

Abstract
Objective
The neuronal ceroid lipofuscinoses (NCL) are genetic degenerative disorders of brain and retina. NCL with juvenile onset (JNCL) is genetically heterogeneous but most frequently caused by mutations of CLN3. Classical juvenile CLN3 includes a rare protracted form, which has previously been linked to autophagic vacuolar myopathy (AVM). Our study investigates the association of AVM with classic, non‐protracted CLN3.
Methods
Evaluation of skeletal muscle biopsies from three, non‐related patients with classic, non‐protracted and one patient with protracted CLN3 disease by histology, immunohistochemistry, electron microscopy, and Sanger sequencing of the coding region of the CLN3 gene.
Results
We identified a novel heterozygous CLN3 mutation (c.1056+34C>A) in one of our patients with classic, non‐protracted CLN3 disease. The skeletal muscle of all CLN3 patients was homogeneously affected by an AVM characterized by autophagic vacuoles with sarcolemmal features and characteristic lysosomal pathology.
Interpretation
Our observations show that AVM is not an exceptional phenomenon restricted to protracted CLN3 but rather a common feature in CLN3 myopathology. Therefore, CLN3 myopathology should be included in the diagnostic spectrum of autophagic vacuolar myopathies.
Introduction
The neuronal ceroid lipofuscinoses (NCL) are a genetically heterogeneous group of progressive neurodegenerative diseases, primarily affecting children. Among the 14 identified different genetic subtypes, now nosologically labeled CLN1 to CLN14, CLN9 is so far genetically unidentified or reclassified.1 The morphological hallmark of all CLN entities is cerebral and extra‐cerebral deposition of lipopigments. Lipopigments are lysosomal residual bodies, that is the end products of prelysosomal and intralysosomal degradation of cellular constituents. Their distribution is almost ubiquitous and, thereby affecting skeletal muscle as well. With increasing mutational spectra of hereditary diseases, also pertaining to the CLN1 to 14, nosological spectra also expand when variants to classical forms become apparent consecutively altering the nosological character of an individual entity. For instance, classical juvenile CLN3, includes a rare protracted form characterized by a prolonged disease course.1, 2 Just recently three patients of protracted CLN3 have been reported3, 4 in whom skeletal muscle was affected by an autophagic vacuolar myopathy (AVM). In order to further expand the nosological spectrum of the NCL and the vacuolar myopathies, that is the lysosomal myopathies, we report lysosomal myopathological findings of four non‐related CLN3 patients suffering from protracted and classic, non‐protracted CLN3 disease.
Material and Methods
Patient data and tissue samples
This study was conducted according to the ethical principles for medical research involving human subjects according to the Declaration of Helsinki. The clinical data were assessed and anonymized for patients’ confidentiality. Informed consent was obtained from all patients. Ethical approval (EA1/215/08) was granted by the institutional ethics board of the Charité Ethics Committee. We investigated the muscle tissue of three patients with classic non‐protracted CLN3 disease and of one patient with protracted CLN3 disease.
Molecular genetics
Genomic DNA was extracted from formalin fixed and paraffin embedded (FFPE) samples using the Qiagen GeneRead DNA FFPE Kit according to the manufacturer′s protocol (Qiagen, Hilden, Germany). Genomic DNA from blood or from cryo‐preserved muscle tissue was isolated according to standard procedures. We performed bidirectional Sanger sequencing of the CLN3 gene. The PCR primers for the genomic regions corresponding to CLN3 exons 2‐16 (exon 1 is non‐coding; transcript: NM_001042432) and the flanking intronic sequences are listed in Table S1. To identify the common 1.02 kb deletion we used a newly designed forward PCR primer (5′‐gatcacgccactgtactcca‐3′) corresponding to introns 7 and 9 and a reverse PCR primer (5′‐ggctatcagagtccagattccg‐3′) that had previously been published.5 The PCR conditions were: 1 × 94°C [4 min], 3 × 94°C [30 s], 61°C [45 s], 72°C [60 s], 3 × 94°C [30 s], 59°C [45 s], 72°C [60 s], 3 × 94°C [30 s], 57°C [45 s], 72°C [60 s], 33 × 94°C [30 s], 55°C [45 s], 72°C [60 s], and finally extension at 72°C [10 min] with AmpliTaq 360 DNA polymerase (Life Technologies, Germany). Sequencing was performed at Eurofins Genomics, Ebersberg, Germany.
Immunohistochemical procedures
Immunohistochemical stainings were performed on a Benchmark XT autostainer (Ventana Medical Systems, Tuscon, AZ) with standard antigen retrieval methods (CC1 buffer, pH8.0; Ventana Medical Systems) using 4 μm FFPE or 7 μm cryo tissue sections. The primary antibodies used are listed in Table S2. The iVIEW DAB Detection Kit (Ventana Medical Systems) was used according to the manufacturer's instructions. Sections were counterstained with hematoxylin, dehydrated in graded alcohol and xylene, mounted and coverslipped. Immunohistochemical stainings were concurrently evaluated by two experienced neuropathologists. In order to corroborate autophagolysosomal morphology in our CLN3 muscle tissues, we added muscle tissue of a patient with adult acid maltase deficiency (Pompe disease, Glycogen storage disease type 2 [GSDII]).
Electron microscopy
Electron microscopy (EM) was performed as described previously.6 Blood lymphocytes were separated using the Ficoll® technique.
Results
Clinical data of the patients with CLN3 disease
Protracted CLN3 disease
Patient B1, was a male, who showed a protracted disease course. His vision became impaired by the age of 5 years, progressing to complete blindness by the age of 18 years due to retinitis pigmentosa. At the age of 11 years, he developed grand mal seizures, preferentially at night during sleep, followed by postictal disorientation and confusion. Recently, at 34 years of age, he showed evidence of cognitive decline and an incipient extrapyramidal movement disorder. The patient was a university student of economics. A cranial magnetic resonance imaging (MRI) with and without contrast enhancement revealed discrete cerebral and substantial global cerebellar atrophy (Figs. S1A, B) and atrophy of both optic nerves. Echocardiography failed to show any abnormalities. The serum creatinine kinase (CK) levels have not been assessed. The preliminary clinical diagnosis of a mitochondrial disease prompted a muscle biopsy of the right vastus lateralis muscle which was cryopreserved.
Classic CLN3 disease
Patient B2, was a male who developed normally until the age of 4 years when a rapid loss of vision was noted and electroretinography evidenced a retinitis pigmentosa. Electron microscopy of isolated blood lymphocytes showed abnormal cellular storage material compatible with the diagnosis of classic CLN3 disease. The boy started regular school but was then transferred to a special school for visually handicapped students where he developed progressive cognitive, language, and emotional problems. Later, epileptic seizures and disturbance of the motor system became apparent. He became helpless in late adolescence; he was cared for in a nursing home where he died from pneumonia and dehydration at the age of 27 years. His CK levels were not elevated on several occasions. Muscle tissue was obtained at autopsy, formalin‐fixed, and paraffin‐embedded.
Patient B3, was a male twin of a healthy brother. He developed normally until the third year of life when a rapid loss of vision was noted. Ophthalmological examination revealed a retinopathy with “bull's eye maculopathy” and an extinguished photopic electroretinogram. Light microscopy of a blood smear revealed 20% of lymphocytes with large vacuoles and electron microscopy of skin and blood cells demonstrated storage material compatible with the diagnosis of classic CLN3 disease. At the age of 6 years, an electroencephalogram was abnormal. He developed severe emotional problems and complete blindness. At the age of 10 years, epilepsy and dementia were evident. Unexplained bradycardia was noted at the age of 14 years and loss of motor abilities ensued. He died from an accident at the age of 21 years. His CK levels were not elevated on several occasions. Muscle tissue was obtained at autopsy, formalin‐fixed and paraffin‐embedded.
Patient B4, an archived CLN3 case,7 was a male who developed a rapid loss of vision and complete blindness by the age of 4 years. Ophthalmological examination revealed a pigmentary degeneration of the retina. At the age of 8 years, an electroencephalogram was abnormal and he developed dementia, epilepsy, ataxia, and behavioral problems. CK levels have not been assessed. A muscle biopsy was taken from the deltoid muscle which was formalin‐fixed and paraffin embedded. He died at the age of 17 years.
Muscle pathology
Light microscopy of protracted CLN3 disease
Patient B1: Cross‐sectioned muscle fibers showed a mildly increased variation in fiber diameters and scattered atrophic, occasionally angular muscle fibers. Non‐rimmed vacuoles were encountered in few muscle fibers with some granular content (Figs. 1A–C, arrowheads) which stained strongly with the PAS stain (Fig. 1D, arrowhead). The content of lipid droplets was within normal limits (data not shown). Furthermore, there was increased autofluorescence in numerous muscle fibers, peripherally scattered as well as across cross‐sectioned muscle fibers (Fig. 1E). Activity of acid phosphatase was strongly enhanced in many muscle fibres (Fig. 1F), the non‐specific esterase activity was also – even though mildly – enhanced (Fig. 1G, arrowhead). Immunohistochemistry revealed multifocal labeling of vacuoles by antibodies against sarcolemmal proteins such as spectrin (Fig. 1H, arrowhead), dysferlin (data not shown), caveolin 3 (Fig. 1I, arrowhead), lysosomal protein (LAMP 2, Fig. 1J, arrowhead) and autophagy‐related proteins (LC3 [Fig. 1K, arrowhead], p62 [Fig. 1L, arrowhead]). The activity of the acetylcholine esterase was mildly enhanced in vacuoles (Fig. 1M, arrowhead). All immunohistochemical stainings to label vacuoles with sarcolemmal features used by us and others are listed in Tables S2 and S3. Muscle fiber types 1, 2A, and 2B were arranged in a normal checker‐board pattern. There was only a small focus suggestive of fiber‐type grouping (data not shown).
Figure 1.
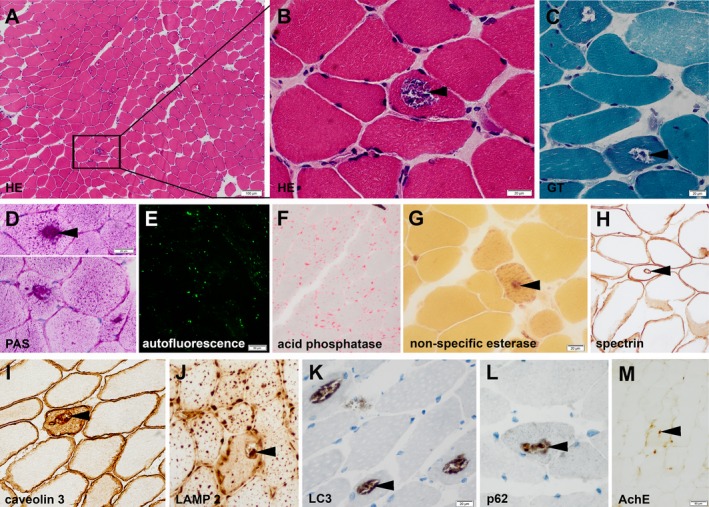
Histology of cross‐sectioned muscle fibers of patient B1. Hematoxylin and Eosin (HE) and Gömöri trichrome (GT) stains demonstrate non‐rimmed vacuoles with some granular content (A–C, arrowheads; solid box [A] indicates magnified area [B]) which stain strongly for PAS (D, arrowhead). Numerous muscle fibers demonstrate increased auto‐fluorescence (E). Acid phosphatase activity (F) is strongly, non‐specific esterase activity (G, arrowhead) is mildly enhanced. Immunohistochemistry reveals multifocal vacuoles with multifocal labeling of sarcolemmal proteins (spectrin (H, arrowhead); caveolin 3 [I, arrowhead]), lysosomal proteins (LAMP 2 [J, arrowhead]) and autophagy‐related proteins (LC3 (K, arrowhead), p62 [L, arrowhead]) inside muscle fibers. The activity of the acetylcholine esterase is mildly enhanced (M, arrowhead).
The skin revealed normal epidermis and unremarkable subepidermal components with dermal appendages, including hair follicles, eccrine sweat glands, and aggregates of smooth muscle cells (data not shown).
Electron microscopy of protracted CLN3 disease
Skeletal muscle, skin, and blood lymphocytes of patient B1 were investigated by EM. Muscle fibers contained regularly arranged sarcomeres, mitochondria were occasionally swollen. Subsarcolemmal, and inside muscle fibers aggregates of lipopigments with rectilinear and curvilinear profiles were recognized (Figs. 2A, B, arrowheads) as lysosomal residual bodies. Likewise, endothelial cells of capillaries contained aggregates of lipopigments which, at higher magnification, showed NCL‐typical multi‐layered fingerprint profiles (Fig. 2C, insert). In addition, vacuoles were filled with debris and lamellar material, often of myelin‐sheath‐like structures (Fig. 2D) and peripheral basal lamina‐like fuzzy material, the ultrastructural equivalent of autophagic or rimmed vacuoles. Occasionally, lipopigments appeared loosely arranged within a somewhat vacuolar space, surrounded by a unit membrane (Fig. 2E, insert, arrowheads). Secretory cells of dermal eccrine sweat glands also contained lipopigments (Fig. 2F, arrowheads). Blood lymphocytes contained several membrane‐bound vacuoles (Fig. 2G, arrowhead), some of which with lipopigments, displaying fingerprint profiles at higher magnification (Fig. 2G, insert, arrow; Fig. 2H, insert). Other lipopigment aggregates appeared as non‐vacuolar inclusions.
Figure 2.
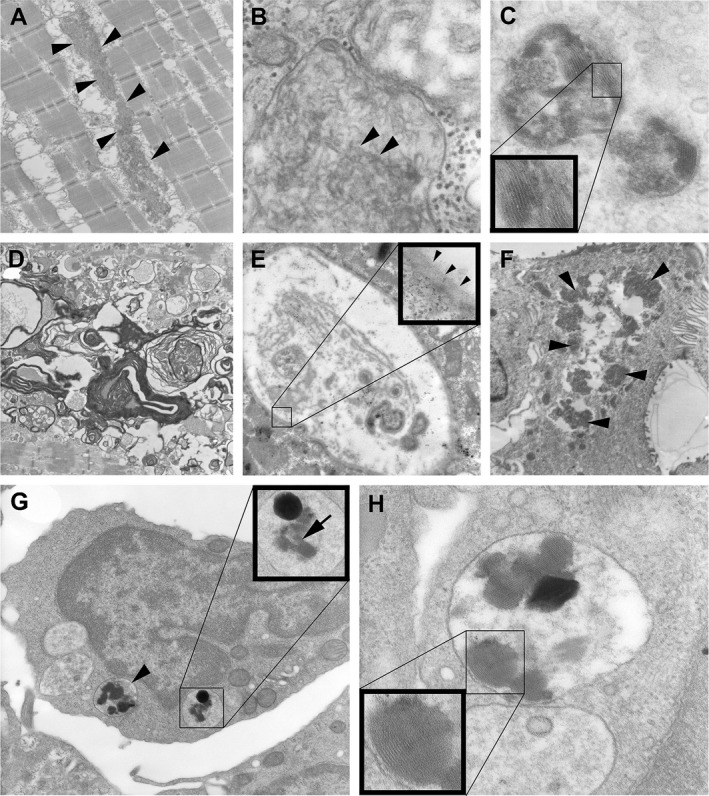
Electron microscopy of skeletal muscle, blood lymphocytes, and skin of patient B1. Aggregates of curvilinear and rectilinear lipopigments are present within the muscle fiber (A, B, arrowheads). Endothelial cells of muscle capillaries demonstrate characteristic fingerprint profiles (C, insert). Ultrastructural equivalent of autophagic or rimmed vacuoles filled with debris and lamellar material, often of myelin‐sheath‐like structures (D) and sometimes covered by a basal lamina (E, insert, arrowheads). Secretory cells of dermal eccrine sweat glands contain lipopigments (F, arrowheads). Blood lymphocyte with several membrane‐bound vacuoles (G, arrowhead), some of which with lipopigments, displaying fingerprint profiles (G, insert, arrow; H, insert).
Light microscopy of classic CLN3 disease
The muscle biopsies presented a significant number of fibers which were reduced in diameter. Gömöri trichrome stains of classic CLN3 patients demonstrated non‐rimmed vacuoles with some granular content (patient B4: Fig. 3A, arrowheads; patients B2 and B3: data not shown). The activity of the phosphatase activity was high (Fig. 3A). Immunohistochemistry demonstrated labeling of vacuoles by antibodies against lysosomal protein (patient B4: LAMP 2, Fig. 3D, arrowheads). Severe autolytic changes were seen in the muscle specimens obtained post mortem (patients B2 and B3, Figs. 3L–O). Nevertheless, the sarcomere structure was fairly well preserved. In all classic CLN3 patients investigated, immunohistochemical labeling of autophagy‐related proteins LC3 (Figs. 3E, F, L, N, arrowheads), and p62 (Figs. 3G, H, M, O) also revealed the presence of autophagic vacuoles and therefore demonstrates that AVM seems to be a common feature in CLN3 myopathology.
Figure 3.
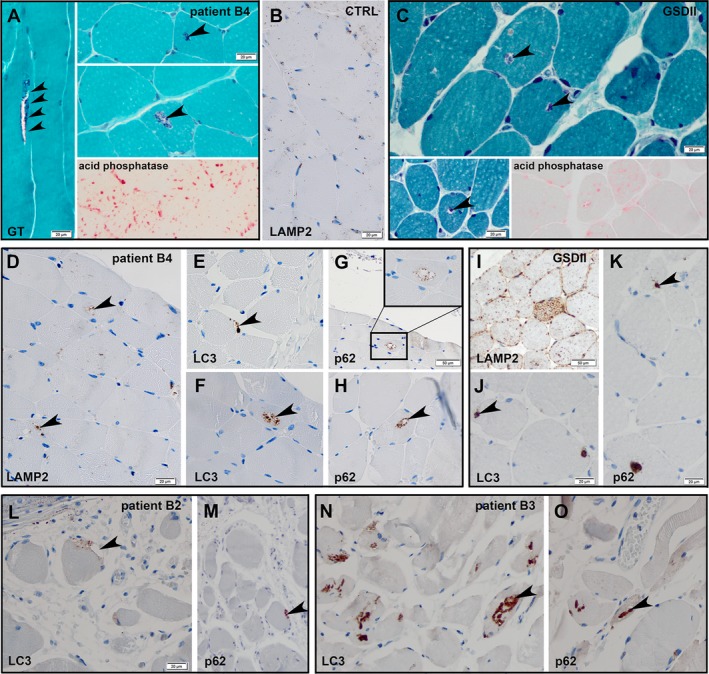
Histology of cross‐sectioned muscle fibers in classic CLN3 disease. Gömöri trichrome stain of an archived case of juvenile CLN3 (patient B4) demonstrates non‐rimmed vacuoles with some granular content (A, arrowheads) and enhanced acid phosphatase activity (A, bottom right corner). Paraffin‐embedded muscle tissue of this case shows lysosomal protein (LAMP 2 (D) and autophagy‐related proteins (LC3 (E, F), p62 (G, H) inside the vacuoles. Paraffin‐embedded muscle tissue of a healthy control (CTRL) was also stained for LAMP2 (B), no significant background staining or vacuoles were detectable. Gömöri trichrome stain of a case of adult Pompe′s disease (GSDII) demonstrates many non‐rimmed vacuoles (C, arrowheads). Acid phosphatase stain reveals intense lysosomal activity (C, bottom right corner). Immunohistochemistry reveals multifocal labeling of lysosomal proteins (LAMP 2 [I]) and autophagy‐related proteins (LC3 (J), p62 [K]) inside the vacuoles. The additional two CLN3 patients (B2 and B3) also revealed non‐rimmed vacuoles with labeling of autophagy‐related proteins (LC3 (L, N), p62 [M, O]).
Genetics
Protracted CLN3 disease
By break point PCR we found a heterozygous 1.02 kb deletion of exons 8 and 9 in the CLN3 gene (Fig. S2), inherited from the mother (Fig. S2). The additional heterozygous missense mutation c.1151T>C het (p.Leu384Pro) of patient B1 was previously reported.8 We used Sanger sequencing of the coding region of the CLN3 gene to confirm the mutation (Fig. 4A, arrow) which was inherited from the father (Fig. 4B, arrow). Other mutations associated with protracted CLN3 reported previously are listed in Table S4.
Figure 4.
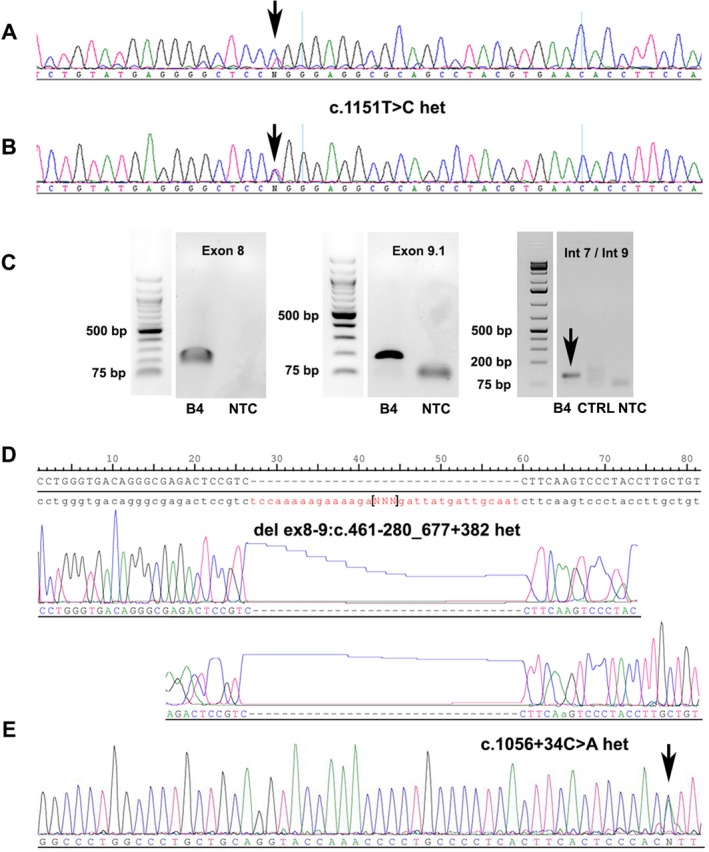
CLN3 mutations. Representative PCR gels and Sanger sequencing chromatograms presenting the different CLN3 mutations found in our patients. For patient B1, we found a heterozygous missense mutation (c.1151T>C het (p.Leu384Pro); A) inherited from the father (B) and a heterozygous 1.02 kb deletion of exons 8 and 9 (please see Supplemental data). For patient B4, the PCR gels show the amplification of the normal exons 8 and 9 (C) and the heterozygous 1.02 kb deletion (break point PCR, C, arrowhead) which was sequenced to determine the exact location of the break points (D). Additionally, we found a new, not previously reported point mutation in the flanking intron of exon 14 (c.1056+34C>A het, [E]). CTRL: control DNA; NTC: no template control.
Classic CLN3 disease
Patients B2 and B3 demonstrated a homozygous 1.02 kb deletion (exons 8 and 9; data not shown), typical for CLN3 disease. Patient B4 presented a heterozygous 1.02 kb deletion of exons 8 and 9 (Figs. 4C, D), which was detected by PCR analysis of the break points (Fig. 4C) and sequenced to determine their exact location (Fig. 4D). The break point PCR (Fig. 4C, arrow) only showed the small product of the mutated allele as we used DNA from formalin‐fixed paraffin embedded tissue which was partly degraded. Therefore, the healthy allele (1.02 kb) was too large to be amplified. Nevertheless, additional PCR showed the products of the full‐length exons 8 and 9 (Fig. 4C, Fig. S3). By Sanger sequencing of the coding region of the CLN3 gene (exons 2‐16), we additionally found a new, not previously reported point mutation in the flanking intron of exon 14 (c.1056+34C>A het; Fig. 4E, arrow) potentially leading to a splicing error. The PCR gels of exons 2 ‐16 are shown in Figure S3.
Discussion
The muscle pathology in protracted juvenile CLN3 has recently been associated with that of AVM showing autophagic vacuoles with sarcolemmal features (AVSF).3, 4 Testing our patient's muscular specimen with antibodies against markers of AVSF provided similarities with the previously reported protracted CLN3 myopathology.3, 4, 9 However, the sarcolemmal features were infrequent. In the previous reports, there was not complete correspondence of tested markers among different muscle specimens from CLN3 and AVM (Table S3). In fact, we clearly show, that not only protracted but also classic CLN3 myopathology is not only lysosomal but also autophagic. Whereas the lysosomal component is marked by increased acid phosphatase activities and transmembranous LAMP2 expression and has been described earlier by others,3, 4, 7 the autophagic changes additionally comprise presence of LC3, p62, and acetyl choline esterase activity.10 AVSF, to our knowledge, have not been reported in classic non‐protracted CLN3 or in other CLN forms, such as CLN1, CLN2, and other late infantile variants (CLN5 and CLN6) – but this may be due to lack of testing. If NCL‐typical lipopigments of muscle fibres may also be detected in AVSF needs to be further investigated by electron microscopy, perhaps, suggesting, two different processes, autophagy and lysosomal degradation, the latter not necessarily following the former in NCL. A pathogenetic connection between autophagy and lysosomal catabolism needs to be further explored in NCL. Whether classical and protracted CLN3 vacuolation of blood lymphocytes represents autophagy, too, is currently a matter of speculation. Involvement of autophagy in the lysosomal degenerative process has been documented before11 in various lysosomal diseases. It seems to be a regular morphological feature in muscle tissues and for that reason we have included additional muscle tissue studies in classical, non‐protracted CLN3 and adult GSDII (Figs. 3C, I–K).
The clinical and laboratory findings of all four patients reported here are compatible with CLN3 disease. In patient B1 who is now in the fourth decade of life, the considerable interval between visual disability and subsequent onset of epilepsy actually suggested a protracted form of CLN3 as reported previously for similar cases (Table S4).3, 4, 9, 12 The diagnosis of CLN3 disease was further supported by the presence of lysosomal vacuoles in blood lymphocytes, which have also been seen in protracted CLN3,3, 4 some of them containing lipopigments with fingerprint profiles, a characteristic lesion in CLN3 disease. Furthermore, the molecular analysis secured a compound heterozygous deletion of exons 8 and 9 (the common 1.02 kb deletion) and a rare heterozygous missense mutation (p.Leu384Pro) in the CLN3 gene, previously reported by Kwon et al.13.
The first symptoms in juvenile NCL usually comprise visual failure, which is common in CLN3 and CLN1 juvenile variant.14 Hence, when loss of vision develops in a juvenile patient a CLN disease ought to be suspected and CLN3, and CLN1 ought to be molecularly analyzed first. Of note is further that protracted NCL appears to be mostly linked to the CLN3 form, but largely not to other CLN forms.15
Only few genetic mutations have been suggested to be associated with protracted CLN3, for example compound heterozygous 1.02 kb and 2.8 kb deletion.16 It was previously reported that patients, compound heterozygous for the 1.02 kb deletion and another mutation, could potentially present a protracted disease course.17 Other larger CLN3 series were unable to confirm such a potential genotype‐phenotype correlation.1, 18 Thus, the genetic spectrum leading to a protracted CLN3 phenotype seems to be larger than previously thought. Therefore, further documentation of the genetic and clinical spectrum of CLN3 disease is needed to consider additional genotype‐phenotype correlations.
The results of our non‐protracted CLN3 muscle tissue studies providing findings identical to those in our patient with protracted disease course expanding the spectrum of autophagy‐related myopathology to non‐protracted, classical CLN3 – and therefore also proved the reliability and feasibility of genetic and morphological investigations of archival FFPE tissues. Our findings further illustrate that muscle biopsy in NCL may be rewarding and may help to find the correct clinical diagnosis. Such studies ought also to be extended for other CLN (CLN1‐14) forms.
Hence, the involvement of enhanced autophagy in muscle fibers in this lysosomal condition does not appear to be a matter of protracted or prolonged duration of CLN3 disease.
Perhaps, size and volume of multinucleated muscle cells allow easier investigation and recognition of the morphology of the autophagy and lysosomal system which also needed to be extended to other cell types in human NCL, foremost neurons.
Author Contributions
JR and HHG drafted and revised the manuscript, designed the study concept, performed data analysis, and participated in data acquisition. RK, EG, LW, MS, AS, AK, CH, and WS performed data analysis, and participated in data acquisition.
Disclosure
The authors have no potential disclosure to report.
Supporting information
Table S1. CLN3 primer (transcript: NM_001042432).
Table S2. Immunohistochemical labeling of vacuoles in patients′ B1 muscle fibers.
Table S3. Immunohistochemical labeling of vacuoles in previous studies and in patient B4.
Table S4. Genetic background in protracted CLN3 disease.
Figure S1. Contrast enhanced brain magnetic resonance imaging (MRI) of patient B1.
Figure S2. CLN3 PCR in patient B1.
Figure S3. CLN3 PCR in patient B4.
Acknowledgments
The authors thank Hanna Plueckhan, Cordula zum Bruch, and Petra Matylewski for excellent technical assistance. JR is a participant of the BIH‐Charité Clinical Scientist Program funded by the Charité –Universitätsmedizin Berlin and the Berlin Institute of Health.
Funding Information
JR is a participant of the BIH‐Charité Clinical Scientist Program funded by the Charité –Universitätsmedizin Berlin and the Berlin Institute of Health.
Funding Statement
This work was funded by Charité –Universitätsmedizin Berlin grant ; Berlin Institute of Health grant .
References
- 1. Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63. [DOI] [PubMed] [Google Scholar]
- 2. Goebel HH, Pilz H, Gullotta F. The protracted form of juvenile neuronal ceroid‐lipofuscinosis. Acta Neuropathol 1976;36:393–396. [DOI] [PubMed] [Google Scholar]
- 3. Cortese A, Tucci A, Piccolo G, et al. Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology 2014;82:2072–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Licchetta L, Bisulli F, Fietz M, et al. A novel mutation af CLN3 associated with delayed‐classic juvenile ceroid lipofuscinois and autophagic vacuolar myopathy. Eur J Med Genet 2015;58:540–544. [DOI] [PubMed] [Google Scholar]
- 5. Jarvela I, Mitchison HM, Munroe PB, et al. Rapid diagnostic test for the major mutation underlying Batten disease. J Med Genet 1996;33:1041–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Preusse C, Allenbach Y, Hoffmann O, et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta Neuropathol Commun 2016;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goebel HH, Zeman W, Pilz H. Significance of muscle biopsies in neuronal ceroid‐lipofuscinoses. J Neurol Neurosurg Psychiatry 1975;38:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Fruscio G, Schulz A, De Cegli R, et al. Lysoplex: an efficient toolkit to detect DNA sequence variations in the autophagy‐lysosomal pathway. Autophagy 2015;11:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taschner PE, Cortese A, Tucci A. Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology 2015;84:632. [DOI] [PubMed] [Google Scholar]
- 10. Stenzel W, Nishino I, von Moers A, et al. Juvenile autophagic vacuolar myopathy – a new entity or variant? Neuropathol Appl Neurobiol 2013;39:449–453. [DOI] [PubMed] [Google Scholar]
- 11. Lieberman AP, Puertollano R, Raben N, et al. Autophagy in lysosomal storage disorders. Autophagy 2012;8:719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wisniewski KE, Zhong N, Kaczmarski W, et al. Compound heterozygous genotype is associated with protracted juvenile neuronal ceroid lipofuscinosis. Ann Neurol 1998;43:106–110. [DOI] [PubMed] [Google Scholar]
- 13. Kwon JM, Adams H, Rothberg PG, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011;77:1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stephenson JB, Greene ND, Leung KY, et al. The molecular basis of GROD‐storing neuronal ceroid lipofuscinoses in Scotland. Mol Genet Metab 1999;66:245–247. [DOI] [PubMed] [Google Scholar]
- 15. Di Giacopo R, Cianetti L, Caputo V, et al. Protracted late infantile ceroid lipofuscinosis due to TPP1 mutations: Clinical, molecular and biochemical characterization in three sibs. J Neurol Sci 2015;356:65–71. [DOI] [PubMed] [Google Scholar]
- 16. Munroe PB, Mitchison HM, O'Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet 1997;61:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauronen L, Munroe PB, Jarvela I, et al. Delayed classic and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis. Neurology 1999;52:360–365. [DOI] [PubMed] [Google Scholar]
- 18. Adams HR, Beck CA, Levy E, et al. Genotype does not predict severity of behavioural phenotype in juvenile neuronal ceroid lipofuscinosis (Batten disease). Dev Med Child Neurol 2010;52:637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. CLN3 primer (transcript: NM_001042432).
Table S2. Immunohistochemical labeling of vacuoles in patients′ B1 muscle fibers.
Table S3. Immunohistochemical labeling of vacuoles in previous studies and in patient B4.
Table S4. Genetic background in protracted CLN3 disease.
Figure S1. Contrast enhanced brain magnetic resonance imaging (MRI) of patient B1.
Figure S2. CLN3 PCR in patient B1.
Figure S3. CLN3 PCR in patient B4.