

Abstract
Objective
We aimed to delineate the distribution of periventricular nodular heterotopia (PNH) in patients with 22q11.2 microdeletion syndrome (22q11.2DS) and place this in the context of other genetic forms of PNH.
Methods
We retrospectively analyzed brain imaging and postmortem data available for adult patients with 22q11.2DS. We included only those with good quality MRI data (n = 29) in addition to two patients with PNH identified through postmortem studies. We also reviewed the pattern of PNH in all genetic conditions reported with this phenotype.
Results
Of the total seven patients (M = 4, F = 3; age: 19–61 years) identified to have PNH, six had a history of seizures, six had schizophrenia, six had variable levels of intellectual disability, and two had obsessive compulsive disorder. In all seven patients, the nodules were located over the dorsal pole of the frontal horn of the lateral ventricles. The nodules were small, noncontiguous, and ranged in number from 1 to 10 per individual. Our review identified 37 genetic conditions associated with PNH. With the cases reported here, 22q11.2DS becomes the fifth most commonly reported genetic condition, and the third most common copy number variation, associated with PNH.
Interpretation
The neuropsychiatric manifestations in our patients with PNH support other data indicating abnormal neurodevelopment as part of the pathogenesis of 22q11.2DS.The location and cellular characteristics of PNH in 22q11.2DS overlaps with a group of migrating postnatal interneurons termed Arc cells, although more research is needed to confirm that PNH in 22q11.2DS represents Arc cells arrested in their migratory pathway.
Introduction
22q11.2 microdeletion syndrome (22q11.2DS), which encompasses conditions formerly known as velocardiofacial syndrome or DiGeorge syndrome, is the most common pathogenic copy number variation (CNV) in humans, affecting about 1 in 4000 people.1 In adults with 22q11.2DS neuropsychiatric disorders such as intellectual disability, seizures and schizophrenia are a significant cause of morbidity.2, 3, 4
The prevalence of recurrent seizure disorder in adults with 22q11.2DS is estimated to be about 11%.4 Seizures may be triggered by hypocalcemia, fever, and/or antipsychotic medications. Epilepsy is reported in patients with epileptogenic structural brain abnormalities, such as periventricular nodular heterotopia (PNH), polymicrogyria, etc. In many cases, however, no additional risk factors can be identified, and the unprovoked, recurrent seizures are thought to result from genetic generalized epilepsy or focal epilepsy.4, 5, 6, 7
Schizophrenia is seen in up to 25% of 22q11.2DS patients.8 Comprising up to 1% of all schizophrenia patients, 22q11.2DS is the most common detectable genetic cause of schizophrenia.9 The pathophysiological mechanisms leading to schizophrenia are believed to be related to abnormal neural development.10
Identifying PNH in the first postmortem pathology of a patient with 22q11.2DS11 prompted us to study the presence of these lesions through high‐resolution brain imaging. In the current, we set out to study the anatomical characteristics of PNH and explore its association with the neuropsychiatric manifestations of 22q11.2DS patients.
Methods
Patients
Adult 22q11.2DS patients were recruited from the epilepsy genetics clinic at Toronto Western Hospital, University of Toronto, and were further screened for the presence of PNH. Of 35 patients who had brain MRI data, 6 were excluded due to poor quality images. We included two additional patients with 22q11.2DS, whose postmortem studies were previously reported by our team.11, 12 Demographic and clinical features of the 31 patients were obtained from chart reviews and included age, sex, and comprehensive neuropsychiatric history. The diagnosis of 22q11.2DS for these patients was previously established by FISH or clinical microarray, and their consent for inclusion in the study was obtained as per research ethics board (REB) protocols at the University Health Network and/or Centre for Addiction and Mental Health.
Imaging
22q11.2DS patients underwent 3 Tesla brain MRI with a dedicated seizure protocol including high‐resolution axial and coronal T1 IR and T2 FLAIR sequences as well as isotropic T1 FSPGR sequences with multiplanar reformats.13 In addition, axial DWI and SWI‐weighted sequences were obtained. Images were reviewed in consensus by a neuroradiologist specialized in epilepsy imaging (TK) and 2 neurologists (AR, DA).
Additional genetic studies
In addition to standard clinical features and the FISH confirmation of typical 22q11.2 deletion in all 7 patients, five patients also had whole genome sequencing (WGS) as part of another study.14 The WGS data was used in this study only to determine if patients with PNH had other structural genomic rearrangements such as CNV or single nucleotide variant (SNV) previously associated with known causes of PNH. A sixth patient had genome‐wide high‐resolution research microarray.
Literature review
We used PubMed (January 1, 1993 to February 20, 2018) to review the literature regarding causes of PNH and its distribution, using the following terms: ((periventricular or ventricular or ventricle or ventricles) (nodular or nodule or nodules) (heterotopia or heterotopic)) AND (deletion or duplication or (copy number) or chromosomal or chromosome or syndrome or syndromic or syndromal or genetic or genomic or gene or mutation).
Results
Of the 29 alive patients ascertained with usable MRI data, five (17.2%) had PNH (Fig. 1A–E). Three of these 5 patients had been reported previously by our team. (patients 1, 2 and 4).6 For one of the two patients in whom PNH was identified through autopsy,11, 12 MRI data had previously been interpreted as normal; however, re‐review of MRI after postmortem findings confirmed the diagnosis of PNH (Fig. 1F). Of the seven patients with PNH studied, three were female. Intellectual disability (ID) was present in six, ranging from mild to severe. Six of these seven patients had a history of acute symptomatic seizures or epilepsy. In these, the median age of seizure onset was 12.5 years. The seizure types included generalized tonic clonic, myoclonic, and/or focal impaired awareness seizures. Six of the seven patients had a diagnosis of schizophrenia and two were diagnosed with obsessive‐compulsive disorder (OCD) (Table 1).
Figure 1.
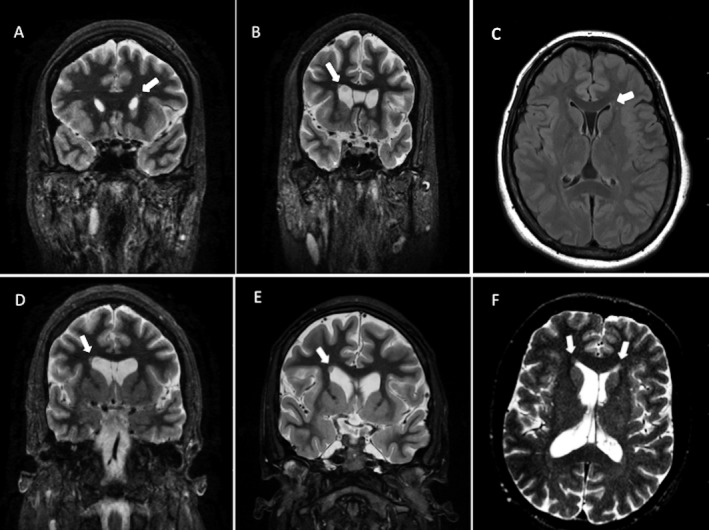
Brain MRIs of six patients with 22q11.2DS. (A) Patient 1: A single, small periventricular nodule (arrow) overlying the dorsal pole of the left frontal horn of the lateral ventricle. (B) Patient 2: single nodule of gray matter adjacent to the dorsal pole of the right frontal horn of the lateral ventricle (arrow). (C) Patient 3: Few small nodules of gray matter over the dorsal pole of the left frontal horn of the lateral ventricle. (D) Patient 4. A single nodule of gray matter adjacent to the dorsal pole of the right frontal horn of the lateral ventricle (arrow). (E) Patient 5: A single nodule of gray matter adjacent to the dorsal pole of the right frontal horn of the lateral ventricle (arrow). (F) Patient 7: Few small nodules of gray matter adjacent to the dorsal poles of the frontal horns of the lateral ventricles (arrows).
Table 1.
Demographic, clinical, and imaging and pathological findings of patients with 22q11.2DS and periventricular nodular heterotopia
| Case | Age included (years) | Sex | Intellect | Seizure disorder | Psychiatric disorder | PNH characteristics in MRI | Pathology |
|---|---|---|---|---|---|---|---|
| 16 | 51 | F | Borderline | FIAS | Schizophrenia, OCD | Single PNH over the left frontal horn. | N/A |
| 26 | 27 | F | Mild impairment | GTCS, MS, FIAS | Schizophrenia, OCD | Single PNH adjacent to the right frontal horn of the lateral ventricle | N/A |
| 3 | 24 | F | Moderate to severe impairment | GTCS, FS | Schizophrenia | Several small PNHs adjacent to the left frontal horn, but distant from subependymal layer. | N/A |
| 46 | 42 | M | Mild impairment | GTCS, MS, FIAS | Schizophrenia | Single nodule of gray matter adjacent to the right frontal horn of the lateral ventricle | N/A |
| 5 | 19 | M | Borderline | Single FS | Schizophrenia, GAD | Single nodule of gray matter adjacent to the right frontal horn of the lateral ventricle | N/A |
| 611 | 44 | M | Borderline | Single GTCS | Schizophrenia | N/A | Bilateral PNHs in the deep frontal white matter adjacent to the dorsal pole of the anterior horn of the lateral ventricles. In total, 7–8 of the nodule on each side, extending over 4 cm and corresponding to approximately 10 cm of overlying frontal cortex. There was ependymal denudation and subependymal fibrosis. There were also multiple spherical noncontiguous nodules that were only seen microscopically in the frontal lobes.11 |
| 712 | 61 | M | Average | None | None | 5–10 small nodules of gray matter adjacent to the frontal horns of the lateral ventricles.12 | Several small, closely‐spaced nodular heterotopia in the deep frontal white matter near the frontal horns of the lateral ventricle.12 |
F, female; FS, Febrile seizure; FIAS, Focal impaired awareness seizures; GAD, Generalized anxiety disorder; GTCS, Generalized tonic clonic seizure; M, male; MS, Myoclonic seizure; N/A, not available; PNH, Periventricular nodular heterotopia.
The PNH nodules detected on MRI and autopsy were small, noncontiguous, and adjacent to the dorsal pole of the anterior (frontal) horn of the lateral ventricles (DPAHLV) in all seven cases (Fig 1A–F and 2A). Nodules were unilateral in five patients and single in four patients (Table 1). Patients with multiple nodules had no more than 10 nodules seen on MRI or macroscopically at autopsy. However, on both autopsy cases, several other small nodules were seen only microscopically.11 For instance, patient 6 had multiple microscopic, spherical noncontiguous nodules, closely spaced around the larger macroscopic nodules (Fig. 2B). In addition, there were “large numbers of individual cortical‐type neurons found in the white matter of the frontal lobe between the heterotopic nodules and the nearest cortical surface” (Fig. 2C).11 The postmortem macroscopic study of patient 7 revealed several small, nodular heterotopia in the deep frontal white matter near the anterior horn of the lateral ventricle (Fig. 2D). Immunohistochemistry studies showed these nodules were formed by mature neurons and GABAergic interneurons‐expressing NeuN, calretinin, and synaptophysin (Fig. 2D–F).12
Figure 2.
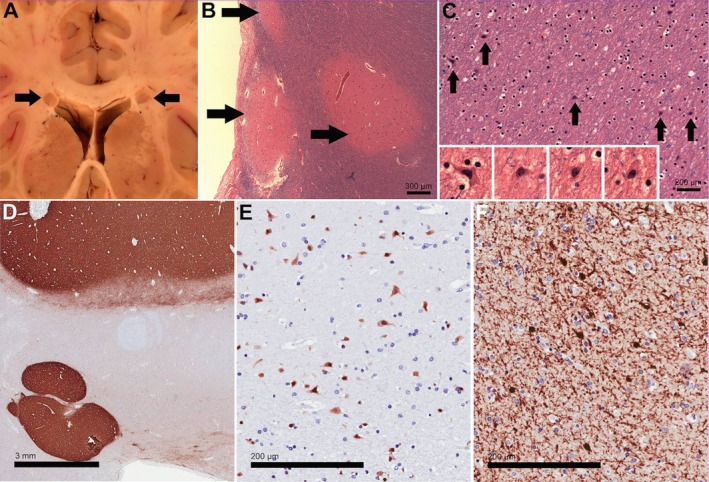
Neuropathologic features in patients with 22q11.2DS (A–C from Patient 6, D–F from Patient 7). (A) Macroscopic image of coronal section through the brain showed bilateral PNH adjacent to the lateral ventricles in the white matter of both frontal lobes. Size of heterotopias was approximately 4 mm. (B) Histologic sections of the PNH (arrows) revealed disorganized aggregates of gray matter containing haphazardly arranged neurons. (C) Frequent individual heterotopic neurons were detected in the white matter surrounding the heterotopias. Magnification (insets) reveals cytologic details, such as pyramidal shapes. (D) Immunohistochemistry for synaptophysin in a section containing two heterotopic nodules underlines the fact that the nodules are composed of gray matter. (E) Immunohistochemistry for NeuN labels the neurons in the nodule, suggesting that they are fully differentiated. (F) Higher magnification of nodule with immunostain for Calretinin, a marker of cortical interneurons.
WGS on patients 1, 2, 4, 6 and 7 did not reveal any other pathogenic SNVs or CNVs known to be associated with PNH Supplemental Table 1).14 Genome‐wide microarray in patient 3 also did not reveal additional CNVs, only the known 22q11.2 microdeletion. To place these 22q11.2DS findings in the context of the overall genetic etiology of PNH, we performed a literature review. Table S1 shows the genes and/or rare CNVs associated with PNH along with a brief description of anatomical distribution of PNH reported. As of February 2018, there were 37 genetic abnormalities associated with various patterns of PNH. The new patients reported here make 22q11.2 microdeletion the third most common CNV and the fifth most common genetic cause described in association with PNH. The details of other CNVs and the characteristics of the PNHs reported are listed in Table S1.
Discussion
Genetics of PNH and its distribution
Although the number of patient is small, the PNH characteristics observed in our patients with 22q11.2DS were consistent: small nodules, noncontiguous, varying in number from 1 to 10, located over the dorsal pole of the anterior horn of the lateral ventricles (DPAHLV), uni‐ or bilaterally. Furthermore, microscopic nodules were present at the tip of the DPAHLV and more distantly along the deep frontal white matter, between the DPAHLV and the cortex in the frontal lobes.
PNH has been previously reported in three children with 22q11.2DS.7, 15 The patient reported by Kim et al., had a single PNH over the DPAHLV.7 The MRIs of the other 2 children demonstrated symmetrical bilateral PNH, lining the temporal horns of the lateral ventricles with limited extension into the frontal horns in one of the patients.15Although the PNH distribution in these two children may still be attributed to the 22q11.2 microdeletion, it is not clear whether these patients had other CNVs or SNVs in other genes that could have accounted for the difference in their distribution, since one of those patients was diagnosed through FISH and the other had a low‐resolution array only. Five of our seven patients had WGS which ruled out the presence of mutations in any other genes previously known to be associated with PNH, as well as the presence of other pathogenic CNVs. No other CNV was detected in patient 3 as per high‐resolution genome‐wide chromosome microarray.
The literature review showed that the majority of patients with PNH and a known genetic cause have heterozygous filamin‐A (FLNA) pathogenic variants.16, 17, 18, 19, 20, 21, 22, 23 This is frequently (but not always) associated with the “classical bilateral PNH” pattern, where nodules are often large, multiple, contiguous, symmetric and abutting the walls of the ventricles. They are distributed bilaterally, along the frontal horns and ventricular bodies, almost always sparing the temporal horns, or with minor temporal involvement, and limited extension into the posterior horns.17, 20, 23 17q21.31 microdeletion represents the second most commonly reported genetic cause of PNH, with small, single, unilateral nodules. Autosomal recessive mutations in the ARFGEF2 gene represent the third most commonly reported genetic etiology of PNH with diffuse bilateral near‐contiguous or contiguous pattern often associated with a microcephaly phenotype.24, 25, 26 6q27 deletion is the fourth most commonly reported cause of PNH with uni or bilateral nodules in the frontal and temporal horns of the lateral ventricles. With the cases reported here, 22q11.2 microdeletion is the fifth most common genetic cause and the third most common CNV reported in association with PNH (see Table S1).
PNH in 22q11.2DS might result from arrested “Arc” migrating cells
Although this study was not designed to determine the origin of PNH in 22q11.2 microdeletion, a remarkable overlap with a recently described population of late migrating neurons called “Arc cells”27 was observed. Therefore, we briefly discuss the hypothesis that PNH in 22q11.2DS may result from arrested migration of these cells.
In postnatal infant human brain an “Arc” of migrating cells moves away from the subventricular zone (SVZ) around the DPAHLV towards the anterior cingulate gyrus and prefrontal cortex (Fig. 3). The migrating neuroblasts are organized in chains or individual cells. In brains studied from 34 gestational weeks to 7 months of life, the neuronal migration process is divided into four tiers with different cell concentrations in different regions at different times. Tier 1 neurons are densely packed in the SVZ around the DPAHLV walls. Tiers 2 and 3 have clusters of neurons that are progressively more distant from the ventricles. Tier 4 neurons are dispersed within areas of the developing white matter, closer to the cortex.27 Given the reported distribution of cells in this Arc, it is possible that in our population of 22q11.2DS patients, the larger PNHs observed on MRI maybe neurons whose migration was arrested in the Arc tiers 1 or 2, while the microscopic nodules and individual neurons observed in the autopsy cases are representative of neurons in the Arc tiers 3 and 4. In addition to the anatomical overlap, both Arc cells and 22q11.2DS neurons studied at autopsy express GABA and calretinin.11, 12, 27 Finally, Arc cells express CXCR4 receptor, a marker of migrating interneurons.27 Interestingly, 22q11.2 deletion leads to a significant decrease in the expression of Cxcr4,28 suggesting that 22q11.2 haploinsufficiency could have a role in the abnormal migration of Arc cells.
Figure 3.
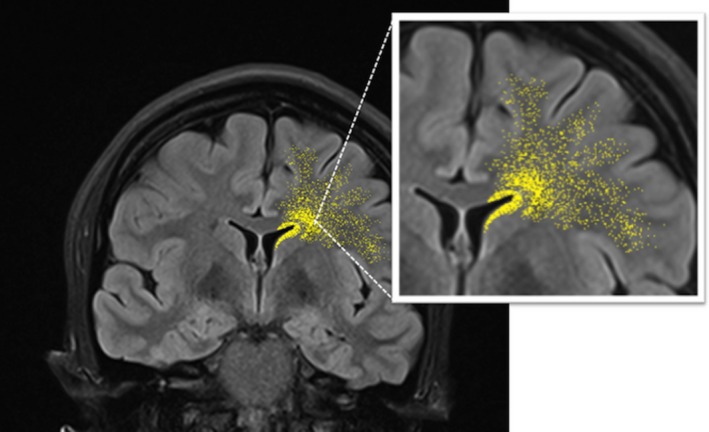
Unilateral schematic representation of Arc cells surrounding the dorsal pole of the anterior horn of the lateral ventricles, with the four tiers of cells migrating toward the cortex.
The Arc is the only migratory pathway described so far that appears to overlap with the PNH findings in patients with 22q.11.2DS. The other group of cells which migrate from the anterior horn of the lateral ventricles, leaves the SVZ through the floor of the ventricles (the “rostral migratory stream” (RMS)).29 Therefore, the arrested migration of RMS cells would be expected to produce PNH around the ventral pole of the anterior horn of the lateral ventricles.
Supporting the hypothesis that PNH is caused by 22q11.2 microdeletion is the fact that six genes located in the 22q11.2 deletion region are relevant for corticogenesis: Cdc45 l and Ranbp1 genes are “specifically and robustly” expressed in VZ/SVZ. Htf9c and Ufd1 l genes are expressed in the VZ/SVZ but also in the developing cortical plate; Hira is weakly distributed throughout the developing cortex and Sept5 is present in the developing cortical plate and absent in the VZ/SVZ.30
Neuropsychiatric symptoms, anatomical and molecular correlation
Although the large majority of patients with 22q11.2DS have the same 3 Mb deletion, not all patients manifest all the phenotypes associated to this syndrome. This is likely due to the complex interplay between the hemizygous deletion, the reminder of the patient's genome and to a certain degree, the environment.
Six out of the 7 patients described here had a clinical diagnosis of schizophrenia, 2 had OCD, 6 had ID, and 6 had seizures. Seizures could be a result of the PNH (which contains epileptogenic cells)31 although patients with 22q11.2DS and no structural abnormalities also have an overall lower seizure threshold.4 Schizophrenia, OCD and ID have been associated with multiple structural abnormalities, including abnormal cortical thickness and connectivity in the cingulate, prefrontal cortex, olfactory bulb, amongst other regions.32, 33, 34, 35, 36 Postmortem studies of non‐22q11.2DS schizophrenic brains have also shown decline in GABAergic interneurons most prominently in pre‐frontal and cingulate association cortices.37, 38 If the cells that form the PNH in our patients are derived from the Arc, and Arc cells were destined to cingulate and pre‐frontal cortex, it is plausible that adult patients with 22q11.2DS and PNH have fewer cells in those regions, which could contribute, at least in part, to their phenotype.
PNH and (possibly) arrested Arc cells migration are not the only mechanism of abnormal neurodevelopment that could underlie the neuropsychiatric manifestations in 22q11.2 patients, but they reflect a disturbed corticogenesis. For instance, 22q11.2DS has been associated with a postnatal generalized microcolumnar cerebral cortical architecture,39 supporting the role of genes in 22q11.2 deletion interval in the process of maturation. Tbx1 gene, which is present in the typical 22q11.2 deletion interval, plays a significant role in prefrontal corticogenesis including migration of inhibitory cells of prefrontal and olfactory bulb from the ganglionic eminence (GE) through RMS.40, 41 A Tbx1 haploinsufficiancy model caused thinner proliferating zones in the mediolateral cortex and GE.41 Animal models of Ranbp1 (another gene in the typical 22q11.2 interval) loss of function show decreased size of GE, decreased proliferation of forebrain progenitor cells, thinned cortex (especially cortical layers 2 and 3) and decreased size of olfactory bulbs.42 Interestingly, RANBP1 gene polymorphisms have been associated with schizophrenia vulnerability in non‐22q11.2DS individuals.43
There are some limitations to this study: Two of the seven patients did not have WGS (although one of these 2 had genome‐wide high‐resolution chromosomal microarray). This retrospective study was not designed to measure cortical thickness reported in 22q11.2DS imaging.44 Furthermore, it is unclear if such a small number of cells that are retained in the PNH would lead to abnormal cortical thickness detectable by current imaging technology. Finally, this was a clinical study, and further experimental studies would be needed to evaluate any relationship between Arc cells migration and 22q11.2DS.
In conclusion, we describe the radiological and pathological characteristics of PNH in 7 adult patients with 22q11.2DS. The anatomical and immunohistochemical features of 22q11.2DS related‐PNH, and additional surrounding microscopic heterotopic neurons found at autopsy suggest that this abnormality could result from arrested migration of Arc cells. Since Arc cells are mainly destined to the prefrontal cortex and anterior cingulate cortex, it is expected that these regions would have subtle cytoarchitectural and physiological abnormalities, which may contribute to the common neuropsychiatric features seen in 22q11.2DS such as epilepsy, schizophrenia, OCD, and ID.
Author Contributions
A.R., and E.B. performed the chart review, analyzed the data, and are the primary authors of the manuscript and tables. T.R.K provided the pathological data of the patients used in the study. He also helped in revising and provided comments on the manuscript. E.W.C. provided the medical chart of one of the patients used in the study. T.K. assisted in MRI interpretation as well as revising and providing comments on the manuscript. A.S.B. provided the medical data and the genetic data of the patients used in the study. She also reviewed and edited the manuscript. D.M.A developed the research question and designed the study. She also reviewed and edited the manuscript.
Conflicts of interest
All the authors declare no financial or intellectual conflicts of interest.
Supporting information
Table S1. Genetic variants reported in patients with periventricular heterotopia (in order of number of patients reported with PNH).
Acknowledgments
We thank Ms. Kelly Mo for her technical help with Figure 3.
Funding information
This research was conducted with the support of EpLink ‐ The Epilepsy Research Program of the Ontario Brain Institute. The Ontario Brain Institute is an independent nonprofit corporation, funded partially by the Ontario government. The funding agency had no role in design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Funding Statement
This work was funded by Ontario Brain Institute grant .
References
- 1. Botto LD, May K, Fernhoff PM, et al. A population‐based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003;112:101–107. [DOI] [PubMed] [Google Scholar]
- 2. Bassett AS, McDonald‐McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr 2011;159:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fung WL, Butcher NJ, Costain G, et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med 2015;17:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wither RG, Borlot F, MacDonald A, et al. 22q11.2 deletion syndrome lowers seizure threshold in adult patients without epilepsy. Epilepsia 2017;58:1095–1101. [DOI] [PubMed] [Google Scholar]
- 5. Kao A, Mariani J, McDonald‐McGinn DM, et al. Increased prevalence of unprovoked seizures in patients with a 22q11.2 deletion. Am J Med Genet A 2004;129:29–34. [DOI] [PubMed] [Google Scholar]
- 6. Andrade DM, Krings T, Chow EW, et al. Hippocampal malrotation is associated with chromosome 22q11.2 microdeletion. Can J Neurol Sci 2013;40:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim EH, Yum MS, Lee BH, et al. Epilepsy and other neuropsychiatric manifestations in children and adolescents with 22q11.2 deletion syndrome. J Clin Neurol 2016;12:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo‐cardio‐facial syndrome. Arch Gen Psychiatry 1999;56:940–945. [DOI] [PubMed] [Google Scholar]
- 9. Bassett AS, Chow EW. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep 2008;10:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pantelis C, Maruff P. The cognitive neuropsychiatric approach to investigating the neurobiology of schizophrenia and other disorders. J Psychosom Res 2002;53:655–664. [DOI] [PubMed] [Google Scholar]
- 11. Kiehl TR, Chow EW, Mikulis DJ, et al. Neuropathologic features in adults with 22q11.2 deletion syndrome. Cereb Cortex 2009;19:153–164. [DOI] [PubMed] [Google Scholar]
- 12. Baharnoori M, Mandell DM, Andrade DM, et al. Periventricular nodular heterotopia and bilateral intraventricular xanthogranulomas in 22q11.2 deletion syndrome. Hum Pathol 2016;9:10–12. [Google Scholar]
- 13. Bano S, Yadav SN, Chaudhary V, Garga UC. Neuroimaging in epilepsy. J Pediatr Neurosci 2011;6:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merico D, Zarrei M, Costain G, et al. Whole‐genome sequencing suggests schizophrenia risk mechanisms in humans with 22q11.2 deletion syndrome. G3: Genes ‐ Genomes ‐ Genetics 2015;5:2453–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Kogelenberg M, Ghedia S, McGillivray G, et al. Periventricular heterotopia in common microdeletion syndromes. Mol Syndromol 2010;1:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ekşioğlu YZ, Scheffer IE, Cardenas P, et al. Periventricular heterotopia: an X‐linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron 1996;16(1):77–87. [DOI] [PubMed] [Google Scholar]
- 17. Sheen VL, Dixon PH, Fox JW, et al. Mutations in the X‐linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet 2001;10:1775–1783. [DOI] [PubMed] [Google Scholar]
- 18. Poussaint TY, Fox JW, Dobyns WB, et al. Periventricular nodular heterotopia in patients with filamin‐1 gene mutations: neuroimaging findings. Pediatr Radiol 2000;30:748–755. [DOI] [PubMed] [Google Scholar]
- 19. Kakita A, Hayashi S, Moro F, et al. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta Neuropathol 2002;104:649–657. [DOI] [PubMed] [Google Scholar]
- 20. Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin a mutations. Brain 2006;129:1892–1906. [DOI] [PubMed] [Google Scholar]
- 21. Clapham KR, Yu TW, Ganesh VS, et al. FLNA genomic rearrangements cause periventricular nodular heterotopia. Neurology 2012;78(4):269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fergelot P, Coupry I, Rooryck C, et al. Atypical male and female presentations of FLNA‐related periventricular nodular heterotopia. Eur J Med Genet 2012;55(5):313–318. [DOI] [PubMed] [Google Scholar]
- 23. Lange M, Kasper B, Bohring A, et al. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis 2015;10:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sheen VL, Ganesh VS, Topcu M, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet 2004;36:69–76. [DOI] [PubMed] [Google Scholar]
- 25. de Wit MC, de Coo IF, Halley DJ, et al. Movement disorder and neuronal migration disorder due to ARFGEF2 mutation. Neurogenetics 2009;10:333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banne E, Atawneh O, Henneke M, et al. West syndrome, microcephaly, grey matter heterotopia and hypoplasia of corpus callosum due to a novel ARFGEF2 mutation. J Med Genet 2013;50:772–775. [DOI] [PubMed] [Google Scholar]
- 27. Paredes MF, James D, Gil‐Perotin S, et al. Extensive migration of young neurons into the infant human frontal lobe. Science 2016;354:aaf7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meechan DW, Tucker ES, Maynard TM, et al. Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc Natl Acad Sci USA 2012. Nov 6;109:18601–18606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanai N, Nguyen T, Ihrie RA, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011;478:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci USA 2009;106:16434–16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aghakhani Y, Kinay D, Gotman J, et al. The role of periventricular nodular heterotopia in epileptogenesis. Brain 2005;128:641–651. [DOI] [PubMed] [Google Scholar]
- 32. Kuperberg GR, Broome MR, McGuire PK, et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry 2003;60:878–888. [DOI] [PubMed] [Google Scholar]
- 33. Narr KL, Woods RP, Thompson PM, et al. Relationships between IQ and regional cortical gray matter thickness in healthy adults. Cereb Cortex 2007;17:2163–2171. [DOI] [PubMed] [Google Scholar]
- 34. Nesvåg R, Lawyer G, Varnäs K, et al. Regional thinning of the cerebral cortex in schizophrenia: effects of diagnosis, age and antipsychotic medication. Schizophr Res 2008;98:16–28. [DOI] [PubMed] [Google Scholar]
- 35. Rotge JY, Guehl D, Dilharreguy B, et al. Meta‐analysis of brain volume changes in obsessive‐compulsive disorder. Biol Psychiatry 2009;65:75–83. [DOI] [PubMed] [Google Scholar]
- 36. Romano A, Cornia R, Moraschi M, et al. Age‐related cortical thickness reduction in non‐demented down's syndrome subjects. J Neuroimaging 2016;26:95–102. [DOI] [PubMed] [Google Scholar]
- 37. Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium‐binding proteins. Biol Psychiatry 2002;52:708–715. [DOI] [PubMed] [Google Scholar]
- 38. Volk DW, Matsubara T, Li S, et al. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am J Psychiatry 2012;169:1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarnat HB, Flores‐Sarnat L. Radial microcolumnar cortical architecture: maturational arrest or cortical dysplasia? Pediatr Neurol 2013. Apr;48:259–270. [DOI] [PubMed] [Google Scholar]
- 40. Hiramoto T, Kang G, Suzuki G, et al. Tbx 1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet 2011;20:4775–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flore G, Cioffi S, Bilio M, Illingworth E. Cortical development requires mesodermal expression of tbx1, a gene haploinsufficient in 22q11.2 deletion syndrome. Cereb Cortex 2017;27:2210–2225. [DOI] [PubMed] [Google Scholar]
- 42. Paronett EM, Meechan DW, Karpinski BA, et al. Ranbp1, deleted in DiGeorge/22q11.2 deletion syndrome, is a microcephaly gene that selectively disrupts layer 2/3 cortical projection neuron generation. Cereb Cortex 2015;25:3977–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu H, Abecasis GR, Heath SC, et al. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci USA 2002;99:16859–16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bearden CE, Van Erp TG, Dutton RA, et al. Mapping cortical thickness in children with 22q11.2 deletions. Cereb Cortex 2007;17:1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genetic variants reported in patients with periventricular heterotopia (in order of number of patients reported with PNH).