

Abstract
Protein disulfide isomerase (PDI) is overexpressed in glioblastoma, the most aggressive form of brain cancer, and folds nascent proteins responsible for the progression and spread of the disease. Herein, we describe a novel, nanomolar PDI inhibitor, pyrimidotriazinedione 35G8, that is toxic in a panel of human glioblastoma cell lines. We performed a medium throughput 20,000-compound screen of a diverse subset of 1,000,000 compounds to identify cytotoxic small molecules. Cytotoxic compounds were screened for PDI inhibition, and, from the screen, 35G8 emerged as the most cytotoxic inhibitor of PDI. Bromouridine-labeling and sequencing (Bru-seq) of nascent RNA revealed that 35G8 induced Nrf2 (nuclear factor-like 2) antioxidant response, endoplasmic reticulum stress response, and autophagy. Specifically, 35G8 upregulated heme oxygenase 1 and SLC7A11 (solute carrier family 7 member 11) transcription and protein expression and repressed PDI target genes such as TXNIP (thioredoxin-interacting protein 1) and EGR1 (early growth response 1). Interestingly, 35G8-induced cell death did not proceed via apoptosis or necrosis, but by a mixture of autophagy and ferroptosis. Cumulatively, our data demonstrate a mechanism for a novel PDI inhibitor as a chemical probe to validate PDI as a target for brain cancer.
Keywords: drug discovery, cancer, oxidoreductases, protein disulfide isomerase, unfolded protein response
Entry for the Table of Contents
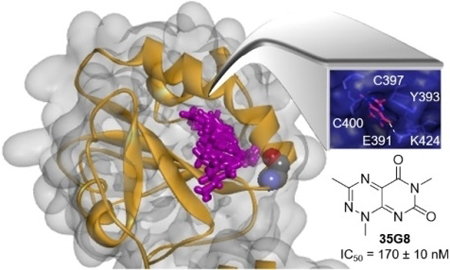
We describe a nanomolar, cytotoxic protein disulfide isomerase inhibitor, 35G8, that is potent in a panel of human glioblastoma cell lines. Bromouridine-labeling and sequencing of nascent RNA revealed that 35G8 induced Nrf2 antioxidant response, ER stress response, and autophagy, and may induce cell death via ferroptosis
Introduction
Glioblastoma is the most common type of malignant central nervous system (CNS) tumor. Prevalence increases with age with peak incidence in individuals aged 60–79 years.[1] Despite the treatment options available – surgical resection followed by chemoradiotherapy and adjuvant chemotherapy (temozolomide) – the five-year survival rate of patients diagnosed with glioblastoma is only 5.0 %.[1, 2] Current treatments are marginally effective and the number of cases is expected to grow with the aging population, emphasizing the urgent need for the development of novel and effective therapies for glioblastoma. Disease recurrence and drug resistance remain the major challenges for a successful cure.
Protein disulfide isomerase (PDI; EC 5.3.4.1) is a 57-kDa endoplasmic reticulum (ER) oxidoreductase of the thioredoxin superfamily that assists protein folding in the ER by catalyzing disulfide rearrangements (isomerase activity), disulfide formation (oxidase activity), and disulfide reduction (reductase activity).[3, 4] PDI is overexpressed in several cancers but most significantly in glioblastoma.[3] Previously, we demonstrated that PDI knockdown by siRNA leads to substantial cytotoxicity in ovarian cancer cells.[5] PDI inhibitors and modulators are being developed to combat cancer and neurological diseases (for a comprehensive review of PDI inhibitors, see ref. 3 and 4a). The PDI inhibitor, bacitracin, inhibits migration and invasion of glioblastoma cells[6] and enhances apoptosis caused by ER stress-inducing agents in melanoma cells.[7] Another class of compounds, T8, are weak inhibitors of PDI and at moderately high concentrations, sensitize several cancer cell lines to etoposide treatment.[8] Recently, a reversible, selective, non-toxic PDI inhibitor, ML359, was developed as a probe to study thrombosis-related diseases.[9] Modulators of PDI have also been shown to be neuroprotective. A reversible PDI modulator, LOC14 (EC50 = 500 nM), has neuroprotective effects in cellular and rat models of Huntington’s disease.[10] Furthermore, PDI inhibitor CCF642 was demonstrated to be effective in a mouse xenograft model of multiple myeloma.[11] Mounting evidence highlights PDI as an important target against several diseases including cancer, emphasizing the need for potent, clinically relevant PDI inhibitors for cancer treatment.
Herein, we report on the development of 35G8 as a novel and potent PDI inhibitor that demonstrates activity in brain cancer cells and has drug-like properties. The activity of 35G8 in a diverse set of robust assays confirmed that the initial observation of activity was not a consequence of its redox cycling status. Results from nascent RNA Bru-seq[12] analysis showed that the transcription of 498 genes increased and 238 genes decreased at least 2-fold following a 4-hour incubation with 35G8 in U87MG glioblastoma cells. Gene set enrichment analysis demonstrated the upregulated genes to be involved in the Nrf2 antioxidant response and the unfolded protein response (UPR). Genes with decreased transcription involved histone and DNA repair pathways. In addition, 35G8 upregulates two key genes, SLC7A11 and HMOX1, and may kill cells through an iron-dependent form of cell death independent of apoptosis and necrosis, called ferroptosis.[13] The alterations in the transcriptional landscape induced by 35G8 provide a more comprehensive understanding of the mechanisms of PDI inhibition in brain cancer therapy.
Results and Discussion
35G8 is a nanomolar inhibitor of PDI
To identify cytotoxic small molecules, we screened a highly diverse library of 20,000 compounds, representing over one million compounds, in the colon cancer cell line HCT116 (Figure 1). From the initial screen, we identified 443 cytotoxic compounds with IC50 values under 10 μM. These 443 compounds were tested for PDI inhibition in an insulin turbidity assay.[14] Eight compounds demonstrated potent inhibition (IC50 < 1.0 μM), and after confirming the activity with re-purchased compound stocks and verifying a dose-dependent response, the most potent compound, 1,3,6-trimethylpyrimido[5,4-e] [1,2,4] triazine-5,7(1H,6H)-dione (35G8), was selected for further analysis and optimization.
Figure 1.
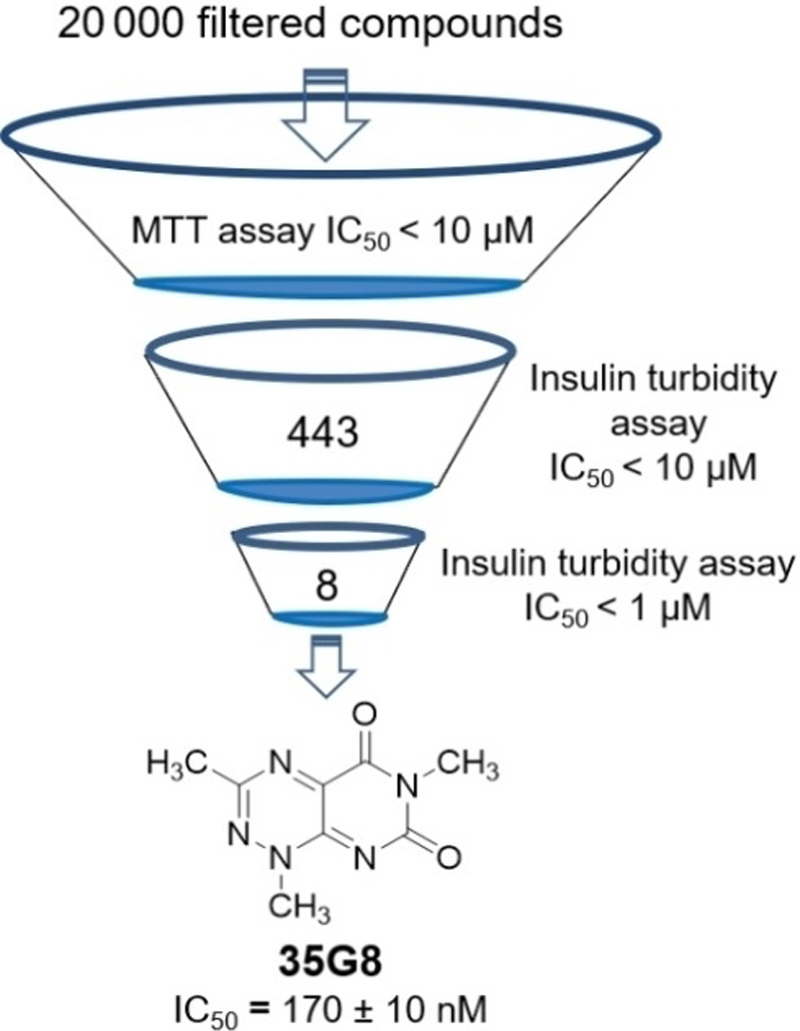
Discovery of 35G8. Work flow summarizing the screening process that identified 35G8 as a potent PDI inhibitor. 20,000 compounds were screened in an MTT assay with HCT116 cells and 443 compounds were cytotoxic in these cells. The 443 compounds were tested further in an insulin turbidity assay; 35G8 had the most potent IC50 value and was taken for further biochemical analysis and optimization.
We next used the thermal shift assay[15] to validate whether 35G8 stabilizes its presumed target, PDI. Intriguingly, 35G8 destabilized PDI, indicated by the decrease in melting temperature of the protein (Figure 2A). The dose-dependence of the negative thermal shifts at all concentrations tested (ΔTm: −3.64 °C at 100 µM; −2.94 °C at 10 µM; −1.43 °C at 1 µM) (Figure 2B) provides further evidence that 35G8 associates with and destabilizes PDI. The melting temperature of a protein shifts positively or negatively in the presence of a ligand, and this change in melting temperature parallels the stability of the protein.[16] These results suggest 35G8 interacts with PDI at a unique site compared to known stabilizing ligands, such as estradiol.[17] To further validate 35G8 binding to PDI, we performed the cellular thermal shift assay (CETSA) and drug affinity responsive target stability (DARTS) assay. 35G8 also destabilized PDI via CETSA (Figure 2C). 35G8 had little effect on a related molecular chaperone, GRP78, but did seem to stabilize the cysteine-containing glutathione-transferase Omega 1 (GSTO1). In the DARTS assay, U87MG cell lysates were subjected to pronase degradation in the presence or absence of PACMA31 or 35G8 (Figure 2D). Both compounds protected PDI from proteolysis, but had no effect on the degradation of GRP78 or GSTO1. These results established 35G8 as a potent, selective inhibitor of PDI.
Figure 2.
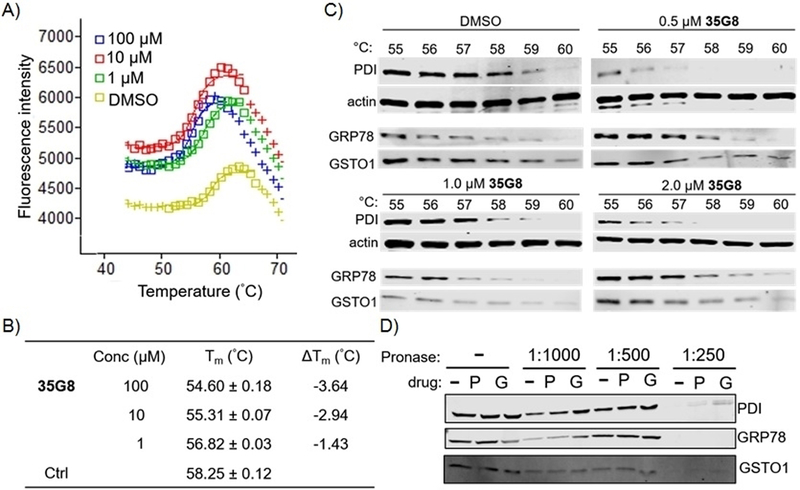
35G8 destabilizes PDI. (A) Thermal shifts observed for recombinant PDI (0.3 mg/ml) with various concentrations of 35G8. DMSO was used as a control. (B) Apparent melting temperatures (Tm) and change in melting temperature derived from ThermoFluor assay (C) Protein expression of PDI, GRP78, GSTO1, and actin (loading control) in the absence or presence of 35G8 at varying temperatures in the cellular thermal shift assay (D) Western blot analysis of DARTS assay with PDI, GRP78, and GSTO1 subjected to 100 μM PACMA31 (P), 100 μM 35G8 (G), or DMSO (−). Samples were subjected to varying concentrations of pronase. Data are means from three independent experiments.
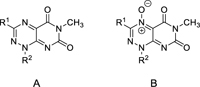
We further validated 35G8 as a bona fide PDI inhibitor by examining several of its close derivatives. Of the 16 analogues reported in the National Cancer Institute (NCI) database (Table S1), we pursued a refined group of eight compounds from the Developmental Therapeutics Program and tested their purity using UPLC-MS. Three of the eight compounds (NSC 67078, 99733 and 280172; hereafter referred to as NC72, NC75, and NC79, respectively) were over 95% pure and were tested in the insulin turbidity assay (Table 1).
Table 1.
PDI inhibitory activity of 35G8 analogues. IC50 values obtained in insulin turbidity assay. Data are means ± standard deviation from three independent experiments.
 |
||||
|---|---|---|---|---|
| Compound | Basic Module | R1 | R2 | IC50 (μM) |
| 35G8 (4a)[a] | A | CH3 | CH3 | 0.17 ± 0.01 |
| 4b[b] | A | CH3 | 0.39 ± 0.03 | |
| 4c[c] | A | CH3 | 0.33 ± 0.04 | |
| 4d[d] | A | CH3 | 0.36 ± 0.05 | |
| 4e[e] | A | CH3 | 0.32 ± 0.01 | |
| 4f[f] | A | CH3 | 0.24 ± 0.04 | |
| 5d[g] | B | CH3 | 0.42 ± 0.07 | |
| NC72 (NSC67078) | A | H | CH3 | 0.105 ± 0.004 |
|
NC75 (NSC99733) |
A | H | H | > 120 |
|
NC79 (NSC280172) |
B | CH3 | CH3 | 6.55 ± 1.19 |
| PACMA31 | - |
- |
- | 5.81 ± 1.23 |
1,3,6-Trimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
1,6-Dimethyl-3-phenylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
3-Benzyl-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
3-(4-Methoxyphenyl)-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
3-(3-Methoxyphenyl)-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
1,6-Dimethyl-3-(4-nitrophenyl)pyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione.
3-(4-Methoxyphenyl)-1,6-dimethyl-5,7-dioxo-1,5,6,7-tetrahydropyrimido[5,4-e][1,2,4]triazine 4-oxide.
We also synthesized several analogues of 35G8 to validate the above findings. The lead compound, 35G8, contains methyl substituents at the three N1, C3, and N6 positions (Figure 1). We incorporated various substituents at the C3 position while maintaining the methyl groups at N1 and N6 due to the efficient introduction of the N1 and N6 methyl groups early in the synthesis (Scheme 1). Nucleophilic attack of methylhydrazine on 6-chloro-3-methyl uracil (1) led to hydrazinylpyrimidine-2,4(1H,3H)-dione (2a).[18] Further condensation with aldehydes furnished the corresponding hydrazones (3a-f). Each hydrazone was cyclized by treatment with sodium nitrite in acetic acid/water to afford a mixture of pyrimidotriazinediones (4a-f) and the corresponding N-oxide derivative (5d).
Scheme 1.
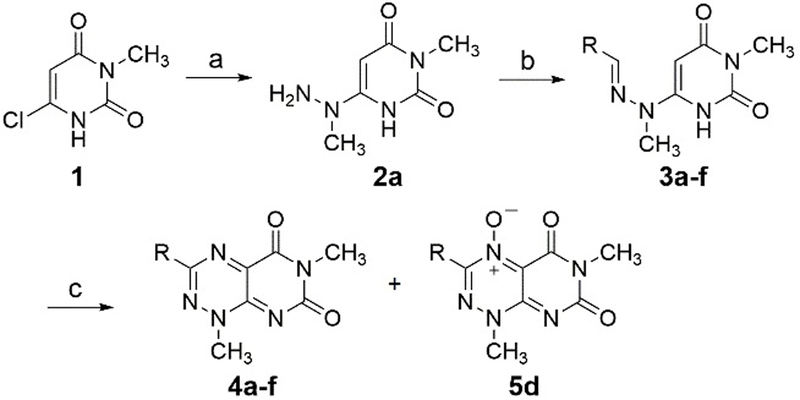
Synthesis of 3-substituted 35G8 analogues. Reagents and conditions: (a) methylhydrazine, EtOH, reflux; (b) aldehyde (R-CHO), anhydrous EtOH, room temperature; (c) NaNO2, AcOH/H2O, room temperature.
All 35G8 analogues had strong PDI inhibitory activity with submicromolar IC50 values, except NC75 (> 120 μM) and NC79 (6.55 ± 1.19 μM) in the insulin turbidity assay (Table 1). The pyrimidotriazinedione compound (35G8, IC50: 0.17 ± 0.01 μM) was more potent than the corresponding N-oxide compound (NC79). A similar trend was observed between 4d (IC50: 0.36 ± 0.05 μM) and 5d (IC50: 0.42 ± 0.07 μM). Among the pyrimidotriazinediones, the compounds containing a methyl group (4a) or no substituent (NC72) at R1 had enhanced activity compared to those with an aromatic moiety (4b-f), likely due to steric effects (Figure S1A). Interestingly, the PDI inhibitory activity was abolished upon removal of the methyl substituent at R2 (NC75: IC50 > 120 µM) compared to NC72 (IC50: 0.105 µM), indicating that the methyl group at R2 may be necessary to retain PDI inhibitory activity (Figure S1B). Furthermore, the removal of PDI inhibitory activity abolished the cytotoxicity of the compound.
35G8 analogues inhibit glioblastoma cell proliferation
All synthesized compounds demonstrated potent cytotoxicity in four glioblastoma cell lines, U87MG, U118MG, A172 and NU04, with IC50 values under 10 μM, except 4c (Table 2). The IC50 value of 35G8 in U87MG cells is 1.1 ± 0.2 μM. NC72 demonstrated the most potent cytotoxicity (IC50 = 0.5 ± 0.1 μM), complementing its potency in the PDI assay (Table 1). NC75 and NC79 had, little effect on cell growth. Interestingly, this suggests that the methyl substituent is important not only for PDI activity (as seen in the dramatic IC50 value increase from NC72 to NC75), but also for cytotoxicity.
Table 2.
In vitro cytotoxicity of 35G8 analogues in a panel of human glioblastoma cell lines. Cytotoxicity measured in the MTT assay. Data are means from at least three independent experiments.
| IC50 (μM) | ||||
|---|---|---|---|---|
| Compound | U87MG | U118MG | NU04 | A172 |
| 35G8 | 1.1 ± 0.2 | 3.9 ± 0.1 | 0.8 ± 0.2 | 2.0 ± 0.6 |
| 4b | 3.0 ± 0.3 | 4.6 ± 0.5 | 3.7 ± 1.2 | 1.8 ± 0.4 |
| 4c | 12.7 ± 3.7 | 24.0 ± 7.4 | > 30 | 8.2 ± 2.5 |
| 4d | 1.2 ± 0.2 | 3.9 ± 0.6 | 0.86 ± 0.04 | 1.5 ± 0.4 |
| 4e | 1.1 ± 0.2 | 2.4 ± 0.6 | 0.76 ± 0.22 | 1.5 ± 0.1 |
| 4f | 1.8 ± 0.7 | 6.2 ± 1.6 | 4.9 ± 1.2 | 1.1 ± 0.2 |
| 5d | 1.9 ± 0.7 | 4.3 ± 0.1 | 1.5 ± 0.7 | 1.7 ± 0.1 |
| NC72 | 0.5 ± 0.1 | - | - | - |
| NC75 | > 100 | - | - | - |
| NC79 | > 100 | - | - | - |
| PACMA31 | 0.13 ± 0.07 | 0.28 ± 0.04 | 0.4 ± 0.1 | 0.12 ± 0.10 |
Pretreatment with Z-VAD-FMK, an irreversible caspase inhibitor,[19] and necrostatin-1, a necroptosis inhibitor,[20] did not protect the cells from 35G8-induced cell death (Figure S2A). These results indicate that neither necrosis nor apoptosis are the main pathways responsible and another pathway may be implicated in cell death.
35G8 may bind in the catalytic site of PDI
The pyrimidotriazinedione class may bind PDI catalytic sites preferentially and interact with the cysteine residues (Figure 3A-C). Interestingly, the compounds are predicted to bind the C-terminal C397 site over the sequence-identical N-terminal C53 site, likely due to the composition of a binding pocket around C397. Docking pose conformation of 35G8 in the C397 catalytic site and the corresponding PoseView26 representation illustrate hydrogen bonding and pi-pi interactions with important residues of the active site: K424, E391 and Y393 (Figure 3D). The synthesized analogues, an additional 409 analogues of 35G8, and the NC compounds also bind in the N-terminal active site pocket, forming similar interactions (Figure S1B). The docking poses of the synthesized analogues (4b-5d) indicate that the larger substitution at R1 is subject to steric hindrance that decreases potency (Figure S1A). NC75 has the lowest docking score for binding to the catalytic site (Figure S1B), in agreement with its inactivity in vitro. This suggests that the presence of a methyl group at the R2 position is important for the hydrophobic interaction in the binding site.
Figure 3.
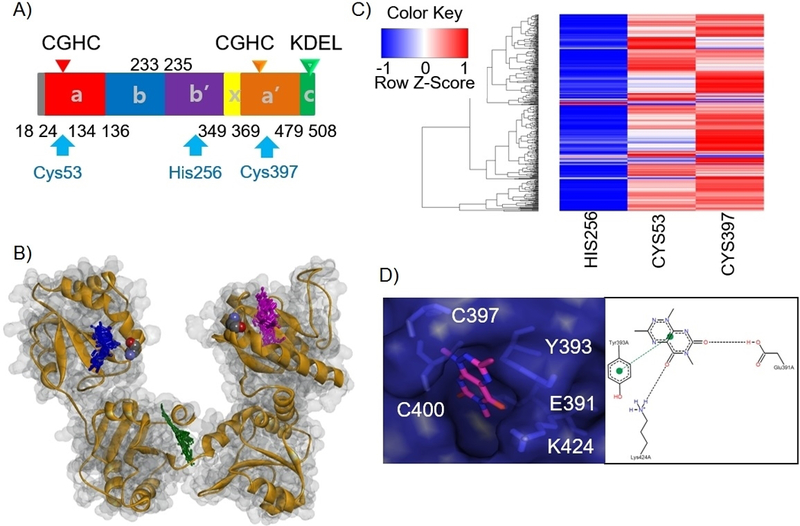
Docking of 35G8 analogues on PDI reveals their interaction with catalytic cysteine 397. (A) Location of the three binding pockets on the domain architecture of PDI. (B) Heat map plot for docking of 409 analogues of 35G8 in three binding pockets of PDI. (C) Structural overview of ten 35G8 analogues docked in PDI binding sites. The catalytic cysteines are colored by atom in a space-filling representation. The rest of the protein is depicted in grey and orange. The docked structures are shown in purple, green and blue for the C53, H256, and C397 site, respectively. (D) Docking pose of 35G8 in the C397 catalytic site of the PDI along with a PoseView representation showing its interactions with the binding site residues.
35G8 induces the Nrf2 antioxidant pathway and ER stress response
To better elucidate the cellular response to the pyrimidotriazinediones, we performed nascent RNA sequencing using the Bru-seq[22] method and analyzed changes in gene transcription rates in response to 35G8 in U87MG cells. Four hours after 35G8 treatment, 498 genes were upregulated at least two-fold and 238 genes were downregulated at least two-fold. Many of the top upregulated genes are implicated in the Nrf2 antioxidant response, ER stress response, and autophagy. We identified the top 20 upregulated and downregulated gene sets (Table S2 and S3) and analyzed the genes that were upregulated or downregulated at least two-fold with IPA (Ingenuity Pathway Analysis) (Figure 4A and Table S4) and GSEA (Gene Set Enrichment Analysis) (Figure 4B, Table S5 and S6). GSEA snapshots of enriched gene sets are reported in Figure S3 and S4. GSEA revealed enrichment of the Nrf2-mediated oxidative stress response upon 35G8 treatment (Figure 4B). Treatment also correlates with KOBAYASHI_EGFR_SIGNALING_24HR_DN gene set, suggesting 35G8 may inhibit EGFR signaling. DAVID (the Database for Annotation, Visualization and Integrated Discovery) analysis identified functional terms related to ER and redox-active disulfide, providing further evidence for PDI inhibition by 35G8 (Figure 4C and4D).
Figure 4.
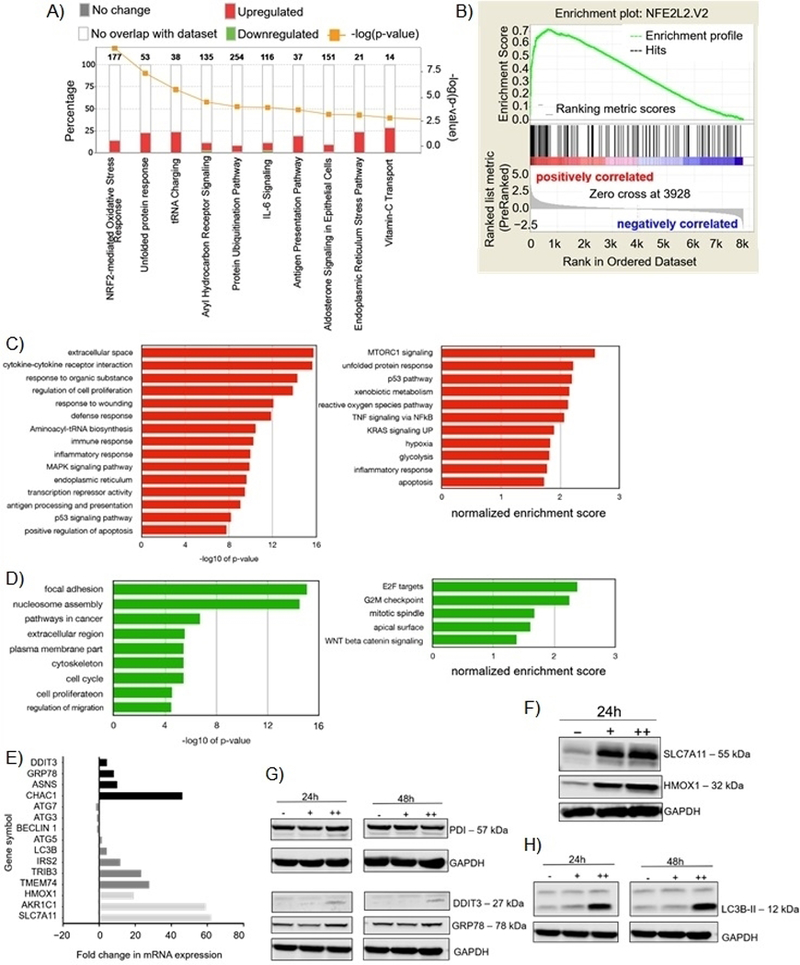
Effects of 35G8 treatment on cellular pathways. (A) Pathways from the Bru-seq analysis of 35G8-treated cells. (B) GSEA for “NFE2L2.V2,” the top gene set matched with upregulated genes from Bru-seq results. Functional terms represented by genes upregulated (C) and downregulated (D) at least 2-fold by 35G8 treatment. Pathway analysis was performed using DAVID (left) and GSEA (right). (E) Histograms of differentially expressed proteins between 35G8-treated and DMSO-treated U87MG cells. Fold change bars are in black for UPR genes, dark grey for autophagy-related genes, and light grey for Nrf2-related genes. (F) Western blot showing Nrf2-regulated proteins SLC7A11 and HMOX1 expression upon 24-hour treatment of U87MG cells with 1 or 2 μM 35G8. GAPDH used as a loading control. (G) Western blot of ER stress-induced proteins DDIT3 and GRP78 expression upon 24-hour treatment of U87MG cells with 1 and 2 μM 35G8. GAPDH used as a loading control. (H) Western blot of autophagy-related proteins LC3B, beclin 1, ATG3, ATG5, and ATG7 expression upon 24-hour treatment of U87MG cells with 1 (+) and 2 (++) μM 35G8. -: vehicle-treated control. GAPDH used as a loading control. Experiments repeated in triplicate. .
The upregulation of Nrf2 response genes, including HMOX1 (19-fold increase), SLC7A11 (63-fold increase), AKR1C1 (59-fold increase), and LOC344887 (23-fold increase), is likely a protective response to the insults caused by 35G8 (Figure 4E). We also confirmed parallel increases in HMOX1 and SLC7A11 protein expression (Figure 4F). The Nrf2 antioxidant pathway mitigates oxidative stress by inducing antioxidant response elements.[23] PDI is vital in the UPR, and inhibiting this key protein disrupts proteostasis, ultimately leading to ER stress and cell death when the cell cannot cope with the accumulation of misfolded proteins. ER stress target genes downstream the PERK-ATF4 ER stress response pathway, CHAC1 (46-fold increase), DDIT3 (4-fold increase), and HSPA5 (8-fold increase) increased as a result of 35G8 treatment (Figure 4E). Protein expression of GRP78 (HSPA5) and DDIT3 increased upon 24-hour treatment of 2 μM 35G8 (Figure 4G); however, CHAC1 protein was undetectable, likely because the CHAC1 protein is rapidly degraded by the proteasome (data not shown).[24] mRNA expression of other downstream targets of the PERK-ATF4 ER stress response pathway, including TRIB3 and ASNS,[25] also increased in response to 35G8 (Figure 4E). These results suggest that brain cancer cells rely on PDI to maintain redox homeostasis, and when PDI is inhibited, cells undergo irremediable ER stress that leads to cell death.
We also identified several autophagic signaling genes that respond to ER stress triggered by 35G8, including TRIB3, IRS2, and TMEM74 (Figure 4E). TRIB3 (23-fold increase), as a downstream target of ATF4, mediates autophagy by inhibiting the mTORC1 pathway.[26] IRS2 (12-fold increase) activation induces protective autophagy to clear unwanted protein aggregates[27] and may also help remove damaged cells. TMEM74 (28-fold increase), a transmembrane protein localized to the lysosome and autophagosome, regulates autophagy.[28] The increased transcription of these autophagy-related genes prompted us to measure protein expression of several autophagy markers (Figure 4H). Cleaved LC3B expression increased significantly after 24-hour treatment with 2 μM 35G8, however expression levels of other autophagy markers, including ATG3, ATG5, ATG7, and beclin 1, did not change (data not shown), suggesting that autophagy may play a more protective role in this case. These results indicate that 35G8 induces the ER stress and Nrf2 response in brain cancer cells to contribute to cell death. GSEA analysis also showed that 35G8 treatment repressed many genes involved in DNA repair pathways such as mismatch repair, homologous recombination, base excision repair and nucleotide excision repair (Figure S5). Even though not all pathways showed significance individually in the GSEA analysis, the fact that all of them were suppressed suggests that the expression of these DNA repair genes is regulated by a common transcription factor that requires PDI-mediated folding for proper activity. These findings open up the interesting possibility that 35G8 could act synergistically with DNA-damaging agents and have therapeutic implications.
Bru-seq analysis identifies novel glioblastoma markers
AKR1C1, IL-6, CHAC1 and TNFSF9 are among the top 20 upregulated genes with significantly decreased expression in brain cancer compared to normal brain tissues (Figure 5A-C). Conversely, genes that were downregulated upon 35G8 treatment, including TXNIP (−7.40-fold change), EGR1 (−5.65-fold change) and ITGA3 (−3.89-fold change) are often overexpressed in brain cancer (Figure 5D-F). Additional genes affected include HMOX1, IRS2, SLC7A11, and mir181A2HG (Figure S6). These data suggest 35G8 inhibits transcription of these mRNA, or inhibits an upstream regulator of ITGA3 and EGR1. The results also indicate a gene such as IL6 may be used as a biomarker of 35G8 inhibition in future studies and EGR1 may be a novel glioblastoma marker.
Figure 5.
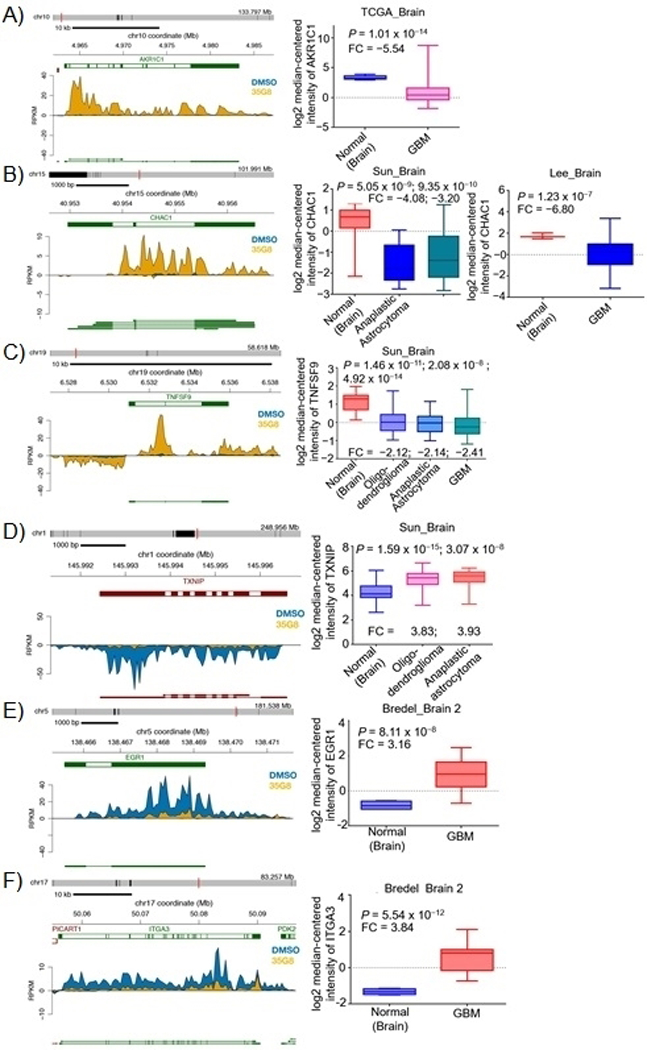
Effect of 35G8 treatment on RNA synthesis in U87MG cells. 35G8 induces transcription of (A) AKR1C1, (B) CHAC1 and (C) TNFSF9 while corresponding box plots show downregulation of these genes in brain cancer. 35G8 inhibits the transcription of (D) TXNIP, (E) EGR1 and (F) ITGA3 while corresponding box plots show upregulation of these genes in brain cancer. FC: fold change; GBM: glioblastoma
35G8 induces ROS formation
Because the cells responded to 35G8 by upregulating the Nrf2-mediated oxidative stress response, we investigated the production of reactive oxygen species (ROS) by 35G8 and its analogues to determine whether the cytotoxicity of these compounds is dependent on ROS induction. We observed significant ROS induction by all 35G8 analogues tested at 5 μM as early as four hours after treatment, except for 4c (Figure 6). ROS accumulation with these compounds was time-dependent. At 24 hours, 5 μM 35G8 treatment achieved maximal ROS induction, comparable to 100 μM hydrogen peroxide treatment (Figure 6C). No change in the fluorescent signal in the samples containing 35G8 without H2DCFDA dye was observed, eliminating the possibility of endogenous fluorescence affecting the assay. N-acetyl-cysteine (NAC) did not affect the the cytotoxicity of 35G8 significantly (Table S7). This suggests 35G8-induced cell death is not solely dependent on ROS induction.
Figure 6.
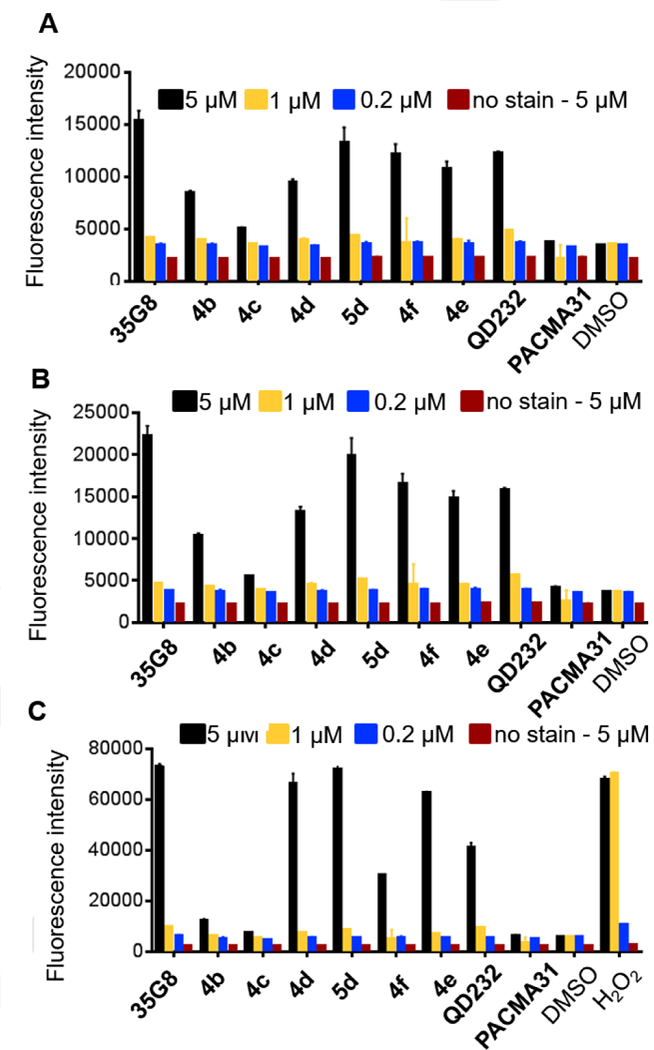
ROS induction activity of synthesized 35G8 analogues at (A) 4 hours, (B) 6 hours, and (C) 24 hours. In (C), hydrogen peroxide concentration is 500, 100, and 20 μM, from left to right. Data are means from three independent experiments; error bars show standard deviation.
35G8 induces ferroptosis
Both transcription and protein expression of HMOX1 and SLC7A11 are highly upregulated by 35G8 (Figure 4E and4F). These proteins have been implicated in the non-apoptotic cell death mechanism, ferroptosis. HMOX1 is necessary for ferroptosis and is a major source of iron in the body.[29] Inhibition of cysteine-glutamate exchange through System xc-, of which SLC7A11 is a component, induces iron-dependent cell death.[30] To determine whether 35G8 induces ferroptosis in U87MG cells, we treated the cells in the presence or absence of deferoxamine (DFO), an iron chelator (Figure S2B).[31] In the presence of DFO, 35G8 is almost three times less potent (IC50 = 5.8 ± 1.0 μM) than when used alone (IC50 = 2.2 ± 0.7 μM). These data suggest that PDI may play an important role in preventing ferroptosis in brain cancer.
35G8 is expected to cross the blood-brain barrier
The likelihood of blood-brain barrier (BBB) permeation, AlogP, water solubility, polar surface area, and number of rotatable bonds of 35G8 and its synthesized analogues were determined with a qualitative model in the ADMET predictor (Version 7.0) (Table S8; Figure S8). The AlogP of the compounds is between −1.1 and 1.1 and the likelihood of BBB permeation is high. The polar surface area of 35G8 is less than 90 Å2, the cutoff for predicted CNS penetration.[32] The average molecular weight of marketed CNS compounds is 310, and the 35G8 analogues range in molecular weight from 207 – 315. Similarly, TMZ has a molecular weight of 194 Da, ClogP of −0.82, and a polar surface area of 108 Å2. These data demonstrate that 35G8 will be able to cross the blood-brain barrier.
Discussion
The screen of 20,000 diverse compounds in a growth inhibition assay produced 35G8 as the most potent inhibitor of proliferation of the colon cancer cell line HCT116. 35G8 destabilizes PDI and blocks its reductase activity. As a consequence, 35G8 likely causes cell death via continuous activation of ER stress and disruption of homeostatic balance, among other factors. 35G8 was validated in orthogonal assays to rule out that activity was not a consequence of its redox-cycling status. 35G8 generates H2O2 in the presence of DTT at the concentrations used in the PDI assay (Figure S9A), however, H2O2 does not interfere with insulin reduction catalyzed by PDI (Figure S9B). The reactive nature of the pyrimidotriazinedione class underlines the importance of testing activity in a wide variety of assays, including non-fluorescent methods, in order to eliminate false positive results. Therefore, we performed several assays with various output methods to test our novel compounds.
The Bru-seq results revealed that 35G8 promoted the activation of the Nrf2 pathway. Of the top 20 upregulated genes following a 4-hour 35G8 treatment, four are implicated in the Nrf2 pathway (SLC7A11, HMOX1, AKR1C1, and LOC344887). Nrf2 is a transcription factor that normally is kept at low levels due to degradation mediated via Keap1.[33] Following exposure to ROS, Keap1 is inactivated and Nrf2 induces transcription of genes, counteracting the oxidative insult.[34] SLC7A11 is part of a cysteine-glutamate transporter (system xc-) that is regulated by Nrf2 as well as ATF4.[35] HMOX1, another Nrf2-regulated gene, increased over 19-fold upon 35G8 treatment. We also found that transcription of the AKR1C1 gene, which is induced by ROS but expressed at low levels in gliomas, increased significantly following 35G8 treatment. Furthermore, the lncRNA LOC344887 has been shown to be activated by Nrf2.[36] Nrf2-regulated genes may be responsible for treatment resistance in glioblastoma, providing further evidence that inhibiting PDI could be a sound strategy to treat glioblastoma. [37]
Several ER stress markers were induced in response to 35G8 treatment, including CHAC1, DDIT3, ASNS, and ATF3. Due to the strong upregulation of CHAC1, a pro-apoptotic marker regulated by ATF4, we hypothesize that the PERK-ATF4-DDIT3 branch of the UPR is likely activated upon PDI inhibition by 35G8 treatment. The ER stress response and autophagy are closely linked, and ER stress may induce autophagy in 35G8-treated cells.
Autophagy is the process of protein and organelle degradation by lysosomes, used as a survival mechanism to provide energy for the cell.[38] The ER stress response protein ATF4 promotes autophagy[39] by upregulating genes like TRIB3.[40] While autophagy can be protective as a survival mechanism, increased autophagic signaling causes cell death. It is still unclear whether TMEM74 is regulated by ATF4, but upregulation of TMEM74 mRNA may lead to autophagic PI3K signaling. The increase of ARG2 expression upon 35G8 treatment may be a result of the activation of the UPR and lower cellular levels of arginine, leading to autophagy.[41] IRS2, a key insulin signaling protein regulated by the UPR and silenced by JNK, is expressed to remove damaged cells.[42] 35G8 treatment initiates a protective response by upregulating the UPR and inducing autophagy to combat ER stress. Ultimately, the unbalanced homeostatic mechanisms overwhelm the cellular machinery, and this leads to cell death.
ROS induction is likely responsible for the increased expression levels of TXNRD1 (9-fold increase) and TXN (2-fold increase). TXNIP inhibits TXN activity and TXNIP expression is significantly inhibited by 35G8 treatment (7.4-fold decrease). ER stress activates the ERK1/2 MAP kinase signaling pathway, repressing TXNIP expression leading to thioredoxin nuclear translocation.[43] Interestingly, TXNIP is overexpressed in brain cancer patients. TXNIP also can bind PDI and increase its activity.[43] Lower TXNIP levels allow TRX to bind ASK1 and prevent apoptosis.[44] Therefore, decreased expression of TXNIP may contribute to the absence of apoptosis signaling observed upon 35G8 treatment.
Another class of genes that were repressed by 35G8 are involved in DNA repair (Figure S5). While this repression was not dramatic, GSEA analysis showed that several genes involved in mismatch repair, homologous recombination, base excision repair and nucleotide excision repair showed reduced transcription following 35G8 treatment. It is possible that these genes share a common transcription factor that requires PDI-assisted protein folding for optimal function. Importantly, these findings suggest that 35G8 may be used in combination with DNA damaging agents or PARP1 inhibitors to augment their therapeutic effectiveness.
The key Nrf2-regulated genes SLC7A11 and HMOX1 are essential markers for iron-dependent, erastin-induced ferroptosis.[29, 30] SLC7A11 is a negative regulator of ferroptosis and upregulation of SLC7A11 occurs as a response to system xc- inhibition.[13] Efforts to treat glioma patients by inhibiting system xc- have failed;[45] however, combining SLC7A11 inhibition with a PDI inhibitor may be a promising new strategy. Importantly, 35G8-induced cell death can be rescued by deferoxamine, suggesting that ferroptosis is occurring.
System xc- imports cystine for glutathione synthesis[13] to maintain intracellular redox balance and the expression of this system is often elevated in several cancers, including gliomas.[46] System xc- inhibitors, in particular sulfasalazine, as single agents for the treatment of gliomas have been unsuccessful,[47] but have been shown to sensitize glioma cells to radiation therapy.[48] Similarly, the ferroptosis inducer erastin sensitizes glioblastoma cells to temozolomide by inhibiting system xc-.[49] These studies provide evidence that system xc- is an important target for combating resistance in brain cancer. Interestingly, Bru-seq analysis of 35G8-treated cells revealed a pattern of gene expression similar to that of erastin-treated cells (Figure S9), including induction of the ER stress response, unfolded protein response, and expression of the erastin-exposure pharmacodynamic marker, CHAC1.[50] This indicates that as a consequence of PDI inhibition, 35G8 is causing blockade of system xc-. However, a link between PDI and SLC7A11 expression has not yet been established and further investigation is warranted.
Conclusions
We identified 35G8 as a markedly potent PDI inhibitor that may have therapeutic potential as a single agent and in combination with SLC7A11 inhibitors or DNA-damaging agents. 35G8 and its analogues demonstrate activity in human brain cancer cells likely through upregulation of ER stress and UPR that leads to autophagy-mediated ferroptosis. Taken together, our data suggest 35G8 is a useful investigational PDI inhibitor, expected to easily cross the blood brain barrier, that can be optimized to develop novel therapeutic agents to treat malignant glioma.
Experimental Section
Chemistry
All commercial chemicals and solvents were reagent grade and were used without further purification unless otherwise specified. Analytical thin layer chromatography was performed on Merck pre-coated plates (silica gel 60 F254) to follow the course of reactions. Proton nuclear magnetic resonance (1H NMR) spectroscopy was performed on Bruker Ascend 400 MHz spectrometer or Varian 500 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) units relative to residual undeuterated solvent. The following abbreviations are used to describe peak splitting patterns when appropriate: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broad singlet), dd (doublet of doublets), dt (doublet of triplets). Coupling constants (J) are expressed in Hertz (Hz). Low-resolution mass spectra were recorded on a Thermo-Scientific LCQ Fleet mass spectrometer or a Micromass LCT time-of-flight instrument utilizing the electro spray ionization (ESI) mode. HPLC was used to determine the purity of biologically tested compounds using the Shimadzu HPLC Test Kit C18 column (3 μm, 4.6 × 50 mm) under the following gradient elution conditions: mobile phase A of acetonitrile/water (10–95%) or mobile phase B of methanol/water (10–95%). The purity of three NCI compounds (NC72, NC75 and NC79) was determined by ultra-performance liquid chromatography (UPLC). UPLC was carried out using Acquity UPLC BEH (C18–1.7 μm, 2.1 mm × 50 mm) with a gradient elution of acetonitrile/water (10–100%). The purity was established by integration of the areas of major peaks detected at 254 nm, and all tested compounds including three NC compounds have ≥ 95% purity.
3-Methyl-6-(1-methylhydrazinyl)pyrimidine-2,4(1H,3H)-dione (2a)
A solution of 6-chloro-3-methyluracil (2.01 g, 12.5 mmol), methylhydrazine (2.87 g, 62.3 mmol) and absolute ethanol (30 mL) was heated at reflux for 3 hours. The reaction mixture was cooled to room temperature, and the precipitate was collected, washed with ethanol, and dried to give 2a as a white solid (819 mg, 39 %). 1H NMR (400 MHz, DMSO-d6) δ: 7.64 (bs, 1H), 4.75 (s, 1H), 3.33 (bs, 2H), 3.03 (s, 3H), 2.43 (s, 3H). MS (ESI) m/z = 171 (M+H)+.
3-Methyl-6-(1-methylhydrazinyl)pyrimidine-2,4(1H,3H)-dione (3a)
To a suspension of 2a (3.18 g, 18.7 mmol) in absolute ethanol (30 mL) was added acetaldehyde (1.65 g, 37.4 mmol) at room temperature with stirring. The reaction mixture was stirred for 2 hours, and the precipitate was filtered off by suction, washed with ethanol, and dried to give 3a as an off-white solid (823 mg, 22 %). 1H NMR (400 MHz, CDCl3) δ: 9.21 (bs, 1H), 7.08 (q, J = 5.2 Hz, 1H), 5.00 (d, J = 2.4 Hz, 1H), 3.31 (s, 3H), 3.16 (s, 3H), 2.09 (s, 3H). MS (ESI) m/z = 197 (M+H)+.
6-(2-Benzylidene-1-methylhydrazinyl)-3-methylpyrimidine-2,4(1H,3H)-dione (3b)
The same procedure for the synthesis of compound 3a was followed using compound 2a (500 mg, 2.94 mmol) and benzaldehyde (636 mg, 5.88 mmol) as reactants to yield 3b as a beige solid (590 mg, 78 %). 1H NMR R (500 MHz, DMSO-d6) δ: 10.64 (s, 1H), 7.97 (s, 1H), 7.96 (d, J = 6.5 Hz, 2H), 7.44 – 7.38 (m, 3H), 5.24 (s, 1H), 3.34 (s, 3H), 3.10 (s, 3H). MS (ESI) m/z = 259 (M+H)+.
3-Methyl-6-(1-methyl-2-(2-phenylethylidene)hydrazinyl)pyrimidine-2,4(1H,3H)-dione (3c)
The same procedure for the synthesis of compound 3a was followed using compound 2a (500 mg, 2.94 mmol) and phenyl acetaldehyde (744 mg, 5.88 mmol) as reactants. The crude compound was further purified by recrystallization from ethanol to yield 3c as a white brilliant solid (427 mg, 53 %). 1H NMR (500 MHz, CDCl3) δ: 9.18 (s, 1H), 7.36 (t, J = 7.3 Hz, 2H), 7.29 (t, J = 7.4 Hz, 1H), 7.21 (d, J = 7.3 Hz, 2H), 7.09 (t, J = 5.7 Hz, 1H), 5.02 (d, J = 2.5 Hz, 1H), 3.70 (d, J = 5.7 Hz, 2H), 3.31 (s, 3H), 3.14 (s, 3H). MS (ESI) m/z = 273 (M+H)+.
6-(2-(4-Methoxybenzylidene)-1-methylhydrazinyl)-3-methylpyrimidine-2,4(1H,3H)-dione (3d)
The same procedure for the synthesis of compound 3a was followed using compound 2a (300 mg, 1.76 mmol) and 4-methoxybenzaldehyde (490 mg, 3.52 mmol) as reactants. The crude compound was further purified by recrystallization from ethanol to yield 3d as a light beige solid (241 mg, 47 %). 1H NMR (500 MHz, CDCl3) δ: 9.18 (s, 1H), 7.66 (s, 1H), 7.63 – 7.60 (m, 2H), 6.98 – 6.94 (m, 2H), 5.10 (d, J = 2.5 Hz, 1H), 3.87 (s, 3H), 3.32 (s, 6H). MS (ESI) m/z = 289 (M+H)+.
6-(2-(3-Methoxybenzylidene)-1-methylhydrazinyl)-3-methylpyrimidine-2,4(1H,3H)-dione (3e)
The same procedure for the synthesis of compound 3a was followed using compound 2a (200 mg, 1.17 mmol) and 3-methoxybenzaldehyde (320 mg, 2.35 mmol) as reactants. The crude compound was further purified by recrystallization from ethanol to yield 3e as an off-white solid (90 mg, 27 %). 1H NMR (500 MHz, CDCl3) δ: 9.16 (s, 1H), 7.66 (s, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.26 – 7.23 (m, 1H), 7.18 – 7.17 (m, 1H), 6.97 (dt, J = 8.2, 1.7 Hz, 1H), 5.13 (d, J = 2.5 Hz, 1H), 3.86 (s, 3H), 3.34 (s, 3H), 3.32 (s, 3H). MS (ESI) m/z = 289 (M+H)+.
3-Methyl-6-(1-methyl-2-(4-nitrobenzylidene)hydrazinyl)pyrimidine-2,4(1H,3H)-dione (3f)
The same procedure for the synthesis of compound 3a was followed using compound 2a (140 mg, 0.82 mmol) and 4-nitrobenzaldehyde (254 mg, 1.65 mmol) as reactants. The crude compound was immediately used in the next step.
1,3,6-Trimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4a)
A stirring solution of the hydrazone 3a (823 mg, 4.19 mmol) in glacial acetic acid (10 mL)/water (0.6 mL) cooled to 0 °C was treated with sodium nitrite (895 mg, 12.6 mmol). The reaction mixture was allowed to warm to room temperature while stirring. Stirring continued until TLC indicated consumption of the starting material, thereby furnishing a mixture of the pyrimidotriazinedione (4a) and the corresponding N-oxide derivative. The reaction mixture was diluted with water and extracted with dichloromethane. The combined organic layers were dried over sodium sulfate, and the solvent was evaporated in vacuo. The resulting residue was chromatographed on silica to afford the product 4a as a brilliant yellow solid (272 mg, 31 %). 1H NMR (500 MHz, CDCl3) δ: 4.11 (s, 3H), 3.49 (s, 3H), 2.75 (s, 3H). MS (ESI) m/z = 208 (M+H)+. HPLC (mobile phase A): purity 99.9%.
1,6-Dimethyl-3-phenylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4b)
The same procedure for the synthesis of compound 4a was followed using the hydrazone 3b (300 mg, 1.16 mmol) and sodium nitrite (247 mg, 3.48 mmol) as reactants to afford the product 4b as an orange solid (112 mg, 36 %). 1H NMR (500 MHz, CDCl3) δ: 8.32 (dd, J = 8.1, 1.6 Hz, 2H), 7.56 – 7.52 (m, 3H), 4.24 (s, 3H), 3.53 (s, 3H). MS (ESI) m/z = 270 (M+H)+. HPLC (mobile phase A): purity 95.0%.
3-Benzyl-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4c)
The same procedure for the synthesis of compound 4a was followed using the hydrazone 3c (300 mg, 1.10 mmol) and sodium nitrite (235 mg, 3.30 mmol) as reactants. The crude product was purified by column chromatography and further recrystallized from ethanol to afford the product 4c as an oil (100 mg, 32 %). 1H NMR (500 MHz, CDCl3) δ: 7.38 – 7.25 (m, 5H), 4.29 (s, 2H), 4.11 (s, 3H), 3.47 (s, 3H). MS (ESI) m/z = 284 (M+H)+. HPLC (mobile phase A): purity 98.6%.
3-(4-Methoxyphenyl)-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4d)
The same procedure for the synthesis of compound 4a was followed using the hydrazone 3d (220 mg, 0.763 mmol) and sodium nitrite (163 mg, 2.29 mmol) as reactants. The crude product was purified by column chromatography and further recrystallized from ethanol to afford the product 4d as a red solid (81 mg, 35 %). 1H NMR (500 MHz, CDCl3) δ: 8.25 (d, J = 8.9 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 4.21 (s, 3H), 3.89 (s, 3H), 3.52 (s, 3H). MS (ESI) m/z = 300 (M+H)+. HPLC (mobile phase A): purity 96.1%.
3-(3-Methoxyphenyl)-1,6-dimethylpyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4e)
The same procedure for the synthesis of compound 4a was followed using the hydrazone 3e (77 mg, 0.267 mmol) and sodium nitrite (70 mg, 0.801 mmol) as reactants to afford the product 4e as a yellow solid (15 mg, 19 %). 1H NMR (500 MHz, CDCl3) δ: 7.91 (d, J = 7.7 Hz, 1H), 7.79 (s, 1H), 7.42 (t, J = 7.9 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 4.23 (s, 3H), 3.90 (s, 3H), 3.52 (s, 3H). MS (ESI) m/z = 300 (M+H)+. HPLC (mobile phase B): purity 97.2%.
1,6-Dimethyl-3-(4-nitrophenyl)pyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (4f)
The same procedure for the synthesis of compound 4a was followed using the crude hydrazone 3f (69 mg, 0.227 mmol) and sodium nitrite (60 mg, 0.683 mmol) as reactants to afford the product 4f as an orange solid (18 mg, 25 %). 1H NMR (500 MHz, CDCl3) δ: 8.51 (d, J = 8.9 Hz, 2H), 8.37 (d, J = 8.9 Hz, 2H), 4.27 (s, 3H), 3.53 (s, 3H). MS (ESI) m/z = 313 (M-H)-. HPLC (mobile phase A): purity 96.8%.
3-(4-Methoxyphenyl)-1,6-dimethyl-5,7-dioxo-1,5,6,7-tetrahydropyrimido[5,4-e][1,2,4]triazine 4-oxide (5d)
Following the same procedure for the synthesis of the pyrimidotriazinedione (4d), the corresponding N-oxide derivative was isolated by column chromatography to afford the product 5d as an orange solid (90 mg, 37 %). 1H NMR (500 MHz, CDCl3) δ: 7.86 (d, J = 9.0 Hz, 2H), 6.99 (d, J = 8.9 Hz, 2H), 4.07 (s, 3H), 3.89 (s, 3H), 3.42 (s, 3H). MS (ESI) m/z = 316 (M+H)+. HPLC (mobile phase A): purity 98.1%.
Reagents
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from Amresco (Solon, OH). N-acetylcysteine (NAC) was purchased from Sigma-Aldrich (St. Louis, MO). Methyl (3S)-5-fluoro-3-[[(2S)-2-[[(2S)-3-methyl-2-(phenylmethoxycarbonylamino) butanoyl] amino] propanoyl] amino]-4-oxopentanoate (Z-VAD-FMK) was purchased from Tocris Bioscience (Bristol, UK). 5-(1H-indol-3-ylmethyl)-3-methyl-2-sulfanylideneimidazolidin-4-one (Necrostatin-1) was purchased from Cayman Chemical Company (Ann Arbor, MI). Phenol red, H2O2, and horseradish peroxidase (HRP) were purchased from Sigma-Aldrich (St. Louis, MO). Hank’s Balanced Salt Solution (HBSS) was purchased from Hyclone, Logan, UT, and sodium hydroxide was purchased from EMD, Gibbstown, NJ.
Cell Culture
The human glioblastoma cells, U87MG, U118MG, NU04 and A172 (ATCC, Manassas, VA), were obtained in 2013, and were maintained in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA) with 10 % fetal bovine serum (Thermo Fisher Scientific, Waltham, MA). Cells were grown as monolayer cultures at 37 °C in a humidified atmosphere of 5 % CO2 and tested for mycoplasma contamination with the Mycoplasma Detection Kit, PlasmoTest (InvivoGen, San Diego, California).
Growth Inhibition Assay
Cell growth inhibition was assessed by MTT assay as previously described.[51] Cells were seeded in duplicate in 96-well plates at 7000 – 10000 cells/well. After overnight incubation at 37 °C and 5 % CO2, cells were treated with indicated compounds for 72 hours. For the combination therapies, NAC was added to the well at the same time as 35G8 (24 hours after plates were seeded), and Z-VAD-FMK and Necrostatin-1 were added to the well 1 hour prior to 35G8 addition. The plates were incubated with drug or vehicle control for 72 hours at 37 °C and 5 % CO2. MTT solution (20 μL 3 mg/mL) was added to the wells, and the cells were incubated for 4 hours at 37 °C. Supernatant was removed and DMSO (100 μl) was added to each well. The plates were shaken for 15 minutes at room temperature and absorbance of the formazan crystals was measured at 570 nm. Cell growth inhibition was assessed by the cell viability rate as [1-(At-Ab)/(Ac- Ab)]×100 (At , Ac and Ab were the absorbance values from cells which were treated with compound, cells which were not treated with compound, and blank, respectively). Cell viability was determined with the MTT assay. U87MG cells were seeded at 5000 cells per well in 96-well plates. Deferoxamine (Sigma Aldrich, St. Louis, MO) was added to cells in a five-point, three-fold dilution series from 400 μM. 35G8 was added immediately after in a five-point, three-fold dilution series from 100 μM. Cells were incubated with compounds for 12 hours at 37 °C, and MTT assay was performed as stated above.
PDI Protein Purification
The expression vector of recombinant human PDI protein with N-terminal His tag was a gift from Dr. Lloyd W. Ruddock (University of Oulu, Oulu, Finland). PDI expression and purification were performed as previously described[5] with slight modifications. In brief, protein production was carried out in Escherichia Coli strain BL21 (DE3) pLysS grown in LB medium with 200 μg/ml ampicillin (EMD Biosciences, La Jolla, CA) at 37°C and incubated at an A600 of 0.5 for 4 hours with 1 mM isopropyl β-D-1-thiogalactopyranoside (GoldBio, St. Louis, MO). Cells were harvested by centrifugation (4000 × g for 15 min) and were re-suspended in one-tenth volume Buffer A (20 mM sodium phosphate, pH 7.3). Cells were lysed by sonication and the cell debris was removed by centrifugation (16000 × g for 30 min). The supernatant was applied to a bed of Ni-nitrilotriacetic acid in a histidine-binding column (Qiagen, Hilden, Germany), equilibrated with 10 ml of Buffer A and incubated at 4 °C, overnight. After incubation, the column was washed in Buffer A and then in Buffer B (20 mM sodium phosphate, 0.5 M sodium chloride and 50 mM imidazole, pH 7.3). His-tagged proteins were eluted using Buffer C (20 mM sodium phosphate and 50 mM EDTA, pH 7.3) and eluent was dialyzed in 100 mM sodium phosphate buffer (pH 7.0) with 2 mM EDTA.
Measurement of PDI Activity
PDI activity was assessed by measuring the PDI-catalyzed reduction of insulin as described previously.[14] In brief, recombinant PDI protein (0.4 μM) was incubated with indicated compounds at 37 °C for 1 hour in sodium phosphate buffer (100 mM sodium phosphate, 2 mM EDTA, 8 μM DTT, pH 7.0). A mixture of sodium phosphate buffer, DTT (500 μM), and bovine insulin (130 μM; Gemini BioProducts, West Sacramento, CA) was added to the incubated PDI protein. The reduction reaction was catalyzed by PDI at room temperature, and the resulting aggregation of reduced insulin B chains was measured at 620 nm. PDI activity was calculated with the formula, PDI activity (%) = [(ODT80[PDI+DTT+compound] - ODT0[PDI+DTT+compound]) - (ODT80[DTT] - ODT0[DTT])] / [(ODT80[PDI+DTT] - ODT0[PDI+DTT]) - (ODT80[DTT] - ODT0[DTT])] × 100 (ODT0 and ODT80 were the absorbance values at 0 min and 80 min after the reduction reaction, respectively).
Thermal Shift Assay
Thermal shift of purified PDI (0.3 mg/ml in 100 mM NaPO4, pH 7.0) in the presence or absence of 35G8 was determined as described.[15] Briefly, 5 µl protein-dye (1,8-ANS, 0.3 mM; Sigma Aldrich, St. Louis, MO) solutions were dispensed in each well of a 384-well microplate (Thermo Scientific, AB1384K) and equal volumes of the test compound solutions were dispensed to each well. Then, 3 µl of silicone oil (Sigma Aldrich, St. Louis, MO) was added to each well to prevent evaporation. DMSO (2 % in buffer) was used as control. Fluorescence emission was detected by measuring light intensity using a CCD camera. The plate was heated at a temperature range from 25 to 90 °C at 1°C/minute in the ThermoFluor instrument (Johnson & Johnson, New Brunswick, NJ). Compounds were replicated three times in a 384-well plate.
Cellular Thermal Shift Assay
The cellular thermal shift assay was performed following previously established procedure.[52] U87MG cells were seeded at 2 × 106 cells/100 mm dish and allowed to attach overnight. Cells were treated with 0.5, 1.0, or 2.0 μM 35G8, or DMSO as the negative control, for 2 hours at 37 °C, 5 % CO2. After treatment, cells were trypsinized, washed with DPBS twice, and suspended in 600 μL DPBS. The cells were split into 100 μL aliquots, heated at indicated temperatures for 3 minutes in the Veriti Thermal Cycler (Applied Biosystems), and incubated for 3 minutes at room temperature. The cells were flash-frozen twice and spun at 14 x g for 20 minutes at 4 °C. Supernatants were collected and loaded onto a 10 % polyacrylamide gel at a volume of 16 μL, with 4 μL 4X SDS loading dye. Subsequently, Western blotting was run following the procedure reported herein.
Drug Affinity Responsive Target Stability
The DARTS assay was performed following previously established procedure.[53] U87MG cells were grown to approximately 80–85% confluence, washed with ice-cold DPBS, and lysed with lysis buffer (150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 50 mM Tris, pH 8.0). Cells were collected and lysis was allowed to occur for 10 minutes on ice. Cells were spun at 18,000 x g for 20 minutes at 4 °C to collect the supernatant. Protein concentration was determined via BCA assay. 100 μM PACMA31 or 35G8 or 1 μL DMSO were incubated with aliquots of cell lysate at 5 mg/ml for 30 minutes with shaking at room temperature. Pronase (Sigma Aldrich, St. Louis, MO) was added to 20 μL aliquots of cell lysates at 0, 1:1000 (0.005 μg/μL), 1:500 (0.01 μg/μL), or 1:250 (0.02 μg/μL) for 30 minutes at room temperature. Digestion was stopped by adding 1X protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO) and incubating the reactions on ice for 10 minutes. SDS-PAGE loading buffer (6 μL of 5X) was added to the samples, and samples were heated for 10 minutes at 70 °C. Samples were spun down briefly and 20 μg of protein was loaded into acrylamide gels (10%) for Western blot analysis.
Docking Study
Docking studies were performed using GOLD, version 4.0 (Cambridge Crystallographic Data Centre).[54] The crystal structure of human PDI in its reduced state (PDB ID: 4EKZ)[55] was used for all calculations. 3D structures of the ligands were created and energy minimized using MMFF94 forcefield implemented in OMEGA 2.5.1.4 (OpenEye Scientific Software, http://www.openeye.com), a systematic, knowledge-based conformer generator.[21] The docking site was defined for all residues within 10 Å around the center, defined as the sulfur atoms of the catalytic residues Cys53 and Cys397 and nitrogen NE2 of His256 for the hydrophobic site. Docking studies were performed using the standard default settings with 100 GA (genetic algorithm) runs on each molecule. Gold Score was used to quantify the interactions between molecules and PDI and the annealing parameters with cutoff values of 3.0 Å for hydrogen bonds and 4.0 Å for van der Waals interactions were used as default. When the top three solutions attained rmsd values within 1.5 Å, docking was terminated. During the docking process, a maximum of 10 conformers was considered for each compound.
Bru-seq Analysis
Bru-seq experiments[12] and analysis were performed as previously reported. Briefly, U87MG cells were placed in dishes on Day 1. Cells were changed to fresh media on Day 5 and treated with DMSO or 35G8 at 1 μM for 4 hours. Bromouridine was added into the media to a final concentration of 2 mM to label newly synthesized nascent RNA in the last 30 minutes of treatment. Cells were then collected in TRIzol (Thermo Fisher Scientific, Waltham, MA) and total RNA was isolated. The bromouridine-containing RNA population was further isolated and sequenced. Sequencing reads were mapped to a reference genome.
Bioinformatic Analysis
Bru-seq data of 35G8 treatment was filtered using the cut off value of gene size > 300 bp and mean (RPKM) > 0.5 and a total of 7,770 genes were ranked based on the fold change values versus control (DMSO). DAVID functional annotation analysis[56] was performed on 460 upregulated and 220 downregulated genes with fold change ≥ 2 and ≤ −2. IPA of Bru-seq data was performed using the IPA web-based application (Ingenuity Systems, Inc.) on the list of 680 up- and downregulated genes (over two-fold change) (Table S4 and S5). Top canonical pathways were ranked based on the P-value of significance and maximum number of genes in the pathway (Figure 4A). Gene Set Enrichment Analysis (GSEA) of Bru-seq data was done on a pre-ranked gene list of 7,770 genes of 35G8 treatment based on the Kolmogorov–Smirnov statistic.[57] Table S3 and S4 show the top 20 gene sets for up- and downregulated genes of the Bru-seq dataset of 35G8 treatment, respectively. The snapshots of the enrichment profiles of these 20 gene sets are provided in Figure S4 and S5.
ROS Detection Assay3
U87MG cells were detached with 0.05% trypsin-EDTA (Thermo Fisher Scientific, Waltham, MA), neutralized, centrifuged and resuspended in cell culture media. Suspension was treated with 20 μM cell-permeable H2DCFDA (Thermo Fisher Scientific, Waltham, MA) for 30 minutes at 37 °C. Cells were centrifuged again and washed with cell culture media to remove excess probe. After washing, cells were placed in a black-wall 384-well plate at 20,000 cells/well, incubated for 30 minutes and treated with compounds at designated conditions. Fluorescent signals were read at 493 nm/523 nm for ROS detection at designated time points (4, 6, and 24 hours).
Western Blot
Primary antibodies for GRP78, HMOX1, CHAC1, CHOP, LC3B, GSTO1, and SLC7A11 and secondary antibodies were purchased from Cell Signaling (Danvers, MA). Primary antibody for P4HB was purchased from Protein Tech ((Rosemont, IL). U87MG cells were treated with DMSO or 2 μM 35G8 for 1, 3, 6, 12, or 24 hours. Cells were harvested with a lysis buffer (25 mM tris(hydroxymethyl)aminomethane, 150 mM NaCl, 17 mM Triton X-100, 3.5 mM SDS, pH 7.4), lysed via sonication, and spun in a centrifuge at 13,500 × g at 4 °C for 10 minutes. Supernatant was collected and protein concentration determined with the BCA assay (Thermo Fisher Scientific, Waltham, MO). Samples were prepared with 50 μg protein and loaded onto 10 % (or 12 % for LC3B and DDIT3) acrylamide (Bio-Rad, Hercules, CA) gels. Protein from gels was electro-transferred to methanol-activated immobilon-FL PVDF membranes (EMD Millipore, La Jolla, CA). Membranes were blocked for 1 hour with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE). Membranes were probed for proteins using primary antibodies (P4HB, 1:1000; GRP78, 1:1000; GSTO1, 1:1000; HMOX1, 1:1000; CHAC1, 1:1000; CHOP, 1:500; LC3B, 1:2000; SLC7A11, 1:2000) overnight at 4 °C. Membranes were incubated with secondary antibodies (anti-rabbit, 1:7500, or anti-mouse, 1:7500) and fluorescence was imaged by Odyssey Imaging Systems (LI-COR Biosciences).
Redox Cycling Assay
The redox cycling assay was adapted from a previously published experiment.[58] In duplicate in a 384-well plate, 20 μL of HBSS buffer, 100 U of catalase, 100 μM H2O2, 100 μM H2O2 + 100 U catalase, 0.5% DMSO, 500 μM DTT, 10 μM 35G8, 10 μM 35G8 + 500 μM DTT, or 10 μM 35G8 + 500 μM DTT + 100 U of catalase was added to a reaction mixture with HBSS to a final volume of 60 μL. The reaction was incubated at room temperature for 30 minutes, and phenol red-HRP detection reagent was added to a final concentration of 100 μg/ml phenol red and 60 μg/ml HRP in each well. The reaction was incubated for an hour at room temperature. Sodium hydroxide (10 μL, 1 N) was added to wells and absorbance was measured at 610 nm.
Statistical Analysis
IC50 values were calculated using GraphPad Prism 7 software (GraphPad Software, Inc.). The error bars indicate mean ± standard deviation.
Supplementary Material
Acknowledgements
The authors would like to thank Michelle Paulsen for running the Bru-seq experiments. The expression vector of recombinant human PDI was a generous gift from Dr. Lloyd W. Ruddock (University of Oulu, Oulu, Finland). This work was supported in part by a grant from NIH (CA193690).
References:
- [1].Ostrom QT, Gittleman H, Liao P, Rouse C, Chen YW, Dowling J, Wolinsky YL, Kruchko C, Barnholtz-Sloan J, Neuro-Oncology 2014, 16, 1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, R. European Organisation for, T. Treatment of Cancer Brain, G. Radiotherapy, G. National Cancer Institute of Canada Clinical Trials, N. Engl. J. Med 2005, 352(10), 987–996. [DOI] [PubMed] [Google Scholar]
- [3].Xu S, Sankar S, Neamati N, Drug Discov. Today 2014, 19(3), 222–240. [DOI] [PubMed] [Google Scholar]
- [4].a) Ellgaard L, Ruddock LW, EMBO Rep 2005, 6(1), 28–32; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shergalis A, Neamati N, in Encyclopedia of Signaling Molecules (Ed.: Choi S), Springer; New York, 2016, pp. 1–12. [Google Scholar]
- [5].Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N, Proc. Natl. Acad. Sci. U S A 2012, 109(40), 16348–16353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Goplen D, Wang J, Enger P, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R, Cancer Res 2006, 66(20), 9895–9902. [DOI] [PubMed] [Google Scholar]
- [7].Lovat PE, Corazzari M, Armstrong JL, Martin S, Pagliarini V, Hill D, Brown AM, Piacentini M, Birch-Machin MA, Redfern CP, Cancer Res 2008, 68(13), 5363–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eirich J, Braig S, Schyschka L, Servatius P, Hoffmann J, Hecht S, Fulda S, Zahler S, Antes I, Kazmaier U, Sieber SA, Vollmar AM, Angew. Chem. Int. Ed. Engl 2014, 53(47), 12960–12965. [DOI] [PubMed] [Google Scholar]
- [9].Khodier C, VerPlank L, Nag PP, Pu J, Wurst J, Pilyugina T, Dockendorff C, Galinski CN, Scalise AA, Passam F, van Hessem L, Dilks J, Kennedy DR, Flaumenhaft R, Palmer MAJ, Dandapani S, Munoz B, Schrieber SL, in Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD: ), 2010. [Google Scholar]
- [10].Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG, Lo DC, Stockwell BR, Proc. Natl. Acad. Sci. U S A 2015, 112(17), E2245–E2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, Parker Y, Lindner DJ, Zhong F, Distelhorst CW, Smith MR, Cotta C, Xu Y, Chilakala S, Kuang RR, Tall S, Reu FJ, Cancer Res 2016, 76(11), 3340–3350. [DOI] [PubMed] [Google Scholar]
- [12].Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M, Methods 2014, 67(1), 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR, Cell 2012, 149(5), 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Khan MM, Simizu S, Lai NS, Kawatani M, Shimizu T, Osada H, ACS Chem. Biol 2011, 6(3), 245–251. [DOI] [PubMed] [Google Scholar]
- [15].Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR, J. Biomol. Screen 2001, 6(6), 429–440. [DOI] [PubMed] [Google Scholar]
- [16].Cimmperman P, Baranauskienė L, Jachimovičiūtė S, Jachno J, Torresan J, Michailovienė V, Matulienė J, Sereikaitė J, Bumelis V, Matulis D, Biophys. J 2008, 95(7), 3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Primm TP, Gilbert HF, J. Biol. Chem 2001, 276(1), 281–286. [DOI] [PubMed] [Google Scholar]
- [18].Daves GD, Cheng CC, Robins RK, J. Am. Chem. Soc 1961, 83(18), 3904–3905; [Google Scholar]; Nagamatsu T, Yamasaki H, Hirota T, Yamato M, Kido Y, Shibata M, Yoneda F, Chem. Pharm. Bull 1993, 41(2), 362–368. [DOI] [PubMed] [Google Scholar]
- [19].Degterev A, Huang ZH, Boyce M, Li YQ, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan JY, Nat. Chem. Biol 2005, 1(2), 112–119. [DOI] [PubMed] [Google Scholar]
- [20].Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J, Nat. Chem. Biol 2008, 4(5), 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT, J. Chem. Inf. Model 2010, 50(4), 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, Chang CW, Tarrier B, Washburn JG, Lyons R, Robinson DR, Kumar-Sinha C, Wilson TE, Ljungman M, Proc. Natl. Acad. Sci. U S A 2013, 110(6), 2240–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y, Biochem. Biophys. Res. Commun 1997, 236(2), 313–322. [DOI] [PubMed] [Google Scholar]
- [24].Oh-hashi K, Nomura Y, Shimada K, Koga H, Hirata Y, Kiuchi K, Mol. Cell. Biochem 2013, 380(1–2), 97–106. [DOI] [PubMed] [Google Scholar]
- [25].Shang YY, Zhong M, Zhang LP, Guo ZX, Wang ZH, Zhang Y, Deng JT, Zhang W, Clin. Exp. Pharmacol. Physiol 2010, 37(1), 51–55; [DOI] [PubMed] [Google Scholar]; Siu F, Bain PJ, LeBlanc-Chaffin R, Chen H, Kilberg MS, J. Biol. Chem 2002, 277(27), 24120–24127. [DOI] [PubMed] [Google Scholar]
- [26].Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vazquez P, Blazquez C, Torres S, Garcia S, Nowak J, Fimia GM, Piacentini M, Cecconi F, Pandolfi PP, Gonzalez-Feria L, Iovanna JL, Guzman M, Boya P, Velasco G, J. Clin. Invest 2009, 119(5), 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yamamoto A, Cremona ML, Rothman JE, J. Cell Biol 2006, 172(5), 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu CF, Wang L, Lv BF, Lu Y, Zeng L, Chen YY, Ma DL, Shi TP, Biochem. Biophys. Res. Commun 2008, 369(2), 622–629. [DOI] [PubMed] [Google Scholar]
- [29].Kwon M-Y, Park E, Lee S-J, Chung SW, Oncotarget 2015, 6(27), 24393–24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR, eLife 2014, 3, e02523-e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gutteridge JMC, Richmond R, Halliwell B, Biochem. J 1979, 184(2), 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA, J. Drug Target 1998, 6(2), 151–165. [DOI] [PubMed] [Google Scholar]
- [33].Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M, Genes Dev 1999, 13(1), 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wondrak GT, Antioxid. Redox Signal 2009, 11(12), 3013–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Bridges RJ, Natale NR, Patel SA, Br. J. Pharmacol 2012, 165, 20–34; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koritzinsky M, Wouters BG, Semin. Radiat. Oncol 2013, 23(4), 252–261. [DOI] [PubMed] [Google Scholar]
- [36].Johnson G, Beaver L, Williams DE, Ho E, Dashwood RH, in Thirteenth Annual AACR International Conference on Frontiers in Cancer Prevention Research, Vol. 8, Can. Prev. Res, New Orleans, LA, 2015. [Google Scholar]
- [37].a) Zhu JH, Wang HD, Fan YW, Lin YX, Zhang L, Ji XJ, Zhou ML, Oncol. Rep 2014, 32(2), 443–450; [DOI] [PubMed] [Google Scholar]; b) Zhu JH, Wang HD, Sun Q, Ji XJ, Zhu L, Cong ZX, Zhou Y, Liu HD, Zhou ML, BMC Cancer 2013, 13, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Levine B, Nature 2007, 446(7137), 745–747. [DOI] [PubMed] [Google Scholar]
- [39].B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A, Nucleic Acids Res 2013, 41(16), 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Salazar M, Lorente M, Orea-Soufi A, Dávila D, Erazo T, Lizcano J, Carracedo A, Kiss-Toth E, Velasco G, Biochem. Soc. Trans 2015, 43(5), 1122–1126. [DOI] [PubMed] [Google Scholar]
- [41].García-Navas R, Munder M, Mollinedo F, Autophagy 2012, 8(11), 1557–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu L, Spinas GA, Niessen M, Horm. Metab. Res 2010, 42(9), 643–651. [DOI] [PubMed] [Google Scholar]
- [43].Ogata FT, Batista WL, Sartori A, Gesteira TF, Masutani H, Arai RJ, Yodoi J, Stern A, Monteiro HP, PLoS One 2013, 8(12), e84588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee S, Kim SM, Lee RT, Antioxid. Redox Signal 2013, 18(10), 1165–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, Vanbelle S, Califice S, Bredel M, Bours V, BMC Cancer 2009, 9(19), 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H, J. Neurosci 2005, 25(31), 7101–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gout PW, Buckley AR, Simms CR, Bruchovsky N, Leukemia 2001, 15(10), 1633–1640. [DOI] [PubMed] [Google Scholar]
- [48].Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, Leiss L, Heggdal JI, Pedersen PH, Wang J, Enger PO, Oncogene 2015, 34(49), 5951–5959. [DOI] [PubMed] [Google Scholar]
- [49].Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y, Oncol. Rep 2015, 33(3), 1465–1474. [DOI] [PubMed] [Google Scholar]
- [50].Dixon SJ, Patel D, Welsch M, Skouta R, Lee E, Hayano M, Thomas AG, Gleason C, Tatonetti N, Slusher BS, Stockwell BR, eLife 2014, 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB, Cancer Res 1987, 47(4), 936–942. [PubMed] [Google Scholar]
- [52].Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Molina DM, Nat. Protoc 2014, 9(9), 2100–2122. [DOI] [PubMed] [Google Scholar]
- [53].Lomenick B, Olsen RW, Huang J, ACS Chem. Biol 2011, 6(1), 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jones G, Willett P, Glen RC, Leach AR, Taylor R, J. Mol. Biol 1997, 267(3), 727–748. [DOI] [PubMed] [Google Scholar]
- [55].Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, Wang CC, Antioxid. Redox Signal 2013, 19(1), 36–45. [DOI] [PubMed] [Google Scholar]
- [56].a) Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA, Genome Biol 2003, 4(5), 3; [PubMed] [Google Scholar]; b) Huang da W S. B. a. L. R., Nat. Protoc 2009, 4(1), 44–57. [DOI] [PubMed] [Google Scholar]
- [57].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Proc. Natl. Acad. Sci. U S A 2005, 102(43),15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Johnston PA, Soares KM, Shinde SN, Foster CA, Shun TY, Takyi HK, Wipf P, Lazo JS, Assay Drug Dev. Technol 2008, 6(4), 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.