pre-warming the RNA in reaction medium for 2 min at 37°C, reactions were initiated by adding 1/10 volume of 400 nM protein. 20 μL portions were withdrawn at different times, and the reaction was stopped by extraction with phenol-CIA followed by centrifugation. The aqueous phase was then mixed 2:1 with formamide loading dye (90% formamide, 50 mM EDTA, and 0.25 mg/mL bromophenol blue and xyan cyanol) and aliquots analyzed by electrophoresis on a denaturing 12% polyacrylamide gel, which was dried and imaged with a phosphorimager (Typhoon FLA 9500, GE Healthcare). The phosphorimager data were quantitated with Imagequant TL 8.1 Toolbox (GE Healthcare) and further analyzed with Microsoft Excel 2016 and Prism 6.0h (Graphpad Software).
CRISPR RNA-Binding Assays
For equilibrium RNA-binding assays, 5’ 32P-labeled CRISPR repeat RNA oligonucleotides (1 nM) was incubated with increasing concentrations of WT or mutant Cas6-RT-Cas1 protein (~1 to 1,500 nM final concentration) in TKN reaction medium. RNA (27 μL) and protein (3 μL) were pre-incubated on ice for 5 min and then for 5 min at 37°C. After binding, 25-μL samples were withdrawn from each tube and filtered through stacked nitrocellulose (Trans Blot, Bio-Rad) and DE-81 (GE Healthcare) filters using a Schleicher and Schuell Minifold 96-well dot blot apparatus. Filters were washed with 75 μl TK buffer (20 mM Tris-HCl, pH 7.5, 100 mM KCl) and then dried and imaged with a phosphorimager (Typhoon FLA 9500, GE Healthcare). The phosphorimager data were quantitated with Imagequant TL 8.1 Array (GE Healthcare) and further analyzed with Microsoft Excel 2016 and Prism 6.0h (Graphpad Software). The radioactivity bound to nitrocellulose reflects the RNA bound to protein, while the radioactivity bound to the DE-81 filter reflects free RNA. For each sample, the fraction of bound RNA is given by the ratio of counts from nitrocellulose relative to the total counts (nitrocellulose + DE-81). Binding curves (fraction RNA bound versus protein concentration) were fit to y= Bmax*x/(Kd+x), where y is the fraction of protein-bound RNA; × is the protein concentration; Bmax is the maximum amount of bound RNA; and Kd is the binding constant determined from the fit. Incubating the 35-nt dC-1 RNA oligonucleotide with the WT Cas6-RT-Cas1 protein at 37°C for different times (5, 10, or 15 min) had no significant effect on the binding curves, indicating that binding had reached equilibrium by 5 min.
For koff assays, RNA-protein complexes were formed by mixing 1 μM Cas6-RT-Cas1 protein with 2 nM 5’ 32P-labeled non-cleavable dC-1 35-nt CRISPR repeat RNA oligonucleotide in 250 μL TKN reaction medium on ice for 5 min. The mixture was then incubated for an additional 5 min at 37°C before adding an equal volume of TK that was pre-warmed to 37°C with or without 2-μM unlabeled dC-1 35-nt repeat RNA oligonucleotide. 20-μL samples were withdrawn at intervals and binding was assayed and quantitated as described above. Data were fit to an exponential function and the off-rate was obtained from the fit. Since the amount of complex differs for various proteins, data were normalized using the sample diluted with buffer alone.
Reverse Transcriptase Assays
RT activity of purified proteins was assayed with poly(rA)/oligo(dT)24 as described previously (Silas et al., 2016). Briefly, pre-annealed poly(rA)/oligo(dT)24 (80 μM and 50 μM, respectively) was pre-incubated for 2 min at 37°C in 20 to 30 μL of reaction medium containing 200 mM KCl, 50 mM NaCl, 10 mM MgCl2, and 20 mM Tris-HCl, pH 7.5; 1 mM unlabeled deoxythymidine triphosphate (dTTP); and 5 μCi [α-32P]-dTTP (3,000 Ci/mmol; PerkinElmer). The reactions were initiated by adding purified WT or mutant Cas6-RT-Cas1 proteins (0.5 μM final concentration) and incubated for up to 30 min. For time-courses, 3 μL portions of each reaction were withdrawn at each time point and added to 10 μL of stop solution (0.5% SDS, 25 mM EDTA). Reaction products were spotted onto Whatman DE81 paper (10 χ 7.5-cm sheets; GE Healthcare Biosciences), which was then washed three times with 0.3 M NaCl and 0.03 M sodium citrate, dried, and scanned with a phosphorimager (Typhoon Trio Variable Mode Imager; GE Healthcare Biosciences) to quantify the bound radioactivity. The first few data points showing pseudo-linearity were used to obtain the initial rate of 32P-dTTP incorporation per second per mole of protein. Data for mutant proteins were normalized to measurements for WT Cas6-RT-Cas1 assayed in parallel. For RT assays in the presence of competitor RNAs (Figure 6), 45 μM of a competing oligonucleotide was added prior to initiating the reaction by adding protein. The reactions were incubated for 10 min at 37 °C, which would be well past the linear range of the reaction without added competitor. The sequence of the spacer RNA oligonucleotide used in Figure 6 was 5’-AGCGUGCGUUCCAGACAUUCAGCCCUCUAGUAGA.
All RT assays were done at least three times, and the mean and standard deviation were calculated in Excel (Microsoft).
In Vitro RNA and DNA Spacer Insertion Assays
Purified Cas6-RT-Cas1 protein (10 μM) was mixed with a two-fold excess of purified Cas2 in reaction medium containing 250 mM KCl, 250 mM NaCl, and 25 mM Tris-HCl pH 7.5, 5 mM DTT, 5 mM BME, and 10% glycerol and incubated on ice for >1 hr prior to use. The MMB-1 CRISPR DNA substrate was a PCR product amplified from cloned CRISPR03 DNA with primers MMB1crisp5b (5’-CACTCGACCGGAATTATCGACGAA) and MMB1crisp3 (5’-TCTGAAACTCTGAATACTAACGAAAAATAG) using Phusion High-Fidelity DNA polymerase according to the manufacturer’s protocol (New England Biolabs or Thermo Fisher). The resulting 268-bp PCR fragment contains 120 bp of the leader, 35 bp of repeat 1, 33 bp of spacer 1, 35 bp of repeat 2, 37 bp of spacer 2, and 8 bp of repeat 3. Internally labeled substrate was prepared by adding 25 μL [α-32P]-dTTP (Perkin Elmer) and 40 mM dTTP to the PCR reactions. The labeled DNA was purified by electrophoresis on a native 6% polyacrylamide gel, cutting out the labeled band, and electroeluting the DNA using midi D-Tube dialyzer cartridges (Novagen). The eluted DNA was extracted with phenol-CIA, ethanol-precipitated, and quantitated using a Qubit dsDNA assay kit (Life Technologies).
CRISPR DNA cleavage-ligation assays contained Cas6-RT-Cas1/Cas2 complex (500 nM final), MMB-1 CRISPR substrate (1 nM), 20 mM Tris-HCl, pH 7.5, and 7.5 mM free MgCl2. DNA or RNA oligonucleotides and an equimolar solution of dNTPs and Mg2+ were added at 2.5 μM and 1 mM final concentrations. Reactions were incubated at 37°C for 1 hr and stopped by adding phenol-CIA. The supernatant was mixed at a 2:1 ratio with loading dye (90% formamide, 20 mM EDTA, and 0.25 mg/ml bromophenol blue and xyan cyanol), and nucleic acids were analyzed on a denaturing 6% polyacrylamide gel. Gels were dried and scanned with a phosphorimager.
Cas6 Cleavage Site Determination by TGIRT-seq
Internally labeled long (1,039 nt; ~300 ng) pre-crRNA in vitro transcript (see above) was incubated with MRF-MMB-1 Cas6-RT-Cas1 protein in TKM buffer at 37°C for 60 min. The protein was removed by phenol-CIA extraction, and the RNA was purified with the Zymo RNA Clean-up and Concentration kit to a final concentration of ~10 ng/μL. About 100 ng RNA was treated with phage T4 polynucleotide kinase (Epicentre) for 30 min at 37°C to remove 3’ phosphates and 2’,3’-cyclic phosphates, followed by clean-up with the Zymo RNA Clean-up and Concentration kit. RNA-seq libraries were made by TGIRT template switching RT using the TGIRT Total RNA-seq method using an initial template-primer substrate consisting of a 34-nt RNA oligonucleotide (R2 RNA), which contains an Illumina Read 2 primer-binding site and a 3’-blocking group (C3 Spacer, 3SpC3; IDT), annealed to a complementary 35-nt DNA primer (R2R DNA) that leaves an equimolar mixture of A, C, G, or T single-nucleotide 3’ overhangs (Nottingham et al., 2016). RNAs were degraded by treatment with NaOH (250 mM, 95°C for 3 min), the sample was neutralized with an equal amount of HCl, and cDNAs were purified by using a MinElute kit (QIAgen). The purified cDNA was ligated to pre-adenylated R1R oligonucleotide (5’ GATCGTCGGACTGTAGAACTCTGAACGTGTAG/3SpC3/) using thermostable 5’ AppDNA/RNA Ligase (65°C for 60 min; New England Biolabs) followed by clean-up with the MinElute kit. The library was then amplified by PCR with Phusion PCR master mix (New England Biolabs) (initial denaturation at 98°C for 5 sec, followed by cycles of 98°C for 5 sec, 65°C for 10 sec, 72°C for 10 sec) (Nottingham et al., 2016), during which Illumina adaptor and index sequences were added. Libraries were size-selected using Ampure XP beads (Beckman-Coulter) and evaluated on an Agilent 2100 Bioanalyzer. Samples were sequenced on the Illumina MiSeq with v3 chemistry in single-read format for 150 cycles. Illumina TruSeq adapters and PCR primer sequences were trimmed from the reads with cutadapt (sequencing quality score cut-off at 20; p-value < 0.01) and reads <15-nt after trimming were discarded. Reads were then mapped to the native CRISPR array sequence with Bowtie2 v2.2.6 with local alignment using command line parameters “--very-sensitive-local -k 1”. Coverage plots were generated without showing soft-clipped nucleotides.
Nucleic Acid Extractions from M. mediterranea MMB-1
This method is a slight modification of a previously published protocol for CRISPR spacer sequencing (Silas et al., 2017a; 2016); we have provided the protocol in entirety for completeness, retaining relevant text from the original protocol. Total RNA for in vivo pre-crRNA processing assays was extracted from 0.3 to 0.5 mL of 15 mL confluent cultures using TRIZOL reagent (Life Technologies) according to manufacturer’s instructions, without any subsequent enzymatic treatments. The remainder of the culture was used for plasmid midiprep. Cells were harvested by centrifugation (4,000 × g, 30 min, 4°C) and homogenized in 300 μL alkaline lysis buffer (40 mM glucose, 10 mM Tris-HCl, pH 7.5, 4 mM EDTA, 0.1 N NaOH, 0.5% SDS) at 50°C by vortexing until clear (10-15 min). Chilled neutralization buffer (600 μL of 3 M CH3COOK, 2 M CH3COOH) was added and lysates were immediately transferred to ice to prevent digestion of genomic DNA. Samples were mixed by inverting and the genomic DNA containing precipitate was removed by centrifugation (20,000 × g, 20 min, 4°C). Clarified lysates were extracted twice with a 1:1 mixture of Tris-saturated-phenol (Life Technologies) and CHCl3 (Fisher Scientific), and once with CHCl3 in Heavy Phaselock Gel tubes (5 Prime). Isopropanol (950 μL) was added and the plasmid DNA was pelleted by centrifugation (16,000 × g, 20 min, 4°C), washed twice in 80% ethanol, and resuspended in 200 μL 1x NEB Cutsmart Buffer. Samples were treated with 50 μg/mL RNase A (Life Technologies) at 37°C for 30 min, linearized with PvuII-HF (New England Biolabs) at 37°C for 60 min (to aid denaturation during PCR), and treated with 200 μg/mL Protease K at 50°C for 30 min. Finally, each digest was purified using a Zymo gDNA Clean and Concentrator column.
In Vivo Cas6 Activity Assay
This assay was published as a stand-alone Bio-protocol (Silas et al., 2018). Total RNA was purified from log phase cultures of MMB-1 (same cultures are used for Spacer acquisition assay) using Trizol reagent according to manufacturer’s instructions. ~5 μg intact RNA was run on a denaturing 6% polyacrylamide gel (Novex) at 180V for 35 min under denaturing conditions. Gel fragments corresponding to 30-80 nt size range were excised, and small RNA fraction was eluted, purified, and prepared for sequencing as described in the Bio-protocol (Silas et al., 2018).
In Vivo Spacer Acquisition Assay
Assays were performed as described previously (Silas et al., 2017a; 2016); we have provided the protocol in entirety for completeness, retaining relevant text from the original protocol. CRISPR spacers were amplified for high-throughput sequencing by two rounds of PCR. We used 1 to 2 ng purified plasmid DNA per μl PCR mix in round 1 using forward primer AF-SS-119 (CGACGCTCTTCCGATCTNNNNNCTGAAATGATTGGAAAAAATAAGG) anchored in the leader sequence and reverse primer AF-SS-121 (ACTGACGCTAGTGCATCACGTGGCGGAGATCTTTAA) in the first native spacer. Phusion High-Fidelity PCR master mix with HF buffer (Fisher Scientific) was used for all reactions. Cycling conditions for round 1 were as follows: one cycle at 98°C for 1 min; two cycles at 98°C for 10 s, 50°C for 20 s, and 72°C for 30 s; 20 cycles at 98°C for 15 s, 65°C for 15 s; and 72°C for 15 s; and one cycle at 72°C for 9 min. The dominant amplicon containing the first native spacer from unmodified CRISPR templates after round 1 was 123 bp. Round 1 amplicons were purified by blind excision of gel slices at 180 to 200 nt after denaturing PAGE (polyacrylamide gel electrophoresis) [pre-run TBE-Urea 10% gels (Novex), 180 V, 80 min in XCell SureLock Mini-Cells (Life Technologies)]. DNA was extracted from the gel slices by pulverizing the gel into fragments and incubating overnight in DNA elution buffer (300 mM NaCl, 1 mM EDTA) at room temperature. Sequencing adaptors were then attached in a second round of PCR with 1/4th of the eluate from the previous reaction mixture as the template, using AF-SS-44:55 (CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCACTGACGCTAGTGCATCA) and AF-KLA-67:74 (AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT), where the (N)8 barcodes correspond to Illumina TruSeq HT indexes D701 to D712 (reverse complemented) and D501 to D508, respectively. Cycling conditions for round 2 were one cycle at 98°C for 1 min; two cycles at 98°C for 10 s, 54°C for 20 s, and 72°C for 30 s; 9 cycles at 98°C for 15 s, 70°C for 15 s, and 72°C for 15 s; and one cycle at 72°C for 9 min. The dominant amplicon containing the first native spacer from unmodified CRISPR templates after round 2 was 241 bp. Sequencing libraries were prepared by blind excision of gel slices at 300 to 320 bp (70 bp above the 241-bp band, consistent with the expected size of an amplicon from an expanded CRISPR array) after agarose electrophoresis (3%, 4.2 V/cm, 2 hours) of the round 2 amplicons. Pooled agarose gel-purified libraries were further PAGE-purified by blind excision of gel slices at 300 to 320 nt (pre-run TBE-Urea 6% gels, 180 V, 90 min as above). The final pooled library was quantified by Qubit and sequenced with Illumina MiSeq v3 kits (150 cycles for read 1; 8 cycles for index 1; 8 cycles for index 2). Spacers were trimmed from reads using a python script and were considered identical if they differed by only 1 nucleotide. Protospacers were mapped using Bowtie 2.0 (-very-sensitive-local alignments). These methods preserve strand information.
Monte Carlo Simulations
We used a Monte Carlo simulation to evaluate a null hypothesis based on random assortment of spacer acquisitions from genomic DNA, with no dependence on gene expression level. Simulations were performed as described previously (Silas et al., 2017a; 2016); for clarity, we provide relevant text from the original method here. For each system, a series of samples of 500 spacers each were randomly chosen in silico from a list of all genes based on the sizes of the individual genes using the stochastic universal sampling algorithm. Sets of 1,000 such trials were used to generate a range of null relationships between gene expression and spacer acquisition. The Monte Carlo bounds (black dotted lines on the respective figures) depict the envelope of such simulated random assortments. Traces above this envelope indicate preferential spacer acquisition from highly expressed genes, whereas traces below the envelope indicate spacer acquisition from poorly expressed genes more often than expected by random chance. M. mediterranea MMB-1 expression data (at NCBI SRA: SRR2914032, SRR2914033) were previously generated by RNAseq (Silas et al., 2016).
Data and Software Availability
The accession number for the MMB-1 Cas6 crystal structure reported in this paper is PDB ID: 6DD5.
The accession number for the sequencing data in Figure S4 is SRP143510.
The accession numbers for the sequencing data in Figures 5, 6 and S7 are as follows: Figure 5B, SAMN06754958; Figure 5D WT, SAMN06754967; Figure 6A WT, SAMN04259748; Figure S7, SAMN04259751; all other data from Figures 5C, 5D, 6A, SRP142733.
Unprocessed and uncompressed imaging data: DOI: 10.17632/cdpyd694d5.1
Quantification and statistical analysis
ImageQuant TL ver. 8.1 (General Electric) was used for quantification of all in vitro Cas6 cleavage, spacer acquisition, RT, and binding assays. Excel ver. 16.16 (Microsoft) was used to determine mean, median and standard deviation values. Prism 6.0h (Graphpad Software) was used for curve fitting of binding, RT, and Cas6 assays in order to determine koff and Kd values.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E. coli Rosetta 2 (DE3) | EMD Millipore | Cat#71400 |
| E. coli Rosetta 2 | EMD Millipore | Cat#71405 |
| E. coli DH5a | Thermo Fisher | Cat#18258-012 |
| Marinomonas mediterranea MMB-1 | ATCC | ATCC 700492 |
| Marinomonas mediterranea MMB-1 III-B operonΔ | Andrew Fire | Silas et al. 2017a |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2216 marine agar | DIFCO | Cat#BD 212185 |
| T4 Polynucleotide Kinase | NEB | Cat#M0201 |
| T4 Polynucleotide Kinase | Epicentre | Cat#P0503K |
| TurboDNase | Ambion | Cat#AM2238 |
| Phusion High-Fidelity DNA polymerase | Thermo Fisher | Cat#F530L |
| Phusion PCR master mix | NEB | Cat#M0531L |
| [γ-32P] ATP (6,000 Ci/mmol) | Perkin-Elmer | Cat#NEG035C005MC |
| [α-32P] UTP (3,000 Ci/mmol) | Perkin-Elmer | Cat#BLU007H001MC |
| [α-32P]-dTTP (3,000 Ci/mmol) | Perkin-Elmer | Cat#BLU005H250UC |
| TGIRT™-III Enzyme | Ingex | Cat#TGIRT10 |
| Critical Commercial Assays | ||
| Gibson Assembly Master Mix | NEB | Cat#E2611S |
| T7 Megascript Kit | Ambion | Cat#AM1334 |
| Oligo Clean and Concentrator kit | Zymo | Cat#D4060 |
| RNA Clean-up and Concentration kit | Zymo | Cat#R1015 |
| Qubit dsDNA assay kit | Life Technologies | Cat#Q32851 |
| MinElute kit | QIAgen | Cat#28004 |
| Ampure XP beads | Beckman-Coulter | Cat#A63881 |
| Deposited Data | ||
| MMB-1 Cas6 Fused to MBP | This paper | PDB ID: 6DD5 |
| Figure S2 RNA-seq data | This paper | SRP143510 |
| Figures 5C, 5D, 6A | This paper | SRP142733 |
| Figure 5B | Silas et al., 2017a | SAMN06754958 |
| Figure 5D WT | Silas et al., 2017a | SAMN06754967 |
| Figure 6A WT | Silas et al., 2016 | SAMN04259748 |
| Figure S5 | Silas et al., 2016 | SAMN04259751 |
| Unprocessed and uncompressed imaging data | This paper | DOI: 10.17632/cdpyd694d5.1 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S2 for primer sequences | ||
| Recombinant DNA | ||
| pMalRF-RTCas1 (WT) | Alan Lambowitz | Silas et al., 2016 |
| pMalRF-RTCas1 Δ307-957 | This paper | N/A |
| pMalRF-RTCas1 H32A | This paper | N/A |
| pMalRF-RTCas1 H33A | This paper | N/A |
| pMalRF-RTCas1 H37A | This paper | N/A |
| pMalRF-RTCas1 R41A | This paper | N/A |
| pMalRF-RTCas1 S46A | This paper | N/A |
| pMalRF-RTCas1 S51A | This paper | N/A |
| pMalRF-RTCas1 S46A S51A (SS) | This paper | N/A |
| pMalRF-RTCas1 R41A S46A S51A (RSS) | This paper | N/A |
| pMalRF-RTCas1 H196A, R197A, D200A (HRD) | This paper | N/A |
| pMalRF-RTCas1 RTΔ | Alan Lambowitz | Silas et al., 2016 |
| pMalRF-RTCas1 E790A | Alan Lambowitz | Silas et al., 2016 |
| pMalRF-RTCas1 E870A | Alan Lambowitz | Silas et al., 2016 |
| pET14b Cas2 | Alan Lambowitz | Silas et al., 2016 |
| Plasmid: CRISPR03 only pKT230 | Andrew Fire | Silas et al. 2017a |
| Plasmid: WT CRISPR adaptation pKT230 | Andrew Fire | Silas et al. 2016 |
| Plasmid: H32A mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: H32A-H33A mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: H37A mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: R41A mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: H196A R197A D200A mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: ∆5-289 mutant CRISPR adaptation pKT230 | This paper | N/A |
| Plasmid: ∆256-289 mutant CRISPR adaptation pKT230 | This paper | N/A |
| Software and Algorithms | ||
| XDS | Kabsch, 2010 | http://xds.mpimfheidelberg.mpg.de/ |
| Aimless | Evans and Murshudov, 2013 | http://www.ccp4.ac.uk/html/aimless.html |
| PHENIX | Adams et al., 2011 | https://www.phenixonline.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrclmb.cam.ac.uk/personal/pemsley/coot/ |
| MUSCLE v 3.7 | Edgar, 2004 | http://www.drive5.com/muscle |
| HHalign v. 1.5.1 | Yu et al., 2015 | http://bioserv.rpbs.univ-parisdiderot.fr/services/HHalign-Kbest/ |
| FastTree v 2.1.4 | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| PSIBLAST v. 2.8.0 | Altschul et al., 1997 | https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-2.html |
| UCLUST v1.2.22q | Edgar, 2010 | http://www.drive5.com/uclust/downloads1_2_22q.html |
| Prism v6.0h | N/A | www.graphpad.com |
| Other | ||
| Archaeal and bacterial complete and draft genome sequences (March 2016) | N/A | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/ |
| Cas6 profiles | Makarova et al., 2015 | ftp://ftp.ncbi.nlm.nih.gov/pub/wolf/_suppl/CRISPR2015/ |
| CDD profiles | Marchler-Bauer et al., 2015 | https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi |
Supplementary Material
Highlights.
A Cas6-RT-Cas1 fusion protein performs both crRNA biogenesis and CRISPR adaptation
Cas6 domain required for RT activity and spacer acquisition from RNA but not DNA
Cas6 domain coevolved with RT domain and regulates RT activity
Free-standing Cas6 stably associated with RT or RT-Cas1 in multiple lineages
Acknowledgments
This work was supported by NIH R01 grants GM37706 to A.Z.F. and GM37949 to A.M.L. S.S. was supported by a Stanford Graduate Fellowship and an HHMI International Student Research Fellowship. K.S.M. and E.V.K. are supported by the Intramural Program of the U.S. Department of Health and Human Services (via funds provided to the National Library of Medicine). A.S.-A. received funding from the Spanish “Ministerio de Economia, Industria y Competitividad” under project BFU2017-85464 supported by FEDER funds. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DEAC02-05CH11231. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung L-W, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillo-Brocal JC, Lucas-Elío P, and Sanchez-Amat A (2013). Identification in Marinomonas mediterranea of a novel quinoprotein with glycine oxidase activity. Microbiologyopen 2, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Christopher RT, Smith JT, Olson S, Barrangou R, Moineau S, Glover CVC, Graveley BR, Terns RM, and Terns MP (2014). The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in Streptococcus thermophilus. Mol. Microbiol. 93, 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, and Terns MP (2008). Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 22, 3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, Richter H, van der Oost J, and White MF (2015). Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 39, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, and Bork P (1998). Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem. Sci. 23, 324–328. [DOI] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JHD, Tjian R, and Doudna JA (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Richter H, Bratovič M, Le Rhun A, and Charpentier E (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. [DOI] [PubMed] [Google Scholar]
- Gesner EM, Schellenberg MJ, Garside EL, George MM, and Macmillan AM (2011). Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat. Struct. Mol. Biol. 18, 688–692. [DOI] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, and Doudna JA (2010). Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, and Doudna JA (2015). Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 40, 58–66. [DOI] [PubMed] [Google Scholar]
- Holm L, and Rosenström P (2010). Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SA, McKenzie RE, Fagerlund RD, Kieper SN, Fineran PC, and Brouns SJJ (2017). CRISPR-Cas: Adapting to change. Science 356, eaal5056. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima KK, and Kanehisa M (2008). Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol. 25, 1395–1404. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, and Koonin EV (2011). Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, and Koonin EV (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, and Koonin EV (2013). The basic building blocks and evolution of CRISPR-cas systems. Biochem Soc. Trans. 41, 1392–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, and van der Oost J (2016). Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147. [DOI] [PubMed] [Google Scholar]
- Mohr S, Ghanem E, Smith W, Sheeter D, Qin Y, King O, Polioudakis D, Iyer VR, Hunicke-Smith S, Swamy S, et al. (2013). Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA 19, 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottingham RM, Wu DC, Qin Y, Yao J, Hunicke-Smith S, and Lambowitz AM (2016). RNA-seq of human reference RNA samples using a thermostable group II intron reverse transcriptase. RNA 22, 597–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPRCas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. USA. 114, E7358–E7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbò P, Makarova KS, Kristensen DM, Wolf YI, and Koonin EV (2017). Reconstruction of the evolution of microbial defense systems. BMC Evol. Biol. 17, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeks J, Sokolowski RD, Graham S, Liu H, Naismith JH, and White MF (2013). Structure of a dimeric crenarchaeal Cas6 enzyme with an atypical active site for CRISPR RNA processing. Biochem. J. 452, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann V, Alkhnbashi OS, Saunders SJ, Scholz I, Hein S, Backofen R, and Hess WR (2017). Structural constraints and enzymatic promiscuity in the Cas6-dependent generation of crRNAs. Nucleic Acids Res. 45, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Jinek M, and Doudna JA (2011). An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat. Struct. Mol. Biol. 18, 680–687. [DOI] [PubMed] [Google Scholar]
- Shao Y, and Li H (2013). Recognition and cleavage of a nonstructured CRISPR RNA by its processing endoribonuclease Cas6. Structure 21, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Jain N, Stadler M, Fu BXH, Sanchez-Amat A, Fire AZ, and Arribere J (2018). A small RNA isolation and sequencing protocol and its application to assay CRISPR RNA biogenesis in bacteria. Bio-protocol 8, e2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Lucas-Elío P, Jackson SA, Aroca-Crevillén A, Hansen LL, Fineran PC, Fire AZ, and Sanchez-Amat A (2017a). Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. Elife 6, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Makarova KS, Shmakov S, Páez-Espino D, Mohr G, Liu Y, Davison M, Roux S, Krishnamurthy SR, Fu BXH, et al. (2017b). On the origin of reverse transcriptase using CRISPR-Cas systems and their hyperdiverse, enigmatic spacer repertoires. Mbio. 8, e00897–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Mohr G, Sidote DJ, Markham LM, Sanchez-Amat A, Bhaya D, Lambowitz AM, and Fire AZ (2016). Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science 351, aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneppen K, Pederson S, Krishna S, Dodd I, and Semsey S (2010). Economy of operon formation: cotranscription minimizes shortfall in protein complexes. MBio. 1, e00177–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano F, Lucas-Elío P, Fernández E, and Sanchez-Amat A (2000). Marinomonas mediterranea MMB-1 transposon mutagenesis: isolation of a multipotent polyphenol oxidase mutant. J. Bacteriol. 182, 3754–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Lentzsch AM, and Lambowitz AM (2017). Structure of a thermostable group II intron reverse transcriptase with template-primer and its functional and evolutionary implications. Mol. Cell 68, 926–939.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, van der Oost J, and Jinek M (2017). Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro N, Martinez-Abarca F, and González-Delgado A (2017). The reverse transcriptases associated with CRISPR-Cas systems. Sci. Rep. 7, 7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro N, Martinez-Abarca F, González-Delgado A, and Mestre MR (2018). On the origin and evolutionary relationships of the reverse transcriptases associated with Type III CRISPR-Cas systems. Front. Microbiol. 9, 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard G, Garrett RA, and Shah SA (2014). CRISPR adaptive immune systems of Archaea. RNA Biol. 11, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Preamplume G, Terns MP, Terns RM, and Li H (2011). Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure 19, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Terns RM, and Terns MP (2015). Cas9 function and host genome sampling in Type II-A CRISPR-Cas adaptation. Genes Dev. 29, 356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Picord G, Tuffery P, and Guerois R (2015). HHalign-Kbest: exploring sub-optimal alignments for remote homology comparative modeling. Bioinformatics 31, 3850–3852. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, and Wu L (2015). An unexplored diversity of reverse transcriptases in bacteria. Microbiol. Spectr. 3, 1253–1269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.