

Abstract
The use of animal models, particularly genetically modified mice, continues to play a critical role in studying the relationship between bile acid metabolism and human liver disease. Over the past 20 years, these studies have been instrumental in elucidating the major pathways responsible for bile acid biosynthesis and enterohepatic cycling, and the molecular mechanisms regulating those pathways. This work also revealed bile acid differences between species, particularly in the composition, physicochemical properties, and signaling potential of the bile acid pool. These species differences may limit the ability to translate findings regarding bile acid-related disease processes from mice to humans. In this review, we focus primarily on mouse models and also briefly discuss dietary or surgical models commonly used to study the basic mechanisms underlying bile acid metabolism. Important phenotypic species differences in bile acid metabolism between mice and humans are highlighted.
Keywords: Liver, intestine, enterohepatic circulation, mouse model, enzyme, transporter
Introduction
Interest in bile acids can be traced back almost three millennia to the widespread use of animal biles in traditional Chinese medicine [1]. By the early 19th century, bile acids as a biliary constituent became the subject of more detailed investigation by chemists and physiologists [2]. Since that time, much has been learned regarding the structure, physicochemical properties, metabolism, and physiological functions of these essential detergents and signaling molecules [3]. Moreover, the study of bile acids and their relationship to the pathophysiology and treatment of human disease continues to be relevant today with the advent of new bile acid-based therapies [4, 5].The use of animal models has played a seminal role in promoting our understanding of bile acid synthesis, metabolism, and role in human disease [6–8]. Bile acids are absent from invertebrates [9], but present in all vertebrate species [10]. Bile acid metabolism has been studied in many vertebrate animal models including lamprey, skate, zebrafish, rat, mouse, hamster, rabbit, prairie dog, and monkey. Of the small animal models, bile acid biosynthesis and metabolism in hamsters appears to be most similar to that of humans [11–13], however over the past 3 decades, mice have become the preferred model for study [14, 15]. This is due to their characteristics such as small size, short gestation period and life span, which facilitated large-scale laboratory breeding and housing, the availability of inbred and specialized strains as genome sequencing technology was rapidly expanding [15], and the similarities between the mouse and human genomes. Moreover, the development of expedient technologies for mouse phenotyping and genome manipulation, and the emergence of a substantial infrastructure of mouse-related research resources, including biorepositories and databases from collaborative consortium and commercial sources, spurred the use of mice as a model organism for the study of human disease [16, 17]. Although invaluable as a research model, mice and humans diverged about 80 million years ago and have adapted to different environments. As such, there are myriad species differences that may impede our ability to translate specific findings with regard to basic biological mechanisms, such as bile acid metabolism (this review) or disease processes from mice to humans [18–20].
In the present review, we focus on the laboratory mouse as a model to study bile acid biosynthesis, enterohepatic cycling, and metabolism. For each section, we introduce the major concepts and highlight similarities and important species differences between mice and humans with regard to bile acid homeostasis. Special emphasis is directed toward knockout mouse models of human genetic disorders impacting bile acid biosynthesis and transport. The regulatory factors that control bile acid homeostasis, such as the nuclear receptor FXR and Fibroblast Growth Factor (FGF)15/19, the interactions of bile acids with the microbiome, and the role of bile acids in the pathogenesis of liver disease are only briefly mentioned and the reader is referred to recent comprehensive reviews on those topics [20–25] and other articles in this series (”Animal Models in Liver Disease”).
2. Mouse Models to Study Bile Acid Biosynthesis and Metabolism
The major mouse genetic models available to study bile acid biosynthesis and metabolism are described in Table 1.
Table 1.
Genetic Mouse Models Used to Study Bile Acid Biosynthesis and Metabolism
| Model Enzyme | Description (Bile acid-related phenotype) |
|---|---|
|
Cyp7a1 KO Cyp7a1 KO |
Constitutive KO (mixed B6/129; UT-Southwestern colony); ↑postnatal mortality; ↓fat & fat-soluble vitamin absorption; ↓BA pool size; ↓fecal BAs, ↓intestinal cholesterol absorption; milder phenotype reported for independent colony [43, 117, 118] Cyp7a1 KO (JAX 002751 B6;129S7-Cyp7a1tm1/Rus/J) backcrossed into C57Bl/6J background; normal survival & growth; ↓BA pool size w/ ↓proportion TCA, ↑TMCA; ↑liver Cyp8b1 & Cyp7b1 expression [36] |
| Cyp7a1 Tg | Tg mouse expressing rat Cyp7a1 driven by apoE hepatic control region (C57Bl/6); ↑BA pool size w/ ↓proportion TCA, ↑TMCA, TCDCA, TUDCA; ↓Cyp8b1 expression; ↑fecal BAs; ↑fecal cholesterol [119] |
| Humanized CYP7A1 Tg | Tg mouse with BAC encompassing human CYP7A1 gene (1 copy) crossed into Cyp7a1 KO background (C57Bl/6J); ↔ BA pool size w/ ↑proportion TCA; human CYP7A1 Tg expression is not induced by cholesterol-feeding [67] |
|
Cyp8b1 KO Cyp8b1 KO9 Cyp8b1 KO |
Constitutive KO replacing Cyp8b1 coding sequence w/ lacZ (mixed B6/129; JAX 018771 B6;129S7-Cyp8b1tm1/Rus/J; ↑BA pool size w/ ↑proportion TMCA, TCDCA, TUDCA; ↑Cyp7a1 expression & activity; ↑fecal BAs, ↑fecal neutral sterols; ↓intestinal cholesterol absorption [121] Constitutive KO replacing Cyp8b1 sequence w/ lacZ (Cyp8b1tm1(KOMP)Vlcg); ↑serum BAs; ↑Cyp7a1 expression; ↓intestinal fat absorption [66] Taconic (C57Bl/6); constitutive KO model #11784; floxed conditional model #12015); ↔BA pool size w/ ↑proportion TMCA, TCDCA; ↓intestinal fat absorption [65] |
|
Cyp27a1 KO Cyp27a1 KO |
Constitutive KO (mixed B6/129; JAX 009106 B6.129-Cyp27a1tm1Elt/ J); ↓BA pool size w/ ↑proportion of side-chain hydroxylated BAs; ↑liver Cyp7a1, Cyp3a11; ↓fecal BAs; ↑fecal neutral sterols; ↓intestinal cholesterol absorption [37, 40, 130] Cyp27a1 KO (JAX 009106 B6.129-Cyp27a1tm1Elt / J) backcrossed into C57Bl/6J background; hepatomegaly; ↑liver Cyp7a1; ↑fecal neutral sterols [281] |
| CYP27A1 Tg | Tg mouse expressing human CYP27A1 under control of actin promoter; no gross morphological changes; ↑serum & liver 27-hydroxycholesterol levels; ↔ biliary BAs [282] |
|
Cyp7b1 KO Cyp7b1 KO |
Constitutive KO (mixed B6/129Sv; JAX 016108 B6.129-Cyp7b1tm1Rus / J); also backcrossed into C57Bl6 background; sex steroid metabolism alterations; ↔ BA pool size or composition; ↑serum & liver 25- and 27-hydroxycholesterol levels [39] Constitutive KO inserting IRES-lacZ; backcrossed into C57Bl/6 background; alterations in neurosteroid and sex steroid metabolism [283, 284] |
|
Cyp2c KO Humanized CYP2C9 Tg |
Constitutive Cyp2c gene cluster KO (C57Bl/6N; Taconic model #9177); blocks synthesis of 6-hydroxylated primary BAs; ↑proportion TCDCA; ↑fecal LCA [47] Targeted replacement of Cyp2c gene cluster with cassette expressing human CYP2C9 under control of albumin promoter; (C57Bl/6N; Taconic model #11746); blocks synthesis of 6-hydroxylated primary BAs; ↑proportion TCDCA; ↑fecal LCA [47] |
2.1. Overview of Bile Acid Biosynthesis and Metabolism
Bile acids are synthesized from cholesterol in pericentral hepatocytes, and this represents the major pathway for cholesterol catabolism [26]. In humans, approximately 0.5 g of cholesterol is converted to bile acids per day (~7 mg per day per kg body weight) to balance fecal loss, and this accounts for almost half of the cholesterol eliminated from the body per day. In mice, the amount of cholesterol converted to bile acids (approximately 1.25 mg of cholesterol per day; ~50 mg per day per kg body weight) is quantitatively greater on a body weight basis, reflecting their higher cholesterol biosynthesis rate and significant daily cholesterol intake (expressed per kg body weight) [27, 28]. However in both species, cholesterol catabolism to bile acids accounts for a similar overall fraction (~45%) of daily cholesterol elimination [28]. The bile acids that are newly synthesized from cholesterol in the hepatocytes are termed primary bile acids. These include cholic acid (CA) and chenodeoxycholic acid (CDCA) in humans, and CA, muricholic acid (MCA), CDCA, and ursodeoxycholic acid (UDCA) in mice (Figure 1). In contrast, the bile acids generated by subsequent modifications carried out by the gut microbiota or host are termed secondary bile acids [29, 30]. Under normal physiological conditions, the primary bile acids are synthesized via two major pathways, the “classical” (neutral) and “alternative” (acidic) pathways [31], with variable contributions by the Yamasaki and 25-hydroxylation pathways in the developing fetus or under pathophysiological conditions [32]. Of the major pathways, the classical (Cholesterol 7α-hydroxylase; CYP7A1) pathway is quantitatively more important and favors the synthesis of CA in humans and mice [33]. The alternative (Oxysterol 7α-hydroxylase; CYP7B1) pathway is also quantitatively significant and favors the synthesis of CDCA in humans and 6-hydroxylated bile acids such as MCA in mice [31, 34]. Since the biosynthetic intermediates in these pathways are often substrates for more than one enzyme and little is known regarding the intracellular trafficking of these compounds in hepatocytes, the mechanisms responsible for metabolic channeling of cholesterol towards CA versus CDCA or MCA remain unclear. Moreover, the specificity is not absolute, with the classical and alternative pathways capable of yielding some CDCA and CA, respectively [35–37].
Figure 1. Bile acids present in humans and mice.
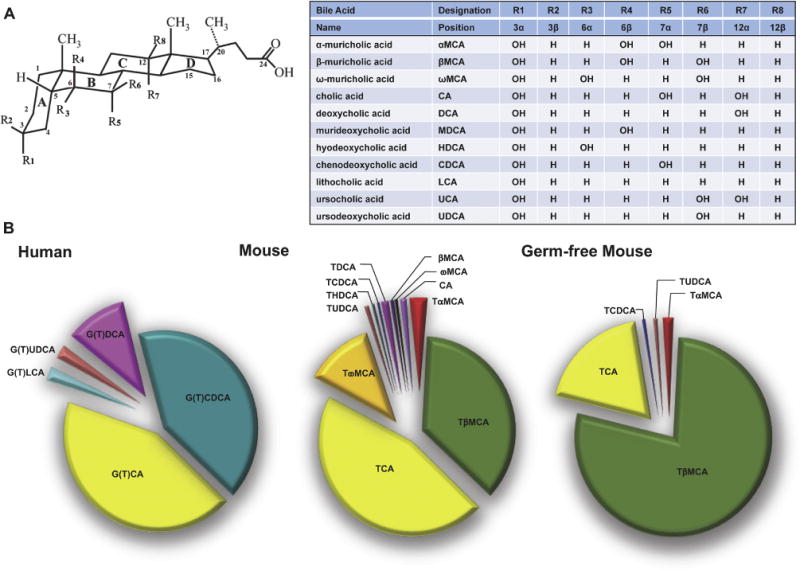
(A) Structure and sites of hydroxylation on the steroid nucleus for bile acid species found in humans and mice. Hydroxyl groups that are oriented in the α-orientation are located below the steroid nucleus and are axial to the plane of the steroid nucleus. Hydroxyl groups that are in the β-orientation are located above the steroid nucleus and are equatorial to the plane of the steroid nucleus. (B) Relative biliary bile acid composition in humans and in conventionalized and germ-free mice (modified from Wahlström et al; [48]).
The overall biosynthetic pathway is complex and involves at least seventeen different enzymes, one or more transporters, and multiple cellular compartments, which includes the cytosol, endoplasmic reticulum, mitochondria, and peroxisomes [38]. In the classical pathway, the microsomal cytochrome P450 (CYP) enzyme cholesterol 7α-hydroxylase (gene symbol: CYP7A1) is the rate-limiting enzyme for bile acid synthesis, whereas the microsomal CYP sterol 12α-hydroxylase (gene symbol, CYP8B1) controls the amount of CA synthesized. In the alternative pathway, the first step is catalyzed by sterol hydroxylases present in liver and extra-hepatic tissues, with the contribution of sterol 27-hydroxylase (gene symbol: CYP27A1) being quantitatively most important [31, 33]. This reaction is followed by an oxysterol 7α-hydroxylation, which is mediated primarily by CYP7B1 in liver, with varying contributions by CYP39A1 [24, 31, 38–41]. The classical and alternative biosynthetic pathways then converge at an isomerization step catalyzed by the enzyme 3β-hydroxy-Δ5-C27-steroid oxidoreductase (HSD3B7). In the next step, 7α-hydroxy-4-cholestene-3-one directly undergoes steroid ring isomerization and saturation or is first converted by CYP8B1 to 7α, 12α-hydroxy-4-cholestene-3-one before proceeding to those steps. The subsequent steroid ring modifications are carried out by Δ4-3-oxosteroid 5β-reductase (aldo-keto reductase 1D1; AKR1D1) and 3α-hydroxysteroid dehydrogenase (aldo-keto reductase 1C4; AKR1C4). Following the steroid ring modifications, CYP27A1 acts on the product of the neutral (CYP7A1) pathway, 5β-cholestan-3α,7α,12α-triol to create a carboxyl group at the C-27 position. The di- and trihydroxycoprostanoic C27 bile acid derivatives are then converted to their corresponding CoA esters by very-long-chain acyl-CoA synthetase (VLCS; SLC27A2; FATP2) and transported by ABCD3 into peroxisomes to undergo side chain β-oxidation and shortening. These intermediates are converted to their respective (25S)-isomers by alpha-methylacyl-CoA racemase (AMACR) or enoyl-CoA hydratase/3-hydroxy CoA dehydrogenase (EHHADH; L-Bifunctional protein; peroxisomal multifunctional enzyme-1), and then oxidized by acyl-coA oxidase 2 (ACOX2). D-bifunctional protein (HSD17B4) then catalyzes the subsequent hydration and dehydrogenation reactions prior to side chain cleavage by thiolase 2/sterol carrier protein 2. Note that there are two enzymes capable of synthesizing bile acid CoA-thioesters [42]. During bile acid biosynthesis, the bile acid-CoA thioester intermediate is synthesized by VLCS (SLC27A2; FATP2). However, for previously synthesized unconjugated bile acids carried back to the liver in the enterohepatic circulation, a distinct bile acid CoA ligase (Bile Acid-CoA Synthetase, BACS; SLC27A5; FATP5) is used. Regardless of the source of the bile acid-CoA thioester, bile acids are then conjugated to taurine or glycine by the bile acid-CoA amino acid N-acyltransferase (BAAT). This last step is remarkably efficient, and more than 98 percent of the bile acids secreted into bile are in taurine or glycine-conjugated form.
Our understanding of rare inherited bile acid biosynthetic defects in patients has been advanced in part through the use of knockout mouse models [43–45]. Conversely, characterization of the clinical presentation of the patients and the phenotype of the corresponding knockout mouse models has yielded insights to human-mouse species differences in bile acid biosynthesis and metabolism. Disease-associated mutations in humans have been described for eleven enzymes and one transporter involved in bile acid biosynthesis, and include CYP7A1, CYP27A1, CYP7B1, HSD3B7, AKR1D1, AMACR, ACOX2, HSD17B4, SCP2, SLC27A5, BAAT, and ABCD3 [32]. The phenotype of knockout mouse models with mutations in many of those genes is described below.
2.2. Bile Acid Biosynthesis and Metabolism: Species Differences Between Humans and Mice
Humans and mice have substantially different bile acid pool compositions (Figure 1), with mice possessing a more hydrophilic complement of bile acid species. These human-mouse species differences are summarized in Figure 2 and reflect: i) synthesis of UDCA as a primary bile acid [46]. ii) Hydroxylation at the 6-position of the bile acid steroid nucleus to generate MCA [47]. Notably this host differences may be accompanied by changes in the gut microbiome bile acid metabolism, which affects the diversity of secondary bile acids [48]. iii) The amino acid specificity for bile acid N-acyl-amidation (conjugation) [10]. iv) Hepatic 7α-rehydroxylation of conjugated deoxycholic acid (DCA) [49], and v) species differences in bile acid detoxification [50, 51]. These species differences are discussed individually below.
Figure 2. Bile acid synthesis and metabolism in humans and mice.
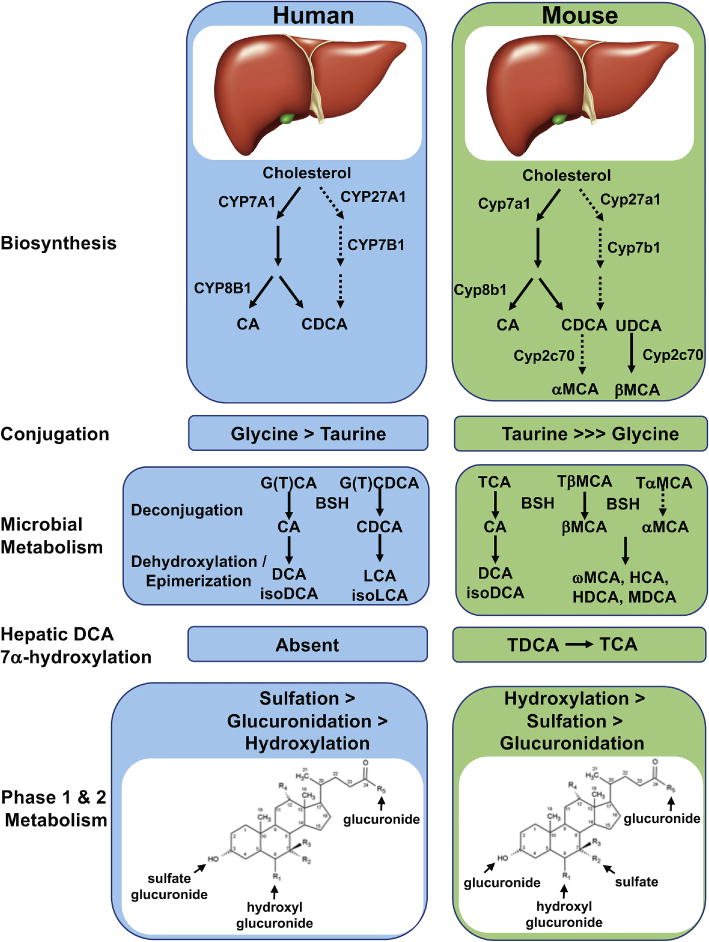
Major pathways for bile acid synthesis and metabolism in humans and mice. (A) The classical pathway for bile acid synthesis begins with cholesterol 7α-hydroxylase (CYP7A1) and intermediates synthesized via this pathway are substrates for the sterol 12α-hydroxylase (CYP8B1), which generates CA. The alternative pathway begins with sterol 27-hydroxylase (CYP27A1) and generates predominantly CDCA. In mice, CDCA and UDCA undergo 6β-hydroxylation by the cytochrome P450, Cyp2c70 to generate αMCA and βMCA. (B) In humans, bile acids in the liver are conjugated (n-acyl-amidated) with glycine (G) or taurine (T), whereas mice use almost exclusively taurine. (C) Primary bile acids are biotransformed by the gut microbiota. These reactions include deconjugation, which is catalyzed by bile salt hydrolases (BSH), epimerization to change the orientation of the hydroxyl groups on the steroid nucleus of the bile acids, and 7-dehydroxylation. (D) After returning to the liver, conjugated DCA can be rehydroxylated at the C-7 position to regenerate conjugated CA. This reaction occurs in mice and other species but is apparently absent in humans. (E) Bile acids undergo Phase 1 and Phase 2 metabolism, including additional hydroxylation, sulfation (sulfonation), and glucuronidation. In humans, sulfation of bile acids such as LCA is a primary pathway for their detoxification, whereas in mice hydroxylation plays a dominant role. The locations of these additional modifications to the bile acid structure are indicated.
2.2.1. Synthesis of UDCA
UDCA is a secondary bile acid in humans and only a very minor component of the bile acid pool. In humans, UDCA is generated from CDCA by the gut microbiota via a 2-step process involving oxidation by a 7α-hydroxysteroid dehydrogenase (7α-HSDH) and stereospecific reduction of the 7-keto functionality by a 7β-HSDH [52] or by hepatic reduction of the bacterial 7α-HSD-derived 7-oxolithocholic by the host liver enzyme 11β-hydroxysteroid dehydrogenase 1 [53]. In mice, studies use germ-free mice demonstrated that UDCA is a primary bile acid synthesized in the absence of the gut microbiota (Figure 1). However, even in the mouse, UDCA is a very minor component, constituting about one percent of the total bile acid pool [46, 54]. Indeed, the significance of this finding may not the abundance of UDCA, but rather that UDCA is an apparent biosynthetic precursor to β-muricholate (βMCA), a 6-hydroxylated bile acid and major bile acid species present in mice (see following section).
2.2.2. Biosynthesis of 6-Hydroxylated Bile Acids
With regard to bile acid metabolism, the most important mouse-human difference is the abundant synthesis of 6-hydroxylated bile acids in mice. In mice, 6-hydroxylated bile acids constitute half or more of the bile acid pool (Figure 1). In contrast, 6-hydroxylated bile acids are typically present at very low concentrations in humans under normal physiological conditions, but can be detected, in fetal and early neonatal development or under pathophysiological conditions such as cholestatic liver disease [55–57]. Hydroxylation at the C-6 position of the steroid nucleus significantly alters the bile acid’s physicochemical and detergent properties, with 6-hydroxylated bile acids being more water-soluble and relatively poor detergents [58–60]. Moreover, 6-hydroxylation of bile acids dramatically alters their signaling properties [46, 61, 62], making them poor activators or even antagonists of FXR. This human-mouse species difference is an important consideration when trying to extrapolate findings in mouse models to bile acid-related diseases in humans [63]. Pathways or interventions that shift the bile acid composition in mouse models toward or away from 6-hydroxylated bile acids have dramatic effects on FXR signaling, and on intestinal cholesterol and lipid absorption, often with profound metabolic effects [61, 64–67]. Since humans typically synthesize very little 6-hydroxylated bile acids, it is unclear (and seemingly unlikely) whether these mechanisms are engaged. On the other hand, the findings raise the interesting question of whether administration of pharmacological doses of exogenous 6-hydroxylated or polyhydroxylated bile acids to humans or stimulating their production may have phenotypic actions comparable to those observed in mouse models.
Although it was known that the synthesis of 6-hydroxylated bile acids involved a cytochrome P450 [59], the identity of the specific enzyme(s) (CYPs) remained unclear. Indirect correlative evidence suggested that Cyp3a11 functioned as the bile acid 6-hydroxylase responsible for the synthesis of the muricholates as a primary bile acid in mice, rats and other species. However that hypothesis turned out to be incorrect [68]. Very recent analysis of knockout mouse models lacking all members of the Cyp1a, Cyp2c, Cyp2d, or Cyp3a families found that only the murine Cyp2c enzymes are required [47, 68]. The murine liver-specific isoform, Cyp2c70 was subsequently shown capable of efficiently 6-hydroxylating CDCA to α-muricholic acid (αMCA) and UDCA to βMCA, whereas the major human CYP2C enzyme, CYP2C9, was unable to hydroxylate bile acids [47]. The identification of Cyp2c enzymes as the major source of 6-hydroxylated primary bile acids in mice was a landmark observation and as discussed below, opens the door to development of new mouse models with “humanized” bile acid pool compositions for the study of liver disease [63]. However, it should be noted that this finding still leaves a number of questions to be answered regarding bile acid biosynthesis and metabolism in mice and humans. For example, Cyp2c70 converts CDCA to αMCA and UDCA to βMCA, with an apparent substrate preference for UDCA [47]. Since conjugated and unconjugated forms of βMCA are the major 6-hydroxylated bile acid species in liver and gallbladder bile of conventional as well as germ-free mice [48], these results suggest that hepatic bile acid biosynthesis in mice preferentially yields UDCA, as an intermediate, which is then efficiently converted -MCA. In this postulated murine pathway, an as yet unidentified hepatic 7α-hydroxysteroid dehydrogenase oxidizes CDCA to 7-oxolithocholic acid [69], which can then be reduced to UDCA by 11-β-hydroxysteroid dehydrogenase 1 [70]. Also, while the findings establish a clear role for Cyp2c enzymes in the biosynthesis of the muricholates, additional studies will be required to determine the role of these enzymes in the murine synthesis of polyhydroxylated bile acids, which are appear under pathophysiological conditions such as bile salt export pump (Bsep; Abcb11)-deficiency [71, 72]. In humans, previous studies suggested that these rehydroxylation reactions involve CYP3A enzymes [73–75], whereas most attention has been focused on Cyp3a11 and Cyp2b10 in mice.
In addition to their hepatic synthesis, bile acids can be modified by the gut bacteria to generate secondary bile acids. In the mouse, their gut microbiota is capable of efficiently epimerizing the 6-hydroxy group of unconjugated βMCA, converting it to the 3α,6α,7β-trihydroxy bile acid, βMCA (Figure 2). Interestingly, in germ-free mice conventionalized using the gut microbiota from human donors, the ability of the humanized gut microbiome to convert βMCA into ωMCA was markedly attenuated [48]. Those results suggest that gut microbiota differences between species likely affect their ability to generate secondary bile acids, an additional factor that should be considered when using murine or other animal models to understand human bile acid metabolism and gut-liver signaling.
2.2.3. Species Differences in Bile Acid Conjugation (N-acyl-amidation)
In most non-mammalian vertebrates, the bile acid side chain is sulfated or N-acyl-amidated to taurine [9, 10, 76]. By contrast in mammals, the side chain is primarily N-acyl-amidated (conjugated) to taurine or glycine, with an amino acid conjugation pattern ranging from almost exclusively glycine in rabbits and guinea pigs to almost exclusively taurine in sheep, dogs, and mice [10, 77–79]. In humans, bile acids are conjugated to glycine or taurine, with glycine conjugates constituting the majority of the biliary bile acids (about 3:1 glycine-to-taurine conjugated species). The selection of taurine or glycine for bile acid conjugation is controlled by the BAAT enzyme [80, 81], and the availability of the taurine precursor [82]. In species such as humans and rats, the BAAT enzyme can utilize either taurine (Km~1 to 2 mM) or glycine (Km~5 to 6 mM). In humans, changes in diet or taurine intake have been shown to influence the proportion of glycine versus taurine-conjugated bile acids [83, 84]. However, in some species such as mice, there is evidence that the BAAT enzyme is particularly inefficient at using glycine for conjugating bile acids. The evidence in mice includes: 1) In the original characterization of the mouse BAAT cDNA by Barnes and colleagues, the recombinant enzyme exhibited significant cholyl-CoA conjugating activity with taurine (Km = 1.9 mM) but had no detectable activity with glycine [81]. Using similar methods, both the human and rat BAAT orthologs utilized taurine and glycine to conjugate cholyl-CoA [80, 85]. 2) Reducing the availability of taurine in mouse liver would be predicted to significantly increase the proportion of bile acid glycine conjugates. However, that has not been the finding in studies using mouse models defective in taurine transport or biosynthesis. In the taurine transporter (taut; Slc6a6) knockout mice, liver taurine content was reduced from ~17.7 μmol/g to 1.6 μmol/g, but the proportion of bile acid glycine conjugates in bile of the taut knockout mice remained at ~3% of the total versus 93% taurine-conjugated and ~3% unconjugated bile acids [86]. In the cysteine dioxygenase (Cdo1) knockout mice, liver taurine levels were reduced to less than 5% of the WT hepatic levels, and the hepatic content of bile acids in those mice was increased by ~30-40% [87]. However, the liver bile acid pool was comprised of ~95% unconjugated bile acids, ~4% taurine-conjugated bile acids, and less than 1% glycine conjugated bile acids, suggesting that the mouse BAAT enzyme is unable to efficiently utilize glycine in place of taurine. 3) The species differences in conjugation are also observed in a chimeric mouse models with a “humanized” liver [88]. Although there was considerable variation in the engraftment with human hepatocytes in that study, the fraction of glycine conjugated bile acids in gallbladder bile increased from ~0.2% to ~7% in mice with the humanized livers. As noted above, the strong preference of the BAAT enzyme for taurine is not unique to the mouse among mammals, and partially purified BAAT enzyme from dog liver yielded bile acid conjugates only with taurine [89, 90], whereas partially purified enzyme from human, pig, and rat liver formed both glycine and taurine bile acid conjugates [90]. Similar to the taurine-restricted genetic mouse models described above, short-term depletion of dietary taurine in a cholic acid perfused dog model leads to an enrichment in biliary output of unconjugated bile acid rather than a shift toward glycine-conjugated species [91]. The evolutionary forces driving selection of the amino acid(s) used for conjugation in a particular species remains a mystery. Both the taurine and glycine-conjugated bile acids are resistant to hydrolysis by the pancreatic peptidases encountered in the intestinal lumen during digestion [92]. However, taurine conjugated bile acids are more water-soluble and have a lower pKa than their respective glycine conjugates. As such, the taurine conjugates will remain ionized over a broader range of pH conditions and their transport across cell membranes is restricted to active or facilitative transport, with little passive diffusion [93, 94]. In studies employing in vitro or in vivo models, the taurine conjugates of bile acids appear to be less cytotoxic than their corresponding glycine-conjugated or unconjugated forms [95–97]. This increased proportion of more hydrophilic taurine-conjugated, 6-hydroxylated, and polyhydroxylated bile acid species is likely an important contributor to the attenuated pathology often observed in mouse models of human liver diseases [25, 97–99].
2.2.4. Deoxycholate 7α-rehydroxylation
Most of the conjugated bile acids that pass into the small intestine are reabsorbed intact. However, a fraction of the bile acid pool is biotransformed by the gut microbiota in the distal small intestine and colon. The initial gateway reaction is carried out by bacterial bile salt hydrolases (BSH), which remove the glycine or taurine moiety. The unconjugated bile acids generated can then undergo additional microbial-mediated reactions. These reactions include modifications of the hydroxy groups on the bile acid steroid nucleus, such as 7α-dehydroxylation, dehydrogenation, epimerization, or elimination to form an unsaturated bile acid (with a double bond in the steroid nucleus) [52]. Among the most important of those reactions is bacterial 7α-dehydroxylation, which in humans converts CA and CDCA to the major secondary bile acids DCA and lithocholic acid (LCA), respectively. In mice, CA is also efficiently converted to DCA. Additional studies suggest that in rats (and most likely also in mice), unconjugated α, β, and ωMCA undergo bacterial 7α or 7β-dehydroxylation to yield hyodeoxycholic acid (HDCA) or murideoxycholic acid (MDCA) [100]. However, based on the low abundance of these bile acid species in murine cecal contents or feces, these reactions appear to be of only minor quantitative importance as compared to the 7α-dehydroxylation of CA [48]. Although there does not appear to be substantial 7α-dehydroxylation, of the muricholates, it should be noted that gut microbial epimerization of the hydroxy group at the C-6 position is a quantitatively important reaction, converting βMCA to ωMCA.
A fraction of the gut microbiome-generated DCA and LCA is absorbed from the colon and returned to the liver in the portal circulation. After uptake by hepatocytes, the DCA and LCA is reconjugated to glycine or taurine and potentially 7α-rehydroxylated to reform conjugated CA and CDCA, respectively. Whereas this reaction may not be important for LCA, which is efficiently sulfated in many species [101], hepatic bile acid 7α-rehydroxylation is an important determinant of the concentration of DCA in the bile acid pool and this activity varies considerably between species. Hepatic bile acid 7α-rehydroxylation activity is relatively high in mice (as well as rats, prairie dogs, and hamsters; species with low biliary DCA concentrations; [102–105]), however the hepatic enzyme activity is apparently lacking in humans (also rabbits, dogs, walruses, and sperm whales) [10, 106, 107]. A CYP enzyme is likely responsible for the hepatic rehydroxylation of TDCA to TCA, as indicated by partial characterization of the purified rat liver enzyme and studies using a hepatic-specific cytochrome P450 reductase knockout mice, which reduces the activity of all liver CYP enzymes by deletion of the shared electron donor enzyme, CYP oxidoreductase [59, 108]. Notably, the earlier purification and characterization studies of the rat liver TDCA 7α-hydroxylase (monooxygenase) and cholesterol 7α-hydroxylase indicated that these are distinct enzymes [108, 109]. However, there has been little additional characterization of the conjugated DCA 7α-hydroxylase activity [110], and the identity of the specific enzyme responsible for this important species bile acid metabolism difference remains to be determined.
2.2.5. Detoxification of Bile Acids by Sulfation and Hydroxylation
Among the phase II detoxification reactions for bile acids is sulfation (sulfonation), where a member of the SULT sulfotransferase family catalyzes the transfer of a sulfonate group (SO3−) preferentially to the C-3 position in humans and to the C-7 position group in mice [49, 50]. In humans, it has been known for many years that sulfation is carried out in the liver by SULT2A1. This reaction serves as a means to detoxify and eliminate bile acids, and sulfation (sulfonation) has profound effects on the physicochemical properties and fate of bile acids. For example, sulfation is particularly important for metabolism of the monohydroxy bile acid, LCA [101]. As compared to humans [50], sulfation is a relatively minor pathway for bile acid metabolism in rats and mice, species that can carry out more extensive phase I hydroxylation of secondary bile acids [51, 93, 99]. In contrast to humans, the major hepatic enzyme responsible for sulfating bile acids in mice was only recently identified [111, 112]. The murine enzyme proved difficult to identify in part because the mouse genome encodes eight Sult2a genes, whereas there is only one SULT2A gene in humans. Bile acid sulfation activity had long been attributed to Sult2a1 or Sult2a2 in rodents, but new evidence such as its high level expression in livers of both male and female mice strongly supports Sult2a8 as the major hepatic bile acid sulfating enzyme [111]. Indeed, Sult2a8 is likely to be particularly important in male mice, which express only low levels of the other Sult2a genes in liver. As indicated above, along with species differences in the quantitative significance of bile acid sulfation, the location of the sulfate group on the steroid nucleus differs between humans and mice. The metabolic and signaling consequences associated with sulfation at the C-3 (humans) versus C-7 (mice) position are not clear, but C-7 sulfates have been reported to be more resistant to hydrolysis and metabolism by the gut microbiota.
Whereas phase II sulfation [50] and to a lesser extent glucuronidation [113] reactions are important pathways for bile acid detoxification in humans, rats and mice are more reliant on hepatic phase I hydroxylation, thought to be mediated in part by rodent Cyp2b10 and Cyp3a11 [51, 114, 115]. The increase synthesis of polyhydroxylated bile acids is perhaps best characterized in Bsep-deficient mice [72, 99]. Overall, these rehydroxylation reactions appear to play a particularly important protective role and likely contribute to the attenuated phenotype often observed in mouse models of human liver disease.
2.3. Mouse Models of Bile Acid Biosynthesis Defects
2.3.1. Cyp7a1 Knockout Mice
Inherited defects in CYP7A1 have been reported in two brothers that are homozygous for a frameshift mutation, who presented with premature gallstone disease and statin-resistant hypercholesterolemia [35]. Loss of CYP7A1 reduced bile acid synthesis by more than 90%, as estimated by measuring fecal bile acid loss using a single 24-h collection from one sibling. However, the subjects exhibited no apparent steatorrhea or fat-soluble vitamin malabsorption. In contrast to the human subjects, the original line of Cyp7a1 knockout mice exhibited a more complex phenotype, with a high rate of postnatal death, malabsorption of fat and fat-soluble vitamins, as well as skin and vision abnormalities in early life [43]. Induction of the alternative pathway for bile acid synthesis at approximately 3 weeks of age effectively compensated for loss of Cyp7a1, and the mice become indistinguishable from their wild-type littermates with regard to their growth and appearance [116]. The original Cyp7a1 knockout mice were not hypercholesterolemic, and exhibited significantly reduced levels of intestinal cholesterol absorption, reflecting a smaller bile acid pool [117]. Subsequently, an independent colony of the Cyp7a1 knockout mice was established from the original cryopreserved mice stored at the Jackson Laboratory. The Cyp7a1 knockout mice in this colony exhibited a mild phenotype, with no apparent increases in postnatal mortality, and were hypercholesterolemic, similar to the CYP7A1-deficient human subjects [118]. Postnatal survival similar to that of wild type littermates was also observed for Cyp7a1 knockout mice after backcrossing the Cyp7a1 null allele into a C57Bl/6J background [36]. As a major determinant of the size and composition of the bile acid pool, Cyp7a1 knockout and rat or human CYP7A1-overexpressing transgenic mice have been used effectively by Chiang and coworkers to explore the relationship of bile acid synthesis to the pathogenesis of liver and metabolic disease. These models have provided important new insights to the molecular mechanisms and pathways that are engaged by changes in the size, composition, hydrophobicity, and signaling potential of the bile acid pool [36, 67, 119, 120].
2.3.2. Cyp8b1 Knockout Mice
No inherited defects in human CYP8B1 have been reported. Cyp8b1 knockout mice have been generated and phenotyped [121]. Loss of Cyp8b1 activity has profound effects on bile acid metabolism, which includes an inability to synthesize CA, shifting the bile acid pool toward more hydrophilic bile acids such as the MCAs and UDCA, and an increased bile acid pool size as a result of increased Cyp7a1 expression. In agreement with the poor detergent properties of 6-hydroxylated bile acids [58] and despite an increase in the whole-body bile acid pool size, intestinal cholesterol absorption is significantly reduced in Cyp8b1 knockout mice [121–123]. In that regard, it is illustrative to compare the effects of inactivation of Cyp7a1, Cyp8b1, and the ileal apical sodium-dependent bile acid transporter (Asbt; Slc10a2) on intestinal cholesterol absorption in mice [117, 121, 124]. The rank ordering for bile acid pool size in these models is: Cyp8b1 knockout (~130% of WT) > Cyp7a1 knockout (~35% of WT) > Asbt knockout (~20% of WT), whereas the rank ordering for intestinal cholesterol absorption is: Asbt knockout (~70% of WT) > Cyp8b1 knockout (~55% of WT) > Cyp7a1 knockout (~10% of WT). Although pool size influences intestinal cholesterol absorption, enrichment of the bile acid pool with the MCAs appears to be particularly important. As such, a smaller and CA-enriched (MCA-deficient) bile acid pool (such as in the Asbt knockout mice) is more effective at promoting intestinal absorption of cholesterol then the substantially larger, but MCA-enriched bile acid pool in Cyp8b1 knockout mice. In Cyp7a1 knockout mice [117], the bile acid pool is both smaller and includes a significant proportion of MCAs, providing a biophysical explanation for the particularly low intestinal cholesterol absorption. The effects of a hydrophilic MCA-enriched bile pool on the detergent solubilization of intestinal luminal contents is not restricted to cholesterol, and more recent studies using the Cyp8b1 knockout mice have also demonstrated inhibitory effects of the MCAs on intestinal fat absorption [65, 66, 123]. Since the bile acid pool in humans is more hydrophobic and largely lacks 6-hydroxylated species, alterations in human CYP8B1 expression are not predicted to yield these profound effects on intestinal lipid absorption or metabolism observed in mouse models. However, more subtle effects on bile acid signaling or bile acid-gut microbiota interactions are possible.
2.3.3. Other Knockout Mouse Models of Bile Acid Biosynthetic Gene Defects
Knockout mouse models have been generated for other enzymes important for bile acid biosynthesis, including CYP27A1, CYP46A1, CH25H, CYP7B1, HSD3B7, AKR1D1, AMACR, ACOX2, HSD17B4, SCP2, SLC27A5, and ABCD3 [38, 125]. As in humans, a single enzyme defect in mice is generally not sufficient to eliminate production of all bile acids because multiple biosynthetic pathways exist. The most commonly reported inherited bile acid biosynthesis defect in humans is HSD3B7-deficiency, and loss of this enzyme affects both the classical and alternative pathways for bile acid biosynthesis. HSD3B7-deficiency is characterized by an early and progressive intrahepatic cholestasis [126], and phenotypic manifestations such as the paucity of primary bile acids, poor growth, and clinical response to bile acid and fat-soluble vitamin supplementation are also evident in the Hsd3b7 knockout mice [45].
In humans, mutations in CYP27A1 caused Cerebrotendinous xanthomatosis (CTX), a rare autosomal recessive disease associated with transient neonatal cholestasis and progressive neurological dysfunction, cerebellar ataxia, premature atherosclerosis, cataracts, and tendinous xanthomas later in life. In humans, the pathophysiology appears to be related to the deposition of cholesterol and oxysterols such as cholestanol in tissues, especially tendons and brain [32]. CYP27A1 is involved in both the acidic and neutral biosynthetic pathways [31]. As such, compensatory increases in CYP7A1 and the microsomal 25-hydroxylase pathways are unable to fully compensate and the bile acid pool size is decreased and enriched in bile acid intermediates. Cyp27a1 knockout mice have been generated and characterized [37, 40]. As compared to CTX patients, Cyp27a1 knockout mice exhibit a milder phenotype with smaller elevations in the levels of oxysterols and bile acid intermediates, and somewhat lower levels of cholestanol in brain and tendons [127]. The attenuated phenotype in the Cyp27a1 knockout mice is thought to be secondary to increased hydroxylation of bile acids and bile acid intermediates by Cyp3a11 and other murine Cyp enzymes [128–130].
In humans, CYP7A1 activity appears to be low in newborns [44]. As such, neonates with a CYP7B1-deficiency have reduced synthesis of bile acids via the classical and alternative pathways. These patients can exhibit severe liver disease [44], which responds favorably to bile acid (CDCA) replacement therapy [131]. Mutations in CYP7B1 are also associated with a rare cause of spastic paraplegia (spastic paraplegia-5A, SPG5A), a recessive neurological disorder [132]. However, the pathogenesis of this progressive motor-neuron degenerative disease is likely due to loss of CYP7B1-dependent metabolism of cholesterol and neurosteroids in the central nervous system rather alterations in bile acid synthesis [133]. Inactivation of Cyp7b1 in mice had little effect on the bile acid pool size and composition, as a result of a compensatory increase in Cyp7a1 expression and bile acid synthesis via the classical pathways. In contrast to the human patients, there was no evidence of liver or neurological disease in the mouse model [39]. In agreement with its role in oxysterol metabolism, both 25- and 27-hydroxycholesterol accumulate in the serum and tissues of Cyp7b1 knockout mice, but not 24-hydroxycholesterol, which is metabolized by Cyp39a1 [134].
2.3.4. Cyp2c Knockout Mice as a Mouse Model with a “Humanized” Bile Acid Pool
The CYP2C enzymes constitute one of the largest subfamilies of the cytochrome P450 enzymes, and includes four CYP2C genes (CYP2C8, CYP2C9, CYP2C19, and CYP2C19) in humans, nine Cyp2c genes in rats, and fifteen (nineteen if the four pseudogenes are counted) in mice. The humans CYP2C isoforms play an important role in metabolism of many drugs and endogenous compounds [135]. However, the murine Cyp2c subfamilies’ complexity and high level of sequence identity between the isoforms has made it difficult to identify the murine orthologs for individual human CYP2C genes. In order to study CYP2C function, Cyp2c gene cluster knockout and CYP2C9 humanized mouse models have been generated [136]. The models either delete fourteen full-length mouse Cyp2c genes (Cyp2c55, 2c65, 2c66, 2c29, 2c38, 2c39, 2c67, 2c68, 2c40, 2c69, 2c37, 2c54, 2c50, 2c70) or replace those fourteen Cyp2c genes with an expression cassette encoding the human CYP2C9 under control of a liver specific albumin promoter. The models are commercially available, viable, and apparently unable to synthesize the muricholates [47, 136]. As such, the use of these models would permit analysis of experimental interventions in the setting of a substantially more human-like bile acid composition. However, it should be noted that there is a paucity of information on these models under different experimental conditions, and the effects on many parameters of bile acid homeostasis such as pool size, enterohepatic cycling, bile acid signaling, and bile acid interactions with the gut microbiota remain to be determined. In the initial characterization of the mice, small increases in serum levels of ALT and AST were observed, along with variable liver histological changes, including a small increased infiltration by lymphocytes and neutrophils [136]. Still, this is a mild phenotype in light of the presumed important detoxification function of these enzymes. Finally, it should be noted that eliminating the murine Cyp2c genes is not predicted to alter the murine taurine-predominance for bile acid conjugation or hepatic TDCA rehydroxylation, which must be carried out by another CYP [47]. However even with those potential limitations, the Cyp2c knockout mice are predicted to be an important new model for studying the relationship between bile acid metabolism and liver disease.
3. Mouse Models to Study Bile Acid Transport
The major mouse genetic models available to study bile acid transport and enterohepatic cycling are described in Table 2.
Table 2.
Genetic Mouse Models Used to Study Bile Acid Biosynthesis and Metabolism
| Model Transporter | Description (Bile acid-related phenotype) |
|---|---|
| Slc10a1 KO (Ntcp) | Constitutive KO (mixed B6/129; Taconic model #TF1367); altered BA phenotype in subset of KO mice: ↑serum BAs; ↑urinary BAs; ↓liver Oatp expression ↓fecal BAs [162] |
|
Abcb11 KO (Bsep) Abcb11 KO |
Constitutive KO (mixed B6/129); backcrossed into FVB/N; ↑liver BAs w/↑proportion polyhydroxylated BAs; ↑serum & urine BAs; ↓liver Cyp8b1; ↓liver Mdr1a/b [72, 99, 174, 184] Abcb11 KO (JAX 002751 B6;129S7-Cyp7a1tm1/Rus/J) backcrossed into C57Bl6/J background; ↔ BA pool size; ↑liver BAs w/ ↑proportion of TMCA, polyhydroxylated BAs; ↑serum & urine BAs; ↓liver Cyp7a1, Cyp7b1, Cyp8b1, Cyp27a1 expression [185] |
| Abcb11 Tg | Tg mouse expressing murine abcb11 under control of transthyretin promoter (FVB/NJ); ↑biliary BA; ↓BA pool w/ ↓proportion TMCA; also backcrossed into C57Bl/6 background [285–287] |
|
Slco1a/1b KO (Oatp1a/1b) Humanized SLCO1B1 Slco1b2 KO (Oatp1b2) |
Constitutive Slco1a/1b gene cluster KO [(FVB.129P2-Del(Slco1b2-Slco1a5)1Ahs]; Taconic model #10707; ↑serum unconjugated BAs; ↑serum conjugated bilirubin [152, 180] Tg mouse expressing human OATP1B1 driven by apoE hepatic control region crossed into Slco1a/1b KO (FVB/129; Taconic model #10708); ↔serum conjugated bilirubin [152] Constitutive Slco1b2 KO (backcrossed into C57Bl/6J); ↑serum unconjugated BAs; ↓hepatic CA clearance [182] |
| Atp8b1 KO | Constitutive KO (Amish PFIC G308V mutation knock-in); ↑BA pool size w/ ↑proportion TMCA; ↑hydroxylation of TDCA to TCA; ↑serum polyhydroxylated BAs; ↔fecal BAs [97, 98, 288] |
| Abcb4 KO (Mdr2) | Constitutive KO (FVB/N; JAX 002539 FVB.129P2-Abcb4tm1Bor/J); ↓↓biliary phospholipid; ↓biliary cholesterol; ↑liver BAs; ↑serum BAs; also backcrossed into Balb.cJ and C57Bl/6J backgrounds [188, 189, 203, 289] |
| Slc10a2 KO (Asbt) | Constitutive KO (129Sv; JAX 005213 129-Slc10a2tm1Pda / J); also backcrossed into C57Bl6 background; ↓ BA pool size w/ ↑proportion TCA; ↑liver Cyp7a1 expression & activity; ↑fecal BAs, ↑fecal neutral sterols; ↓intestinal cholesterol absorption [124, 290] |
|
Slc51a KO (Ostα) Slc51a KO |
Constitutive KO (mixed B6/129; JAX 009082 B6.129S6-Slc51atm1Pda / J); also back- crossed into C57Bl/6J background; altered ileal morphology; ↓BA pool size; ↔liver Cyp7a1 expression & activity; ↔fecal BAs, ↑fecal neutral sterols; ↓intestinal cholesterol absorption [218, 224] Constitutive KO (C57Bl/6); ↓BA pool size; ↓liver Cyp7a1 expression; ↔fecal BAs [225] |
3.1. Overview of Bile Acid Transport – Major pathways
The enterohepatic circulation of bile acids is summarized in Figure 3. Bile acids are secreted into bile and pass into the small intestine where the majority (> 90%) is reabsorbed from the distal small intestine and returned to the liver via the portal venous circulation. For conjugated bile acids, there is efficient first pass clearance by hepatocytes and resecretion into bile [137]. The unconjugated bile acids returning from the intestine in the portal circulation are also taken up by hepatocytes, reconjugated to taurine or glycine, and resecreted into bile. Because these processes are very efficient, less then 10% of the bile acids secreted into bile are newly synthesized, and most have previously undergone enterohepatic cycling. In hepatocytes, bile acids are taken up across the sinusoidal membrane by sodium-dependent and independent mechanisms involving the Na+-taurocholate cotransporting polypeptide (NTCP; SLC10A1) and members of the Organic Anion Transport Protein (OATP) family. After internalization, monovalent bile acids are secreted into bile across the canalicular membrane by BSEP, which exhibits greater affinity for conjugated (amidated) versus unconjugated bile acids [138]. Bile acids in the hepatocyte that have been modified by the addition of sulfate or glucuronide are secreted into bile by the multidrug resistance-associated protein-2 (MRP2; ABCC2) and possibly the breast cancer related protein (BCRP; ABCG2), whereas bile acids modified by additional hydroxylations are secreted into bile by MRP2, P-glycoprotein (MDR1; ABCB1A), and BSEP [139]. In the distal small intestine, bile acids are efficiently taken up across the enterocyte brush border membrane by the apical sodium-dependent bile acid transporter (ASBT; SLC10A2). The bile acids then bind to the Ileal bile acid binding protein (IBABP; FABP6) and are shuttled to the basolateral membrane for export into the portal circulation by the heteromeric transporter, OSTα-OSTβ (SLC51A, SLC51B) [29].
Figure 3. Enterohepatic circulation of bile acids showing the major transport proteins.
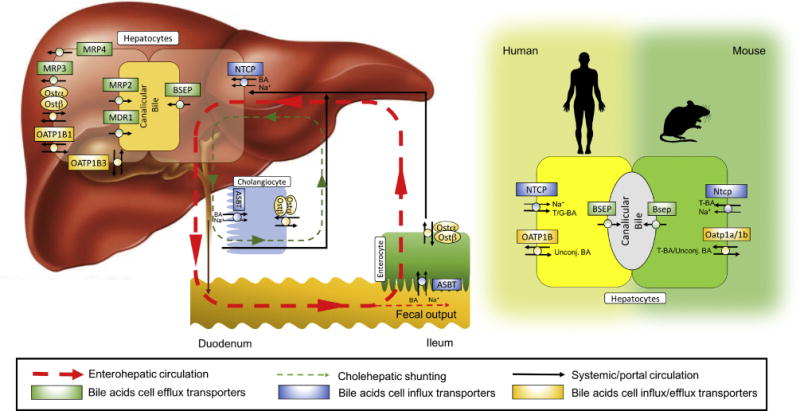
(Left panel) After their synthesis, conjugated monovalent bile acids are secreted into bile canaliculi by the bile salt export pump (BSEP), whereas modified (sulfated, glucuronidated and polyhydroxylated) bile acids can be secreted by the canalicular transporters, MRP2 and MDR1. Alternatively, monovalent or modified bile acids can be effluxed across the basolateral (sinusoidal) membrane of the hepatocyte by OSTα-OSTβ, MRP3, MRP4, and possibly other transporters as a mechanism to protect the hepatocytes from bile acid overload. A fraction of the bile acids secreted into bile undergo “Cholehepatic Shunting” (green dotted line), whereby the bile acids are prematurely absorbed in the biliary tract and returned directly to the liver. Unconjugated bile acids or bile acid analogs such as norUDCA undergo passive absorption, whereas conjugated bile acids can be absorbed by cholangiocytes via the ASBT and exported by OSTα-OSTβ for return to the hepatocyte in the periductular circulation. Ultimately, the bile acids secreted into bile move through the biliary tract, empty into the intestinal lumen, and are passively absorbed along the length of the intestine and actively absorbed in the ileum. The bile acids are then carried back to the liver in the portal circulation for hepatocellular reuptake and resecretion into bile (red dotted line). (Right panel) The transporters that maintain the enterohepatic circulation of bile acids are largely conserved between humans in mice. In mice but not humans, members of the Oatp family appear to play an important secondary role in hepatic uptake of conjugated bile acids.
3.1.1 Overview of Bile Acid Transport – Sinusoidal Membrane Bile Acid Efflux and Hepatocyte Hopping
In the liver, unconjugated, conjugated, or modified (sulfated, glucuronidated, and polyhydroxylated) bile acids can be backfluxed across the hepatocyte sinusoidal membrane into the space of Disse by OSTα-OSTβ, MRP3, multidrug resistance-associated protein-4 (MRP4; ABCC4), and possibly other carriers [140–144]. This is thought to be a protective mechanism to prevent bile acid overload in a cycle analogous to the “hepatocyte-hopping” observed for conjugated bilirubin and certain drugs such as sorafenib [145, 146]. In this iterative fashion, bile acids are backfluxed across the sinusoidal membrane of periportal hepatocytes and carried in sinusoidal blood to more pericentral hepatocytes for reuptake and secretion into bile. This process dynamically recruits additional hepatocytes within the liver lobule to maintain the clearance and secretion of bile acids into bile, while safeguarding vulnerable zone 1 periportal hepatocytes, particularly under cholestatic conditions [147]. The backflux of bile acids has been modeled recently in human subjects using an innovative 11C-bile acid PET tracer [148]. In healthy subjects, the rate constant for 11C-cholylsarcosine transport from hepatocytes to bile was approximately 20 times greater than the rate constant for backflow (hepatocytes into blood). But in cholestatic subjects, hepatocyte transport of 11C-cholylsarcosine into bile was reduced, and backflux into blood was increased. As a result, the probability of 11C-cholylsarcosine secretion into bile was reduced about 90%, and the mean hepatocyte residence time was increased more than 2.5-fold [148].
The relative contribution in humans and mice of the different bile acid efflux transporters remains to be determined. However current results suggest significant roles for MRP4, which cotransports glutathione along with bile acids, and possibly OSTα-OSTβ [142, 144]. For MRP3, it should be noted that there are important rodent-human substrate specificity differences with regard to bile acids. Along with glucuronidated substrates, amidated (taurine and glycine-conjugated) and sulfated bile acids are transported with high affinity by rat [149] but not human MRP3 [150]. Based on MRP3’s strong preference for glucuronidated substrates, as well as results from knockout mice, MRP3 is not thought to be a significant contributor to the sinusoidal efflux of monovalent bile acids [151].
3.2. Bile Acid Transport: Species Differences Between Humans and Mice
The enterohepatic circulation of bile acids and role of the transporters controlling that process are remarkably similar in humans and mice, despite differences in the composition of their bile acid pools. The human-mouse species differences in bile acid transport include: i) a greater reliance on members of the OATP family in mice for hepatic clearance of conjugated bile acids [152]; ii) OATP gene family differences due to gene duplication [153]; iii) differences in hepatic expression of select transporters [154], and iv) increased intestinal passive absorption of bile acids in humans versus mice, reflecting the increased proportion of glycine-conjugated and hydrophobic bile acids in humans [29, 155, 156].
3.2.1. Hepatocyte Sinusoidal Membrane Bile Acid Uptake
NTCP is responsible for transporting the majority of glycine/taurine conjugated bile acids across the hepatic sinusoidal membrane [157, 158]. In humans, serum bile acid levels are significantly elevated in patients with an inherited NTCP deficiency [159, 160], and to a lesser extent in healthy human subjects after administration of the NTCP inhibitor, myrcludex B [161]. In contrast, serum bile acid levels were not consistently elevated in Ntcp knockout mice. In that model, more than half of the adult mice are normocholanemic (serum bile acid levels < 20 μM) and only a subset of mice were markedly hypercholanemic (serum bile acid levels as high as 1000 μM) [162]. The findings for Ntcp knockout mice suggest the presence of compensatory transport mechanisms and follow from mathematical simulations of hepatic clearance and enterohepatic cycling of bile acids that were carried out by Hofmann and colleagues [163]. In those modeling studies, significant reductions in unidirectional hepatic uptake of bile acids had little effect on plasma bile acid concentrations, which were predicted to rise dramatically only after the majority of hepatic bile acid capacity is lost [152]. As such, in the absence of NTCP, small quantitative changes in the residual secondary hepatic bile acid clearance mechanisms or in the bile acid pool size can tip the balance, resulting in dramatic rises in serum bile acid levels (hypercholanemia). Notably, the hypercholanemic Ntcp knockout mice expressed lower hepatic levels of several bile acid transporting OATPs, particularly Oatp1a1 [162]. A subsequent study using knockout mice lacking the entire group of Oatp1a/Oatp1b (Slco1a/1b) transporters and pharmacological inhibition of Ntcp convincingly demonstrated that both Ntcp and members of the Oatp family participate in the hepatic clearance of conjugated bile acids in mice [152].
3.2.3. Hepatocyte Canalicular Membrane Bile Acid Export
The bile salt export pump (BSEP, ABCB11) mediates hepatocyte secretion of bile acids across the canalicular membrane [164], and loss of BSEP activity is responsible for progressive familial intrahepatic cholestasis type 2 (PFIC2) and benign recurrent intrahepatic cholestasis type 2 (BRIC2) [165]. Hallmarks of the disease include hepatocellular bile acid accumulation and liver injury, which can progress to cirrhosis, hepatic failure, hepatocellular carcinoma, and death [97, 166–168]. For these patients, the extent of BSEP impairment correlates with the severity of disease [169]. Mutations in BSEP also contribute as predisposing factors to intrahepatic cholestasis of pregnancy [170, 171], and BSEP inhibition has been proposed as a mechanism for drug-induced liver injury [172, 173]. In contrast to humans, Bsep-deficient mice (in a mixed genetic or FVB background) do not exhibit progressive liver injury [99, 164]. The compensatory mechanisms underlying this important species difference is thought to be related to the substantial hepatic phase I bile acid rehydroxylation activity in rodents. These CYP-mediated reactions generate polyhydroxylated bile acids, which are less injurious and are exported into the canalicular space by Mdr1a/1b and Mrp2 [99, 139, 164]. In support of the concept of a compensatory role for Mdr1, mice lacking Bsep and Mdr1a/1b (triple knockout) exhibit cholestatic injury [174].
3.3. Mouse Models of Bile Acid Transporter Defects
3.3.1. Ntcp Knockout Mice
Slijepcevic and coworkers reported the first phenotypic characterization of an Ntcp (Slc10a1) knockout mouse [162]. As described above, the serum bile acid levels were within a normal range for ~70% of the experimental Ntcp knockout mice (1-20 μM; normocholanemic), and dramatically elevated (>1000 μM; hypercholanemic) in the remaining ~30%, with no mice found with intermediate serum bile acid levels. Morphologically, the Ntcp knockout mice were similar to wild type mice, with the exception of reduced body weights, which were more evident in the hypercholanemic mice. Bile acid synthesis and enterohepatic cycling appear to be unperturbed in normocholanemic mice, but were altered in the hypercholanemic mice, which exhibit changes in bile flow, biliary bile acid secretion, and fecal and urinary bile acid excretion [162]. Analysis of the normocholanemic mice suggested that hepatic Oatps were compensating for loss of Ntcp. In contrast, hepatic expression of several Oatps were down-regulated in the hypercholanemic mice, perhaps secondary to increased ileal Fgf15 production and enterohepatic signaling [152, 160]. In addition to its role as a bile acid transporter, human NTCP was recently identified as the entry receptor for hepatitis B (HBV) and hepatitis D virus for liver cells [175]. This breakthrough finding and subsequent studies provided an explanation for the hepatocyte and species-specificity for HBV infection. For example, studies have used in vitro cell-based assays to compare the ability of human NTCP versus NTCP from cynomologous monkey, rhesus monkey, pig, mouse, rat, and dog to promote cellular entry of HBV or HDV. Notably, human NTCP was the only homologue that efficiently mediated HBV and HDV entry, establishing its importance as one of the host factors required for establishment of viral infection [176]. Furthermore, myrcludex B, a synthetic preS1 lipopeptide derived from the HBV L-protein was shown to block viral entry and NTCP-mediated bile acid uptake [162, 177]. Thus, in addition to providing insight to bile acid metabolism, the use of NTCP knockout and human NTCP transgenic mice is predicted to contribute to the study of HBV and HDV hepatotropism [162, 178].
3.3.2. Oatp-Knockout Mice
The OATPs are polytopic membrane glycoproteins that mediate sodium-independent transport of bile acids and other organic compounds, such as steroids, thyroid hormone, drugs, and toxins. The OATP super gene family can be subdivided into 6 subgroups, which includes eleven human and sixteen rodent genes [145]. Mice encode only a single OATP1B ortholog (Oatp1b2; gene symbol: Slco1b2), whereas humans encodes two genes, OATP1B1 and OATP1B3, which arose in primates by duplication after divergence from rodents [179]. Conversely, the human genome encodes only one member of the OATP1A family (OATP1A2), whereas the rodent genome encodes five Oatp1a orthologs (Oatp1a1, Oatp1a3, Oatp1a4, Oatp1a5, Oatp1a6). These arose by gene duplication and are clustered in the same chromosomal region (chromosome 6G2 in mouse). In humans, the liver expresses OATP1B1, OATP1B3, and OATP2B1, and the intestine expresses OATP1A2 and OATP2B1. With regard to bile acid metabolism in humans, OATP1B1 and OATP1B3 are thought to account for the majority of hepatic Na+-independent unconjugated bile acid clearance [180], and loss of both SLCO1B1 and SLCO1B3, adjacent genes encoded on chromosome 12p12.1, is responsible for Rotor Syndrome, an autosomal recessive disorder characterized by elevated plasma levels of conjugated bilirubin and unconjugated bile acids [180]. In mice, several bile acid-transporting Oatps are expressed on the hepatic sinusoidal membrane, including Oatp1a1, Oatp1a4, and Oatp1b2 [181], with Oatp1b2 thought to be particularly important for clearance of unconjugated bile acids [182]. Several mouse models have been developed to investigate the role of the Oatps in drug disposition and bile acid metabolism. These models include single gene knockouts for Oatp1a1, Oatp1a4, Oatp1b2, deletion of the entire Oatp1a/1b locus (which includes Oatp1a1, 1a3, 1a4, 1a5, 1a6, 1b2), and transgenic mice expressing human OATP1A2, OATP1B1, OATP1B3, or OATP1B1 plus OATP1B3 in liver [183]. Due to functional redundancy and gene duplication in humans and rodents, the mouse model deleting the entire Oatp1a/1b locus has proven to be especially useful for studying the in vivo role of the OATPs [145, 152, 180].
3.3.3. Bsep-Knockout Mice
To study the mechanisms by which loss of BSEP induces liver injury and causes PFIC2, a Bsep knockout mouse was generated in 2001 [99]. Notably, the liver phenotype of the Bsep knockout mice (in mixed genetic or FVB backgrounds) was markedly attenuated as compared to PFIC2 patients. Further analysis revealed that Bsep knockout mice continue to secrete polyhydroxylated bile acids into bile [99, 164], and did not show significant evidence of liver injury or inflammation unless fed CA in the diet [184]. In contrast to the attenuated phenotype associated with Bsep-deficiency in mice with a mixed genetic or FVB background, Bsep knockout mice backcrossed into a C57Bl/6J background exhibited progressive liver injury, with a phenotype more similar to PFIC2 patients [185]. In those mice, the increased generation of polyhydroxylated bile acids and induction of alternative efflux transporters was not sufficient to protect the liver. Instead, the results suggested that bile acid accumulation induces metabolic dysregulation, which contributes to defective fatty acid oxidation, and synergizes with the elevated intracellular bile acids to induce liver damage [185].
3.3.4. Mdr2 Knockout Mice
The multidrug resistance gene (MDR3 in humans, Mdr2 in mice; gene symbol: ABCB4) encodes a canalicular membrane flippase that is responsible for secretion of phospholipids (mainly phosphatidylcholine) into bile [186, 187]. Loss of ABCB4 effectively blocks phospholipid secretion into bile, dramatically reducing the formation of less toxic bile acid mixed micelles, thereby leaving high concentrations of bile acid micelles and increased levels of non-micellar bile acids, which damage the biliary epithelium [187–189]. The subsequent loss of plasma membrane and tight junction functional integrity by cholangiocytes allows bile to leak into the portal tract, where it can induce inflammation, ductular proliferation, and ultimately peribiliary deposition of fibrotic tissue [190]. Human cholestatic liver diseases associated with ABCB4 defects include progressive familial intrahepatic cholestasis type 3 (PFIC3), intrahepatic cholestasis of pregnancy, and low phospholipid-associated cholelithiasis [191–193]. An Mdr2 (Abcb4) knockout mouse was generated in 1993 [188]. Similar to PFIC3 patients, the Mdr2 knockout mice are unable to secrete phospholipid into bile. In the absence of phospholipid, bile acids damaged the bile canaliculi and ducts, and eventually causing a destructive cholangitis [188]. As one of the few models of a clearly defined “toxic bile”-associated liver injury [25], the Mdr2 knockout mouse has proven to especially useful for exploring the role of bile acids in the pathogenesis human cholestatic liver disease [187, 193–199]. The use of Mdr2 knockout mice as a model for the study of liver disease has been the subject of recent reviews [20, 200].
3.3.5. Atp8b1 and Atp11c Knockout Mice
ATP8B1 (ATPase, Class I, type 8B, member 1; formally FIC1) is not a bile acid transporter, but rather a P4-type ATPase that flips aminophospholipids from the outer to the inner leaflet of the plasma membrane to maintain the asymmetric lipid distribution [201–203]. This lipid asymmetry and enrichment with cholesterol/sphingomyelin of specialized membranes such as the hepatocyte canalicular membrane plays an important protective role and makes the membrane more resistant to hydrophobic bile acids [97]. In addition to the canalicular membrane, ATP8B1 is expressed on apical membrane of the biliary and intestinal epithelium, as well as in other tissues [98, 168]. Studies of Dutch and Amish patients revealed that BRIC type 1 (BRIC1) and PFIC type 1 (PFIC1) are caused by mutations in ATP8B1 [168]. As compared to patients with BSEP mutations, patients with ATP8B1-deficiency typically presents with somewhat milder liver damage, but also extrahepatic complications, such as secretory diarrhea, pancreatitis and hearing loss, which are not alleviated by liver transplantation [204–206]. In 2004, mice were generated that are homozygous for the ATP8B1 G308V point mutation, which had been reported in Amish PFIC patients [98, 168]. Unlike PFIC1 patients, the Atp8b1 G308V mice did not develop cholestatic liver disease [98], likely as a result of their more hydrophilic bile acid pool and enhanced ability to rehydroxylate secondary bile acids.
ATP11C is also a phospholipid-flipping P4-ATPase, and mutations in this X-linked gene were originally reported in mouse models with a heritable B-cell deficiency [207]. Subsequently, the Atp11c mutant mouse lines were also shown exhibit to hyperbilirubinemia and an unconjugated hypercholanemia [208, 209], a Rotor-syndrome-like phenotype [180]. In contrast to the canalicular (apical) membrane localization for ATP8B1, ATP11C is expressed on the sinusoidal (basolateral) membrane of central hepatocytes in the liver lobule. In the Atp11c-deficient mice, sinusoidal membrane uptake of unconjugated bile acids is impaired secondary to reduced expression and basolateral membrane localization of Oatp1a1, Oatp1a4, Oatp1b2, and Ntcp in central hepatocytes [209]. Despite these deficiencies, the Atp11c-deficient mice are viable with an apparently normal lifespan. In humans, a male patient was recently described with a mild hemolytic anemia and a missense mutation (c.1253C>A; Thr418Asn) in ATP11C that was associated with reduced erythrocyte phosphatidylserine flipping activity [210]. The liver phenotype and serum bile acids were not analyzed in this patient, although conjugated bilirubin appeared to be normal. Additional characterization of this patient and identification of others patients with potentially more severe mutations in ATP11C will be required to determine if the phenotypic changes in the Atp11c-mutant mice are also evident in humans.
Finally, the plasma membrane routing and activity of the phospholipid flipping P4-ATPases, ATP8B1 and ATP11C, require an accessory protein, CDC50A (Tmem30A) [209, 211]. Whole body knockout of Tmem30a was embryonic lethal [212]. However, liver-specific inactivation of Tmem30a results in hyperbilirubinemia, hypercholanemia, reduced bile formation, impaired expression and localization of bile acid transporters, and increased susceptibility to CA-induced liver injury [213].
3.3.6. Asbt Knockout Mice
Mutations in the ASBT (SLC10A2) gene are responsible for primary bile acid malabsorption. This congenital diarrheal disorder is associated with steatorrhea, failure to thrive, interruption of the enterohepatic circulation of bile acids, and reduced plasma cholesterol levels [214, 215]. Asbt knockout mice were generated in 2003 and have been used to study the mechanisms controlling bile acid homeostasis and bile acid-related disease [124, 216–218]. Similar to patients with ASBT mutations, the Asbt knockout mice exhibit impaired intestinal bile acid absorption, induction of hepatic bile acid synthesis, and increased fecal bile acid loss in the absence of ileal histological or ultrastructural changes [124]. With regard to species differences, mice may be more dependent upon Asbt-mediated intestinal bile acid absorption than humans. In humans, a significant fraction of the glycine conjugates and unconjugated bile acids are absorbed from the intestine by passive diffusion across the apical brush border membrane [155, 219, 220]. In contrast, the hydrophilic and taurine-conjugated bile acid pool in mice is more reliant on carrier-mediated transport. Indeed, feeding Asbt knockout mice (129SvEv background) a diet containing cholestyramine, a bile acid sequestrant that blocks both passive and active mechanisms for intestinal bile acid absorption, did not further increase fecal loss of bile acids, supporting a predominant role for the Asbt [124]. In Asbt knockout mice, new hepatic bile acid synthesis is unable to compensate for fecal loss. As a result, the bile acid pool size is reduced by almost 80% and the whole-body bile acid pool composition becomes more hydrophobic, with an increased proportion of taurine-conjugated CA and DCA and reduced amounts of βMCA. The shift away from 6-hydroxylated species in these mice enhances the detergent properties of the bile acid pool, partially attenuating the effects of the smaller bile acid pool size on intestinal cholesterol and fat absorption [221]. Reducing the return of bile acids to the liver in the enterohepatic circulation is predicted to affect the development of liver disease. In that regard, pharmacological approaches employing small molecule inhibitors of the ASBT have been used in Mdr2 knockout mice and in mouse models of fatty liver disease [6, 222, 223]. The use of Asbt knockout mouse, a complementary genetic model, is predicted to provide additional mechanistic insight to the relationship of bile acids to the pathophysiology of liver disease.
3.3.7. Ostα Knockout Mice
OSTα-OSTβ (gene symbols: SLC51A and SLC51B) is a heteromeric transporter expressed in small intestine, colon, liver, biliary tract, kidney, brain, and adrenal gland [154]. In the small intestine, OSTα-OSTβ is expressed on the basolateral membrane of enterocytes and responsible for the export of bile acids into the portal circulation to maintain their enterohepatic circulation [154]. However, it should be noted that OSTα-OSTβ functions as a bidirectional facilitative transporter and potential substrates include a variety of steroids and organic solutes in addition to bile acids. As such, OSTα-OSTβ may also be involved in the mediating the uptake of these solutes under certain physiological or pathophysiological conditions [152, 154]. Ostα knockout mice were developed to study the in vivo functions of this unusual transporter. Since stable expression and trafficking of OSTα and OSTβ from the endoplasmic reticulum requires co-expression of both subunits, levels of Ostβ protein are reduced to almost undetectable levels in the Ostα knockout mice [224]. Unlike Asbt knockout mice, which have a classical bile acid malabsorption phenotype [124], Ostα knockout mice exhibit a complex phenotype that includes a paradoxical reduction in hepatic bile acid synthesis [224, 225]. The changes in bile acid metabolism in Ostα knockout mice are due to altered gut-liver bile acid signaling through the FXR-FGF15/19-FGFR4 pathway [226]. The ileal hypertrophy and altered morphology observed in Ostα knockout mice are typical of that associated with epithelial damage and subsequent healing. Recent evidence indicates that these ileal morphological changes are secondary to enterocyte injury caused by bile acid accumulation [218]. Disease-causing mutations in human OSTα have not yet been reported, however a family with two affected brothers suffering from an inherited OSTβ (SLC51B) deficiency were recently identified. The clinical presentation of these pediatric patients included congenital diarrhea, fat-soluble vitamin deficiency, and mildly elevated liver serum chemistries [227]. It is unclear at this time whether the phenotypic differences between the Ostα knockout mice and these patients are due to OSTα versus OSTβ deficiency or to mouse-human species differences. However, the generation and characterization of an Ostβ knockout mouse model should provide additional insight with regard to those questions.
4. Mouse Models with “Humanized” Livers
In addition to the animal models discussed above, mouse models with “humanized” livers are being used to study bile acid metabolism, although only in a limited fashion [88, 228, 229]. These models are distinct from the genetically engineered mice where individual or small groups of genes are replaced by their human orthologs [230, 231]. In chimeric or humanized liver mouse models, the population of endogenous mouse hepatocytes are selectively damaged and replaced with human hepatocytes. The approach involves engrafting human hepatocytes from donors with specific features, such as from healthy donors to model normal metabolism or from patients to model different forms of liver disease [232], or from hepatocytes derived from human induced pluripotent stem cells (iPSCs) [233–235]. In these models, repopulation of the mouse liver with more than 95% human hepatocytes has been achieved. There are several approaches for generating mice with humanized livers, but the underlying principles are similar. The mouse models are severely immune deficient, e.g. NOD/Shi-scid Il2rg−/− (NOG), NOD/LtSz-scid Il2rg−/− (NSG), or Balb/c Rag2−/−Il2rg−/− (BRG), to prevent rejection of the transplanted human cells [232] and use schemes to damage the endogenous murine hepatocytes such as expression of a human transgene expressing urokinase plasminogen activator, expression of herpes simplex virus thymidine kinase, or knockout of the murine enzyme fumaryl acetoacetate hydrolase (FAH) [236, 237]. Such models have recently been used to study bile acid metabolism and may provide additional insights to human-mouse species differences [228, 229]. For example, CYP7A1 expression and bile acid synthesis is down-regulated in both human and mouse hepatocytes in response to human FGF19, a critical regulator of bile acid homeostasis. However, in the liver of humanized mice, CYP7A1 expression is elevated, reflecting an apparent inability of the human hepatocytes to respond to the murine ortholog, Fgf15 [229]. This work, which utilized an innovative model harboring a human FGF19 transgene in the immune-deficient FAH−/− mice, also demonstrated the importance of the circulating bile acid pool as a determinant of liver size [229].
The human liver chimeric mouse models also have a number of limitations. These include the need to use immune-deficient hosts for the human transplanted cells, and variable or limited human liver chimerism [236]. The remaining murine hepatocytes can greatly complicate the interpretation of findings from these models and has led investigators to attempt new strategies such as conditionally knocking out specific murine genes in an immune-deficient background to mitigate the murine contribution to the experimental endpoints. For example, a mouse with a conditional knockout of NADPH-P450 oxidoreductase (Por) in a Rag2−/−Il2rg−/−FAH−/− background was recently generated to reduce the background contribution of residual murine tissue to hepatic drug metabolism in the human hepatocyte-transplanted chimeric mice [238]. At this time for the study of bile acid metabolism, it is unclear if similar improved human liver chimeric mouse models can be generated and applications for the existing models appear to be more qualified.
5. Non-Genetic Models Used to Study Bile Acid Metabolism
The non-genetic models used to study bile acid biosynthesis, bile acid metabolism and the roles of bile acids in human liver disease are described in Table 3.
Table 3.
Non-Genetic Mouse Models Used to Study Bile Acid Metabolism
| Model | Description (Bile acid-related phenotype) |
|---|---|
| Bile Duct Ligation | ↓ BA pool; ↑ liver BAs; ↑ urinary BAs; ↑ polyhydroxylated & sulfated BAs; [144, 291] |
| CA Feeding | Typically 0.5 - 1.0% (w/w) in diet; ↑ Biliary CA and ↓ UDCA in bile; ↑ serum, hepatic, biliary, fecal & urinary total BAs; ↑ liver cholesterol levels [249] [49, 292, 293] |
| LCA Feeding | Typically 0.5-1.0% (w/w) in diet to induce rapid hepatotoxicity; ↑ serum, hepatic, biliary fecal & urinary total BAs; ↑ BA hydroxylation & sulfation; ↑ liver cholesterol levels [249, 261, 294] |
| UDCA Feeding | Typically 05. – 1.0% (w/w) in diet; ↑ Biliary BA output; ↑ serum BA level. ↑ liver BAs [49, 196, 295] |
| BA Sequestrant Feeding | Typically 2% (w/w) in diet; ↔ or ↓ BA pool size; ↓ plasma BAs; ↑ fecal BAs; ↑ TCA synthesis; ↓ liver BAs; ↓ BA biliary output, ↔ bile flow [280] [296] |
5.1. Surgical Models
Bile duct ligation (BDL) is a widely used experimental procedure for the study of bile acid metabolism and cholestatic liver disease in mice [239, 240]. This surgical procedure is simple, reproducible, associated with high survival rates (over 95%), and employs a sham operative group with only midventral dissections as a necessary control. The procedure was first performed on rats, and then successfully adapted for mice [241]. The hepatocellular and cholangiolar changes induced by BDL is highly reproducible for long-term bile duct ligation in rodents and yields morphological changes comparable to that observed with acute obstructive biliary lesions in patients [240]. The typical features of BDL surgical model include extensive ductular reaction, proliferation of cholangiocytes and oval cells (hepatocytes progenitor), obstructive jaundice and cholestasis, portal inflammation, rapid biliary fibrosis, and even long-term cholestasis-driven tumorigenesis [239, 242, 243]. On the other hand, the rapid time (up to 30 days) [244] of cirrhosis development in BDL models clearly does not reflect the slow progression of most human chronic cholestatic liver diseases, which can take decades. Furthermore, the acute obstructive lesions caused by BDL are rarely seen in human chronic cholestatic liver diseases, except in the case of biliary atresia or choledocolithiasis [239, 242, 245]. In the mouse BDL model, high mortality is typically observed after approximately 7 to 10 weeks due to liver failure [242].
5.2. Feeding Bile Acids or Bile Acid Sequestrants
Bile acids cause liver cell injury by inducing plasma membrane damage, mitochondrial damage, oxidative stress, and inflammation [246, 247], with a potency order that generally follows their hydrophobicity: LCA>DCA>CDCA>CA>UDCA [248]. However, that rank order is not absolute. For example, DCA feeding has been reported to be more hepatotoxic than LCA in rats [249, 250], which may reflect a greater metabolic ability to detoxify LCA by hydroxylation or sulfation. Depending upon the research questions being asked, different bile acid species, doses, and feeding periods have been used and are critical considerations affecting the severity of the hepatotoxicity and phenotype observed. However, there are few studies where side-by-side comparisons were carried out. In one such example, different type and concentrations of bile acid were fed for one week to wild type mice. That study found that the lowest concentration of dietary bile acid that induces hepatotoxicity in mice is 0.3% for CA and CDCA, 0.1% for DCA, and 0.03% for LCA [251]. In this case, the potency for inducing hepatotoxicity generally followed hydrophobicity, with a rank order of LCA>DCA>CDCA>CA>UDCA [251]. As discussed in the following sections, diets containing CA or LCA are typically fed to induce jury, whereas feeding UDCA is hepatoprotective in animal models used study the pathogenesis of liver disease.
5.2.1. CA Feeding
Dietary CA feeding is a widely used model used to induce alterations in bile acid homeostasis and cholestasis. In wild type mice, expression of Cyp7a1, Cyp7b1, and the sinusoidal membrane bile acid uptake transporters Ntcp and Oatp1a1 are down-regulated by CA feeding [49, 252], whereas expression of the canalicular membrane transporters Bsep, Mrp2, and Mdr1a/b is up-regulated [252]. Liver morphological changes induced by CA feeding includes disseminated hepatocyte necrosis, increased cell size, increased number of mitotic figures, and dilation of interlobular bile ducts [252]. Feeding CA (typically ~0.5%) to mice with genetic defects in bile acid biosynthetic enzymes or transporters is typically used to unmask susceptibilities to liver injury. For example, feeding CA to Mdr2 (Abcb4) knockout mice produces liver damage, significantly elevated serum liver enzymes and bilirubin, bile duct proliferation, peribiliary fibroblast activation, and fibrosis [253]. Bsep knockout mice typically exhibit features of mild cholestasis, in contrast to the clinical presentation of PFIC2 patients. However, after feeding CA, Bsep knockout mice develop a severe cholestatic phenotype, characterized by jaundice, weight loss, elevated plasma bile acids, elevated transaminases, cholangiopathy, liver necrosis, and high mortality [184]. For the recently generated liver-specific Tmem30a (Cdc50a)-knockout mouse, short-term feeding a diet containing 0.5% CA for twenty days exacerbated liver injury, as evident by significant increases in ALT, AST, ALP. Over this time course, body weight and survival of the wild type mice was unaffected, whereas the liver-specific Tmem30a knockout mice exhibited significant reductions in body weight with more than half of the knockout mice dying within the first twelve days [213].
5.2.2. LCA Feeding
LCA-feeding as a model to induce hepatotoxicity and cholestatic liver injury in rodents was first reported in early 1960’s [254]. The well-accepted pathogenesis of LCA-induced cholestasis in rodents includes biochemical alterations of the bile canalicular membrane [255, 256], crystalline plugs in bile canaliculi due to the poor solubility of LCA [257, 258], and impaired trafficking or increased retrieval of transporters from the canalicular membrane [259, 260]. In mice, LCA-feeding yields bile duct obstruction and a suppurative cholangitis-induced periductal fibrosis [261].
5.2.3. UDCA Feeding
UDCA is used clinically for the treatment of cholestatic disorders such as primary biliary cholangitis (PBC), intrahepatic cholestasis of pregnancy, and cystic fibrosis-associated liver disease [262–264]. In mouse models, UDCA is reported to improve portal inflammation, ductular proliferation, and fibrosis in Mdr2 knockout mice [265, 266]. However, the choleresis generated by UDCA in bile duct ligated mice model aggravates liver injury. UDCA feeding in BDL mice increased biliary pressure, leading to rupture of cholangioles (canals of Hering), exacerbating hepatocyte necrosis [194, 252]. UDCA also stimulates expression of hepatic, renal, and intestinal ABC transporters independent of FXR without inducing liver toxicity [267].
5.2.4. BA Sequestrant Feeding
Bile acid sequestrants (BAS) are non-absorbable positively charged polymeric compounds that bind negatively charged bile acids in the intestinal lumen [268]. Clinically, BAS deplete the bile acid pool by approximately 40% by diverting bile acids from enterohepatic circulation, stimulating bile acid synthesis, and increasing fecal bile acid loss [269]. The increase of BA synthesis reduces liver cholesterol levels, stimulating LDL receptor expression and reducing LDL cholesterol levels [269, 270]. As such, BAS were in the treatment of dyslipidemias such as atherosclerotic cardiovascular disease and coronary heart disease, particularly prior to development of the HMG CoA reductase inhibitors [271]. In addition to their use as a cholesterol-lowering agent, BAS have been used to and to treat bile acid diarrhea [272, 273]. With regard to liver disease, attempts to treat different forms of cholestasis-associated pruritus with BAS has met with inconsistent success [274–276], and the use of BAS in the treatment of a limited number of patients with benign recurrent intrahepatic cholestasis [277] and a patient with PSC [278] has also been described. BAS approved for clinical use include cholestyramine, cholestipol, colestimide (in Japan), and colesevelam [279]. For mouse model studies, bile acid sequestrants are typically administered in the diet at concentrations from 1 to 2% (w/w) and have been shown to effectively block intestinal bile acid reabsorption [280]. The effects of BAS on mouse bile acid metabolism is summarize in Table 3.
6. Conclusions
Experimental animal models, and particularly mouse models, have played a critical role in elucidating the major pathways responsible for bile acid biosynthesis, enterohepatic cycling, and metabolism. Among the challenges faced by attempts to use these mouse models to study the role of bile acids in the pathogenesis of human liver are important species differences with regard to the physicochemical and signaling properties of the bile acid pool. As highlighted in this review, new insights to the murine pathways responsible for production of hydrophilic and less injurious bile acids will facilitate the generation of mouse models with more a “human-like” bile acid pool. It is predicted that these models will exhibit more “human-like” responses to the pathophysiological changes in cholestasis, with the potential to facilitate pre-clinical development new bile acid-based therapeutics.
Highlights.
Mouse models have been used to elucidate the core pathways controlling bile acid homeostasis
Important species differences may limit “translatability” of the findings to humans
Mouse model with “humanized” bile acid compositions are predicted to offer critical new insights to the role of bile acids in liver disease
Acknowledgments
This work was supported by the National Institutes of Health [DK047987]. We thank Dr. Stephan Barnes (University of Alabama at Birmingham) for enlightening discussions regarding mammalian species differences with regard to bile acid N-acyl-amidation and the amino acid specificity of the human, rat and mouse BAAT enzymes. We also thank Dr. Anu Rao (Emory University) for her thoughtful comments and critical reading of the manuscript.
Abbreviations
- ABC
ATP binding cassette
- ASBT
apical sodium-dependent bile acid transporter
- BAS
bile acid sequestrant
- BCRP
breast cancer related protein
- BDL
bile duct ligation
- BRIC
Benign recurrent intrahepatic cholestasis type 2
- BSEP
bile salt export pump
- BSH
bile salt hydrolase
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CTX
Cerebrotendinous xanthomatosis
- CYP
cytochrome P450
- DCA
deoxycholic acid
- HBV
hepatitis B virus
- IBABP
ileal bile acid binding protein
- LCA
lithocholic acid
- MCA
muricholic acid
- MDR
multidrug resistance protein
- MRP
multi-drug resistance-associated protein
- NTCP
Na+-taurocholate transporting polypeptide
- OATP
organic anion transporting polypeptide
- OST
organic solute transporter
- PBC
Primary biliary cholangitis
- PET
positron emission tomography
- PFIC
Progressive familial intrahepatic cholestasis
- PSC
Primary sclerosing cholangitis
- UDCA
ursodeoxycholic acid
- SULT
sulfotransferase
- VLCS
very long chain synthetase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang DQ, Carey MC. Therapeutic uses of animal biles in traditional Chinese medicine: an ethnopharmacological, biophysical chemical and medicinal review. World J Gastroenterol. 2014;20(29):9952–75. doi: 10.3748/wjg.v20.i29.9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chemistry, Physiology, and Metabolism. Vol. 1. New York: Plenum Press; 1971. The Bile Acids; p. 372. [Google Scholar]
- 3.Hofmann AF, Hagey LR. Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res. 2014;55(8):1553–95. doi: 10.1194/jlr.R049437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beuers U, et al. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. 2015;62(1 Suppl):S25–37. doi: 10.1016/j.jhep.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 5.Trauner M, et al. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology. 2017;65(4):1393–1404. doi: 10.1002/hep.28991. [DOI] [PubMed] [Google Scholar]
- 6.Baghdasaryan A, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64(3):674–81. doi: 10.1016/j.jhep.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 7.Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol. 2012;18(36):4985–93. doi: 10.3748/wjg.v18.i36.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen PL, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology. 2017;65(2):722–738. doi: 10.1002/hep.28965. [DOI] [PubMed] [Google Scholar]
- 9.Haslewood GA. Bile salt evolution. J Lipid Res. 1967;8(6):535–50. [PubMed] [Google Scholar]
- 10.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res. 2010;51(2):226–46. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34(10):1637–59. [PubMed] [Google Scholar]
- 12.Gurantz D, Hofmann AF. Influence of bile acid structure on bile flow and biliary lipid secretion in the hamster. Am J Physiol. 1984;247(6 Pt 1):G736–48. doi: 10.1152/ajpgi.1984.247.6.G736. [DOI] [PubMed] [Google Scholar]
- 13.van Golen RF, et al. The pathophysiology of human obstructive cholestasis is mimicked in cholestatic Gold Syrian hamsters. Biochim Biophys Acta. 2018;1864(3):942–951. doi: 10.1016/j.bbadis.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9(9):993–9. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- 15.Eppig JT. Mouse Genome Informatics (MGI) Resource: Genetic, Genomic, and Biological Knowledgebase for the Laboratory Mouse. ILAR J. 2017;58(1):17–41. doi: 10.1093/ilar/ilx013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meehan TF, et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat Genet. 2017;49(8):1231–1238. doi: 10.1038/ng.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, et al. Joint mouse-human phenome-wide association to test gene function and disease risk. Nat Commun. 2016;7:10464. doi: 10.1038/ncomms10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Scheidt M, et al. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017;25(2):248–261. doi: 10.1016/j.cmet.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perlman RL. Mouse models of human disease: An evolutionary perspective. Evol Med Public Health. 2016;2016(1):170–6. doi: 10.1093/emph/eow014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariotti V, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta. 2018;1864(4 Pt B):1254–1261. doi: 10.1016/j.bbadis.2017.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massafra V, van Mil SWC. Farnesoid X receptor: A "homeostat" for hepatic nutrient metabolism. Biochim Biophys Acta. 2018;1864(1):45–59. doi: 10.1016/j.bbadis.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Wahlstrom A, et al. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016;24(1):41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Copple BL, Li T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21. doi: 10.1016/j.phrs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang JY. Bile acid metabolism and signaling. Compr Physiol. 2013;3(3):1191–212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fickert P, Wagner M. Biliary bile acids in hepatobiliary injury - What is the link? J Hepatol. 2017;67(3):619–631. doi: 10.1016/j.jhep.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 26.Twisk J, et al. Heterogeneous expression of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase genes in the rat liver lobulus. J Clin Invest. 1995;95(3):1235–43. doi: 10.1172/JCI117773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turley SD, et al. Gender-related differences in bile acid and sterol metabolism in outbred CD-1 mice fed low- and high-cholesterol diets. Hepatology. 1998;28(4):1088–94. doi: 10.1002/hep.510280425. [DOI] [PubMed] [Google Scholar]
- 28.Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277(6):3801–4. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- 29.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56(6):1085–99. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ridlon JM, et al. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. doi: 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 32.Vaz FM, Ferdinandusse S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol Aspects Med. 2017;56:10–24. doi: 10.1016/j.mam.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50 Suppl:S120–5. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Botham KM, Boyd GS. The metabolism of chenodeoxycholic acid to beta-muricholic acid in rat liver. Eur J Biochem. 1983;134(1):191–6. doi: 10.1111/j.1432-1033.1983.tb07550.x. [DOI] [PubMed] [Google Scholar]
- 35.Pullinger CR, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110(1):109–17. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrell JM, et al. Cholesterol 7alpha-hydroxylase-deficient mice are protected from high-fat/high-cholesterol diet-induced metabolic disorders. J Lipid Res. 2016;57(7):1144–54. doi: 10.1194/jlr.M064709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosen H, et al. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem. 1998;273(24):14805–12. doi: 10.1074/jbc.273.24.14805. [DOI] [PubMed] [Google Scholar]
- 38.Dawson PA. Bile Acid Metabolism. Biochemistry of Lipids. Lipoproteins and Membranes. 2015:359. [Google Scholar]
- 39.Li-Hawkins J, et al. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J Biol Chem. 2000;275(22):16536–42. doi: 10.1074/jbc.M001811200. [DOI] [PubMed] [Google Scholar]
- 40.Repa JJ, et al. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem. 2000;275(50):39685–92. doi: 10.1074/jbc.M007653200. [DOI] [PubMed] [Google Scholar]
- 41.Lund EG, et al. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278(25):22980–8. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- 42.Hubbard B, et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology. 2006;130(4):1259–69. doi: 10.1053/j.gastro.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Ishibashi S, et al. Disruption of cholesterol 7alpha-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J Biol Chem. 1996;271(30):18017–23. doi: 10.1074/jbc.271.30.18017. [DOI] [PubMed] [Google Scholar]
- 44.Setchell KD, et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J Clin Invest. 1998;102(9):1690–703. doi: 10.1172/JCI2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shea HC, et al. Analysis of HSD3B7 knockout mice reveals that a 3alpha-hydroxyl stereochemistry is required for bile acid function. Proc Natl Acad Sci U S A. 2007;104(28):11526–33. doi: 10.1073/pnas.0705089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sayin SI, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(2):225–35. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi S, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57(12):2130–2137. doi: 10.1194/jlr.M071183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wahlstrom A, et al. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J Lipid Res. 2017;58(2):412–419. doi: 10.1194/jlr.M072819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Klaassen CD. Effects of feeding bile acids and a bile acid sequestrant on hepatic bile acid composition in mice. J Lipid Res. 2010;51(11):3230–42. doi: 10.1194/jlr.M007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alnouti Y. Bile Acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci. 2009;108(2):225–46. doi: 10.1093/toxsci/kfn268. [DOI] [PubMed] [Google Scholar]
- 51.Marschall HU, et al. Fxr(−/−) mice adapt to biliary obstruction by enhanced phase I detoxification and renal elimination of bile acids. J Lipid Res. 2006;47(3):582–92. doi: 10.1194/jlr.M500427-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 53.Odermatt A, et al. Hepatic reduction of the secondary bile acid 7-oxolithocholic acid is mediated by 11beta-hydroxysteroid dehydrogenase 1. Biochem J. 2011;436(3):621–9. doi: 10.1042/BJ20110022. [DOI] [PubMed] [Google Scholar]
- 54.Selwyn FP, et al. Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab Dispos. 2015;43(10):1544–56. doi: 10.1124/dmd.115.065276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bathena SP, et al. The profile of bile acids and their sulfate metabolites in human urine and serum. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;942–943:53–62. doi: 10.1016/j.jchromb.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 56.Setchell KD, et al. Hepatic bile acid metabolism during early development revealed from the analysis of human fetal gallbladder bile. J Biol Chem. 1988;263(32):16637–44. [PubMed] [Google Scholar]
- 57.Lee CS, et al. Prognostic roles of tetrahydroxy bile acids in infantile intrahepatic cholestasis. J Lipid Res. 2017;58(3):607–614. doi: 10.1194/jlr.P070425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang DQ, et al. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am J Physiol Gastrointest Liver Physiol. 2003;285(3):G494–502. doi: 10.1152/ajpgi.00156.2003. [DOI] [PubMed] [Google Scholar]
- 59.Kunne C, et al. Defective bile salt biosynthesis and hydroxylation in mice with reduced cytochrome P450 activity. Hepatology. 2013;57(4):1509–17. doi: 10.1002/hep.26133. [DOI] [PubMed] [Google Scholar]
- 60.Montet JC, et al. Beta-Muricholic acid; potentiometric and cholesterol-dissolving properties. Biochim Biophys Acta. 1987;918(1):1–10. doi: 10.1016/0005-2760(87)90002-6. [DOI] [PubMed] [Google Scholar]
- 61.Jiang C, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125(1):386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez FJ, Jiang C, Patterson AD. An Intestinal Microbiota-Farnesoid X Receptor Axis Modulates Metabolic Disease. Gastroenterology. 2016;151(5):845–859. doi: 10.1053/j.gastro.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudling M. Understanding mouse bile acid formation: Is it time to unwind why mice and rats make unique bile acids? J Lipid Res. 2016;57(12):2097–2098. doi: 10.1194/jlr.C072876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang C, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertaggia E, et al. Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. Am J Physiol Endocrinol Metab. 2017;313(2):E121–E133. doi: 10.1152/ajpendo.00409.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaur A, et al. Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes. 2015;64(4):1168–79. doi: 10.2337/db14-0716. [DOI] [PubMed] [Google Scholar]
- 67.Li T, et al. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem. 2012;287(3):1861–73. doi: 10.1074/jbc.M111.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wahlström A, et al. Cyp3a11 is not essential for the formation of murine bile acids. Biochemistry and Biophysics Reports. 2017;10:70–75. doi: 10.1016/j.bbrep.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amuro Y, et al. Partial purification and characterization of 7 alpha-hydroxysteroid dehydrogenase from rat liver microsomes. Biochim Biophys Acta. 1987;917(1):101–7. doi: 10.1016/0005-2760(87)90289-x. [DOI] [PubMed] [Google Scholar]
- 70.Penno CA, et al. Impaired oxidoreduction by 11beta-hydroxysteroid dehydrogenase 1 results in the accumulation of 7-oxolithocholic acid. J Lipid Res. 2013;54(10):2874–83. doi: 10.1194/jlr.M042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hrycay E, et al. Hepatic bile acid metabolism and expression of cytochrome P450 and related enzymes are altered in Bsep (−/−) mice. Mol Cell Biochem. 2014;389(1–2):119–32. doi: 10.1007/s11010-013-1933-y. [DOI] [PubMed] [Google Scholar]
- 72.Fuchs CD, et al. Metabolic preconditioning protects BSEP/ABCB11(−/−) mice against cholestatic liver injury. J Hepatol. 2017;66(1):95–101. doi: 10.1016/j.jhep.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 73.Araya Z, Wikvall K. 6alpha-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes. Biochim Biophys Acta. 1999;1438(1):47–54. doi: 10.1016/s1388-1981(99)00031-1. [DOI] [PubMed] [Google Scholar]
- 74.Deo AK, Bandiera SM. Identification of human hepatic cytochrome p450 enzymes involved in the biotransformation of cholic and chenodeoxycholic acid. Drug Metab Dispos. 2008;36(10):1983–91. doi: 10.1124/dmd.108.022194. [DOI] [PubMed] [Google Scholar]
- 75.Hayes MA, et al. CYP3A Specifically Catalyzes 1beta-Hydroxylation of Deoxycholic Acid: Characterization and Enzymatic Synthesis of a Potential Novel Urinary Biomarker for CYP3A Activity. Drug Metab Dispos. 2016;44(9):1480–9. doi: 10.1124/dmd.116.070805. [DOI] [PubMed] [Google Scholar]
- 76.Haslewood GA. Recent developments in our knowledge of bile salts. Physiol Rev. 1955;35(1):178–96. doi: 10.1152/physrev.1955.35.1.178. [DOI] [PubMed] [Google Scholar]
- 77.Moschetta A, et al. A phylogenetic survey of biliary lipids in vertebrates. J Lipid Res. 2005;46(10):2221–32. doi: 10.1194/jlr.M500178-JLR200. [DOI] [PubMed] [Google Scholar]
- 78.Coleman R, et al. Membranes and bile formation. Composition of several mammalian biles and their membrane-damaging properties. Biochem J. 1979;178(1):201–8. doi: 10.1042/bj1780201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alnouti Y, I, Csanaky L, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;873(2):209–17. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Falany CN, et al. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J Biol Chem. 1994;269(30):19375–9. [PubMed] [Google Scholar]
- 81.Falany CN, et al. Cloning, expression, and chromosomal localization of mouse liver bile acid CoA:amino acid N-acyltransferase. J Lipid Res. 1997;38(6):1139–48. [PubMed] [Google Scholar]
- 82.Stephan ZF, Armstrong MJ, Hayes KC. Bile lipid alterations in taurine-depleted monkeys. Am J Clin Nutr. 1981;34(2):204–10. doi: 10.1093/ajcn/34.2.204. [DOI] [PubMed] [Google Scholar]
- 83.Fitzpatrick WJ, Zentler-Munro PL, Northfield TC. Ileal resection: effect of cimetidine and taurine on intrajejunal bile acid precipitation and lipid solubilisation. Gut. 1986;27(1):66–72. doi: 10.1136/gut.27.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardison WG, Grundy SM. Effect of bile acid conjugation pattern on bile acid metabolism in normal humans. Gastroenterology. 1983;84(3):617–20. [PubMed] [Google Scholar]
- 85.He D, Barnes S, Falany CN. Rat liver bile acid CoA:amino acid N-acyltransferase: expression, characterization, and peroxisomal localization. J Lipid Res. 2003;44(12):2242–9. doi: 10.1194/jlr.M300128-JLR200. [DOI] [PubMed] [Google Scholar]
- 86.Warskulat U, et al. Chronic liver disease is triggered by taurine transporter knockout in the mouse. FASEB J. 2006;20(3):574–6. doi: 10.1096/fj.05-5016fje. [DOI] [PubMed] [Google Scholar]
- 87.Stipanuk MH, et al. Identification of Taurine-Responsive Genes in Murine Liver Using the Cdo1-Null Mouse Model. Adv Exp Med Biol. 2017;975:475–495. doi: 10.1007/978-94-024-1079-2_38. [DOI] [PubMed] [Google Scholar]
- 88.Ellis EC, et al. Mice with chimeric livers are an improved model for human lipoprotein metabolism. PLoS One. 2013;8(11):e78550. doi: 10.1371/journal.pone.0078550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Czuba B, Vessey DA. Identification of a unique mammalian species of cholyl-CoA: amino acid N-acyltransferase. Biochim Biophys Acta. 1981;665(3):612–4. doi: 10.1016/0005-2760(81)90278-2. [DOI] [PubMed] [Google Scholar]
- 90.Kwakye JB, et al. A comparative study of bile acid CoA:amino acid:N-acyltransferase (BAT) from four mammalian species. Comp Biochem Physiol B. 1991;100(1):131–6. doi: 10.1016/0305-0491(91)90095-u. [DOI] [PubMed] [Google Scholar]
- 91.O’Maille ER, Richards AH, Short TG. Acute taurine depletion and maximal rates of hepatic conjugation and secretion of cholic acid in the dog. J Physiol. 1965;180(1):67–79. [PMC free article] [PubMed] [Google Scholar]
- 92.Huijghebaert SM, Hofmann AF. Pancreatic carboxypeptidase hydrolysis of bile acid-amino conjugates: selective resistance of glycine and taurine amidates. Gastroenterology. 1986;90(2):306–15. doi: 10.1016/0016-5085(86)90925-x. [DOI] [PubMed] [Google Scholar]
- 93.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65(16):2461–83. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kamp F, et al. Movement of fatty acids, fatty acid analogues, and bile acids across phospholipid bilayers. Biochemistry. 1993;32(41):11074–86. doi: 10.1021/bi00092a017. [DOI] [PubMed] [Google Scholar]
- 95.Sharma R, et al. Bile acid toxicity structure-activity relationships: correlations between cell viability and lipophilicity in a panel of new and known bile acids using an oesophageal cell line (HET-1A) Bioorg Med Chem. 2010;18(18):6886–95. doi: 10.1016/j.bmc.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 96.Woolbright BL, et al. Bile Acid-Induced Toxicity in HepaRG Cells Recapitulates the Response in Primary Human Hepatocytes. Basic Clin Pharmacol Toxicol. 2016;118(2):160–7. doi: 10.1111/bcpt.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paulusma CC, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44(1):195–204. doi: 10.1002/hep.21212. [DOI] [PubMed] [Google Scholar]
- 98.Pawlikowska L, et al. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum Mol Genet. 2004;13(8):881–92. doi: 10.1093/hmg/ddh100. [DOI] [PubMed] [Google Scholar]
- 99.Wang R, et al. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A. 2001;98(4):2011–6. doi: 10.1073/pnas.031465498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eyssen HJ, De Pauw G, Van Eldere J. Formation of hyodeoxycholic acid from muricholic acid and hyocholic acid by an unidentified gram-positive rod termed HDCA-1 isolated from rat intestinal microflora. Appl Environ Microbiol. 1999;65(7):3158–63. doi: 10.1128/aem.65.7.3158-3163.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36(3–4):703–22. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 102.Subbiah MT, Kuksis A, Mookerjea S. Secretion of bile salts by intact and isolated rat livers. Can J Biochem. 1969;47(9):847–54. doi: 10.1139/o69-133. [DOI] [PubMed] [Google Scholar]
- 103.Wang DQ, Paigen B, Carey MC. Genetic factors at the enterocyte level account for variations in intestinal cholesterol absorption efficiency among inbred strains of mice. J Lipid Res. 2001;42(11):1820–30. [PubMed] [Google Scholar]
- 104.Schoenfield LJ, Sjovall J. Identification of bile acids and neutral sterols in guinea pig bile. Bile acids and steroids 163. Acta Chem Scand. 1966;20(5):1297–303. doi: 10.3891/acta.chem.scand.20-1297. [DOI] [PubMed] [Google Scholar]
- 105.Kuroki S, et al. Sex differences in gallbladder bile acid composition and hepatic steroid 12 alpha-hydroxylase activity in hamsters. J Lipid Res. 1983;24(12):1543–9. [PubMed] [Google Scholar]
- 106.Washizu T, et al. Bile acid composition of dog and cat gall-bladder bile. Nippon Juigaku Zasshi. 1990;52(2):423–5. doi: 10.1292/jvms1939.52.423. [DOI] [PubMed] [Google Scholar]
- 107.Carulli N, et al. Review article: effect of bile salt pool composition on hepatic and biliary functions. Aliment Pharmacol Ther. 2000;14 Suppl 2:14–8. doi: 10.1046/j.1365-2036.2000.014s2014.x. [DOI] [PubMed] [Google Scholar]
- 108.Murakami K, Okuda K. Purification and characterization of taurodeoxycholate 7 alpha-monooxygenase in rat liver. J Biol Chem. 1981;256(16):8658–62. [PubMed] [Google Scholar]
- 109.Ogishima T, Deguchi S, Okuda K. Purification and characterization of cholesterol 7 alpha-hydroxylase from rat liver microsomes. J Biol Chem. 1987;262(16):7646–50. [PubMed] [Google Scholar]
- 110.Yamashita H, Kuroki S, Nakayama F. Deoxycholate 7 alpha-hydroxylase in the hamster: substrate specificity and effect of phenobarbital. J Lipid Res. 1989;30(5):711–8. [PubMed] [Google Scholar]
- 111.Feng L, et al. Identification and characterization of a novel PPARalpha-regulated and 7alpha-hydroxyl bile acid-preferring cytosolic sulfotransferase mL-STL (Sult2a8) J Lipid Res. 2017;58(6):1114–1131. doi: 10.1194/jlr.M074302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dawson PA, Setchell KDR. Will the real bile acid sulfotransferase please stand up? Identification of Sult2a8 as a major hepatic bile acid sulfonating enzyme in mice. J Lipid Res. 2017;58(6):1033–1035. doi: 10.1194/jlr.C077420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hofmann AF. Why bile acid glucuronidation is a minor pathway for conjugation of endogenous bile acids in man. Hepatology. 2007;45(4):1083–4. doi: 10.1002/hep.21576. author reply 1084-5. [DOI] [PubMed] [Google Scholar]
- 114.Vu DD, et al. Pathogenesis of lithocholate-induced intrahepatic cholestasis: role of glucuronidation and hydroxylation of lithocholate. Biochim Biophys Acta. 1992;1126(1):53–9. doi: 10.1016/0005-2760(92)90216-i. [DOI] [PubMed] [Google Scholar]
- 115.Lee J, et al. Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology. 2001;121(6):1473–84. doi: 10.1053/gast.2001.29608. [DOI] [PubMed] [Google Scholar]
- 116.Schwarz M, et al. Disruption of cholesterol 7alpha-hydroxylase gene in mice. II. Bile acid deficiency is overcome by induction of oxysterol 7alpha-hydroxylase. J Biol Chem. 1996;271(30):18024–31. doi: 10.1074/jbc.271.30.18024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schwarz M, et al. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J Lipid Res. 2001;42(10):1594–603. [PubMed] [Google Scholar]
- 118.Erickson SK, et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7A1-deficient mice. J Lipid Res. 2003;44(5):1001–9. doi: 10.1194/jlr.M200489-JLR200. [DOI] [PubMed] [Google Scholar]
- 119.Li T, et al. Overexpression of cholesterol 7alpha-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011;53(3):996–1006. doi: 10.1002/hep.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li T, et al. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology. 2013;58(3):1111–21. doi: 10.1002/hep.26427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li-Hawkins J, et al. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J Clin Invest. 2002;110(8):1191–200. doi: 10.1172/JCI16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Slatis K, et al. Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J Lipid Res. 2010;51(11):3289–98. doi: 10.1194/jlr.M009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonde Y, Eggertsen G, Rudling M. Mice Abundant in Muricholic Bile Acids Show Resistance to Dietary Induced Steatosis, Weight Gain, and to Impaired Glucose Metabolism. PLoS One. 2016;11(1):e0147772. doi: 10.1371/journal.pone.0147772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dawson PA, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278(36):33920–7. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 125.Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids--from mouse models to human diseases. FEBS J. 2012;279(9):1516–33. doi: 10.1111/j.1742-4658.2011.08432.x. [DOI] [PubMed] [Google Scholar]
- 126.Cheng JB, et al. Molecular genetics of 3beta-hydroxy-Delta5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003;88(4):1833–41. doi: 10.1210/jc.2002-021580. [DOI] [PubMed] [Google Scholar]
- 127.Bavner A, et al. On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J Lipid Res. 2010;51(9):2722–30. doi: 10.1194/jlr.M008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Honda A, et al. Differences in hepatic levels of intermediates in bile acid biosynthesis between Cyp27(−/−) mice and CTX. J Lipid Res. 2001;42(2):291–300. [PubMed] [Google Scholar]
- 129.Honda A, et al. Side chain hydroxylations in bile acid biosynthesis catalyzed by CYP3A are markedly up-regulated in Cyp27−/− mice but not in cerebrotendinous xanthomatosis. J Biol Chem. 2001;276(37):34579–85. doi: 10.1074/jbc.M103025200. [DOI] [PubMed] [Google Scholar]
- 130.Goodwin B, et al. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A. 2003;100(1):223–8. doi: 10.1073/pnas.0237082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dai D, et al. Liver disease in infancy caused by oxysterol 7 alpha-hydroxylase deficiency: successful treatment with chenodeoxycholic acid. J Inherit Metab Dis. 2014;37(5):851–61. doi: 10.1007/s10545-014-9695-6. [DOI] [PubMed] [Google Scholar]
- 132.Tsaousidou MK, et al. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am J Hum Genet. 2008;82(2):510–5. doi: 10.1016/j.ajhg.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stiles AR, et al. CYP7B1: one cytochrome P450, two human genetic diseases, and multiple physiological functions. J Biol Chem. 2009;284(42):28485–9. doi: 10.1074/jbc.R109.042168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li-Hawkins J, et al. Expression cloning of an oxysterol 7alpha-hydroxylase selective for 24-hydroxycholesterol. J Biol Chem. 2000;275(22):16543–9. doi: 10.1074/jbc.M001810200. [DOI] [PubMed] [Google Scholar]
- 135.Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br J Clin Pharmacol. 2001;52(4):349–55. doi: 10.1046/j.0306-5251.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Scheer N, et al. Generation and characterization of novel cytochrome P450 Cyp2c gene cluster knockout and CYP2C9 humanized mouse lines. Mol Pharmacol. 2012;82(6):1022–9. doi: 10.1124/mol.112.080036. [DOI] [PubMed] [Google Scholar]
- 137.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340–57. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mita S, et al. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G550–6. doi: 10.1152/ajpgi.00364.2005. [DOI] [PubMed] [Google Scholar]
- 139.Megaraj V, et al. Hepatobiliary disposition of 3alpha, 6alpha, 7alpha, 12alpha-tetrahydroxy-cholanoyl taurine: a substrate for multiple canalicular transporters. Drug Metab Dispos. 2010;38(10):1723–30. doi: 10.1124/dmd.110.033480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zollner G, et al. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm. 2006;3(3):231–51. doi: 10.1021/mp060010s. [DOI] [PubMed] [Google Scholar]
- 141.Belinsky MG, et al. Analysis of the in vivo functions of Mrp3. Mol Pharmacol. 2005;68(1):160–8. doi: 10.1124/mol.104.010587. [DOI] [PubMed] [Google Scholar]
- 142.Mennone A, et al. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43(5):1013–21. doi: 10.1002/hep.21158. [DOI] [PubMed] [Google Scholar]
- 143.Boyer JL, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1124–30. doi: 10.1152/ajpgi.00539.2005. [DOI] [PubMed] [Google Scholar]
- 144.Soroka CJ, et al. Mouse organic solute transporter alpha deficiency enhances renal excretion of bile acids and attenuates cholestasis. Hepatology. 2010;51(1):181–90. doi: 10.1002/hep.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van de Steeg E, et al. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest. 2010;120(8):2942–52. doi: 10.1172/JCI42168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vasilyeva A, et al. Hepatocellular Shuttling and Recirculation of Sorafenib-Glucuronide Is Dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 2015;75(13):2729–36. doi: 10.1158/0008-5472.CAN-15-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wagner M, Zollner G, Trauner M. New molecular insights into the mechanisms of cholestasis. J Hepatol. 2009;51(3):565–80. doi: 10.1016/j.jhep.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 148.Orntoft NW, et al. Hepatobiliary transport kinetics of the conjugated bile acid tracer 11C-CSar quantified in healthy humans and patients by positron emission tomography. J Hepatol. 2017;67(2):321–327. doi: 10.1016/j.jhep.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 149.Hirohashi T, et al. ATP-dependent transport of bile salts by rat multidrug resistance-associated protein 3 (Mrp3) J Biol Chem. 2000;275(4):2905–10. doi: 10.1074/jbc.275.4.2905. [DOI] [PubMed] [Google Scholar]
- 150.Zeng H, et al. Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res. 2000;60(17):4779–84. [PubMed] [Google Scholar]
- 151.Zelcer N, et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J Hepatol. 2006;44(4):768–75. doi: 10.1016/j.jhep.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 152.Slijepcevic D, et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology. 2017;66(5):1631–1643. doi: 10.1002/hep.29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609(1):1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 154.Dawson PA, Hubbert ML, Rao A. Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim Biophys Acta. 2010;1801(9):994–1004. doi: 10.1016/j.bbalip.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Krag E, Phillips SF. Active and passive bile acid absorption in man. Perfusion studies of the ileum and jejunum. J Clin Invest. 1974;53(6):1686–94. doi: 10.1172/JCI107720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fiamoncini J, et al. Determinants of postprandial plasma bile acid kinetics in human volunteers. Am J Physiol Gastrointest Liver Physiol. 2017;313(4):G300–G312. doi: 10.1152/ajpgi.00157.2017. [DOI] [PubMed] [Google Scholar]
- 157.Hagenbuch B, et al. Functional expression cloning and characterization of the hepatocyte Na+/bile acid cotransport system. Proc Natl Acad Sci U S A. 1991;88(23):10629–33. doi: 10.1073/pnas.88.23.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stieger B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb Exp Pharmacol. 2011;(201):205–59. doi: 10.1007/978-3-642-14541-4_5. [DOI] [PubMed] [Google Scholar]
- 159.Vaz FM, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology. 2015;61(1):260–7. doi: 10.1002/hep.27240. [DOI] [PubMed] [Google Scholar]
- 160.Dawson PA. Hepatic bile acid uptake in humans and mice: Multiple pathways and expanding potential role for gut-liver signaling. Hepatology. 2017;66(5):1384–1386. doi: 10.1002/hep.29325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Blank A, et al. The NTCP-inhibitor Myrcludex B: Effects on Bile Acid Disposition and Tenofovir Pharmacokinetics. Clin Pharmacol Ther. 2018;103(2):341–348. doi: 10.1002/cpt.744. [DOI] [PubMed] [Google Scholar]
- 162.Slijepcevic D, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology. 2015;62(1):207–19. doi: 10.1002/hep.27694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cravetto C, et al. Computer simulation of portal venous shunting and other isolated hepatobiliary defects of the enterohepatic circulation of bile acids using a physiological pharmacokinetic model. Hepatology. 1988;8(4):866–78. doi: 10.1002/hep.1840080428. [DOI] [PubMed] [Google Scholar]
- 164.Lam P, Wang R, Ling V. Bile acid transport in sister of P-glycoprotein (ABCB11) knockout mice. Biochemistry. 2005;44(37):12598–605. doi: 10.1021/bi050943e. [DOI] [PubMed] [Google Scholar]
- 165.Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflugers Arch. 2007;453(5):611–20. doi: 10.1007/s00424-006-0152-8. [DOI] [PubMed] [Google Scholar]
- 166.Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011;31(1):3–10. doi: 10.1055/s-0031-1272831. [DOI] [PubMed] [Google Scholar]
- 167.Strautnieks SS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20(3):233–8. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 168.Bull LN, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18(3):219–24. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 169.Lam P, et al. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Physiol Cell Physiol. 2007;293(5):C1709–16. doi: 10.1152/ajpcell.00327.2007. [DOI] [PubMed] [Google Scholar]
- 170.Dixon PH, et al. Contribution of variant alleles of ABCB11 to susceptibility to intrahepatic cholestasis of pregnancy. Gut. 2009;58(4):537–44. doi: 10.1136/gut.2008.159541. [DOI] [PubMed] [Google Scholar]
- 171.Dixon PH, et al. A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am J Gastroenterol. 2014;109(1):76–84. doi: 10.1038/ajg.2013.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dawson S, et al. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos. 2012;40(1):130–8. doi: 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- 173.Aleo MD, et al. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology. 2014;60(3):1015–22. doi: 10.1002/hep.27206. [DOI] [PubMed] [Google Scholar]
- 174.Wang R, et al. Compensatory role of P-glycoproteins in knockout mice lacking the bile salt export pump. Hepatology. 2009;50(3):948–56. doi: 10.1002/hep.23089. [DOI] [PubMed] [Google Scholar]
- 175.Yan H, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lempp FA, et al. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology. 2017;66(3):703–716. doi: 10.1002/hep.29112. [DOI] [PubMed] [Google Scholar]
- 177.Donkers JM, et al. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter. NTCP Sci Rep. 2017;7(1):15307. doi: 10.1038/s41598-017-15338-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.He W, et al. Hepatitis D Virus Infection of Mice Expressing Human Sodium Taurocholate Co-transporting Polypeptide. PLoS Pathog. 2015;11(4):e1004840. doi: 10.1371/journal.ppat.1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447(5):653–65. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 180.van de Steeg E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122(2):519–28. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cheng X, et al. Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps) Drug Metab Dispos. 2005;33(7):1062–73. doi: 10.1124/dmd.105.003640. [DOI] [PubMed] [Google Scholar]
- 182.Csanaky IL, et al. Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology. 2011;53(1):272–81. doi: 10.1002/hep.23984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Durmus S, van Hoppe S, Schinkel AH. The impact of Organic Anion-Transporting Polypeptides (OATPs) on disposition and toxicity of antitumor drugs: Insights from knockout and humanized mice. Drug Resist Updat. 2016;27:72–88. doi: 10.1016/j.drup.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 184.Wang R, et al. Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatology. 2003;38(6):1489–99. doi: 10.1016/j.hep.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Y, et al. Abcb11 deficiency induces cholestasis coupled to impaired beta-fatty acid oxidation in mice. J Biol Chem. 2012;287(29):24784–94. doi: 10.1074/jbc.M111.329318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Oude Elferink RP, et al. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95(1):31–8. doi: 10.1172/JCI117658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fickert P, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127(1):261–74. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 188.Smit JJ, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75(3):451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 189.Popov Y, et al. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43(6):1045–54. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 190.Mariotti V, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbadis.2017.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.de Vree JM, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95(1):282–7. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. 2007;27(1):77–98. doi: 10.1055/s-2006-960172. [DOI] [PubMed] [Google Scholar]
- 193.Fickert P, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130(2):465–81. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 194.Fickert P, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123(4):1238–51. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 195.Jones H, et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(−/−) mice. Hepatology. 2016;64(4):1202–1216. doi: 10.1002/hep.28704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Halilbasic E, et al. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2−/− mice. Hepatology. 2009;49(6):1972–81. doi: 10.1002/hep.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005;33(1):181–4. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- 198.Katzenellenbogen M, et al. Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol Cancer Res. 2007;5(11):1159–70. doi: 10.1158/1541-7786.MCR-07-0172. [DOI] [PubMed] [Google Scholar]
- 199.Mu X, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125(10):3891–903. doi: 10.1172/JCI77995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fickert P, et al. Characterization of animal models for primary sclerosing cholangitis (PSC) J Hepatol. 2014;60(6):1290–303. doi: 10.1016/j.jhep.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ujhazy P, et al. Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology. 2001;34(4 Pt 1):768–75. doi: 10.1053/jhep.2001.27663. [DOI] [PubMed] [Google Scholar]
- 202.Tang X, et al. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996;272(5267):1495–7. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- 203.Groen A, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. 2011;141(5):1927–37. e1–4. doi: 10.1053/j.gastro.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 204.Bull LN, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity. Hepatology. 1997;26(1):155–64. doi: 10.1002/hep.510260121. [DOI] [PubMed] [Google Scholar]
- 205.Knisely AS. Progressive familial intrahepatic cholestasis: a personal perspective. Pediatr Dev Pathol. 2000;3(2):113–25. doi: 10.1007/s100240050016. [DOI] [PubMed] [Google Scholar]
- 206.Chen HL, et al. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low gamma-glutamyltranspeptidase levels. J Pediatr. 2002;140(1):119–24. doi: 10.1067/mpd.2002.119993. [DOI] [PubMed] [Google Scholar]
- 207.Siggs OM, et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol. 2011;12(5):434–40. doi: 10.1038/ni.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Siggs OM, et al. X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc Natl Acad Sci U S A. 2011;108(19):7890–5. doi: 10.1073/pnas.1104631108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.de Waart DR, et al. ATP11C targets basolateral bile salt transporter proteins in mouse central hepatocytes. Hepatology. 2016;64(1):161–74. doi: 10.1002/hep.28522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Arashiki N, et al. ATP11C is a major flippase in human erythrocytes and its defect causes congenital hemolytic anemia. Haematologica. 2016;101(5):559–65. doi: 10.3324/haematol.2016.142273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Paulusma CC, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47(1):268–78. doi: 10.1002/hep.21950. [DOI] [PubMed] [Google Scholar]
- 212.Zhang L, et al. Loss of Tmem30a leads to photoreceptor degeneration. Sci Rep. 2017;7(1):9296. doi: 10.1038/s41598-017-09506-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liu L, et al. Hepatic Tmem30a Deficiency Causes Intrahepatic Cholestasis by Impairing Expression and Localization of Bile Salt Transporters. Am J Pathol. 2017;187(12):2775–2787. doi: 10.1016/j.ajpath.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 214.Balistreri WF, Heubi JE, Suchy FJ. Bile acid metabolism: relationship of bile acid malabsorption and diarrhea. J Pediatr Gastroenterol Nutr. 1983;2(1):105–21. [PubMed] [Google Scholar]
- 215.Heubi JE, et al. Treatment of bile acid amidation defects with glycocholic acid. Hepatology. 2015;61(1):268–74. doi: 10.1002/hep.27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Out C, et al. Gut microbiota inhibit Asbt-dependent intestinal bile acid reabsorption via Gata4. J Hepatol. 2015;63(3):697–704. doi: 10.1016/j.jhep.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lan T, Haywood J, Dawson PA. Inhibition of ileal apical but not basolateral bile acid transport reduces atherosclerosis in apoE(-)/(-) mice. Atherosclerosis. 2013;229(2):374–80. doi: 10.1016/j.atherosclerosis.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ferrebee CB, et al. Organic Solute Transporter α-β Protects Ileal Enterocytes From Bile Acid–Induced Injury. Cellular and Molecular Gastroenterology and. Hepatology. 2018;5(4):499–522. doi: 10.1016/j.jcmgh.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Aldini R, et al. Intestinal absorption of bile acids in the rabbit: different transport rates in jejunum and ileum. Gastroenterology. 1996;110(2):459–68. doi: 10.1053/gast.1996.v110.pm8566593. [DOI] [PubMed] [Google Scholar]
- 220.Aldini R, et al. Bile acid active and passive ileal transport in the rabbit: effect of luminal stirring. Eur J Clin Invest. 1992;22(11):744–50. doi: 10.1111/j.1365-2362.1992.tb01439.x. [DOI] [PubMed] [Google Scholar]
- 221.Dawson PA. Impact of Inhibiting Ileal Apical versus Basolateral Bile Acid Transport on Cholesterol Metabolism and Atherosclerosis in Mice. Dig Dis. 2015;33(3):382–7. doi: 10.1159/000371691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Miethke AG, et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. 2016;63(2):512–23. doi: 10.1002/hep.27973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rao A, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med. 2016;8(357):357ra122. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rao A, et al. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105(10):3891–6. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ballatori N, et al. Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am J Physiol Gastrointest Liver Physiol. 2008;295(1):G179–G186. doi: 10.1152/ajpgi.90319.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lan T, et al. Mouse organic solute transporter alpha deficiency alters FGF15 expression and bile acid metabolism. J Hepatol. 2012;57(2):359–65. doi: 10.1016/j.jhep.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sultan M, et al. Organic Solute Transporter-beta (SLC51B) Deficiency in Two Brothers with Congenital Diarrhea and Features of Cholestasis. Hepatology. 2017 doi: 10.1002/hep.29516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chow EC, et al. Disrupted Murine Gut-to-Human Liver Signaling Alters Bile Acid Homeostasis in Humanized Mouse Liver Models. J Pharmacol Exp Ther. 2017;360(1):174–191. doi: 10.1124/jpet.116.236935. [DOI] [PubMed] [Google Scholar]
- 229.Naugler WE, et al. Fibroblast Growth Factor Signaling Controls Liver Size in Mice With Humanized Livers. Gastroenterology. 2015;149(3):728–40e15. doi: 10.1053/j.gastro.2015.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Scheer N, Roland Wolf C. Xenobiotic receptor humanized mice and their utility. Drug Metab Rev. 2013;45(1):110–21. doi: 10.3109/03602532.2012.738687. [DOI] [PubMed] [Google Scholar]
- 231.Scheer N, Wolf CR. Genetically humanized mouse models of drug metabolizing enzymes and transporters and their applications. Xenobiotica. 2014;44(2):96–108. doi: 10.3109/00498254.2013.815831. [DOI] [PubMed] [Google Scholar]
- 232.Scheer N, Wilson ID. A comparison between genetically humanized and chimeric liver humanized mouse models for studies in drug metabolism and toxicity. Drug Discov Today. 2016;21(2):250–63. doi: 10.1016/j.drudis.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 233.Nagamoto Y, et al. Efficient Engraftment of Human Induced Pluripotent Stem Cell-Derived Hepatocyte-Like Cells in uPA/SCID Mice by Overexpression of FNK, a Bcl-xL Mutant Gene. Cell Transplant. 2015;24(6):1127–38. doi: 10.3727/096368914X681702. [DOI] [PubMed] [Google Scholar]
- 234.Takebe T, et al. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc. 2014;9(2):396–409. doi: 10.1038/nprot.2014.020. [DOI] [PubMed] [Google Scholar]
- 235.Zhu S, et al. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature. 2014;508(7494):93–7. doi: 10.1038/nature13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bissig KD, et al. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest. 2010;120(3):924–30. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wilson EM, et al. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014;13(3 Pt A):404–12. doi: 10.1016/j.scr.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Barzi M, et al. A novel humanized mouse lacking murine P450 oxidoreductase for studying human drug metabolism. Nat Commun. 2017;8(1):39. doi: 10.1038/s41467-017-00049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tag CG, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 201596 doi: 10.3791/52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65(3):305–11. [PMC free article] [PubMed] [Google Scholar]
- 241.Krstulovic B, Van D, Desmet VJ. Comparative histochemical study of rat liver in bile-duct ligation and in alpha-napthyl isothiocyanate (ANIT) intoxication. Am J Pathol. 1968;52(2):423–36. [PMC free article] [PubMed] [Google Scholar]
- 242.Geerts AM, et al. Comparison of three research models of portal hypertension in mice: macroscopic, histological and portal pressure evaluation. Int J Exp Pathol. 2008;89(4):251–63. doi: 10.1111/j.1365-2613.2008.00597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Aller MA, et al. Bile duct ligation: step-by-step to cholangiocyte inflammatory tumorigenesis. Eur J Gastroenterol Hepatol. 2010;22(6):651–61. doi: 10.1097/MEG.0b013e32832e0a2f. [DOI] [PubMed] [Google Scholar]
- 244.Marques TG, et al. Review of experimental models for inducing hepatic cirrhosis by bile duct ligation and carbon tetrachloride injection. Acta Cir Bras. 2012;27(8):589–94. doi: 10.1590/s0102-86502012000800013. [DOI] [PubMed] [Google Scholar]
- 245.Popov Y, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298(3):G323–34. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178(1):175–86. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15(14):1677–89. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30(5):719–30. [PubMed] [Google Scholar]
- 249.Delzenne NM, et al. Comparative hepatotoxicity of cholic acid, deoxycholic acid and lithocholic acid in the rat: in vivo and in vitro studies. Toxicol Lett. 1992;61(2–3):291–304. doi: 10.1016/0378-4274(92)90156-e. [DOI] [PubMed] [Google Scholar]
- 250.Tsuda H, et al. Promotive effect of primary and secondary bile acids on the induction of gamma-glutamyl transpeptidase-positive liver cell foci as a possible endogenous factor for hepatocarcinogenesis in rats. Gan. 1984;75(10):871–5. [PubMed] [Google Scholar]
- 251.Song P, Zhang Y, Klaassen CD. Dose-response of five bile acids on serum and liver bile Acid concentrations and hepatotoxicty in mice. Toxicol Sci. 2011;123(2):359–67. doi: 10.1093/toxsci/kfr177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fickert P, et al. Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transporter expression in mouse liver. Gastroenterology. 2001;121(1):170–83. doi: 10.1053/gast.2001.25542. [DOI] [PubMed] [Google Scholar]
- 253.Lamireau T, et al. Dietary lecithin protects against cholestatic liver disease in cholic acid-fed Abcb4- deficient mice. Pediatr Res. 2007;61(2):185–90. doi: 10.1203/pdr.0b013e31802d7780. [DOI] [PubMed] [Google Scholar]
- 254.Palmer RH, Ruban Z. Production of bile duct hyperplasia and gallstones by lithocholic acid. J Clin Invest. 1966;45(8):1255–67. doi: 10.1172/JCI105432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kakis G, I, Yousef M. Pathogenesis of lithocholate- and taurolithocholate-induced intrahepatic cholestasis in rats. Gastroenterology. 1978;75(4):595–607. [PubMed] [Google Scholar]
- 256.Kakis G, Phillips MJ, Yousef IM. The respective roles of membrane cholesterol and of sodium potassium adenosine triphosphatase in the pathogenesis of lithocholate-induced cholestasis. Lab Invest. 1980;43(1):73–81. [PubMed] [Google Scholar]
- 257.Miyai K, et al. Subcellular pathology of rat liver in cholestasis and choleresis induced by bile salts. 1. Effects of lithocholic, 3beta-hydroxy-5-cholenoic, cholic, and dehydrocholic acids. Lab Invest. 1977;36(3):249–58. [PubMed] [Google Scholar]
- 258.Bonvicini F, et al. Cholesterol in acute cholestasis induced by taurolithocholic acid. A cytochemical study in transmission and scanning electron microscopy. Lab Invest. 1978;38(4):487–95. [PubMed] [Google Scholar]
- 259.Kubitz R, et al. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology. 2004;126(2):541–53. doi: 10.1053/j.gastro.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 260.Beuers U, et al. Taurolithocholic acid exerts cholestatic effects via phosphatidylinositol 3-kinase-dependent mechanisms in perfused rat livers and rat hepatocyte couplets. J Biol Chem. 2003;278(20):17810–8. doi: 10.1074/jbc.M209898200. [DOI] [PubMed] [Google Scholar]
- 261.Fickert P, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168(2):410–22. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Trauner M, I, Graziadei W. Review article: mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment Pharmacol Ther. 1999;13(8):979–96. doi: 10.1046/j.1365-2036.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 263.Poupon R, Poupon RE. Ursodeoxycholic acid therapy of chronic cholestatic conditions in adults and children. Pharmacol Ther. 1995;66(1):1–15. doi: 10.1016/0163-7258(94)00073-c. [DOI] [PubMed] [Google Scholar]
- 264.Beuers U, Boyer JL, Paumgartner G. Ursodeoxycholic acid in cholestasis: potential mechanisms of action and therapeutic applications. Hepatology. 1998;28(6):1449–53. doi: 10.1002/hep.510280601. [DOI] [PubMed] [Google Scholar]
- 265.Van Nieuwkerk CM, et al. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology. 1996;111(1):165–71. doi: 10.1053/gast.1996.v111.pm8698195. [DOI] [PubMed] [Google Scholar]
- 266.van Nieuwerk CM, et al. The role of bile salt composition in liver pathology of mdr2 (−/−) mice: differences between males and females. J Hepatol. 1997;26(1):138–45. doi: 10.1016/s0168-8278(97)80020-7. [DOI] [PubMed] [Google Scholar]
- 267.Zollner G, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. 2003;39(4):480–8. doi: 10.1016/s0168-8278(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 268.Hermankova E, et al. Polymeric bile acid sequestrants: Review of design, in vitro binding activities, and hypocholesterolemic effects. Eur J Med Chem. 2017;144:300–317. doi: 10.1016/j.ejmech.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 269.Catapano AL, et al. ESC/EAS Guidelines for the management of dyslipidaemias The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Atherosclerosis. 2011;217(1):3–46. doi: 10.1016/j.atherosclerosis.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 270.Insull W., Jr Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J. 2006;99(3):257–73. doi: 10.1097/01.smj.0000208120.73327.db. [DOI] [PubMed] [Google Scholar]
- 271.The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251(3):351–64. doi: 10.1001/jama.1984.03340270029025. [DOI] [PubMed] [Google Scholar]
- 272.Hofmann AF, Poley JR. Cholestyramine treatment of diarrhea associated with ileal resection. N Engl J Med. 1969;281(8):397–402. doi: 10.1056/NEJM196908212810801. [DOI] [PubMed] [Google Scholar]
- 273.Borghede MK, et al. Bile acid malabsorption investigated by selenium-75-homocholic acid taurine ((75)SeHCAT) scans: causes and treatment responses to cholestyramine in 298 patients with chronic watery diarrhoea. Eur J Intern Med. 2011;22(6):e137–40. doi: 10.1016/j.ejim.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 274.Kuiper EM, et al. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: results of a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52(4):1334–40. doi: 10.1002/hep.23821. [DOI] [PubMed] [Google Scholar]
- 275.Kronsten V, Fitzpatrick E, Baker A. Management of cholestatic pruritus in paediatric patients with alagille syndrome: the King’s College Hospital experience. J Pediatr Gastroenterol Nutr. 2013;57(2):149–54. doi: 10.1097/MPG.0b013e318297e384. [DOI] [PubMed] [Google Scholar]
- 276.Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129(3):894–901. doi: 10.1053/j.gastro.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 277.Bijleveld CM, et al. Benign recurrent intrahepatic cholestasis: a long-term follow-up study of two patients. Hepatology. 1989;9(4):532–7. doi: 10.1002/hep.1840090404. [DOI] [PubMed] [Google Scholar]
- 278.Polter DE, et al. Beneficial effect of cholestyramine in sclerosing cholangitis. Gastroenterology. 1980;79(2):326–33. [PubMed] [Google Scholar]
- 279.Out C, Groen AK, Brufau G. Bile acid sequestrants: more than simple resins. Curr Opin Lipidol. 2012;23(1):43–55. doi: 10.1097/MOL.0b013e32834f0ef3. [DOI] [PubMed] [Google Scholar]
- 280.Herrema H, et al. Bile salt sequestration induces hepatic de novo lipogenesis through farnesoid X receptor- and liver X receptor alpha-controlled metabolic pathways in mice. Hepatology. 2010;51(3):806–16. doi: 10.1002/hep.23408. [DOI] [PubMed] [Google Scholar]
- 281.Dubrac S, et al. Role of CYP27A in cholesterol and bile acid metabolism. J Lipid Res. 2005;46(1):76–85. doi: 10.1194/jlr.M400219-JLR200. [DOI] [PubMed] [Google Scholar]
- 282.Meir K, et al. Human sterol 27-hydroxylase (CYP27) overexpressor transgenic mouse model. Evidence against 27-hydroxycholesterol as a critical regulator of cholesterol homeostasis. J Biol Chem. 2002;277(37):34036–41. doi: 10.1074/jbc.M201122200. [DOI] [PubMed] [Google Scholar]
- 283.Rose K, et al. Neurosteroid hydroxylase CYP7B: vivid reporter activity in dentate gyrus of gene-targeted mice and abolition of a widespread pathway of steroid and oxysterol hydroxylation. J Biol Chem. 2001;276(26):23937–44. doi: 10.1074/jbc.M011564200. [DOI] [PubMed] [Google Scholar]
- 284.Omoto Y, et al. Early onset of puberty and early ovarian failure in CYP7B1 knockout mice. Proc Natl Acad Sci U S A. 2005;102(8):2814–9. doi: 10.1073/pnas.0500198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Figge A, et al. Hepatic overexpression of murine Abcb11 increases hepatobiliary lipid secretion and reduces hepatic steatosis. J Biol Chem. 2004;279(4):2790–9. doi: 10.1074/jbc.M307363200. [DOI] [PubMed] [Google Scholar]
- 286.Henkel AS, et al. Hepatic overexpression of abcb11 promotes hypercholesterolemia and obesity in mice. Gastroenterology. 2011;141(4):1404–11. 1411e1–2. doi: 10.1053/j.gastro.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Henkel AS, et al. Hepatic overexpression of Abcb11 in mice promotes the conservation of bile acids within the enterohepatic circulation. Am J Physiol Gastrointest Liver Physiol. 2013;304(2):G221–6. doi: 10.1152/ajpgi.00322.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Groen A, et al. Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology. 2008;134(7):2091–100. doi: 10.1053/j.gastro.2008.02.097. [DOI] [PubMed] [Google Scholar]
- 289.Lammert F, et al. Spontaneous cholecysto- and hepatolithiasis in Mdr2−/− mice: a model for low phospholipid-associated cholelithiasis. Hepatology. 2004;39(1):117–28. doi: 10.1002/hep.20022. [DOI] [PubMed] [Google Scholar]
- 290.Raufman JP, et al. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis. 2015;36(10):1193–200. doi: 10.1093/carcin/bgv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Li J, et al. Sortilin 1 Loss-of-Function Protects Against Cholestatic Liver Injury by Attenuating Hepatic Bile Acid Accumulation in Bile Duct Ligated Mice. Toxicol Sci. 2018;161(1):34–47. doi: 10.1093/toxsci/kfx078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Murphy C, et al. Cholic acid as key regulator of cholesterol synthesis, intestinal absorption and hepatic storage in mice. Biochim Biophys Acta. 2005;1735(3):167–75. doi: 10.1016/j.bbalip.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 293.Soroka CJ, et al. Ostalpha depletion protects liver from oral bile acid load. Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G574–9. doi: 10.1152/ajpgi.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Woolbright BL, et al. Lithocholic acid feeding results in direct hepato-toxicity independent of neutrophil function in mice. Toxicol Lett. 2014;228(1):56–66. doi: 10.1016/j.toxlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Fickert P, et al. Differential effects of norUDCA and UDCA in obstructive cholestasis in mice. J Hepatol. 2013;58(6):1201–8. doi: 10.1016/j.jhep.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Out C, et al. Liver receptor homolog-1 is critical for adequate up-regulation of Cyp7a1 gene transcription and bile salt synthesis during bile salt sequestration. Hepatology. 2011;53(6):2075–85. doi: 10.1002/hep.24286. [DOI] [PubMed] [Google Scholar]