

Summary
Obesity is a major driver of cancer, especially hepatocellular carcinoma (HCC). The prevailing view is that non-alcoholic steatohepatitis (NASH) and fibrosis or cirrhosis are required for HCC in obesity. Here, we report that NASH and fibrosis and HCC in obesity can be dissociated. We show that the oxidative hepatic environment in obesity inactivates the STAT-1 and STAT-3 phosphatase T cell protein tyrosine phosphatase (TCPTP) and increases STAT-1 and STAT-3 signaling. TCPTP deletion in hepatocytes promoted T cell recruitment and ensuing NASH and fibrosis as well as HCC in obese C57BL/6 mice that normally do not develop NASH and fibrosis or HCC. Attenuating the enhanced STAT-1 signaling prevented T cell recruitment and NASH and fibrosis but did not prevent HCC. By contrast, correcting STAT-3 signaling prevented HCC without affecting NASH and fibrosis. TCPTP-deletion in hepatocytes also markedly accelerated HCC in mice treated with a chemical carcinogen that promotes HCC without NASH and fibrosis. Our studies reveal how obesity-associated hepatic oxidative stress can independently contribute to the pathogenesis of NASH, fibrosis, and HCC.
Keywords: hepatocellular carcinoma, non-alcoholic steatohepatitis, nonalcoholic fatty liver disease, fibrosis, obesity, oxidative stress, protein tyrosine phosphatase, STAT-1, STAT-3, T cells, PTPN2
Graphical Abstract
Highlights
-
•
Obesity promotes hepatic STAT-1 and STAT-3 signaling
-
•
Obesity promotes STAT-1-dependent T cell-infiltration, NASH, and fibrosis
-
•
Obesity promotes NASH-independent STAT-3-dependent HCC
Independent pathways contribute to development of NASH and hepatocellular carcinoma, disentangling the pathologies and providing insight for cancer treatments distinct from liver disease.
Introduction
Primary liver cancer is one of the world’s deadliest cancers and the third most common cause of cancer death. Hepatocellular carcinoma (HCC) represents 85%–90% of primary liver cancers and is refractory to nearly all currently available anti-cancer therapies. Over the last 20 years, the incidence of HCC in developed countries has been increasing, doubling in the United States (Font-Burgada et al., 2016). The obesity epidemic and the accompanying development of non-alcoholic fatty liver disease (NALFD), evident in >85% of all obese individuals, are thought to be key contributors to the development of HCC (Younes and Bugianesi, 2018). Indeed, obesity, the metabolic syndrome, and NAFLD account for 30%–40% of the increase in HCC in developed countries, and the risk of mortality from HCC in men with a body mass index (BMI) of 35–40 kg/m2 is 4.5-fold times greater than in patients with normal body weight (Calle et al., 2003).
NAFLD has an estimated worldwide prevalence of 25.2% (Younossi et al., 2016) and encompasses a broad spectrum of liver conditions ranging from simple steatosis, or non-alcoholic fatty liver (NAFL), to the more severe and progressive disease, non-alcoholic steatohepatitis (NASH). NASH is characterized by overt hepatic inflammation and tissue damage and ensuing reparative responses that result in fibrosis and ultimately cirrhosis, the principal cause of liver-related morbidity and mortality (Font-Burgada et al., 2016). Obesity-associated NASH is currently the third leading cause for liver transplantation and is expected to surpass hepatitis C as the principal cause for liver transplantation in the developed world (Shaker et al., 2014).
The mechanisms that underpin the progression from NAFL to NASH are complex and involve multiple insults and contributions from genetic modifiers that influence disease severity and progression (Font-Burgada et al., 2016). Lipotoxicity and oxidative stress are considered central to the transition to NASH and fibrosis (Font-Burgada et al., 2016). Several processes produce reactive oxygen species (ROS), including hepatic anaplerotic/cataplerotic reactions heightened in obesity, fatty acid oxidation, endoplasmic reticulum (ER) stress, inflammation, and the induction of ROS-producing NAD(P)H oxidases (Anderson et al., 2009, Bettaieb et al., 2015, Nakagawa et al., 2014, Perry et al., 2015, Satapati et al., 2015, Xu et al., 2005). Such oxidative stress is thought to promote hepatocyte cell death and the ensuing inflammatory and reparative responses that lead to fibrosis and if unresolved, cirrhosis (Font-Burgada et al., 2016, Ringelhan et al., 2018). ROS have been detected in liver biopsies from patients with chronic hepatitis (Tanaka et al., 2008) and in the liver parenchyma in rodent models of obesity (Gurzov et al., 2014, Park et al., 2010). Moreover, genetic studies in mice provide evidence for the increased ROS contributing to NASH and the genesis of liver cancer (Elchuri et al., 2005, Luedde et al., 2007, Neumann et al., 2003). In rodents, drugs that attenuate ROS production prevent NASH (Perry et al., 2015). At least one mechanism by which ROS may drive disease progression is through the induction of protein tyrosine kinase (PTK) signaling by oxidizing and inactivating protein tyrosine phosphatases (PTPs) (Tiganis, 2011). Our previous studies have shown that hepatic PTPs can be extensively oxidized in the livers of obese mice with NAFL (Gurzov et al., 2014), but the extent to which this might be pertinent to NASH remains unknown.
A key feature of NASH is the activation of resident immune cells, in particular Kupffer cells, and the recruitment of macrophages, B cells, natural killer (NK) cells, and CD4+ and CD8+ T cells that contribute to the inflammation and the persistent cycle of tissue damage and repair (Font-Burgada et al., 2016, Ringelhan et al., 2018). The importance of liver inflammatory macrophages is underscored by studies showing that tumor necrosis factor (TNF) from macrophages is essential for NASH and fibrosis in MUP-Upa mice that exhibit increased steatosis and progress to NASH and fibrosis and HCC when fed a high-fat obesity-promoting diet (Nakagawa et al., 2014). The importance of infiltrating T cells is illustrated by the reliance on antigen presentation via MHC class I and the recruitment of activated CD8+ T cells for the development of NASH and pericellular fibrosis in mice fed a choline-deficient high fat diet (Wolf et al., 2014). The molecular mechanisms driving the recruitment and activation of immune cells contributing to NASH pathogenesis have remained unresolved.
HCC can develop in a subset of obese NASH patients, but the precise mechanisms that give rise to HCC in some NASH patients and not others remain unresolved (Font-Burgada et al., 2016). Moreover, while HCC is typically accompanied by cirrhosis or severe fibrosis (Font-Burgada et al., 2016), there are a growing number of NAFLD patients without cirrhosis or advanced fibrosis that develop HCC (Alexander et al., 2013, Baffy et al., 2012, Mittal et al., 2015, Paradis et al., 2009, Rahman et al., 2013, Takuma and Nouso, 2010, Younes and Bugianesi, 2018). How HCC develops under these disparate conditions remains unclear. Over 28,000 somatic mutations have been identified in HCC (Schulze et al., 2015). These mutations influence the activation of tumor-promoting signaling pathways, including interleukin (IL)-6 and/or Janus-activated kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathways (Schulze et al., 2015). STAT-3 signaling is particularly important in driving the transformation of tumor progenitors and the development of HCC in rodents (He et al., 2010, He et al., 2013, Naugler et al., 2007, Park et al., 2010). Furthermore, STAT-3 is activated in the majority of human HCCs, positively correlating with tumor aggressiveness and negatively correlating with prognosis (Calvisi et al., 2006, He et al., 2010).
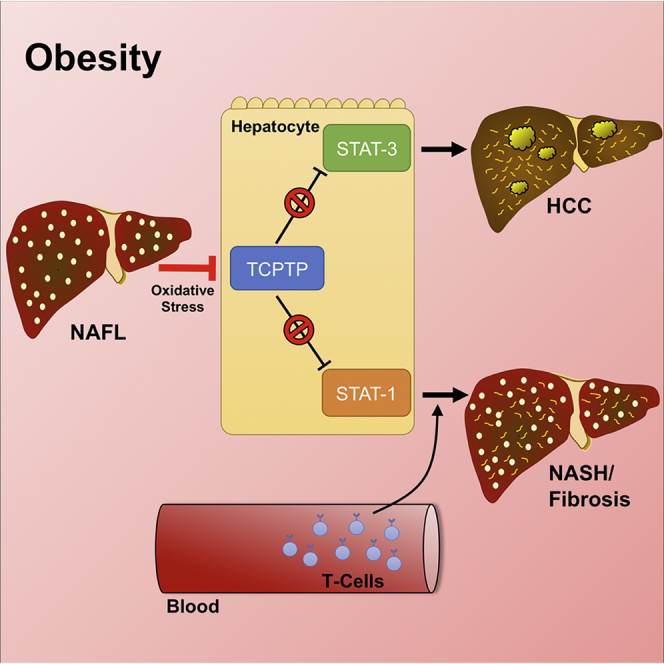
We report that the inactivation of negative regulators of STAT-1 and STAT-3 signaling in obesity can drive the development of NASH and HCC. The oxidation and inactivation of the STAT-1 and STAT-3 phosphatase TCPTP and heightened STAT-1 and STAT-3 signaling were evident in NASH in both obese mice and humans. While heightened STAT-1 signaling was responsible for the recruitment of activated cytotoxic T cells and the ensuing NASH and fibrosis, this was not essential for HCC. Rather, TCPTP inactivation promoted HCC in obesity via STAT-3, independent of T cell recruitment and NASH and fibrosis. Our results shed light on mechanisms that may underpin the growing incidence of HCC in non-cirrhotic livers in the setting of NAFLD.
Results
PTP Oxidation in NAFLD
Our previous studies have established that PTPs can be extensively oxidized in the livers of high-fat-fed mice that develop NAFL but not NASH (Gurzov et al., 2014). The extent to which PTP oxidation may be evident and contribute to the progression to NASH in obesity and the development of HCC remains unknown. To assess hepatic PTP oxidation in NASH we fed C57BL/6 mice (1) a standard chow diet (4% fat) that does not promote obesity, NAFL, or NASH, (2) a high fat (23% fat) diet (HFD) that promotes obesity, insulin resistance and NAFL, but not NASH, or (3) a choline-deficient high fat diet (CD-HFD; 23.5% fat) that promotes obesity, insulin resistance, and the progression from NAFL to NASH (Wolf et al., 2014). To monitor the oxidation status of PTPs, we took advantage of a monoclonal antibody (PTPox) that was raised against a PTP1B signature motif peptide with the catalytic Cys oxidized to the irreversible sulfonic acid. This antibody can detect virtually all tyrosine-specific PTPs when oxidized to the sulfonic state (Karisch et al., 2011). To monitor the extent of PTP oxidation in NASH, C57BL/6 mice were administered a chow diet versus an HFD or CD-HFD for 20 weeks (Figure 1A). Livers were homogenized in an anaerobic chamber and processed for analysis by PTPox immunoblotting. As noted previously (Gurzov et al., 2014), the oxidation of several PTPs was increased in the livers of mice fed an HFD (Figure 1A). As seen previously (Gurzov et al., 2014), PTPox species exhibiting increased oxidation in NAFL included those co-migrating with PTP1B and TCPTP (Figure 1A). Importantly, we found that the oxidation status of such PTPs was evident and/or increased further in mice fed a CD-HFD (Figure 1A) that became obese (Figures S1A and S1B) and developed NASH and fibrosis (Figure S1C). To explore the extent to which PTP oxidation may occur in humans with NAFLD we processed liver core needle biopsies from obese patients (BMI >35) with no steatosis (NAFLD activity score [NAS] 0) versus those with NAFLD (NAS 2–4) (Table S1). We found that the oxidation of several PTPs including those co-migrating with PTP1B and TCPTP was increased in the livers of patients with NAFLD versus those with no steatosis (Figure 1B). These findings establish the potential for hepatic PTPs to be oxidized in vivo in mice and humans in the context of NAFL and NASH and raise the possibility that such oxidation may contribute to the progressive development of NAFLD.
Figure 1.

Increased Hepatic PTP Oxidation and Elevated STAT Signaling in NAFL and/or NASH
(A) 8-week-old male C57BL/6 mice were fed a chow diet, an HFD, or a CD-HFD for 20 weeks. Livers from individual mice were processed for immunoblot analysis for total PTP oxidation.
(B) Liver core biopsies from individual obese humans with no steatosis (NAS = 0) or with NAFLD (NAS 2–4) were processed for immunoblot analysis for total PTP oxidation.
(C) Murine liver extracts immunoblotted for STAT-1 Y701 (p-STAT-1), STAT-3 Y705 (p-STAT-3), or STAT-5 Y694 (p-STAT-5) phosphorylation.
(D) Human livers biopsies processed for immunoblotting. Results are representative of at least three independent experiments.
See also Figure S1.
Figure S1.

Mice Fed a CD-HFD Do Not Become More Obese Than Mice Fed an HFD but Develop NASH, Related to Figure 1
(A–C) Ten-week-old C57BL/6 male mice were fed a HFD or a CD-HFD for 20 weeks and (A) body weights and (B) epididymal white adipose tissue (WAT) weights were assessed. (C) Livers were extracted and processed for histology monitoring for steatosis and lymphocytic infiltrates (Hematoxylin and Eosin) and fibrosis (Picrosirius red).
STAT-1 and STAT-3 Activation in NASH
PTP1B and TCPTP are key negative regulators of JAK/STAT signaling. PTP1B dephosphorylates JAK-2 and Tyk-2 whereas TCPTP dephosphorylates JAK-1 and JAK-3 (Tiganis and Bennett, 2007). TCPTP additionally dephosphorylates STAT family members, including STAT-1, -3, and -5 in the nucleus (Loh et al., 2011, ten Hoeve et al., 2002, Wiede et al., 2017). Accordingly, the oxidation and inactivation of PTP1B and TCPTP in obesity and NAFLD might be expected to promote STAT-1, STAT-3, and STAT-5 signaling. We found that basal STAT-1 Y701 phosphorylation (p-STAT-1) and STAT-3 Y705 phosphorylation (p-STAT-3) were increased in the livers of mice that had been fed an HFD for 20 weeks to promote obesity and NAFL but not NASH, and increased yet further in mice had been fed a CD-HFD for 20 weeks to promote obesity and the progression from NAFL to NASH (Figure 1C). By contrast, basal STAT5 Y694 phosphorylation was not overtly increased in the livers of mice fed an HFD or a CD-HFD for 20 weeks (Figure 1C). Accordingly, we hereon focused our attention on STAT-1 and STAT-3. As in mice, we found that p-STAT-1 and p-STAT-3 were also increased in the livers of obese patients (BMI >35) with NAFLD (NAS 2–4) (Table S1) versus those from non-obese patients (Figure 1D). Thus, the inactivation of hepatic JAK/STAT PTPs in obese mice and humans with NAFLD and/or NASH is accompanied by increased STAT-1 and STAT-3 signaling.
TCPTP Inactivation Promotes NASH and Fibrosis in Obesity
As TCPTP (Loh et al., 2011, ten Hoeve et al., 2002) but not PTP1B can directly dephosphorylate STAT-1 and -3 in the nucleus, and TCPTP was increasingly oxidized in the livers of obese mice with NASH versus NAFL (Figure 1A), we focused on TCPTP and assessed the impact of deleting TCPTP in the hepatocytes (Alb-Cre;Ptpn2fl/fl) of C57BL/6 mice fed an HFD for up to 40 weeks. We have reported previously that juvenile 5- to 6-week-old Alb-Cre;Ptpn2fl/fl mice fed an HFD for 12 weeks exhibit increased adiposity, hepatic steatosis, and insulin resistance (Gurzov et al., 2014). This was attributed to perturbations in the growth hormone (GH)-insulin-like growth factor (IGF)-1 pituitary axis, as a consequence of increased insulin-induced and p-STAT-5-mediated Igf-1 expression in the liver and the suppression of GH release from the pituitary (Gurzov et al., 2014). By contrast, high-fat feeding adult male Ptpn2fl/fl versus Alb-Cre;Ptpn2fl/fl mice from 10–12 weeks of age for periods of up to 40 weeks did not lead to changes in body weight or adiposity (Figures S2A and S2B), nor overt alterations in STAT-5 signaling (Figure S2C) or Igf-1 expression in the livers of 4-hr fasted mice (Figure S2D). This discordance in age-related phenotypes is in keeping with the increased levels of IGF-1 and GH that occur in early development and their ability to influence body weight primarily during pubertal adolescence (Berryman et al., 2008, Lichanska and Waters, 2008). Importantly, high-fat feeding adult male Alb-Cre;Ptpn2fl/fl mice for 40 weeks did not alter hepatic steatosis, as monitored by histology (Figure S2E), and the expression of fatty acid uptake/synthesis and lipogenesis genes (Figures S2F and S2G). Consistent with unaltered steatosis, triacylglycerol (TAG), diacylglycerol (DAG), and ceramide levels were unaltered in the livers of 20-week high-fat-fed Alb-Cre;Ptpn2fl/fl male mice (Figure S2H). Hepatic TCPTP deficiency also did not affect whole-body insulin sensitivity and glucose metabolism (Figures S2I and S2J).
Figure S2.

Unaltered Steatosis and Glucose Metabolism In High-Fat-Fed Alb-Cre;Ptpn2fl/fl Mice, Related to Figure 2
(A–D) Ten-twelve-week-old male liver-specific TCPTP-deficient mice (Alb-Cre;Ptpn2fl/fl) and Ptpn2fl/fl littermate controls were fed a HFD for up to 40 weeks. (A) Incremental body weights and (B) body composition at 40 weeks high fat feeding, as assessed by Dual-energy X-ray absorptiometry (DEXA). Livers were extracted from mice fed a HFD for 20 weeks and processed for (C) immunoblot analysis and (D) quantitative (ΔΔCt) real-time PCR to monitor for p-STAT-5 and Igf1 expression respectively.
(E–G) Livers extracted from mice fed a HFD for 40 weeks and processed for (E) histology (Hematoxylin and Eosin), (F) real-time PCR to monitor for fatty acid synthase (FAS; encoded by Fasn), sterol regulatory element-binding protein (SREBP-1c; encoded by Srebf1), stearoyl-CoA desaturase 1 (SCD-1; encoded by Scd1), fatty acid transporter CD36 (Cd36) or peroxisome proliferator-activated receptor γ (PPARγ; encoded by Pparg) and (G) immunoblotting to monitor for steatosis and the expression lipid synthesis genes.
(H–K) Ten-week-old male liver-specific TCPTP-deficient mice (Alb-Cre;Ptpn2fl/fl) and Ptpn2fl/fl littermate controls were fed a HFD for 20 weeks. (H) Livers were extracted and processed for analysis of triacylglyceride (TAG), diacylglyceride (DAG) and ceramide content. (I) Mice were fasted for 4-6 h and subjected to insulin (0.75 mU/g) and glucose (2 mg/g) tolerance tests. (J) Fed and fasted (12 h) blood glucose and plasma insulin levels were measured. (K) Ten-twelve week-old male Alb-Cre;Ptpn2fl/fl and Ptpn2fl/fl littermate controls were fed a CDAA diet for 12 weeks and body weights and epididymal white adipose tissue (WAT) and liver weights determined. Representative and quantified results (means ± SEM) are shown for the indicated number of mice with significance determined using a Student’s t test.
Although C57BL/6 mice fed a high fat diet become obese and steatotic, they do not develop NASH (Nakagawa et al., 2014, Wolf et al., 2014). Strikingly 40-week high-fat-fed male Alb-Cre;Ptpn2fl/fl mice, but not Ptpn2fl/fl mice, developed NASH (Figures 2A–2I), with many of the key diagnostic features of human NASH. This included hepatocyte ballooning (Figure 2A), a form of hepatocyte degeneration, accompanied by apoptotic and/or necrotic hepatocytes, as assessed by TUNEL staining (Figure 2B). High-fat-fed Alb-Cre;Ptpn2fl/fl mice also exhibited lymphocytic infiltrates as assessed by histology (Figures 2A and 2C), another key hallmark of NASH. This included the sporadic formation of ectopic lymphoid-like structures (Pitzalis et al., 2014), with clear T cell and B cell zones as assessed by immunohistochemistry (Figures 2C and 2D); immune cell infiltrates and ectopic lymphoid-like structures were not evident in Alb-Cre;Ptpn2fl/fl mice fed a chow diet for 40 weeks (data not shown). Ectopic lymphoid-like structures can develop at sites of inflammation and influence disease progression including cancer and have been shown to function as microniches for the formation of HCC progenitor cells in rodents and humans (Finkin et al., 2015). Consistent with the increased immune cell infiltrates and ectopic lymphoid-like structure formation, we noted that the mRNA expression of pro-inflammatory cytokines including IL-6 (Il6), TNF (Tnf), and interferon (IFN)γ (Ifng) and that of acute phase response genes, including Saa1, Saa3, and Crp, were elevated in the livers of high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 2E). Therefore TCPTP-deficiency in the livers of high-fat-fed mice results in marked hepatic inflammation and hepatocyte ballooning, key hallmarks of NASH.
Figure 2.

Hepatic TCPTP Deficiency Promotes NASH
(A–I) 10- to 12-week-old male Alb-Cre;Ptpn2fl/fl and Ptpn2fl/fl mice were fed an HFD for 40 weeks. Livers were processed for histology monitoring for (A) ballooning hepatocytes (black arrow) and lymphocytic infiltrates or (C) ectopic lymphoid-like structures (blue arrows). Alternatively, livers were processed for immunohistochemistry monitoring for (B) apoptotic and/or necrotic hepatocytes by TUNEL staining, or (D) for the presence of CD3ε+ T cells or B220+ B cells. (E) Inflammatory cytokine, chemokine, and acute phase reactant genes assessed by real-time qPCR. (F) Liver CD4+ and CD8+ T cells, including activated (CD69hi, CD25hi, IFNγ+, TNF+) effector memory (CD44hiCD62Llo) T cells, B cells, and immunosuppressive IgA+PD1hi cells assessed by flow cytometry. Liver fibrosis and liver damage assessed by (G) histology, (H) the expression of fibrotic genes, and (I) the presence of hepatic AST and ALT in serum.
(J and K) Alb-Cre;Ptpn2fl/fl;Rag1–/– and Ptpn2fl/fl;Rag1–/– littermate controls were fed an HFD for 40 weeks. Livers were analyzed for (J) fibrosis or (K) Acta2 and Tgfb1 expression.
(L–P) Alb-Cre;Ptpn2fl/fl and Ptpn2fl/fl mice were fed a CDAA diet for 12 weeks. Livers were processed for (L) histology, to monitor for steatosis, immune infiltrates, and fibrosis, or for (M, O, and P) real-time PCR to monitor for the expression of (M) lipid synthesis, (O) inflammatory, and (P) fibrotic genes, or (N) analyzed for TAG content. Representative and quantified results (means ± SEM) results are shown for the indicated number of mice.
See also Figures S2 and S3.
A requisite feature of NASH is the recruitment of immune cells that contribute to the inflammation and the accompanying tissue damage and repair. Accordingly, we characterized the immune cell subsets recruited to the livers of 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice by flow cytometry. The increase in lymphocytic infiltrates was predominated by CD4+ and CD8+ T cells that had an effector-memory phenotype (CD44hiCD62Llo) and expressed markers of activated T cells (CD25hiCD69hi), including cytotoxic CD8+ T cells (IFNγhiTNFhi) (Figures 2F and S3A). We also found a small but significant increase in B cells (Figure 2F), but no alterations in NKT cells, NK cells, macrophages/monocytes, granulocytes, myeloid-derived suppressor cells, T helper 17 cells, or immunosuppressive IL-10 expressing CD4+ T cells and regulatory T cells (Figures S3B–S3F). Recent studies have shown that inflammation in NAFLD is accompanied by the accumulation of liver-resident immunoglobulin-A producing (IgA+) cells that express programmed death ligand-1 (PD-L1) and IL-10 and suppress the anti-tumor activity of cytotoxic CD8+ T cells and thereby contribute to the formation of HCC (Shalapour et al., 2017). We found increased IgA+ cells that expressed high levels of PD-L1 in the livers of 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 2F). However, whereas hepatic CD8+ T cells in 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice constituted 15% of the total lymphocyte pool, IgA+ cells constituted 0.1% of the total lymphocyte pool. An expected outcome of cytotoxic T cell recruitment and activation would be tissue damage and ensuing reparative responses leading to fibrosis. In keeping with this we found that 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl had increased liver damage, as indicated by the increased collagen deposition and fibrosis (Picro Sirius Red or Mason’s trichrome staining; Figure 2G), the increased expression of fibrotic genes, including those encoding α-smooth muscle actin (Acta2) and transforming growth factor β (Tgfb) (Figure 2H), and the increased levels of the liver enzymes alanine transaminase (ALT) and aspartate aminotransferase (AST) in serum (Figure 2I), a sign of liver damage. By contrast, high-fat-fed Alb-Cre;Ptpn2fl/fl mice crossed onto the Rag1–/– background, so that they lacked mature T cells and B cells, did not exhibit overt fibrosis (Figures 2J and 2K). These results are consistent with TCPTP deletion, or otherwise oxidation and inactivation, driving T cell recruitment and the progression from NAFL to NASH and fibrosis.
Figure S3.

Unaltered Myeloid Cell and CD4+ IL-10-Expressing Immunosuppressive and Regulatory T Cell Recruitment in High-Fat-Fed Alb-Cre;Ptpn2fl/fl Mice, Related to Figure 2
(A) Gating strategy for liver T cells. Lymphocytes isolated from the livers of Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks were stained with fluorochrome-conjugated antibodies for CD8, CD44, CD62L, CD69, CD25, intracellular IFN-γ and intracellular TNF and analyzed by flow cytometry. CD8+ T cells were gated for CD62LloCD44hi effector-memory T cells, recently activated CD69hiCD44hi memory T Cells, CD25hiCD44hi memory T cells and cytotoxic CD44hiIFN-γ+ or CD44hiTNF+ T cells.
(B) Lymphocytes isolated from the livers Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed an HFD for 40 weeks were stained with α-galactosylceramide-loaded CD1d tetramers (CD1d/αGC) and α-TCRβ or NK1.1 and α-CD3 or α-CD11b and α-Gr-1 and NKT cells (CD1d/αGC)+/α-TCRβ+), NK cells (NK1.1+CD3-), CD11b+ myeloid lineage subsets including macrophages/monocytes (CD11b+Gr-1loSSClo/CD11b+Gr-1medSSClo), eosinphils (CD11b+Gr-1medSSChi) and neutrophils (CD11b+Gr-1hiSSCmed) were quantified by flow cytometry.
(C) Lymphocytes isolated from the livers of Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks were stained with fluorochrome-conjugated antibodies for CD11b and CD11c and CD11c+ dendritic cell subsets determined by flow cytometry.
(D) Lymphocytes isolated from the livers of Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks were stained with fluorochrome-conjugated antibodies for CD11b, F4/80, Ly6G and MHC-II and MCH-IIlo and MCH-IIhi monocytic myeloid-derived suppressor (MO-MDSC; CD11bhi/F4/80hiLy6Glo) and granulocytic myeloid-derived suppressor (G-MDSC; CD11bhi/F4/80–Ly6Ghi) were quantified by flow cytometry.
(E) Lymphocytes isolated from the livers of Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks were stained with fluorochrome-conjugated antibodies for CD4, CD44 and intracellular IL-17A or IL-10 and the relative proportion of T helper 17 (Th17) cells CD4+CD44hiIL-17A+ and immunosuppresive IL-10 expressing CD4+ T cells (CD4+CD44hiIL-10+) CD4+CD44hiIL-10+ T cells was determined by flow cytometry.
(F) Lymphocytes isolated from the livers of Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks were stained with fluorochrome-conjugated antibodies for CD4, CD25, and intracellular FoxP3 and CD4+CD25+FoxP3+ or CD4+CD25–FoxP3+ regulatory T cells were quantified by flow cytometry. Representative results (means ± SEM) and (A) representative cytometry profiles from at least two independent experiments are shown.
To further explore the impact of TCPTP-deficiency on NASH pathogenesis, Ptpn2fl/fl versus Alb-Cre;Ptpn2fl/fl mice were fed a choline-deficient amino acid-defined (CDAA) diet that promotes NASH and fibrosis in C57BL/6 mice without promoting obesity (Merry et al., 2016). Hepatic TCPTP-deficiency in CDAA-fed mice did not alter body weight or adiposity (Figure S2K) and did not alter the development of steatosis, as assessed by (1) histology (Figure 2L), (2) the expression of lipogenic and fatty acid synthesis genes (Figure 2M), and (3) by measuring TAGs (Figure 2N). However, TCPTP-deficiency resulted in increased lymphocytic infiltrates (Figure 2L), hepatic inflammation (Figure 2O), and fibrosis (Figures 2L and 2P). These results point toward the oxidation and inactivation of TCPTP in obesity contributing to the exacerbation of NASH and fibrosis (Figures 2L and 2P).
TCPTP Inactivation Promotes HCC in Obesity
HCC can develop in a subset of NAFLD patients, particularly in patients where persistent inflammation and reparative responses drive the development of fibrosis or cirrhosis (Font-Burgada et al., 2016, Ringelhan et al., 2018). Such fibrotic/cirrhotic livers may contain hepatocyte-derived HCC progenitors that can undergo malignant transformation and give rise to HCC (Finkin et al., 2015, He et al., 2013). Moreover, the development of HCC in NAFLD can also involve the recruitment of immunosuppressive cells that subvert anti-tumor immunity (Shalapour et al., 2017). Consistent with the development of NASH and fibrosis and the increased abundance of IgA+PD-L1hi immunosuppressive cells, 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice developed nodular tumors of variable size in approximately one-third of mice (Figures 3A–3C). Tumor formation did not occur (Figures 3A–3C) or was evident at a low incidence in high-fat-fed Ptpn2fl/fl control mice. Tumors were not evident in Ptpn2fl/fl or Alb-Cre;Ptpn2fl/fl mice fed a standard chow diet consistent with tumor formation being associated with obesity (Figure 3A; data not shown). The tumors that developed in high-fat-fed Alb-Cre;Ptpn2fl/fl mice were clearly defined from the adjacent parenchyma and were steatohepatitic (Figure 3D), but did not exhibit any changes in the abundance of enzymes associated with lipogenesis (Figure S4A). Tumors in high-fat-fed Alb-Cre;Ptpn2fl/fl mice completely lacked any reticular structure as reflected by the absence of reticulin staining and were disorganized and multi-layered (Figure 3E), consistent with these being carcinomas. Tumors, but not the adjacent parenchyma, exhibited increased Ki67 staining (Figure 3F), indicative of proliferating cells within tumor lesions, as occurs in human HCC. Further immunohistological assessment indicated that tumors were positive for EpCAM and cytokeratin 19 (Figure 3F), which are associated with poor prognosis and invasion in human HCC (Yamashita et al., 2008). Tumors also expressed other markers of aggressive disease, including GRP78 (Figure 3F), whose expression is negatively correlated with tumor grade and positively associated with invasion (Su et al., 2010), and phosphorylation of the mitogen-activated protein kinases (MAPKs) ERK-1 and ERK-2 (p-ERK1 and p-ERK2; Figure 3F), which are activated in the majority of human HCCs and drive growth (Calvisi et al., 2006). The increase in ERK-1 and ERK-2 signaling was substantiated by immunoblot analysis monitoring for p-ERK-1/2 in tumor versus tumor-adjacent tissue (Figure 3G); JNK MAPK signaling that is required for DEN-induced HCC in mice (Sakurai et al., 2006) was also increased (Figure 3G), whereas there were no overt increases in p38 MAPK (Figure S4B) or phosphatidylinositol 3-kinase (PI3K)-AKT signaling (Figure S4C). As seen in human HCC, tumors in high-fat-fed Alb-Cre;Ptpn2fl/fl mice expressed higher levels of the epidermal growth factor receptor (EGFR) PTK (Figure 3G). By contrast, c-MET and insulin receptor (IR) PTK levels were unaltered or decreased (Figure S4D). These results indicate that TCPTP-deficiency in hepatocytes drives tumor development in obesity with many of the characteristic features of aggressive HCCs seen in humans.
Figure 3.

Hepatic TCPTP Deficiency Promotes HCC
(A–C) Mice were fed an HFD or a chow diet for 40 weeks. Livers were extracted and analyzed for (A) nodular tumors (black arrows). (B) Tumor incidence and sizes. (C) Tumor incidence in 5 independent cohorts.
(D–G) Livers and tumors analyzed by (D and E) histology, (F) immunohistochemistry, or (G) immunoblotting. Representative and quantified results (means ± SEM) results are shown for the indicated number of mice (B) or cohorts (C).
See also Figure S4.
Figure S4.

The Development of NASH and HCC in High-Fat-Fed Alb-Cre;Ptpn2fl/fl Mice Is Not Accompanied by Increased PTK and PI3K/AKT/mTOR Signaling, Related to Figure 3
(A–G) Ten-twelve week-old male Alb-Cre;Ptpn2fl/fl and Ptpn2fl/fl littermate controls were fed a HFD for 40 weeks and liver tissue and tumors extracted from individual mice for (A)–(E), (G) immunoblot analysis with the indicated antibodies [including those for c-MET, IR β subunit (IRβ), Ser-473 phosphorylated AKT (p-AKT), Thr-180/Tyr-182 phosphorylated p38 (p-p38), Ser-2448 phosphorylated mammalian target of rapamycin (p-mTOR), Thr-37/46 phosphorylated eukaryotic translation initiation factor 4E-binding protein 1 (p-4E-BP1), Ser-235/6 phosphorylated ribosomal protein S6 (p-S6), Tyr-1022/Tyr-1023 phosphorylated JAK-1 (p-JAK-1), Tyr-1007/Tyr-1008 phosphorylated JAK-2 (p-JAK-2), Tyr-418 phosphorylated SFKs (p-SFK)], or (F) real time PCR. Representative and quantified results (means ± SEM) are shown for the indicated number of mice with significance determined using a Student’s t test.
TCPTP Inactivation Promotes a STAT-1 and STAT-3 Molecular Phenotype in NASH
To explore the mechanisms by which TCPTP inactivation in hepatocytes might drive the development of NASH and HCC, we performed an unbiased transcriptome analysis by RNA sequencing (RNA-seq). Total RNA was isolated from the livers (excluding tumors) of 40-week high-fat-fed Ptpn2fl/fl versus Alb-Cre;Ptpn2fl/fl mice and used to generate libraries for sequencing on a Illumina HiSeq1500. Differential gene expression was analyzed by supervised clustering with altered pathways determined using Ingenuity pathway analysis (Figures 4A and S5A). Only 24 genes were differentially expressed between the genotypes, with all but two genes showing increased expression (Figure 4A); 6/24 genes were also assessed by real-time qPCR in an independent cohort and shown to be upregulated (Figures 4B and S5B). Strikingly, 18 of the differentially expressed genes were bona fide or predicted targets of IFN signaling and/or transcriptional targets of STAT-1 and/or STAT-3 and were significantly upregulated (Figure 4A). The top-ranked upregulated gene was Cxcl9 (Figure 4A) a STAT-1 transcriptional target that encodes the T cell chemoattractant CXCL9 (Semba et al., 2013, Tacke et al., 2011). Other differentially expressed genes included those encoding the acute-phase reactants SAA-1/2 and ORM2 (Figure 4A) and those encoding the IFN-γ-induced guanylate-binding proteins (GBPs)-2/3/7/10 and the immunity-related GTPases TGTP1 and IGTP (Figure 4A). Consistent with this, Ingenuity analyses identified acute-phase response, JAK/STAT, and cytokine signaling pathways among the top-pathways altered by TCPTP-deficiency (Figure S4A). These findings are in keeping with the increased hepatic inflammation and T cell infiltration, NASH, and liver damage evident in high-fat-fed Alb-Cre;Ptpn2fl/fl mice. Indeed, other differentially expressed STAT-1 and STAT-3 target genes included those encoding fibrinogen-like 1 (FGL-1) and lipocalin-2 (LCN2) (Figures 4A and 4B), hepatokines elevated in the livers or serum of patients with NALFD (Auguet et al., 2013, Semba et al., 2013, Tacke et al., 2011, Wu et al., 2013, Yu et al., 2009, Zhao et al., 2014). Increased Cxcl9 gene expression was also evident in hepatocytes from chow-fed Alb-Cre;Ptpn2fl/fl mice (Figure 4C) consistent with the increased Cxcl9 expression being hepatocyte cell intrinsic and potentially an early event in disease progression. By contrast Fgl1 and Lcn2 were not elevated in hepatocytes from chow-fed Alb-Cre;Ptpn2fl/fl mice (Figure 4C), indicating that their expression is reliant on obesity and might reflect a later stage in disease progression. Moreover in keeping with studies showing that circulating CXCL9, FGL-1, and LCN2 levels are elevated in rodents and/or humans with NAFLD (Auguet et al., 2013, Semba et al., 2013, Tacke et al., 2011, Wu et al., 2013), we found that CXCL9, FGL-1, and LCN2 levels were significantly increased in the sera of high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 4D).
Figure 4.

A STAT-1 and STAT-3 Molecular Phenotype in Obesity-Associated NASH/HCC
(A–D) Mice were fed an HFD for 40 weeks.
(A) Liver tissue was processed for RNA-seq and a heatmap representing the normalized gene expression values (log2 fold-change from mean) generated. Genes differentially expressed between genotypes, ordered by smallest to largest p value (top to bottom). Genes tested by real-time PCR in independent samples are indicated with an asterisk. Known and putative STAT-1 and STAT-3 transcriptional targets are highlighted.
(B) Liver gene expression in an independent cohort assessed by real-time PCR.
(C) Gene expression in hepatocytes isolated from chow-fed mice.
(D) Serum levels of CXCL9, LCN2, and FGL-1.
(E) Human liver core biopsies from obese male or female patients (BMI 40–74 kg/m2) were processed for histology and those with non-steatotic livers (NAS 0), simple steatosis, and no fibrosis (NAS 1–2) or overt NASH with fibrosis (NAS ≥ 5) were identified. Representative histology micrographs. Biopsies were processed for real-time PCR.
(F and G) Mice were fed an HFD for 40 weeks. Liver and tumor tissues were extracted for (F) immunoblotting or (G) immunohistochemistry.
(H) Mice were fed a CDAA diet for 12 wks. Livers were extracted and processed for immunoblotting. Representative and quantified results (means ± SEM) results are shown for the indicated number of mice or human liver biopsies.
Figure S5.

RNA-Seq Defines a STAT-1 and STAT-3 Molecular Phenotype in High-Fat-Fed Alb-Cre;Ptpn2fl/fl Mice, Related to Figure 4
(A) Ten-twelve week-old male Alb-Cre;Ptpn2fl/fl and Ptpn2fl/fl littermate controls were fed a HFD for 40 weeks. Liver tissues from six Ptpn2fl/fl and six Alb-Cre;Ptpn2fl/fl mice were processed for transcriptome analysis by RNaseq. Stacked bar-chart depicting the overlap between the top 119 differentially expressed hepatic genes in Alb-Cre;Ptpn2fl/fl mice by RNA-seq (log fold-change > 0.2 and p value < 0.01) and annotated Canonical Pathways in the Ingenuity Knowledge Base. Numbers (top) indicate the total number of genes included in the pathway. Red, green and clear bars indicate overlap with upregulated genes, overlap with downregulated genes, and genes not in the regulated set, respectively (left-hand axis). p values for the extent of overlap of gene sets (orange line; scaled to negative log10, see right-hand axis) were calculated using a right-tailed Fisher’s exact test.
(B) An independent cohort of ten week-old male Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice were fed a HFD for 40 weeks and liver tissues processed for quantitative real time PCR. Quantified results (means ± SEM) are shown for the indicated number of mice with significance determined using a Student’s t test.
Our results demonstrate that TCPTP inactivation in hepatocytes can drive the STAT-1 and STAT-3-mediated expression of genes that might play important roles in recruiting T cells, shaping the immune response and promoting the reparative responses of the liver that underpin the development of NASH and fibrosis. Accordingly, we asked whether such genes might be differentially expressed in obese humans with NAFL versus NASH. The expression of CXCL9, FGL1, and LCN2 was assessed in liver core biopsies from obese patients (BMI 36–61) with normal livers without steatosis (NAS 0) versus obese patients (BMI 36–61) with NAFL (NAS 1–2), without immune cell infiltrates or fibrosis, or in obese patients (BMI 47–74) with overt NASH (NAS ≥5) and fibrosis (Figure 4E; Table S1). We found that the hepatic expression of all three human genes was increased in the livers of obese patients with NAFL when compared to those with normal livers (Figure 4E), consistent with studies implicating such factors in human NAFLD (Tacke et al., 2011, Wu et al., 2013). Strikingly, we found FGL1 and CXCL9 expression increased yet further in the livers of obese patients with NASH (Figure 4E). These findings identify FGL1 and CXCL9 as potential discriminatory factors for NASH versus NAFL. Also, these studies indicate that the molecular processes underpinning NASH in high-fat-fed Alb-Cre;Ptpn2fl/fl mice may reflect those occurring in human patients developing NASH.
TCPTP Inactivation Promotes STAT-1-Dependent NASH
As STAT-1 and STAT-3 can serve as direct nuclear substrates for TCPTP (Loh et al., 2011, ten Hoeve et al., 2002), we assessed their phosphorylation in the livers of 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 4F). TCPTP deletion enhanced p-STAT-1 and p-STAT-3 in tumor adjacent and tumor tissue in the livers of high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 4F). The promotion of STAT-1 and STAT-3 signaling was cell-intrinsic, as indicated by the increased p-STAT-1 and p-STAT-3 in the nuclei of hepatocytes examined by immunohistochemistry (Figure 4G). No overt increases were evident in the tyrosine phosphorylation and activation of PTKs that normally phosphorylate STAT-1 and STAT-3 including JAK-1, JAK-2, and Src family kinases (SFKs) (Figures S4B and S4E) indicating that TCPTP deficiency may elicit its effects at the level of STAT-1 and STAT-3. Indeed, JAK-1 phosphorylation was decreased in the livers and tumors of 40-week high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure S4B) and this was accompanied by increased expression of the gene encoding the JAK PTK inhibitor and STAT transcriptional target, suppressor of cytokine signaling (SOCS)-1 (Figure S4F); the expression of other negative regulators of JAK/STAT signaling including SOCS-3 and the tyrosine phosphatases PTP1B (Ptpn1), SHP-1 (Ptpn6), and SHP-2 (Ptpn11) was unaltered (Figures S4F and S4G). Nonetheless, consistent with TCPTP-deficiency increasing STAT-1 and STAT-3 signaling, we found that the exacerbation of NASH in Alb-Cre;Ptpn2fl/fl mice fed a CDAA-diet was also accompanied by the increased tyrosine phosphorylation of STAT-1 and STAT-3 (Figure 4H).
To determine the extent to which the heightened hepatic STAT-1 and STAT-3 signaling might drive NASH and HCC in high-fat-fed Alb-Cre;Ptpn2fl/fl mice, we sought to correct the enhanced STAT-1 or STAT-3 signaling. To this end, we crossed Alb-Cre;Ptpn2fl/fl mice onto the Stat-1fl/+ or Stat-3fl/+ backgrounds to reduce STAT-1 or STAT-3 by 50% in hepatocytes so that hepatic p-STAT-1 and p-STAT-3 might approximate that in Ptpn2fl/fl controls. Stat-1fl/+ or Stat-3fl/+ heterozygosity reduced STAT-1 and STAT-3 protein levels in liver homogenates by ∼50% (Figures 5A and 6A). Importantly, STAT-1 heterozygosity partially corrected the enhanced p-STAT-1 (Figure 5A), whereas STAT-3 heterozygosity completely corrected the enhanced p-STAT-3 (Figure 6A). Strikingly, we found that repressing p-STAT-1 corrected the hepatic inflammation, as reflected by the expression of inflammatory and acute-phase reactant genes (Figure 5B). Conditional Stat1 heterozygosity also corrected the enhanced hepatic expression of the STAT-1 target gene Cxcl9 (Figure 5B), which we had identified as the top hit in our transcriptome analysis (Figure 4A). Stat1 heterozygosity also largely corrected the enhanced hepatic expression of STAT-1 target gene Lcn2 (Figure 5B). By contrast, conditional Stat1 heterozygosity did not alter the expression of Il6, Fgl1, or Ccl5 that were also elevated in the livers of high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 5B), consistent with these being targets of STAT-3 (He et al., 2013, Kovacic et al., 2010, Yu et al., 2009). Nonetheless, attenuating the enhanced p-STAT-1 corrected the increased immune cell recruitment (Figures 5C and 5D). In particular, Stat1 heterozygosity corrected the increased recruitment of effector-memory CD4+ and CD8+ T cells expressing markers of activated and/or cytotoxic T cells (Figure 5D). Moreover, Stat1 heterozygosity corrected the increased fibrosis (Figures 5B and 5C). By contrast, correcting the enhanced STAT-3 signaling resulting from TCPTP-deletion in hepatocytes reduced the expression of the STAT-3 target genes Il6, Ccl5, and Fgl1 (Figure 6B) and reduced the expression of Ifng, Tnf, and Saa1 (Figure 6B). However, Stat3 heterozygosity neither altered the expression of Cxcl9 or Lcn2 (Figure 6B), nor did it attenuate the accumulation of activated T cells (Figures 6C and 6D) or fibrosis (Figures 6B and 6C). These results indicate that although both the enhanced STAT-1 and STAT-3 signaling accompanying TCPTP deletion in hepatocytes contribute to inflammation, it is the enhanced STAT-1 signaling that is responsible for the T cell accumulation and the development of NASH and fibrosis in high-fat-fed Alb-Cre;Ptpn2fl/fl mice.
Figure 5.

STAT-1 Is Required for Inflammation, T Cell Recruitment, and Fibrosis
(A–C) Ptpn2fl/fl, Alb-Cre;Ptpn2fl/fl, and Alb-Cre;Ptpn2fl/fl;Stat-1fl/+ mice were fed an HFD for 40 weeks. Liver tissue was processed for (A) immunoblotting, (B) real-time PCR, and (C) histology monitoring for immune cell infiltrates and fibrosis (Picro Sirius Red).
(D) The recruitment of liver T cells, including activated effector-memory T cells, was assessed by flow cytometry. Representative and quantified results (means ± SEM) results are shown for the indicated number of mice.
Figure 6.

STAT-3 Is Required for Inflammation, but Not for T Cell Recruitment or Fibrosis
(A–C) Ptpn2fl/fl, Alb-Cre;Ptpn2fl/fl, and Alb-Cre;Ptpn2fl/fl;Stat-3fl/+ mice were fed an HFD for 40 weeks. Liver tissue was processed for (A) immunoblotting, (B) real-time PCR, and (C) histology monitoring for immune cell infiltrates and fibrosis (Mason’s Trichrome stain).
(D) The recruitment of liver T cells, including activated effector-memory T cells, was assessed by flow cytometry. Representative and quantified results (means ± SEM) are shown for the indicated number of mice.
TCPTP Inactivation Promotes STAT-3-Dependent HCC
An expectation of attenuating the development of NASH in high-fat-fed Alb-Cre;Ptpn2fl/fl mice might be the prevention of HCC. This expectation emanates from studies indicating that the development of HCC in humans occurs primarily in the context of NASH with advanced fibrosis or cirrhosis (Font-Burgada et al., 2016). Remarkably, although Stat1 heterozygosity in hepatocytes repressed the accumulation of activated T cells and prevented inflammation and fibrosis, it did not prevent tumors forming in high-fat-fed Alb-Cre;Ptpn2fl/fl mice (Figure 7A). These results indicate that tumor development in high-fat-fed Alb-Cre;Ptpn2fl/fl mice occurs independently of NASH and fibrosis. To explore the mechanisms that drive HCC in high-fat fed Alb-Cre;Ptpn2fl/fl mice, we turned our attention on STAT-3. STAT-3 is frequently activated in human HCC and is essential for HCC in DEN-treated mice (He et al., 2010). Accordingly, we assessed whether STAT-3 heterozygosity might suppress the development of HCC in high-fat-fed Alb-Cre;Ptpn2fl/fl mice. Although Stat3 heterozygosity in hepatocytes did not influence the recruitment of activated effector T cells and the development of fibrosis (Figures 6C and 6D), it completely repressed the increased tumor incidence (Figure 7B). These results point toward TCPTP-deficiency and the promotion of STAT-3 signaling driving the development of HCC in obesity.
Figure 7.

Tumor Development Occurs Independently of NASH
(A and B) Ptpn2fl/fl, Alb-Cre;Ptpn2fl/fl, and either (A) Alb-Cre;Ptpn2fl/fl;Stat-1fl/+ or (B) Alb-Cre;Ptpn2fl/fl;Stat-3fl/+ mice were fed an HFD for 40 weeks. Livers were analyzed for nodular tumors and tumor incidence and sizes recorded.
(C–F) DEN-treated mice were fed a chow diet and processed at 40 weeks of age.
(C) Liver tissue extracted for immunoblotting.
(D–F) Livers were analyzed for nodular tumors (D) and livers weighed (E) and tumor incidence and sizes (F) determined.
(G and H) Tumor tissue was processed for real-time PCR to monitor proliferation (G) and glycolytic metabolism genes (H).
(I–L) HCC cells were isolated and transduced with GFP or GFP and Cre-encoding retroviruses to generate isogenic cell lines with and without TCPTP. (I) Soft-agar growth. (J) Gene expression.
(K and L) Tumor cells were xenografted into the flanks of BALB/c nu/nu mice. (K) Tumor volumes and weights and (L) immunohistochemistry. Representative and quantified results (means ± SEM) are shown for the indicated number of mice or experimental repeats.
See also Figures S6 and S7.
To further explore whether the enhanced p-STAT-3 in obese Alb-Cre;Ptpn2fl/fl mice might promote tumorigenicity independently of NASH and fibrosis, we assessed the impact of TCPTP-deficiency in hepatocytes on the development of HCC in mice treated with the chemical carcinogen diethylnitrosamine (DEN). DEN promotes the development of liver tumors in mice with many of the key characteristics of HCC in humans (Maeda et al., 2005). However, although DEN-induced tumorigenicity is dependent on inflammation and IL-6 and/or STAT-3 signaling (He et al., 2010, Park et al., 2010), tumors develop in the absence of NASH or fibrosis (Park et al., 2010). We found that TCPTP deletion in hepatocytes promoted the development of HCCs in DEN-treated mice (Figure S6A). The increased tumorigenicity resulting from TCPTP-deficiency was accompanied by an increase in p-STAT-3 in tumor and non-tumor tissue alike (Figures S6B and S6C). Tumors were highly proliferative as noted by an increase in Ki67 staining (Figure S6D) and an increase in EGFR and cyclin D1 protein and gene expression (Figures S6E and S6F). Tumors also exhibited increased markers of glycolytic metabolism, including increased expression of the gene encoding HIF-1α (Figure S6F), a transcriptional target of STAT-3 that can promote glycolysis by driving GLUT1, PDK1, and PKM2 expression (Demaria and Poli, 2012). Consistent with the increase in Hif1α, we found that Glut1, Pdk1, and PKM2 levels were also elevated in TCPTP-deficient tumors (Figures S6E and S6F). Importantly, we found that conditional Stat3 heterozygosity in Alb-Cre;Ptpn2fl/fl mice (Figure 7C) completely repressed the enhanced tumorigenicity otherwise associated with TCPTP deficiency (Figures 7D–7F) and corrected the increased expression of genes associated with cellular proliferation and glycolysis (Figures 7G and 7H). These findings provide causal evidence for TCPTP inactivation in hepatocytes driving HCC in a STAT-3-dependent manner and substantiate the assertion that the inactivation of hepatic TCPTP in obesity may drive HCC independent of NASH and fibrosis.
Figure S6.

TCPTP Deletion in Hepatocytes Promotes DEN-Induced HCC, Related to Figure 7
14 day-old male Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl neonates were injected with DEN (25 mg/kg) and weaned mice fed a standard chow diet.
(A) At 40 weeks of age body weights were recorded and livers extracted, weighed and analyzed for nodular tumors. Tumor incidence and sizes are shown for the indicated number of mice.
(B) Liver and tumor homogenates were processed for immunoblotting monitoring for STAT-3 signaling.
(C and D) Tumors were processed for immunohistochemistry monitoring for p-STAT-3 and Ki67.
(E and F) Tumor homogenates were processed for immunoblotting and real time PCR monitoring for the expression of cellular proliferation and glycolytic metabolism genes. Representative and quantified results (means ± SEM) are shown for the indicated number of mice with significance determined using a Student’s t test or a one-way ANOVA.
TCPTP Deletion in HCC Cells Promotes Tumorigenicity
TCPTP inactivation might not only contribute to the transformation of progenitors and development of HCC in obesity, but also elicit cell autonomous effects on the growth of HCC once established. To explore this, we isolated HCC cells from DEN-induced tumors in Ptpn2fl/fl mice (Figure S7A). Ptpn2fl/fl HCC cells were transduced with retroviruses expressing GFP, or GFP and Cre, and sorted to generate isogenic HCC cell lines with or without TCPTP (Figure S7B). TCPTP was deleted in two Ptpn2fl/fl HCC cell lines expressing Cre recombinase and this was accompanied by the promotion of basal and IL-6-induced STAT-3 signaling (Figures S7B and S7C; data not shown); TCPTP-deficiency also increased p-STAT-3 in HCC cell lines derived from tumors arising in DEN-treated Alb-Cre;Ptpn2fl/fl versus Ptpn2fl/fl mice (Figure S7D). TCPTP deletion did not alter PI3K-AKT or ERK-1 and ERK-2 signaling (Figure S7C) or cellular proliferation, migration, or anoikis (data not shown), but significantly increased anchorage-independent growth in soft agar (Figure 7I). The increased growth in soft agar was accompanied by the increased expression of STAT-3-target genes encoding c-Myc and cyclin D1 (Figure 7J) that drive tumor growth. Finally, we found that TCPTP-deficiency in HCC cells increased tumor xenograft growth in nude mice (Figure 7K) accompanied by increased p-STAT-3 and proliferation as assessed by Ki67 staining (Figure 7L). Taken together, these results point toward TCPTP inactivation in HCC cells eliciting cell autonomous effects on HCC growth.
Figure S7.

TCPTP Deletion in HCC Cells Promotes STAT3 Signaling, Related to Figure 7
(A–C) Tumors from DEN-treated Ptpn2fl/fl mice were dissociated and HCC cell lines established. (A) Ptpn2fl/fl HCC cells, primary hepatocytes from chow-fed C57BL/6 mice and MCF10A immortalized mammary epithelial cells were processed for immunofluorescence microscopy monitoring for the presence of the hepatocyte marker albumin, or the liver cancer marker α-fetoprotein. Ptpn2fl/fl HCC cells were transduced with retroviruses encoding GFP alone (MSCV-GFP) or GFP and Cre recombinase (MSCV-GFP-CRE) and sorted twice for GFP and either processed for (B) immunofluorescence microscopy monitoring for GFP and TCPTP, or (C) serum starved and stimulated with IL-6 (1 ng/ml) and then processed for immunoblotting monitoring for STAT-3 signaling. Quantified results (means ± SEM) are shown for the indicated number of experiments with significance determined using a Student’s t test.
(D) Tumors from DEN-treated Ptpn2fl/fl or Alb-Cre;Ptpn2fl/fl were dissociated and HCC cell lines established. HCC cells were serum starved and processed for immunoblotting.
Discussion
The incidence of overweight and obese individuals and the accompanying development of type 2 diabetes and the metabolic syndrome have ballooned to epidemic proportions. The obesity and type 2 diabetes epidemics are major drivers of NAFLD (Font-Burgada et al., 2016). Patients with NAFLD are at risk of developing HCC and this typically occurs in the setting of NASH and advanced fibrosis or cirrhosis (Font-Burgada et al., 2016). This coincidence has yielded the widely held belief that HCC in NAFLD must be predicated by the development of NASH with advanced fibrosis or cirrhosis. Indeed, American and European surveillance programs for HCC have been limited to those individuals with cirrhosis. However, in the last few years, it has become apparent that in roughly half of all cases where HCC develops in the context of NASH, this is evident before the onset of cirrhosis (Alexander et al., 2013, Mittal et al., 2015, Paradis et al., 2009, Rahman et al., 2013, Takuma and Nouso, 2010, Younes and Bugianesi, 2018). Indeed, in patients with metabolic syndrome, HCC might even occur in the context of simple steatosis in the absence of any NASH and inflammation or fibrosis (Alexander et al., 2013, Baffy et al., 2012). The mechanisms that contribute to the development of HCC in NAFLD in the absence of NASH and/or overt fibrosis or cirrhosis have remained unexplored. Our studies indicate that development of NASH and fibrosis versus HCC in obesity can be independent events, driven by the respective engagement of hepatocyte STAT-1 and STAT-3 transcriptional programs that promote T cell recruitment and liver damage versus tumorigenicity.
In this study we have shown that STAT-1 signaling is progressively elevated in the livers of mice fed NAFL- versus NASH-promoting diets and increased in the livers of obese patients with NAFLD. Moreover, we have shown that the STAT-1 phosphatase TCPTP (ten Hoeve et al., 2002) is oxidized in the livers of obese mice with NAFL or NASH and obese humans with NAFLD. Furthermore, the deletion of TCPTP increased STAT-1 signaling in hepatocytes to promote T cell accumulation, inflammation, NASH, and fibrosis in C57BL/6 mice fed a high fat diet that normally develop steatosis, but not NASH and fibrosis (Park et al., 2010). RNA-seq analyses demonstrated that known and/or putative hepatic STAT-1 and STAT-3 target genes were elevated in the livers of high-fat-fed obese TCPTP-deficient mice. Although some of the elevated hepatic STAT-1 and STAT-3 target genes might reflect contributions from infiltrating immune cells, the expression of the STAT-1 target gene Cxcl9 was hepatocyte intrinsic, whereas the STAT-3 target genes Il6, Fgl1, and Ccl5 were elevated independently of NASH and T cell recruitment. Cxcl9 is produced by parenchymal and non-parenchymal cells and is elevated in the Shionogi mouse model, an inbred mouse strain that spontaneously develops NASH and HCC (Semba et al., 2013). Cxcl9 expression is also elevated in humans with NAFLD and correlates with worsening fibrosis (Wasmuth et al., 2009). Similarly Lcn2 is highly expressed by myeloid cells and hepatocytes, especially under conditions of stress, and may recruit immune cells such as neutrophils to sites of injury and inflammation (Li et al., 2018). Increased circulating LCN-2 has been reported in human NALFD (Auguet et al., 2013) and may contribute to NAFLD pathogenesis (Ye et al., 2016). Our studies are consistent with a STAT-1 gene signature being of functional relevance to the development of NASH. However, whether the promotion of STAT-1 signaling occurs solely as a consequence of the oxidation and inactivation of TCPTP, or whether this involves the oxidation of other phosphatases that antagonize JAK/STAT signaling, or indeed even alternate mechanisms, remains to be resolved.
We found that several STAT-3 target genes were also elevated in the livers of high-fat-fed liver-TCPTP-deficient mice. These included Fgl1, a hepatocyte-derived factor whose circulating levels are increased in human NAFLD (Wu et al., 2013) and acute phase reactant genes. Although such STAT-3 target genes may contribute to inflammatory processes in NAFLD, they were not essential for the T cell recruitment or activation, nor the development of fibrosis. Irrespective, consistent with STAT-1 and STAT-3 signaling being important in NAFLD pathogenesis, we found that the STAT-1 and STAT-3 targets genes Cxcl9, Lcn2, and Fgl1 were also elevated in the livers of obese humans with NAFL when compared to those without steatosis. Notably, CXCL9 and FGL1 were increased yet further in the livers of obese humans with NASH and fibrosis. These findings reaffirm the importance of STAT-1 and STAT-3 signaling in NAFLD pathogenesis and substantiate our mouse model as one that appropriately reflects the NAFL to NASH and fibrosis transition in humans.
Strikingly, our studies indicate that the recruitment of T cells and the development of NASH and fibrosis per se are not essential for the development of HCC in obesity. Although STAT-1 heterozygosity in hepatocytes in high-fat-fed liver-TCPTP-deficient mice corrected the recruitment of T cells and prevented fibrosis, it did not prevent tumor formation. Rather, obesity and the enhancement of STAT-3 signaling were instrumental in driving HCC, as tumors were not evident in chow-fed mice and tumor development in high-fat-fed mice was prevented when the increased STAT-3 signaling was corrected. STAT-3 is activated in nearly 60% of HCCs that arise in the context alcoholic- and hepatitis B/C-driven cirrhosis (Calvisi et al., 2006, He et al., 2010) and is essential for DEN-induced HCC in rodents (He et al., 2010, He et al., 2013, Naugler et al., 2007). Hepatocyte STAT-3 signaling is also essential for the exacerbation of DEN-induced HCC by obesity (Park et al., 2010), whereas an IL-6-STAT-3 autocrine loop is required for the malignant progression of HCC progenitors in high-fat-fed MUP-Upa mice (He et al., 2013). We found that Il6 expression was increased in the livers of high-fat-fed liver-TCPTP-deficient mice and this was corrected by STAT-3 heterozygosity. Therefore, TCPTP inactivation in obesity may promote the development of HCC by increasing STAT-3 signaling and exacerbating an IL-6-STAT-3 autocrine loop to drive the transformation of HCC progenitors. These results are consistent with others implicating TCPTP as a tumor suppressor (Kleppe et al., 2010, Shields et al., 2013).
The results of this study point toward the promotion of STAT-3 signaling being sufficient to drive tumor development in obesity and NAFLD independently of NASH and fibrosis. Our findings argue for the oxidative and inflammatory environments in NAFLD being principally important in the promotion of tumorigenesis, rather than NASH and fibrosis per se. Our results shed light on the mechanisms that may be contributing to the growing incidence of HCC in NAFLD patients without advanced fibrosis or cirrhosis, a phenomenon likely to rise with the growing incidence of obesity and type 2 diabetes and the alarming rise in childhood obesity and NAFLD.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin, clone ACTN05, mouse mAb | Thermo Fisher Scientific | Cat# MS-1295; RRID:AB_63314 |
| GAPDH, clone 6C5, mouse mAb | Thermo Fisher Scientific | Cat# AM4300; RRID:AB_2536381 |
| Vinculin, clone E1E9V, rabbit mAb | Cell Signaling | Cat# 13901; RRID:AB_2728768 |
| Tubulin, clone B-5-1-2, mouse mAb | Sigma-Aldrich | Cat# T 5168; RRID:AB_477579 |
| TCPTP, clone 3E2, mouse mAb | M. Tremblay, McGill University | N/A |
| TCPTP, clone 6F3, mouse mAb | M. Tremblay, McGill University | N/A |
| TCPTP, clone CF4, mouse mAb | N. K. Tonks, Cold Spring Harbor Laboratory, NY | N/A |
| PTP1B, polyclonal goat Ab | R&D Systems | Cat# AF3954; RRID:AB_2174947 |
| Oxidized PTP active site, clone 335636, mouse mAb | R&D Systems | Cat# MAB2844; RRID:AB_664170 |
| Phospho-Stat1 (Tyr701), clone 58D6, rabbit mAb | Cell Signaling | Cat# 9167; RRID:AB_561284 |
| Stat1, polyclonal rabbit Ab | Cell Signaling | Cat# 9172; RRID:AB_561284 |
| Phospho-Stat3 (Tyr705), clone D3A7, rabbit mAb | Cell Signaling | Cat# 9145; RRID:AB_561284 |
| Stat3, clone 124H6, mouse mAb | Cell Signaling | Cat# 9139; RRID:AB_331757 |
| Phospho-Stat5 (Tyr694), clone C11C5, rabbit mAb | Cell Signaling | Cat# 9359; RRID:AB_823649 |
| Stat5, clone D2O6Y, rabbit mAb | Cell Signaling | Cat# 94205; RRID:AB_2737403 |
| Fatty Acid Synthase (FAS), clone C20G5, mouse mAb | Cell Signaling | Cat# 3180; RRID:AB_2100796 |
| SREBP-1, rabbit polyclonal Ab | Santa Cruz | Cat# sc-366; RRID:AB_2194229 |
| PPARγ, clone C26H12, rabbit mAb | Cell Signaling | Cat# 2435; RRID:AB_2166051 |
| SCD1 clone C12H5, rabbit mAb | Cell Signaling | Cat# 2794; RRID:AB_2183099 |
| CD3ε clone D7A6E, rabbit mAb | Cell Signaling | Cat# 85061; RRID:AB_2721019 |
| B220 (CD45R), clone RA3-6B2, rat mAB | BioLegend | Cat# 103202; RRID:AB_312987 |
| EpCAM, clone E6V8Y, rabbit mAb | Cell Signaling | Cat# 93790 |
| GRP-78 (BiP), clone C50B12, rabbit mAb | Cell Signaling | Cat# 3177; RRID:AB_2119845 |
| Keratin19, rabbit polyclonal Ab | Abcam | Cat# ab15463; RRID:AB_2281021 |
| Phospho- Erk1/2 (Thr202/Tyr204), polyclonal rabbit Ab | Cell Signaling | Cat# 9101; RRID:AB_331646 |
| Erk 2, clone D2, mouse mAb | Santa Cruz | Cat# sc-1647; RRID:AB_627547 |
| Ki-67, clone D3B5, rabbit mAb | Cell Signaling | Cat# 12202; RRID:AB_2620142 |
| c-MET, clone D1C2, rabbit mAb | Cell Signaling | Cat# 8198; RRID:AB_10860590 |
| Insulin Receptor β clone CT-3, mouse mAb | Thermo Fisher Scientific | Cat# MS-636; RRID:AB_142262 |
| Phospho-p38 MAPK, clone 28B10, mouse mAb | Cell Signaling | Cat# 9216; RRID:AB_331296 |
| Phospho-Jak1(Tyr1022/1023), polyclonal rabbit Ab | Cell Signaling | Cat# 3331; RRID:AB_2265057 |
| Phospho-Jak2 (Tyr1007/1008), polyclonal rabbit Ab | Cell Signaling | Cat# 3771; RRID:AB_330405 |
| Jak2 (C20), polyclonal rabbit Ab | Santa Cruz | Cat# sc-294; RRID:AB_631854 |
| Phospho-Src Family (Y416), polyconal rabbit Ab | Cell Signaling | Cat# 2101; RRID:AB_331697 |
| Phospho-Akt (Ser473), clone D9E, rabbit mAb | Cell Signaling | Cat# 4060; RRID:AB_2315049 |
| Akt, clone 40D4, mouse mAb | Cell Signaling | Cat# 2920; RRID:AB_1147620 |
| mTOR, clone 7C10, rabbit mAb | Cell Signaling | Cat# 2983; RRID:AB_2105622 |
| Phospho-S6 Ribosomal Protein, clone 2F9 rabbit mAb | Cell Signaling | Cat# 4856; RRID:AB_2181037 |
| Phospho-4E-BP1 (Thr37/46), clone 236B4, rabbit mAb | Cell Signaling | Cat# 2855; RRID:AB_560835 |
| SHP-1, clone C14H6, rabbit mAb | Cell Signaling | Cat# 3759; RRID:AB_2173694 |
| SHP-2, polyclonal rabbit Ab | Cell Signaling | Cat# 3752; RRID:AB_2300607 |
| EGF Receptor clone D38B1, rabbit mAb | Cell Signaling | Cat# 4267; RRID:AB_2246311 |
| PKM2, polyclonal rabbit Ab | Cell Signaling | Cat# 3198; RRID:AB_2252325 |
| Cyclin D1, clone92G2, rabbit mAb | Cell Signaling | Cat# 2978; RRID:AB_2259616 |
| Albumin, clone 188835, mouse mAb | R&D Systems | Cat# MAB1455; RRID:AB_2225797 |
| α-Fetoprotein/AFP, polyclonal rabbit Ab | Thermo Fisher Scientific | Cat# PA5-21004; RRID:AB_11157055 |
| Fluorescein isothiocyanate (FITC)-conjugated CD44 (clone IM7) | BD Biosciences | Cat#561859; RRID:AB_10894581 |
| Pacific Blue (PB)-conjugated CD69 (clone H1.2F3) | BioLegend | Cat#104523; RRID:AB_2260064 |
| Phycoerythrin (PE)-conjugated CD3 (clone 145-2C11) | BD Biosciences | Cat#553063; RRID:AB_394596 |
| Allophycocyanin (APC)-Cyanine 7 (Cy7)-conjugated TCR-β (clone H57-597) | BD Biosciences | Cat#560656; RRID:AB_1727574 |
| Phycoerythrin-Cyanine 7 (PE-Cy7)-conjugated CD4 (clone RM4-5) | BD Biosciences | Cat#561099; RRID:AB_2034007 |
| Pacific Blue (PB)-conjugated CD8 (clone 53-6.7) | BD Biosciences | Cat#558106; RRID:AB_397029 |
| Allophycocyanin (APC)-conjugated CD8 (clone 53-6.7) | BD Biosciences | Cat#553035; RRID:AB_398527 |
| Alexa 647-conjugated CD11c (clone N418) | BD Biosciences | Cat#565587 |
| Fluorescein isothiocyanate (FITC)-conjugated CD11b (clone M1/70) | BD Biosciences | Cat#557396; RRID:AB_396679 |
| Phycoerythrin (PE)-conjugated Ly-6G/Ly-6C (Gr-1) (clone RB6-8C5) | BD Biosciences | Cat#553128; RRID:AB_396679 |
| Phycoerythrin (PE)-conjugated Ly-6G (clone 1A8) | BD Biosciences | Cat#551461; RRID:AB_394208 |
| Allophycocyanin (APC)-Cyanine 7 (Cy7)-conjugated Ly6C (clone AL-21) | BD Biosciences | Cat#560596; RRID:AB_1727555 |
| Phycoerythrin-Cyanine 7 (PE-Cy7)-conjugated I-A/I-E (clone M5/114.115.2) | BioLegend | Cat#107630; RRID:AB_2069376 |
| Allophycocyanin (APC)-conjugated F4/80 (clone BM8) | BioLegend | Cat#123116; RRID:AB_893481 |
| Phycoerythrin (PE)-conjugated NK1.1 (clone PK136) | BD Biosciences | Cat#557391; RRID:AB_396674 |
| Allophycocyanin (APC)-Cyanine 7 (Cy7)-conjugated B220 (clone RA3-6B2) | BD Biosciences | Cat#552094; RRID:AB_394335 |
| Phycoerythrin-Cyanine 7 (PE-Cy7)-conjugated CD19 (clone 1D3) | BD Biosciences | Cat#552854; RRID:AB_394495 |
| Phycoerythrin (PE)-conjugated CD25 (clone PC61) | BD Biosciences | Cat#553866; RRID:AB_395101 |
| Allophycocyanin (APC)-Cyanine 7 (Cy7)-conjugated CD25 (clone PC61) | BD Biosciences | Cat#557658; RRID:AB_396773 |
| Allophycocyanin (APC)-conjugated CD62L (clone MEL-14) | BD Biosciences | Cat#553152; RRID:AB_398533 |
| Allophycocyanin (APC)-conjugated CD138 (clone 281-2) | BD Biosciences | Cat#558626; RRID:AB_1645216 |
| Biotinylated CD274 (clone MIH5) | eBioscience | Cat#13-5982-81; RRID:AB_466836 |
| Fluorescein isothiocyanate (FITC)-conjugated IgA (clone mA-6E1) | eBioscience | Cat#11-4204-81; RRID:AB_465220 |
| Phycoerythrin (PE)-conjugated IL-17A (clone TC11-18H10) | BD Biosciences | Cat#559502; RRID:AB_397256 |
| BD Horizon BV421-conjugated IL-17A (clone TC11-18H10) | BD Biosciences | Cat#566286; RRID:AB_2739660 |
| Phycoerythrin (PE)-conjugated IL-10 (clone JES5-16E3) | BD Biosciences | Cat#554467; RRID:AB_395412 |
| Phycoerythrin (PE)-conjugated IFN-γ (clone XMG1.2) | BD Biosciences | Cat#554412; RRID:AB_395376 |
| Phycoerythrin-Cyanine 7 (PE-Cy7)- conjugated IFN-γ (clone XMG1.2) | BD Biosciences | Cat#557649; RRID:AB_396766 |
| Allophycocyanin (APC)-conjugated TNF (clone MP6-XT22) | BD Biosciences | Cat#554420; RRID:AB_398553 |
| BD Horizon BV421-conjugated TNF (clone MP6-XT22) | BD Biosciences | Cat#563387; RRID:AB_2738173 |
| V450-conjugated FoxP3 (clone MF23) | BD Biosciences | Cat#561293; RRID:AB_10611728 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Triton X-100 | Sigma-Aldrich | Cat#T8787 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#D6750 |
| SDS | Sigma-Aldrich | Cat#71729 |
| NaCl | Merck-Millipore | Cat#1064060500 |
| Glycerol | Sigma-Aldrich | Cat#G5516 |
| MgCl2 | Calbiochem | Cat#442615 |
| EGTA | Sigma-Aldrich | Cat#E3889 |
| NaF | Sigma-Aldrich | Cat#201154 |
| EDTA | Sigma-Aldrich | Cat#E9884 |
| Leupeptin | Sigma-Aldrich | Cat#L8511 |
| Pepstatin A | Sigma-Aldrich | Cat#P5318 |
| Benzamadine | Sigma-Aldrich | Cat#12072 |
| Phenylmethysulfonyl fluoride | Sigma-Aldrich | Cat#P7626 |
| Sodium vanadate | Sigma-Aldrich | Cat#S6508 |
| NP40 | Sigma-Aldrich | Cat#NP40S |
| N-Ethylmaleimide | Sigma-Aldrich | Cat#E3876 |
| Tris-EDTA | Sigma-Aldrich | Cat#93283 |
| Ionomycin | Sigma-Aldrich | Cat#I0634 |
| PMA | Sigma-Aldrich | Cat#79346 |
| HEPES | Sigma-Aldrich | Cat#H3375 |
| BSA | Sigma-Aldrich | Cat#A7906 |
| FBS | GIBCO Life Technologies | Cat#10270 |
| Dulbecco’s modified Eagle’s medium | GIBCO Life Technologies | Cat#11885-084 |
| Liberase blend | Roche | Cat#5401020001 |
| HEPES for tissue culture | GIBCO Life Technologies | Cat#15630-080 |
| Penicillin/Streptomycin | GIBCO Life Technologies | Cat#15140-122 |
| L-Glutamine | GIBCO Life Technologies | Cat#35050-061 |
| Sodium pyruvate | GIBCO Life Technologies | Cat#11360-070 |
| Bovine insulin | Sigma-Aldrich | Cat#I05-16 |
| Nystatin | Sigma-Aldrich | Cat#N6261 |
| Human recombinant EGF | GIBCO Life Technologies | Cat#PHG0311 |
| Murine recombinant IL-6 | Peprotech | Cat#216-16 |
| Paraformaldehyde | Sigma-Aldrich | Cat#P6148 |
| Oligomycin | Sigma-Aldrich | Cat#O48-76 |
| 2-Deoxy-D-glucose | Sigma-Aldrich | Cat#D8375 |
| Collagenase A | Roche | Cat#10103578001 |
| HBBS | GIBCO Life Technologies | Cat#14025092 |
| D-PBS | Sigma-Aldrich | Cat#D8537 |
| M199 medium | GIBCO Life Technologies | Cat#11150-067 |
| SeahorseXF medium | Agilent Technologies | Cat#103334-100 |
| Horse serum | GIBCO Life Technologies | Cat#16050122 |
| Cholera toxin | Sigma-Aldrich | Cat#C8052 |
| Percoll | GE Healthcare | Cat#17-0891-01 |
| Red Blood Cell Lysing Solution | Sigma-Aldrich | Cat#R7757 |
| BD GolgiStop Protein Transport Inhibitor | BD Biosciences | Cat#554724 |
| BD GolgiPlug Protein Transport Inhibitor | BD Biosciences | Cat#555029 |
| Experimental Models: Cell Lines | ||
| Mammary epithelial MCF10A cells | ATCC, Manassas, VA | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ptpn2fl/fl (C57BL/6) | Wiede et al., 2011 | N/A |
| Mouse: Alb-Cre;Ptpn2fl/fl (C57BL/6) | Gurzov et al., 2014 | N/A |
| Mouse: Rag1–/– (C57BL/6) | Animal House Kew Bioservices, Walter & Eliza Hall Institute of Medical Research (Kew, VIC, Australia) | N/A |
| Mouse: Ptpn2fl/fl;Rag1–/– (C57BL/6) | This paper | N/A |
| Mouse: Alb-Cre;Ptpn2fl/fl;Rag1–/– (C57BL/6) | This paper | N/A |
| Mouse: Stat1fl/fl (C57BL/6) | Wallner et al., 2012 | N/A |
| Mouse: Stat3fl/fl (C57BL/6) | Alonzi et al., 2001 | N/A |
| Mouse: Alb-Cre;Ptpn2fl/fl;Stat1fl/+ (C57BL/6) | This paper | N/A |
| Mouse: Alb-Cre;Ptpn2fl/fl;Stat3fl/+ (C57BL/6) | This paper | N/A |
| Mouse: BALBC/c nu/nu | Animal Resources Centre (Canning Vale, WA, Australia) | N/A |
| Critical Commercial Assays | ||
| SureSelect Strand Specific RNA Library Prep | Agilent | Cat#G9691A |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| QuantiNova SYBR Green PCR Kit | QIAGEN | Cat# 208056 |
| Dako EnVision + Dual Link System-HRP (DAB+) | Agilent | Cat#K4065 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| RNAIzol Reagent | Sigma-Aldrich | Cat# R4533 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat#43044347 |
| Fisher Chemical Permount Mounting Medium | Thermo Fisher Scientific | Cat#SP15-500 |
| DeadEnd Calorimetric TUNEL System Kit | Promega | Cat#G7360 |
| ELISA Kit for Mouse Fibrinogen Like Protein 1 (FGL1) | USCN Life Science | Cat#E3022Mu |
| CXCL9 Mouse ELISA Kit | Thermo Fisher Scientific | Cat#EMCCXCL9 |
| NGAL Mouse ELISA Kit | Thermo Fisher Scientific | Cat#EMLCN2 |
| FGL1 Mouse ELISA Kit | USCN Life Science | Cat#E3022Mu |
| Rat insulin RIA kit | Merck Millipore | Cat#RI-13K |
| Transaminase CII kit | Wako Pure Chemical Industries | Cat#431-30901 |
| BD Cytofix/Cytoperm Kit | BD Biosciences | Cat#554714 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Oligonucleotides | ||
| RT-PCR Primers for TaqMan Gene Expression Assays | Thermo Fisher Scientific | STAR Methods; Method Details (See Table S2) |
| Software and Algorithms | ||
| Ingenuity Pathway Analysis (IPA) software | QIAgen Bioinformatics | N/A |
| Other | ||
| Growth factor-reduced Matrigel | BD Biosciences | Cat#356230 |
| Peridinin Chlorophyll-Cyanine 5.5 (PerCP-Cy5.5)-conjugated Streptavidin | BD Biosciences | Cat#551419 |
| BD Horizon V500-conjugated Streptavidin | BD Biosciences | Cat#561419 |
| Allophycocyanin (APC)-labeled, α-galactosylceramide (αGalCer)-loaded CD1 tetramers | Laboratory of Dale I. Godfrey, University of Melbourne, Australia | N/A |
Contact for Reagent and Resources Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tony Tiganis, Ph.D. (Tony.Tiganis@monash.edu).
Experimental Model and Subject Details
Mice
Mice were maintained on a 12 h light-dark cycle in a temperature-controlled pathogen-free facility with free access to food and water. Age- and sex-matched mice were used for experiments. Ptpn2fl/fl (C57BL/6) and Alb-Cre;Ptpn2fl/fl (C57BL/6) mice have been described previously (Gurzov et al., 2014, Wiede et al., 2011). Alb-Cre;Ptpn2fl/fl mice were mated with Rag1–/– (C57BL/6) mice to generate Ptpn2fl/fl;Rag1–/– (C57BL/6) and Alb-Cre;Ptpn2fl/fl;Rag1–/– (C57BL/6) mice. Stat1fl/fl (C57BL/6) mice (Wallner et al., 2012) and Stat3fl/fl (C57BL/6) mice (Alonzi et al., 2001) were mated with Alb-Cre;Ptpn2fl/fl mice to generate Alb-Cre;Ptpn2fl/fl;Stat1fl/+ (C57BL/6) and Alb-Cre;Ptpn2fl/fl;Stat1fl/+ (C57BL/6) mice respectively. BALBC/c nu/nu mice were purchased from the Animal Resources Centre (Canning Vale, WA, Australia) and Rag1–/– (C57BL/6) mice from Animal House Kew Bioservices, Walter & Eliza Hall Institute of Medical Research. Mice were fed a breeding diet (8.5% fat; Barastoc, Ridley AgriProducts, Australia), or where indicated a standard chow diet (20% protein, 4.8% fat, and 4.8% crude fiber; Specialty Feeds), a high-fat diet [HFD; 18.4% protein, 23.5% fat (46% energy from fat), 4.7% fiber; Specialty Feeds #SF04-027), a choline-deficient, L-amino acid-defined (CDAA; 14% protein, 14.5% fat, and 4.7% crude fiber; Specialty Feeds #SF13-103) diet, or a choline-deficient HFD [CD-HFD; no added choline chloride, 22.6% protein, 23.5% fat (43% energy from fat), 5.4% fiber; Specialty Feeds #SF15-078]. Where indicated male mice were injected intraperitoneally with 25 mg/kg DEN on postnatal day 14 and tumor development assessed after 36-40 weeks. Experiments were approved by the Monash University School of Biomedical Sciences Animal Ethics Committee and performed in accordance with the NHMRC Australian Code of Practice for the Care and Use of Animals.
Human liver biopsies
The use of human tissue was approved by the Monash University Human Research Ethics Committee (CF12/2339-2012001246; CF15/3041-2015001282). All subjects gave their written consent before participating in this study. Normal tumor-adjacent liver biopsies from non-obese HCC patients (gender and ages not available), or liver core biopsies from obese Class II (BMI 35-39.9) and Class III (BMI ≥ 40) men (n = 8, ages 28-61 years old) and women (n = 20, ages 23-64 years old) undergoing bariatric surgery taken on the day of surgery were fixed in formalin and processed for histology or snap frozen in liquid nitrogen for biochemical assessment. Depending on availability, liver biopsies from either male or female subjects were processed and used in experiments. Due to the low frequency of suitable donors analyses on gender differences were not performed.
Cultured Cells
For isolating HCC cells, tumor tissues from DEN-treated male Ptpn2fl/fl mice were rinsed with ice-cold Dulbecco’s modified Eagle’s medium (DMEM; GIBCO Life Technologies, Carlsbad, CA), minced with scalpel blades under aseptic conditions and digested enzymatically at 37°C with 50 μg/ml Liberase blend (Roche, Penzberg, Germany) in Hank’s Balanced Salt Solution (HBSS; GIBCO Life Technologies, Carlsbad, CA) containing 10 mM HEPES pH 7.4. Samples were agitated for a further 90 min at 100 rpm on an orbital shaker and disaggregated cell suspensions filtered through a 100 μm cell strainer (BD Biosciences, San Jose, CA), washed with HBSS and resuspended in DMEM supplemented with 20% (v/v) heat-inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 0.5mM sodium pyruvate, 1 nM bovine insulin (Sigma-Aldrich, ST Louis, MO), 10 μg/ml hydrocortisone (Sigma-Aldrich, ST Louis, MO), 0.25% (v/v) nystatin (10,000 unit/mL) and 20 ng/ml human recombinant EGF (GIBCO Life Technologies, Carlsbad, CA). Cells were incubated in culture flasks at 37°C, 5% CO2 for 2 days and thereon medium was changed twice weekly and cells passaged at 70%–80% confluency. Ptpn2fl/fl HCC cells grew as adherent monolayers with an epithelial morphology and expressed albumin and α-fetoprotein (AFP).
To generate isogenic HCC cell lines with and without TCPTP, Ptpn2fl/fl HCC cells isolated from DEN treated male mice were retrovirally transduced with GFP (MSCV-GFP)- or GFP and Cre recombinase (MSCV-GFP-Cre)-expressing retroviruses generated as described previously (Shields et al., 2013) and sorted for GFP using a BD Influx Cell Sorter (BD Biosciences).
Hepatocytes were isolated by the two-step collagenase A (0.05% w/v) perfusion method described previously (Gurzov et al., 2014). Perfused livers from adult male mice were removed and minced with scalpel blades under aseptic conditions, washed extensively with HBBS and cultured in M199 medium (GIBCO Life Technologies, Carlsbad, CA) containing 10% (v/v) heat inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 1 nM insulin for no more than 2 days at 37°C, 5% CO2.
Mammary epithelial MCF10A cells (ATCC, Manassas, VA) originally derived from a 36 years old Caucasian female were cultured in DMEM/Ham’s F12 medium (GIBCO Life Technologies, Carlsbad, CA) containing 5% (v/v) horse serum, 100 ng/ml cholera toxin (Sigma-Aldrich, ST Louis, MO), 0.5 mg/ml hydrocortisone (Sigma-Aldrich, ST Louis, MO), 20 ng/ml human recombinant EGF (GIBCO Life Technologies, Carlsbad, CA) and 10 μg/ml bovine insulin (Sigma-Aldrich, ST Louis, MO).
Methods Details
IL-6 signaling
For IL-6 stimulation assays HCC cells were serum-starved in DMEM containing 0.1% (v/v) FBS for 6 h and stimulated with 1 ng/ml murine recombinant IL-6 (Peprotech, Rehovot, Israel) for the indicated times and processed for immunoblotting as described previously (Shields et al., 2013).
Immunofluorescence microscopy
For immunofluorescence microscopy, cells grown on glass coverslips were fixed in 3.2% (w/v) paraformaldehyde and processed as described previously (Loh et al., 2011).
Metabolic and blood measurements
Mouse body weights were monitored weekly and body composition was assessed by dual-energy X-ray absorptiometry (DEXA; Lunar PIXImus2, GE Healthcare). Insulin and glucose tolerance tests were performed as described previously (Loh et al., 2009). Blood was collected from the retro-orbital sinus and fed (9 am) and fasted (12 h overnight fast) plasma insulin levels determined using a Rat insulin RIA kit (Merck Millipore, CA) and the corresponding blood glucose levels determined with an Accu-Check glucometer. Fed serum ALT and AST levels were determined using the Transaminase CII kit from Wako Pure Chemical Industries (Osaka, Japan) as described previously (Wiede et al., 2011) and serum CXCL9 (CXCL9 Mouse ELISA Kit, Thermo Fisher Scientific, Waltham, WA), FGL1 (USCN Life Science, Wuhan, China) and LCN2 (NGAL Mouse ELISA Kit, Thermo Fisher Scientific, Waltham, WA) levels measured by ELISA according to the manufacturers’ instructions.
Biochemical analyses
Livers and tumor tissues were snap frozen in liquid nitrogen. Samples were mechanically homogenized in 10-20 volumes of ice-cold RIPA lysis buffer (50 mM HEPES [pH 7.4], 1% (v/v) Triton X-100, 1% (v/v) sodium deoxycholate, 0.1% (v/v) SDS, 150 mM NaCl, 10% (v/v) glycerol, 1.5 mM MgCl2, 1 mM EGTA, 50 mM NaF, leupeptin (5 μg/ml), pepstatin A (1 μg/ml), 1 mM benzamadine, 2 mM phenylmethysulfonyl fluoride, 1 mM sodium vanadate) and clarified by centrifugation at 50,000 g for 20 min at 4°C. Clarified tissue/tumor homogenates were resolved by SDS-PAGE and transferred to PVDF and immunoblotted.
For analysis of total PTP oxidation livers were processed as described previously (Gurzov et al., 2014) and oxidized PTPs detected by immunoblotting using the PTPox antibody that detects virtually all classical tyrosine-specific PTPs when their catalytic Cys residues are oxidized to the -SO3H state (Karisch et al., 2011). Briefly, liver samples were mechanically homogenized under anaerobic conditions in degassed, ice-cold PTPox lysis buffer (20 mM HEPES pH 6.5, 150 mM NaCl, 10% glycerol, 1%NP40, 20 mM NaF) containing 10 mM N-ethylmaleimide and incubated for 1 hr at 4°C to prevent any post-lysis Cys oxidation and to alkylate any reduced and active PTPs. Homogenates were then clarified by centrifugation (50,000 g, 20 min at 4°C) and the buffer exchanged (NAP-5 columns, GE Healthcare) into 20 mM HEPES containing 10 mM DTT to reduce oxidized Cys before further buffer exchange into 20 mM HEPES containing 100 mM pervanadate to hyper-oxidise reduced PTPs to the -SO3H state for analysis by SDS-PAGE and PTPox immunoblotting.
Liver TAGs, DAGs and ceramide were extracted and quantified as described previously (Loh et al., 2009)
Histology and immunohistochemistry
Livers and tumor tissues were fixed overnight in a neutral-buffered formalin solution and then transferred to 70% ethanol. Tissue processing and embedding were performed by the Monash Histology Platform (Monash University, Clayton Campus, Australia) and sections were stained with Hematoxylin-Eosin, PicoSirius Red or Mason's Trichrome using standard procedures. Immunohistochemistry was performed using the EnVision detection system (Dako; Agilent Technologies; Santa Clara, CA, USA) as per the manufacturer’s instructions with modifications. Briefly, sections were de-paraffinised with xylene and rehydrated with three successive changes in ethanol. Heat-induced epitope retrieval was performed by placing slides in a pressure cooker at 120°C for 9 min with sections immersed in Tris-EDTA (pH 8.0) followed by endogenous peroxidase blocking (Dako REAL Peroxidase-Blocking Solution). Non-specific antibody binding was blocked with 1% (w/v) BSA and primary antibodies were incubated at 4°C in a humidified chamber overnight. The appropriate horseradish peroxidase (HRP)-conjugated secondary antibody polymer was incubated for 1 h at room temperature. Immunoreactivity was visualized by diaminobenzidine tetrahydrochloride staining. Slides were counterstained with hematoxylin, dehydrated and mounted in a slide-mounting medium (Permount, Thermo Fisher Scientific, Carlsbad, CA, USA). Primary antibodies used for immunohistochemistry are listed in the resources table. Image acquisition was performed using an Aperio Scanscope AT Turbo slide scanner with 40x magnification and 0.25 μm/pixel resolution. Quantification of positive staining per mm2 was performed using an automated cell counter script in Aperio ImageScope (Aperio Technologies, Vista, CA, USA).
NASH scoring
The NAFLD Activity Score, the unweighted sum of the steatosis (0-3), lobular inflammation (0-3) and hepatocellular ballooning (0-2) scores, and the fibrosis score (0-4) according to the Kleiner classication were determined for liver biopsies from obese patients.
Xenograft and soft agar assays
HCC cells were processed for soft-agar assays as described previously (Shields et al., 2013). Xenograft studies were performed as described previously (Shields et al., 2013). Briefly HCC cells were detached with 10 mM EDTA–PBS and 1x106 cells resuspended in 100 μl in a 1:1 mixture of PBS and growth factor-reduced Matrigel (BD Biosciences) and injected subcutaneously into the flanks of male BALB/c nu/nu mice under isoflurane anesthesia [2% (v/v) isoflurane in 250 mL/min oxygen]. Palpable tumors were measured twice weekly using digital calipers (tumor volume = length x width2/2) and growth curves (up to 1000 mm3) determined. Tumors were dissected and weighted and processed for immunohistochemistry or biochemical analyses.
Flow cytometry
Hepatic lymphocytes were isolated from liver tissue and mashed through a wire mesh (200 micron pore size). Hepatocytes and cell debris were removed by Percoll 33.75% (v/v) (GE Healthcare) gradient centrifugation at room temperature. Contaminating red blood cells were lysed using a 0.17 M hypotonic ammonium chloride buffer (Red Blood Cell Lysing Solution, Sigma-Aldrich).
For surface staining, cells (1x106/10 μl) were resuspended in D-PBS supplemented with 2% (v/v) FBS containing the antibody cocktail in 96-well microtiter plates (Falcon, BD Biosciences) for 20 min on ice in the dark and analyzed using a LSRII or Fortessa or Symphony (BD Biosciences). For detection of intracellular FoxP3 the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used according to the manufacturer’s instructions. For the detection of intracellular cytokines, lymphocytes were stimulated with ionomycin (1 μg/ml) and PMA (20 ng/ml) in the presence of Golgi Stop and Golgi Plug (BD Biosciences) for 4 h in complete T cell medium at 37°C. Cells were harvested, fixed and permeablized with the BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Cells were stained with the specified antibodies at room temperature for 30 min and processed for flow cytometry.
The following antibodies from BD Biosciences (San Jose, CA) or BioLegend (San Diego, CA) or eBioscience (San Diego, CA) were used for flow cytometry: Fluorescein isothiocyanate (FITC)-conjugated CD44 (IM7), Pacific Blue (PB)-conjugated CD69 (H1.2F3), phycoerythrin (PE)-conjugated CD3 (145-2C11), allophycocyanin (APC)-Cy7-conjugated TCR-β (H57-597), phycoerythrin-cyanine 7 (PE-Cy7)-conjugated CD4 (RM4-5), PB- or allophycocyanin (APC)-conjugated CD8 (53-6.7), Alexa 647-conjugated CD11c (N418), FITC-conjugated CD11b (M1/70), PE-conjugated Ly-6G/Ly-6C (RB6-8C5), PE-conjugated Ly-6G (1A8), APC-Cy7-conjugated Ly6C (AL-21), PE-Cy7-conjugated I-A/I-E (M5/114.115.2), APC-conjugated F4/80 (BM8), PE-conjugated NK1.1 (PK136), APC-Cy7-conjugated B220 (RA3-6B2), PE-Cy7-conjugated CD19 (1D3), PE- or APC-Cy7-conjugated CD25 (PC61), APC-conjugated CD62L (MEL-14), APC-conjugated CD138 (281-2), biotinylated CD274 (MIH5), FITC-conjugated IgA (mA-6E1), PE- or BD Horizon BV421-conjugated IL-17A (TC11-18H10), PE-conjugated IL-10 (JES5-16E3), PE- or PE-Cy7-conjugated IFN-γ (XMG1.2), APC- or BD Horizon BV421-conjugated TNF (MP6-XT22), V450-conjugated FoxP3 (MF23). PerCP-Cy5.5- or BD Horizon V500-conjugated Streptavidin was used to detect cells stained with biotinylated antibodies. APC-labeled, α-galactosylceramide (αGalCer)-loaded CD1 tetramers were a gift of Dale I. Godfrey (Peter Doherty Institute, Melbourne, Australia).
Quantitative PCR
RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA) or RNAZol (Sigma-Aldrich, ST Louis, MO) and RNA quality and quantity was determined using a NanoDrop 3300 (Thermo Scientific, Wilmington, DE, USA) and by gel electrophoresis. mRNA was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and processed for quantitative real-time PCR using the TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) (Table S2). Reactions were run in technical duplicate on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Gene expression was normalized to Gapdh or Rn18s (mouse) or 18 s (human). Relative quantification was achieved using the ΔΔCT method.
Table S2.
TaqMan Gene Expression Assays
| Fasn | Mm00662319_m1 |
| Srebp-f1 | Mm00550338_m1 |
| Pparg | Mm00440940_m1 |
| Cd36 | Mm00432403_m1 |
| Scd1 | Mm00772290_m1 |
| Igf1 | Mm00439560_m1 |
| Il6 | Mm00446190_m1 |
| Tnf | Mm00443258_m1 |
| Ifng | Mm01168134_m1 |
| Cxcl9 | Mm00434946_m1 |
| CXCL9 | Hs00171065_m1 |
| Saa1 | Mm00656927_g1 |
| Saa3 | Mm00441203_m1 |
| Crp | Mm00432680_g1 |
| Acta2 | Mm01546133_m1 |
| Tgfb | Mm01178820_m1 |
| Socs1 | Mm00782550_s1 |
| Socs3 | Mm00545913_s1 |
| Ptpn1 | Mm00448427_m1 |
| Ptpn6 | Mm00469153_m1 |
| Ptpn11 | Mm00448434_m1 |
| Fgl1 | Mm01198706_m1 |
| FGL1 | Hs00189514_m1 |
| Lcn2 | Mm01324470_m1 |
| LCN2 | Hs01008571_m1 |
| Gbp2 | Mm00494576_g1 |
| Rcan2 | Mm00472671_m1 |
| Shisa5 | Mm00458793_g1 |
| Ccl5 | Mm01302427_m1 |
| Hif1a | Mm00468869_m1 |
| Ccnd1 | Mm00432359_m1 |
| Egfr | Mm01187858_m1 |
| Pdk1 | Mm00554300_m1 |
| Glut1 | Mm00441480_m1 |
| Myc | Mm00487804_m1 |
| 18 s | Hs99999901_s1 |
| Gapdh | Mm99999915_g1 |
Transcriptome analysis
Mouse liver transcriptome analysis was performed using Agilent SureSelect Strand Specific RNA Library Preparation Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. Briefly, RNA was isolated using TRIzol and quantity and integrity of RNA was analyzed using an RNA 6000 Pico LabChip on an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). 3 μg of RNA from each sample was used to prepare the sequencing library through fragmentation of the RNA followed by first- and second-strand synthesis of cDNA. After adenylation of the cDNA 3′end sequencing adaptors were ligated and the adaptor-ligated cDNA amplified with indexing primers by PCR (37°C for 15 min, 95°C for 2 min followed by 11 cycles of 95°C for 30 s, 65°C for 30 s and 72°C for 1 min and a final extension at 72°C for 5 min). Purified cDNA libraries were analyzed for sample integrity and quantity using a high sensitivity DNA chip on an Agilent Bioanalyzer 2100. RNA sequencing was performed on an Illumina HiSeq1500 (with a 50-base single-end read mode; > 7 × 106 reads) sequencing platform at the Kazusa DNA Research Institute (Kisarazu, Chiba, Japan). Raw sequencing reads were trimmed for adaptor contamination using Cutadapt (adaptor sequence AGATCGGAAGAGC; minimum overlap 3 nucleotides; maximum mismatches 10%) and for base-call quality with Trimmomatic, before being mapped to the mouse genome, including annotated or novel splice junctions, using STAR. Uniquely mapped reads were assigned to genes using HTSeq to produce gene-wise read-counts for each sample. Genes having less than 10 reads in all samples were filtered out, then EdgeR was used to identify expressed RNAs that were differentially abundant between Ptpn2fl/fl and Alb-Cre;Ptpn2fl/fl mice fed a HFD for 40 weeks, normalizing for library size by the Trimmed Mean of M-values method. Relationships between regulated genes and functional groups of genes were searched for using the Ingenuity Pathway Analysis (IPA) software and knowledgebase (QIAgen Bioinformatics, Redwood City, CA).
Quantification and Statistical Analysis
All data are expressed as the mean ± the standard error of the mean (SEM). All statistical tests were performed in GraphPad Prism 7. For all experiments where comparisons were made for more than two populations, statistically significance was assessed by one-way or two way ANOVA as appropriate with corrections for multiple comparisons; these included Figures 4E, 5A, 5B, 6A, 6B, 7A–7C, 7E–7H, and 7K. For all experiments where comparisons were made between two populations, statistically significance was assessed using unpaired Student’s t tests; these include Figure 2E, 2H, 2I, 2K, 2O, 2P, 4B–4D, 4F, 4H, and 7I–7L. For statistical evaluation of the immunological flow cytometry data in Figures 2F, 5D, and 6D a non-parametric 2-tailed Mann-Whitney U Test was used. All values for n are indicated within each figure panel. With the exception of Figures 3C, 4C, 7I, and 7J all values for n are for individual mice or individual patient samples. For Figures 3C, 4C, 7I, and 7J values for n are for independent experiments. A minimum of 5 mice per genotype was used to investigate effects of obesity on NASH and a minimum of 15 mice was used to investigate effects of obesity on HCC development. p < 0.05 was considered significant: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001 and ∗∗∗∗ p < 0.0001.
Acknowledgments
This work was supported by the NHMRC of Australia T.T. (1103037), T.T. and C.A.M. (1047060), and M.J.W. (107703 and 1061278), Cancer Council Victoria T.T. and C.A.M. (1141137), Association for International Cancer Research, United Kingdom to T.T. and C.A.M. (12-1172), and the Austrian Science Fund, Vienna Austria to M.M. (FWF SFB F6101 and F6106).
Author Contributions
T.T. conceived, conceptualized, and designed the study, wrote the manuscript, and interpreted the data. The other authors performed and/or analyzed experiments and/or contributed to the reviewing and editing of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 25, 2018
Footnotes
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.cell.2018.09.053.
Supplemental Information
References
- Alexander J., Torbenson M., Wu T.T., Yeh M.M. Non-alcoholic fatty liver disease contributes to hepatocarcinogenesis in non-cirrhotic liver: a clinical and pathological study. J. Gastroenterol. Hepatol. 2013;28:848–854. doi: 10.1111/jgh.12116. [DOI] [PubMed] [Google Scholar]
- Alonzi T., Maritano D., Gorgoni B., Rizzuto G., Libert C., Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell. Biol. 2001;21:1621–1632. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E.J., Lustig M.E., Boyle K.E., Woodlief T.L., Kane D.A., Lin C.T., Price J.W., 3rd, Kang L., Rabinovitch P.S., Szeto H.H. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguet T., Terra X., Quintero Y., Martínez S., Manresa N., Porras J.A., Aguilar C., Orellana-Gavaldà J.M., Hernández M., Sabench F. Liver lipocalin 2 expression in severely obese women with non alcoholic fatty liver disease. Exp. Clin. Endocrinol. Diabetes. 2013;121:119–124. doi: 10.1055/s-0032-1331696. [DOI] [PubMed] [Google Scholar]
- Baffy G., Brunt E.M., Caldwell S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J. Hepatol. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- Berryman D.E., Christiansen J.S., Johannsson G., Thorner M.O., Kopchick J.J. Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm. IGF Res. 2008;18:455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A., Jiang J.X., Sasaki Y., Chao T.I., Kiss Z., Chen X., Tian J., Katsuyama M., Yabe-Nishimura C., Xi Y. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015;149:468–480. doi: 10.1053/j.gastro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle E.E., Rodriguez C., Walker-Thurmond K., Thun M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- Calvisi D.F., Ladu S., Gorden A., Farina M., Conner E.A., Lee J.S., Factor V.M., Thorgeirsson S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Demaria M., Poli V. PKM2, STAT3 and HIF-1α: the Warburg’s vicious circle. JAK-STAT. 2012;1:194–196. doi: 10.4161/jkst.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchuri S., Oberley T.D., Qi W., Eisenstein R.S., Jackson Roberts L., Van Remmen H., Epstein C.J., Huang T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- Finkin S., Yuan D., Stein I., Taniguchi K., Weber A., Unger K., Browning J.L., Goossens N., Nakagawa S., Gunasekaran G. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015;16:1235–1244. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font-Burgada J., Sun B., Karin M. Obesity and Cancer: The oil that feeds the flame. Cell Metab. 2016;23:48–62. doi: 10.1016/j.cmet.2015.12.015. [DOI] [PubMed] [Google Scholar]
- Gurzov E.N., Tran M., Fernandez-Rojo M.A., Merry T.L., Zhang X., Xu Y., Fukushima A., Waters M.J., Watt M.J., Andrikopoulos S. Hepatic oxidative stress promotes insulin-STAT-5 signaling and obesity by inactivating protein tyrosine phosphatase N2. Cell Metab. 2014;20:85–102. doi: 10.1016/j.cmet.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Yu G.Y., Temkin V., Ogata H., Kuntzen C., Sakurai T., Sieghart W., Peck-Radosavljevic M., Leffert H.L., Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Dhar D., Nakagawa H., Font-Burgada J., Ogata H., Jiang Y., Shalapour S., Seki E., Yost S.E., Jepsen K. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–396. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karisch R., Fernandez M., Taylor P., Virtanen C., St-Germain J.R., Jin L.L., Harris I.S., Mori J., Mak T.W., Senis Y.A. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppe M., Lahortiga I., El Chaar T., De Keersmaecker K., Mentens N., Graux C., Van Roosbroeck K., Ferrando A.A., Langerak A.W., Meijerink J.P. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010;42:530–535. doi: 10.1038/ng.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic J.C., Gupta R., Lee A.C., Ma M., Fang F., Tolbert C.N., Walts A.D., Beltran L.E., San H., Chen G. Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J. Clin. Invest. 2010;120:303–314. doi: 10.1172/JCI40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Feng D., Cai Y., Liu Y., Xu M., Xiang X., Zhou Z., Xia Q., Kaplan M.J., Kong X., Gao B. Hepatocytes and neutrophils cooperatively suppress bacterial infection by differentially regulating lipocalin-2 and neutrophil extracellular traps. Hepatology. 2018 doi: 10.1002/hep.29919. Published online April 10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichanska A.M., Waters M.J. How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet. 2008;24:41–47. doi: 10.1016/j.tig.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B.J., Skiba B., Ooms L.M. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh K., Fukushima A., Zhang X., Galic S., Briggs D., Enriori P.J., Simonds S., Wiede F., Reichenbach A., Hauser C. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011;14:684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T., Beraza N., Kotsikoris V., van Loo G., Nenci A., De Vos R., Roskams T., Trautwein C., Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maeda S., Kamata H., Luo J.L., Leffert H., Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Merry T.L., Tran M., Dodd G.T., Mangiafico S.P., Wiede F., Kaur S., McLean C.L., Andrikopoulos S., Tiganis T. Hepatocyte glutathione peroxidase-1 deficiency improves hepatic glucose metabolism and decreases steatohepatitis in mice. Diabetologia. 2016;59:2632–2644. doi: 10.1007/s00125-016-4084-3. [DOI] [PubMed] [Google Scholar]
- Mittal S., Sada Y.H., El-Serag H.B., Kanwal F., Duan Z., Temple S., May S.B., Kramer J.R., Richardson P.A., Davila J.A. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. 2015;13:594–601.e1. doi: 10.1016/j.cgh.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H., Umemura A., Taniguchi K., Font-Burgada J., Dhar D., Ogata H., Zhong Z., Valasek M.A., Seki E., Hidalgo J. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler W.E., Sakurai T., Kim S., Maeda S., Kim K., Elsharkawy A.M., Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Neumann C.A., Krause D.S., Carman C.V., Das S., Dubey D.P., Abraham J.L., Bronson R.T., Fujiwara Y., Orkin S.H., Van Etten R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- Paradis V., Zalinski S., Chelbi E., Guedj N., Degos F., Vilgrain V., Bedossa P., Belghiti J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. 2009;49:851–859. doi: 10.1002/hep.22734. [DOI] [PubMed] [Google Scholar]
- Park E.J., Lee J.H., Yu G.Y., He G., Ali S.R., Holzer R.G., Osterreicher C.H., Takahashi H., Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.J., Zhang D., Zhang X.M., Boyer J.L., Shulman G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347:1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzalis C., Jones G.W., Bombardieri M., Jones S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014;14:447–462. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- Rahman R., Hammoud G.M., Almashhrawi A.A., Ahmed K.T., Ibdah J.A. Primary hepatocellular carcinoma and metabolic syndrome: An update. World J. Gastrointest. Oncol. 2013;5:186–194. doi: 10.4251/wjgo.v5.i9.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringelhan M., Pfister D., O’Connor T., Pikarsky E., Heikenwalder M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018;19:222–232. doi: 10.1038/s41590-018-0044-z. [DOI] [PubMed] [Google Scholar]
- Sakurai T., Maeda S., Chang L., Karin M. Loss of hepatic NF-κ B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satapati S., Kucejova B., Duarte J.A., Fletcher J.A., Reynolds L., Sunny N.E., He T., Nair L.A., Livingston K.A., Fu X. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Invest. 2015;125:4447–4462. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K., Imbeaud S., Letouzé E., Alexandrov L.B., Calderaro J., Rebouissou S., Couchy G., Meiller C., Shinde J., Soysouvanh F. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba T., Nishimura M., Nishimura S., Ohara O., Ishige T., Ohno S., Nonaka K., Sogawa K., Satoh M., Sawai S. The FLS (fatty liver Shionogi) mouse reveals local expressions of lipocalin-2, CXCL1 and CXCL9 in the liver with non-alcoholic steatohepatitis. BMC Gastroenterol. 2013;13:120. doi: 10.1186/1471-230X-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker M., Tabbaa A., Albeldawi M., Alkhouri N. Liver transplantation for nonalcoholic fatty liver disease: new challenges and new opportunities. World J. Gastroenterol. 2014;20:5320–5330. doi: 10.3748/wjg.v20.i18.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S., Lin X.J., Bastian I.N., Brain J., Burt A.D., Aksenov A.A., Vrbanac A.F., Li W., Perkins A., Matsutani T. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017;551:340–345. doi: 10.1038/nature24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields B.J., Wiede F., Gurzov E.N., Wee K., Hauser C., Zhu H.J., Molloy T.J., O’Toole S.A., Daly R.J., Sutherland R.L. TCPTP regulates SFK and STAT3 signaling and is lost in triple-negative breast cancers. Mol. Cell. Biol. 2013;33:557–570. doi: 10.1128/MCB.01016-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R., Li Z., Li H., Song H., Bao C., Wei J., Cheng L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer. 2010;10:20. doi: 10.1186/1471-2407-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F., Zimmermann H.W., Berres M.L., Trautwein C., Wasmuth H.E. Serum chemokine receptor CXCR3 ligands are associated with progression, organ dysfunction and complications of chronic liver diseases. Liver Int. 2011;31:840–849. doi: 10.1111/j.1478-3231.2011.02504.x. [DOI] [PubMed] [Google Scholar]
- Takuma Y., Nouso K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J. Gastroenterol. 2010;16:1436–1441. doi: 10.3748/wjg.v16.i12.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Fujita N., Sugimoto R., Urawa N., Horiike S., Kobayashi Y., Iwasa M., Ma N., Kawanishi S., Watanabe S. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer. 2008;98:580–586. doi: 10.1038/sj.bjc.6604204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hoeve J., de Jesus Ibarra-Sanchez M., Fu Y., Zhu W., Tremblay M., David M., Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol. Sci. 2011;32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Tiganis T., Bennett A.M. Protein tyrosine phosphatase function: the substrate perspective. Biochem. J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner B., Leitner N.R., Vielnascher R.M., Kernbauer E., Kolbe T., Karaghiosoff M., Rülicke T., Decker T., Müller M. Generation of mice with a conditional Stat1 null allele. Transgenic Res. 2012;21:217–224. doi: 10.1007/s11248-011-9519-5. [DOI] [PubMed] [Google Scholar]
- Wasmuth H.E., Lammert F., Zaldivar M.M., Weiskirchen R., Hellerbrand C., Scholten D., Berres M.L., Zimmermann H., Streetz K.L., Tacke F. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology. 2009;137:309–319. doi: 10.1053/j.gastro.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiede F., Shields B.J., Chew S.H., Kyparissoudis K., van Vliet C., Galic S., Tremblay M.L., Russell S.M., Godfrey D.I., Tiganis T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Invest. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiede F., Dudakov J.A., Lu K.-H., Dodd G.T., Butt T., Godfrey D.I., Strasser A., Boyd R.L., Tiganis T. PTPN2 regulates T cell lineage commitment and αβ versus γδ specification. J. Exp. Med. 2017;214:2733–2758. doi: 10.1084/jem.20161903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M.J., Adili A., Piotrowitz K., Abdullah Z., Boege Y., Stemmer K., Ringelhan M., Simonavicius N., Egger M., Wohlleber D. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Wu H.T., Lu F.H., Ou H.Y., Su Y.C., Hung H.C., Wu J.S., Yang Y.C., Wu C.L., Chang C.J. The role of hepassocin in the development of non-alcoholic fatty liver disease. J. Hepatol. 2013;59:1065–1072. doi: 10.1016/j.jhep.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Xu Z., Chen L., Leung L., Yen T.S., Lee C., Chan J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA. 2005;102:4120–4125. doi: 10.1073/pnas.0500660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T., Forgues M., Wang W., Kim J.W., Ye Q., Jia H., Budhu A., Zanetti K.A., Chen Y., Qin L.X. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- Ye D., Yang K., Zang S., Lin Z., Chau H.T., Wang Y., Zhang J., Shi J., Xu A., Lin S., Wang Y. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016;65:988–997. doi: 10.1016/j.jhep.2016.05.041. [DOI] [PubMed] [Google Scholar]
- Younes R., Bugianesi E. Should we undertake surveillance for HCC in patients with NAFLD? J. Hepatol. 2018;68:326–334. doi: 10.1016/j.jhep.2017.10.006. [DOI] [PubMed] [Google Scholar]
- Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- Yu H.T., Yu M., Li C.Y., Zhan Y.Q., Xu W.X., Li Y.H., Li W., Wang Z.D., Ge C.H., Yang X.M. Specific expression and regulation of hepassocin in the liver and down-regulation of the correlation of HNF1α with decreased levels of hepassocin in human hepatocellular carcinoma. J. Biol. Chem. 2009;284:13335–13347. doi: 10.1074/jbc.M806393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P., Elks C.M., Stephens J.M. The induction of lipocalin-2 protein expression in vivo and in vitro. J. Biol. Chem. 2014;289:5960–5969. doi: 10.1074/jbc.M113.532234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.