

Abstract
Immunotoxins are proteins containing a cell-targeting element linked to a toxin that are under investigation for next-generation cancer treatment. However, these agents are difficult to synthesize, chemically heterogeneous, expensive, and show toxicity toward healthy cells. In this work, we describe the synthesis and characterization of a new type of immunotoxin that showed exquisite selectivity toward targeted cells. In our construct, targeting molecules were covalently attached or genetically fused to oligomeric pore-forming toxins. The activity of the immunotoxin was then caged by fusing a soluble protein to the transmembrane domain and activated via cleavage with furin, which is a protease that is overexpressed in many cancer cells. During the several coupling steps, directed evolution allowed the efficient synthesis of the molecules in E. coli cells, as well as selection for further specificity toward targeted cells. The final construct showed no off-target activity, while acquiring an additional degree of specificity toward the targeted cells upon activation. The pore-forming toxins described here do not require internalization to operate, while the many protomeric subunits can be individually modified to refine target specificity.
Introduction
Protein-based drugs have become increasingly important in the pharmaceutical industry. In the period from 2011 to 2016, the FDA approved 62 proteins as drugs,1 most of which contain monoclonal antibodies (mAb). These agents recognize molecular targets on cancer cell surfaces, blocking their biological function, or, most often, marking the cells for the body’s immune system.2 An advantage of this approach is that drugs can be developed to recognize specifically a complex biological signature in malignant cells. However, mAbs are complex molecules that cannot be synthesized chemically and are manufactured in living organisms.3 Furthermore, they often require complex post-translational modifications that can only be introduced when heterologous expression systems are used.4 In addition, since the products are synthesized by cells or organisms, their chemical modification is not straightforward, and complex purification processes are involved.5,6 Finally, because their large size (up to 150 kDa), they have limited tumor penetration,7 and they are often recognized by the host immune system.8
In next-generation targeted cancer therapy,9 mAbs were, e.g., conjugated to a drug (antibody-drug conjugates),1,10,11 to a toxin (immunotoxin or IT),12,13 to a cytokine,14 or to a radioactive particle.15 In such constructs, the antibody recognizes a specific cell target, which allows the deadly cargo to be delivered to the diseased tissue. In immunotoxins, mAbs or growth factors are either chemically or genetically fused to a potent protein toxin, which inhibit protein synthesis, such as diphtheria toxin16 or pseudomonas exotoxin A.17 These are very efficient toxins, because they act catalytically on their cytosolic targets, hence, at very low concentrations. However, ITs must be internalized into the cell and not every target has a sufficient internalization rate, allowing sufficient accumulation of toxophore to effectively kill cancer cells.18−20 Hence, highly potent payload drugs must be used frequently, which, in turn, can produce life-threatening toxicities.21 In fact, the high potency of the payload requires a highly selective expression of membrane targets in cancer cells compared to healthy cells,22 and the number of suitable targets may be limited to just a few dozen.23
Alternative hybrid molecules might be built from membrane-acting toxins, which assemble into unregulated oligomeric pores in the membrane of targeted cells. Different hemolytic toxins from sea anemones, bacteria, or humans24−26 have been used to target different cell lines, including immature T lymphocytes,27 leukemic cells,26 breast cancer cells,28 lung cancer cells,29 or colon cancer cells.30 Since hundreds of pores might be necessary to obtain a cytotoxic effect,31 pore-forming toxins are much less potent than intracellular toxins commonly used in ITs. However, toxicity may be complemented or regulated by using other drugs, which are preferably internalized into the cells permeabilized by the pore-forming toxin.32 In addition, each monomer of the pore could be fused to different targeting elements, which, in turn, should bring a higher level of control of the targetability of the drug. However, the main limitation of most pore-forming immunotoxins is their basal toxicity toward most cells, including red blood cells, which, in turn, prevents their pharmacologic use.
In nature, many toxins are synthesized as protoxin and activated by proteolytic removal of a polypeptide segment at either terminus. Cancer cells often overexpress specific tumor-associated proteases, which are important for the invasion and metastasis of cancer cells.33,34 In one approach, to reduce the toxicity toward healthy cells, immunotoxins have been prepared to specifically cleave the linker between the targeting moiety and the payload by intracellular cancer-associated proteases, hence activating the toxin in situ.35,36 Pore-forming toxins have also been inactivated by genetic fusion with a polypeptide trigger and then proteolytically activated by cancer-associated proteases.37,38
In this work, we describe the preparation of pore-forming immunotoxins consisting of a chemical or protein-based targeting element, a pore-forming toxin, and a protease trigger (see Figure 1). Crucially, each fusion step is optimized by directed evolution, which allowed the toxicity to be tuned toward the target cells, as well as efficient synthesis of the protein complexes in E. coli cells. We show a construct that recognizes a molecular target on cancer cells and is selectively activated by a cancer-associated protease, while displaying no off-target activity on other cells.
Figure 1.
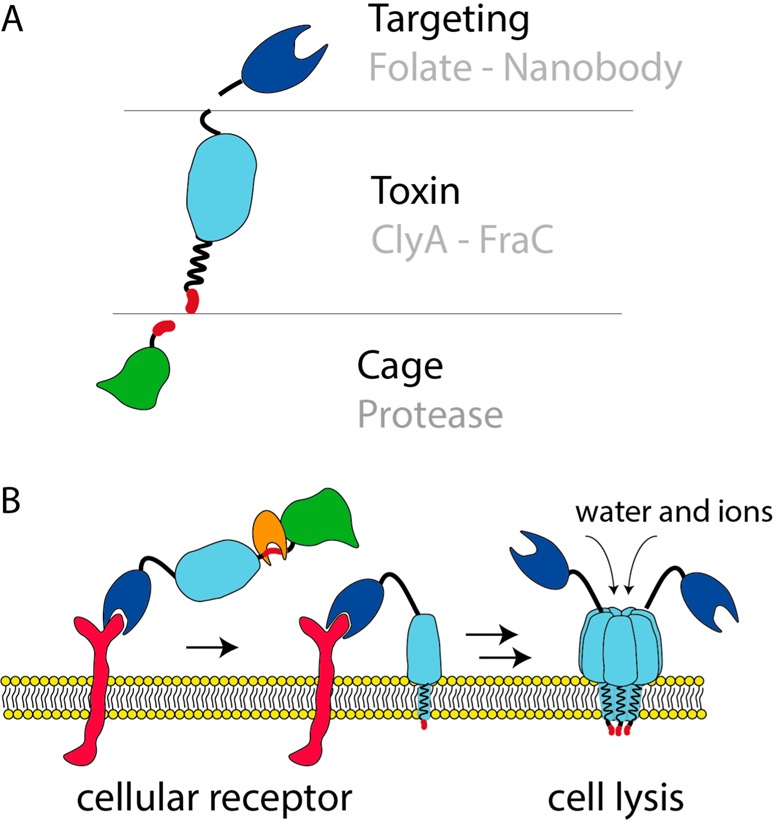
Modular nanopore immunotoxins. (A) Schematic representation of different modules used to build up a pore-forming immunotoxin with caged activity. The central point is the toxin. In this study, we used the membrane-acting toxins ClyA and FraC. The C-terminus of the toxin was conjugated to different targeting molecules, including folate and an anti-EGFR nanobody, to direct the toxin to cancer cells. For site-specific activation of the toxin, DHFR was fused to the N-terminus of the toxin via a cancer protease sensitive linker. Activation of the toxin by proteolysis is necessary to enable pore formation and, thus, cell killing activity. (B) Schematic representation of the pore formation of the designed protein drug. Soluble toxin is recruited to the host membrane by interactions of the targeting module and the corresponding receptor. Subsequent proteolytic cleavage at the specific protease site is necessary for activation of the toxin. Finally, the toxin can insert into the membrane. Pore formation changes membrane permeability, ultimately leading to cell death.
Results and Discussion
Immunotoxin Preparation by Targeted Chemical Modification
Cytolysin A (ClyA) is a protein toxin synthesized as soluble 34 kDa monomeric protein, which assembles into a dodecameric pore, causing the lysis of cell membranes rich in cholesterol.39 In our first effort to prepare an immunotoxin, we conjugated folate to a cysteine residue introduced at position 272 in a previously engineered cysteine-less ClyA (ClyA-AS) from Salmonella typhi.40 Folate was covalently attached to ClyA-AS-S272C monomers via a PEG-5K linker bearing a maleimide moiety (see Figure 2A). SDS-PAGE revealed that ∼50% of the ClyA-AS-S272C was conjugated (see Figure 2B). The construct was purified by size exclusion chromatography (SEC) and tested for cytotoxic activity on KB cells overexpressing folate receptors (FR). A MTT assay, which assesses the cell metabolism by measuring the mitochondrial potential, revealed that conjugation to folate increased toxicity toward FR+ cells by ∼2-fold [IC50(ClyA-folate) = 5.45 nM vs IC50(ClyA) = 13.5 nM] (see Figure 2C). In order to confirm that ClyA-folate induces preferential cell lysis, we added 10 nM of ClyA-folate to FR+ and FR– cells containing culture medium with a standard folate concentration (0.002 mM), and we assessed the integrity of the cell membrane using a propidium iodide assay. Under these conditions, we found that ClyA-folate causes increased cell death in FR+ cells but not in FR– cells (see Figure 2D). This difference is abolished when folate concentration in the medium is increased 250-fold (see Figure 2D). This suggests that cell death mediated by ClyA-folate is receptor-mediated. Thus, conjugation of ClyA with folate increases its specificity to FR+ cells, making this a possible candidate for a targeted therapy approach against FR-overexpressing tumors.
Figure 2.

ClyA targeting cancer cells. (A) Schematic representation of ClyA (blue, PDB: 2WCD) conjugated to folate. Folate was covalently attached to ClyA-AS-S272C via a disulfide bond and a PEG linker. (B) ClyA folate conjugation examined by 12% SDS-PAGE electrophoresis. Lanes 1 and 2: ClyA-AS-S272C modified with folate-5k PEG-maleimide showing ∼50% modification. The arrow indicates modified ClyA-folate, the band below the arrow is unmodified ClyA, while additional bands above the arrow most likely represent additional incorporations of PEG-folate molecules reacting to lysine residues in ClyA-AS. Lanes 3 and 4: ClyA-AS-S272C prior modification. (C) Comparison of the IC50 values of ClyA and fol-ClyA in FR-positive KB cells. (D) Comparison of cell death of FR-positive cells (KB) and FR-negative cells (A549) by 10 nM Fol-ClyA, in a standard medium (containing 0.002 mM of folate) and in a medium supplemented with 250-fold higher folate concentration (0.5 mM). (E) Schematic representation of ClyA conjugated to nanobody at the C-terminus. ClyA is shown in blue the anti-EGFR nanobody 7D12 is shown in red (PDB: 4KRL). (F) Representative dose–response curves of ClyA-AS and ClyA-Nb in the absence of EGF. Conjugation to the anti-EGFR nanobody increases toxicity toward EGFR overexpressing A431 cells. (G) Comparison of the IC50 values of ClyA and ClyA-Nb, in the presence and absence of EGF.
A Genetically Encoded Immunotoxin
In a second approach, the anti-EGFR nanobody 7d1241,42 was genetically attached to the C-terminus of ClyA-AS via a 16 amino acid long linker (see Figure 2E). EGFR overexpression in cells is associated with different cancer types and is an indication of especially aggressive breast cancer.43,44 The ClyA-AS-nanobody construct (ClyA-Nb) was overexpressed in E. coli cells and purified by Ni-NTA affinity chromatography. Conjugation to the nanobody preserved hemolytic activity of ClyA toward sheep red blood cells (see Figure 3E, as well as Figure S2D in the Supporting Information). To test whether nanobody attachment improved toxicity toward EGFR-overexpressing cells, we measured the mitochondrial activity of A431 epidermoid carcinoma cells, overexpressing EGFR,45 at increasing concentration of immunotoxin. The cell viability experiments showed a 2-fold reduced IC50 of ClyA-Nb [IC50(ClyA-Nb) = 7.2 ± 1.1 nM], compared to ClyA-AS [IC50(ClyA) = 17.1 ± 2.9 nM] (see Figure 2F). Incubation with EGF abolished this difference (see Figure 2G, as well as Figures S1A, S1B, and S3A in the Supporting Information). Thus, as observed for ClyA-folate conjugation, the attachment of the targeting unit increased toxicity to the target cells by 2-fold, making ClyA-Nb a possible candidate for a targeted therapy approach against EGFR-overexpressing tumors.
Figure 3.

Directed evolution of ClyA-Nb. (A) Schematic representation of ClyA conjugated to nanobody. The amino acids at the N-terminus are not observed in the crystal structure of ClyA, suggesting that they do not have a well-defined secondary structure. Amino acids 5 and 6, which were mutated to improve toxin properties, are shown in red. (B) Hemolytic activity of a part of the ClyA-Nb library. Hemolysis rates are presented as a percentage of ClyA-Nb activity. (C) Part of the ClyA-Nb library analyzed by a 4%–20% blue-native PAGE.40 Lane 1, protein ladder; lane 2, ClyA-Nb; lane 3, ClyA-Nb with 0.2% SDS; lane 4, SE-ClyA-Nb; lane 5, SE-ClyA-Nb with 0.2% SDS; lane 6, FR-ClyA-Nb; lane 7, FR-ClyA-Nb with 0.2% SDS; lane 8, QR-ClyA-Nb; lane 9, QR-ClyA-Nb with 0.2% SDS; lane 10, GG-ClyA-Nb; and lane 11, GG-ClyA-Nb with 0.2% SDS. (D) ClyA-nanobody fusion purification examined by 12% SDS-PAGE. Lane 1, protein ladder; lane 2, ClyA; lane 3, ClyA-Nb; and lane 4, SE-ClyA-Nb. (E) Comparison of the hemolysis percentage of ClyA and ClyA-Nb constructs. Fusion of ClyA to the Nb reduces hemolytic activity and the mutation I5S and F6E of ClyA reduces activity even further. (F) Toxicity of ClyA constructs toward EGFR overexpressing A431 cells, showing that both the nanobody and the additional mutation at the N-terminus improve toxicity toward the cells. (G) Toxicity of SE-ClyA-Nb in the presence and absence of 15 nM EGF, showing that the effect of the increased toxicity of SE-ClyA-Nb is due to the nanobody. (H) Comparison of the IC50 values of ClyA, ClyA-Nb, and SE-ClyA-Nb in the presence and absence of EGF, showing the high specificity of the SE-ClyA-Nb construct.
Protein Engineering Allows Efficient Synthesis in E. coli and Improves Targetability toward Cancer Cells
To increase specificity of ClyA toward the membranes of target cancer cells, we performed random mutagenesis using degenerate primers targeting isoleucine 5 and phenylalanine 6 in ClyA-Nb (see Figure 3A). The two residues are hydrophobic and are located just before the transmembrane region of ClyA. Thus, we hypothesized they are important for membrane binding. An initial negative screening on red blood cells was performed, where toxins which showed slower hemolysis rates than ClyA-Nb were selected. This step was intended to reduce the affinity of the toxin for nontarget cell membrane. Moderate active variants were then purified by Ni-NTA affinity chromatography and tested for expression and purity by blue native polyacrylamide gel electrophoresis (BN-PAGE; see Figure 3C, as well as Figure S2A in the Supporting Information). Variants, which were highly expressed and showed no preoligomerization (i.e., they do not oligomerize in the absence of membranes or surfactants) were then selected and tested on A431 cells. Among the variants tested, SE-ClyA-Nb could be purified with reasonably high yields (∼1 mg from 300 mL culture), showed low tendency to preoligomerize, and showed a 3-fold slower hemolytic activity, compared to ClyA-AS and ClyA-Nb [t50(ClyA-AS) = 12 min, t50(ClyA-Nb) = 14 min, and t50(SE-ClyA-Nb) = 48 min for 1 μg toxin] (see Figure S2D in the Supporting Information). Crucially, the toxicity on A431 cells improved 2-fold more, compared to ClyA-Nb [IC50(SE-ClyA-Nb) = 4.1 ± 0.5 nM] (see Figure 3F). In the presence of 15 nM, EGF the toxicity reduced 4-fold [IC50(SE-ClyA-Nb, EGF) = 16.0 ± 0.7 nM] (see Figure 3G, as well as Figures S3B and S3C in the Supporting Information) to the level observed for ClyA [IC50(ClyA, EGF) = 18.5 ± 3.1 nM] (see Figure 3H), indicating that the additional toxicity of SE-ClyA-Nb is due to the specific interaction of the nanobody with the cancer cell receptors.
Toxins Can Be Exchanged
The cytotoxic efficiency of ClyA toxins is dependent on the membrane composition of the targeted cell line. Thus, to refine and generalize our approach, we exchanged the ClyA nanopore toxin for the actinoporin Fragaceatoxin C (FraC). Similarly to ClyA, FraC is a pore-forming toxin that is expressed as a water-soluble monomer and self-assembles into (octameric) transmembrane pores. However, the N-terminal membrane spanning region of FraC interacts tightly with three sphingomyelin molecules per monomer (see Figure S4A in the Supporting Information). Thus, FraC requires sphingomyelin to cause cell damage.46 The anti-EGFR nanobody containing the 18-amino acid linker used to prepare ClyA-Nb was fused to the C-terminus of S-FraC to prepare S-FraC-Nb. S-FraC carries the mutation W112S, compared to wild-type (WT) FraC that we found increased its expression in E. coli cells. S-FraC-Nb could be purified at high yield in E. coli cells and exhibited hemolytic activity that was similar to that of S-FraC (see Figure S4C in the Supporting Information), indicating that the protein fusion did not impair protein activity. As observed for ClyA, the fusion to the nanobody increases the toxicity of S-FraC by 2-fold toward EGFR-overexpressing cells [IC50(S-FraC) = 135.5 ± 22.7 nM, vs IC50(S-FraC-Nb) = 63.9 ± 16.4 nM] (see Figure S4D in the Supporting Information). Predictably, 15 nM of EGF reduced the toxicity of S-FraC-Nb [IC50(S-FraC-Nb, EGF) = 197.9 ± 15.5 nM], but showed no effect on S-FraC [IC50(S-FraC, EGF) = 138.5 ± 28.7 nM (see Figures S4E, S4F, and S4G in the Supporting Information)].
A Triggered Toxin Improved by Directed Evolution
Although directed evolution could increase the affinity of SE-ClyA-Nb for A431 cells while decreasing the toxicity toward red blood cells, the latter could not be completely abolished. Many tumor cells secrete proteases such as furin,34,47,48 urokinase plasminogen activator,49 or cathepsin B,50 which, in turn, activates other proteases, eventually promoting tumor metastasis by helping tumor cells digest the extracellular matrix and penetrate the basal lamina. Therefore, we planned to design an immunotoxin that is activated by furin. To silence the toxicity of ClyA, we fused dihydrofolate reductase (DHFR, 19 kDa) to the N-terminus of the ClyA-Nb and FraC-Nb toxins via an 18-amino-acid-long linker containing a furin cleavage site (see Figure 4A, as well as Figure S5A in the Supporting Information). DHFR was selected because it is a relatively small protein that shows high water solubility, is expressed well in E. coli cells, and is nontoxic to human cells. The digestion of the constructs by furin will then remove DHFR, thereby activating the toxin. Surprisingly, however, DHFR-SE-ClyA showed similar hemolytic activity as SE-ClyA-Nb, indicating that DHFR-SE-ClyA-Nb can oligomerize on membranes, despite the large protein on the transmembrane helix (Figure S5C in the Supporting Information). By contrast, the DHFR-FraC construct was much less hemolytic active. However, the construct suffered from a small background activity on red blood cells, and it could not be purified at high concentrations, because of aggregation.
Figure 4.

Caging FraC toxin. (A) Schematic representation of FraC conjugated to DHFR. FraC (purple, PDB: 4TSY) fused N-terminal to DHFR (green, PDB: 1RH3) by an 18-amino-acid-long linker, including a protease cleavage site for furin. (B) Comparison of the hemolytic activity of different DHFR-FraC mutants resulted from directed evolution. Fusion of FraC to DHFR deactivates the toxin and therefore no hemolysis of red blood cells can be observed. Activation with the protease furin regains hemolytic activity of all mutants. (C) Representative dose–response curves of MD-DHFR-FraC and proteolyzed MD-DHFR-FraC on A431 cells. Therefore, conjugation to DHFR deactivates the toxin, and no cell killing is observed for MD-DHFR-FraC. Proteolysis with furin triggers pore formation of the toxin and recovers toxicity toward A431 cells. (D) Representative dose–response curves of MD-DHFR-FraC (DHFR-(furin)-FraC) and proteolyzed MD-DHFR-FraC on Calu-6 cells. Calu-6 cells produce furin, which activates MD-DHFR-FraC, resulting in cell killing. (E) Representative dose–response curves of DHFR-(TEV)-FraC and proteolyzed DHFR-(TEV)-FraC on Calu-6 cells. Conjugation to DHFR deactivates the toxin and Calu-6 cells do not express TEV proteases; therefore, no cell killing is observed for DHFR-(TEV)-FraC. Proteolysis with TEV triggers pore formation of the toxin and recovers toxicity toward Calu-6 cells.
To improve the E. coli synthesis of the DHFR-FraC construct, we performed three rounds of random mutagenesis on the entire construct. Libraries were generated by error-prone PCR and screened for hemolytic activity in the presence and absence of protease. Variants were selected that showed low or no background activity but were hemolytic active after proteolytic cleavage. The best three variants from the third round were purified by Ni-NTA affinity chromatography and compared using hemolytic activity. MD-DHFR-FraC (displaying the T50M mutation on the DHFR sequence and the N325D mutation in the FraC sequence), C-DHFR-FraC (R312C at the interface of FraC protomers), and HC-DHFR-FraC (R102H in the active site of DHFR and R312C at the interface of FraC protomers) did not induce red blood cell lysis after the addition of 10 μg of the purified proteins (see Figure 4B, as well as Figure S6A in the Supporting Information). However, upon incubation with furin (3 h, 37 °C), all mutants lysed red blood cells within minutes [t50(C-DHFR-FraC) = 4.8 min, t50(HC-DHFR-FraC) = 5 min, and t50(MD-DHFR-FraC) = 2.8 min for 10 μg toxin] (see Figure 4B). MD-DHFR-FraC was selected for further cytotoxic characterization on A431 cells. The addition of up to 600 nM DHFR-FraC caused no cell death, while an equal concentration of proteolyzed MD-DHFR-FraC showed cytotoxicity comparable to S-FraC [IC50(cut FraC) = 98.9 ± 11.9 nM] (see Figure 4C).
In order to test whether a DHFR-FraC construct can be activated by a cell-secreted protease, the MD-DHFR-FraC was tested on Calu-6 cells, which is a cell line expressing the protease furin,51,52 the latter is a protease that is overexpressed by many cancer cells.34,51 Rewardingly, MD-DHFR-FraC showed cytotoxicity on Calu-6 cells [IC50 = 212.0 ± 35.4 nM] that was comparable to the preproteolyzed MD-DHFR-FraC [IC50 = 142.5 ± 30.5 nM] (see Figure 4D). In contrast, when up to 820 nM of a caged immunotoxin containing a TEV (DHFR-(TEV)-FraC) rather than a furin cleavage site was used, no cytotoxic activity was observed (see Figure 4E), indicating that the activation of FraC by Calu-6 cells was induced by furin and not by other nonspecific proteases.
A Triggered Immunotoxin
In the last step, the nanobody was genetically fused to the C-terminus of evolved MD-DHFR-FraC (see Figure 5A). Although the size of the protein increased to 56 kDa, no additional directed evolution was required, as the construct successfully expressed well in E. coli cells (∼1 mg from 300 mL culture) and could be efficiently purified in one-step by Ni-NTA affinity chromatography (see Figure 5B). The full construct (DHFR-FraC-Nb) exhibited no hemolytic activity, while incubation with furin induced red blood cell lysis within a few minutes [t50(DHFR-FraC-Nb) = 4.6 min for 10 μg toxin] (see Figure 5C). As expected, DHFR-FraC-Nb was not cytotoxic toward A431 cells (24 h incubation) (see Figure 5D), but caused cell death after incubation with furin [IC50 = 146 ± 18.5 nM] (see Figure S7B in the Supporting Information). Cytotoxicity was reduced when EGF was added to the growth medium [IC50 = 283.9 ± 15.1 nM] (see Figure S7C in the Supporting Information), confirming that the interaction with the cell membrane receptor increased the immunotoxin activity. In situ activation of DHFR-FraC-Nb was tested, using the furin-expressing cell line Calu-6.51,52 As expected, DHFR-FraC-Nb, which was not active on A431 cells, induced cell death toward Calu-6 cells [IC50 = 95.2 ± 14.4 nM] (see Figure 5D, as well as Figures S7B and S7D in the Supporting Information). The addition of 15 nM EGF reduced the toxicity of DHFR-FraC-Nb by almost 2-fold [IC50 = 148.2 ± 3.6 nM] (see Figures S7C and S7E in the Supporting Information). Most likely, the effect of the nanobody on Calu-6 cells is less pronounced because this cell line expresses less EGFR than A431 cells.53
Figure 5.

A caged pore-forming immunotoxin. (A) Schematic representation of FraC conjugated to DHFR and nanobody. FraC is shown in purple, DHFR is shown in green, and the C-terminal of anti-EGFR nanobody 7D12 is shown in red. (B) 12% SDS-PAGE to examine DHFR-FraC-nanobody fusion and its proteolysis. Lane 1, protein ladder; lane 2, DHFR-FraC-Nb; and lane 3, DHFR-FraC-Nb proteolyzed by furin (1:100 ratio). Only ∼50% of DHFR-FraC-Nb was proteolytically activated. The arrows indicate the proteins of interest, while additional bands most likely correspond to partial proteolytic products or impurity in the commercial furin sample. (C) Comparison of the hemolytic activity of DHFR-FraC-Nb and proteolyzed DHFR-FraC-Nb. Only proteolyzed DHFR-FraC-Nb is active. (D) Representative dose–response curves of DHFR-FraC-Nb toward A431 cells or Calu-6 cells, showing that DHFR-(furin)-FraC-Nb is only active on Calu-6 cells, which is the only cell line expressing furin necessary for activation.
Discussion
Since the introduction of the first recombinant protein therapeutic, human insulin, 35 years ago, proteins have long remained a rarely used subset of medical treatments. In recent years, protein therapeutics have increased dramatically in number and frequency of use, and they now have a significant role in almost every field of medicine, despite this role is still only in its infancy. In this work, we describe a method that uses directed evolution to prepare protein-based therapeutic agents consisting of a membrane-targeting element covalently linked to a caged toxin.
We tested ClyA from Salmonella typhi and FraC from Actinia fragacea pore-forming toxins as cytotoxic elements. Both toxins are expressed as soluble monomers that form oligomeric pores into lipid membranes. In order to direct protein toxicity toward selected cells, we explored two approaches. A small molecule folate was chemically attached to ClyA via a 5 kDa PEG linker. The latter was used to mediate the interaction distance with the receptor and to facilitate the purification of the conjugate. This approach is fast and might allow the screening of hundreds of molecules. However, the bioorthogonal chemical linkages can be instable, and they can produce low reaction yields that might result in heterogeneous samples. Thus, we tested a second approach, where the variable antibody domain of a single-domain camelid antibodies, called nanobodies (Nb), conjugated to the C-terminus of the toxin. Nanobodies are small and stable,54 penetrate tissues efficiently,55 show low immunogenicity,56 and can be easily produced in bacteria. For both approaches, we found that ClyA and FraC toxins conjugated to the targeting unit induced a 2-fold increase in IC50 toward cancer cell lines overexpressing the cognate receptor.
In order to improve the soluble expression of the immunotoxin, increase target affinity, and reduce the toxicity toward blood cells, we resorted to a directed evolution experiment in which the hydrophobic residues at the N-terminus of the ClyA toxin were randomized. The rationale was that a reduced affinity of the immunotoxin for nontargeted membranes would be compensated by the nanobody-mediated binding to a specific membrane target, leading to a greater difference in toxicity between cell types. Using this approach, we identified pore-forming immunotoxins with an additional 2-fold-increased IC50 toward cells expressing EGFR and a 3-fold-reduced off-target hemolytic activity. Simultaneously, the production yield in E. coli cells was more efficient.
In the final step, we sought to completely suppress off-target activity. In nature, many toxins are synthesized as protoxins that are proteolytically activated extracellularly. Borrowing from nature, we introduced a polypeptidic segment at the transmembrane N-terminus of FraC and ClyA to cap the activity of the nanopore toxins. The conjugate was preceded by a cleaving sequence for furin, which is a cancer-associated protease involved in degrading the extracellular matrix in tumor formation and metastasis.57 We found that ClyA was fully active on red blood cells,58 while the toxicity of FraC was reduced but not completely abolished. Gladly, the toxicity of the caged FraC-Nb toxin could be suppressed by three rounds of directed evolution, allowing one to identify a construct that was completely inactive toward cellular membranes but fully active upon in situ protease activation by a cancer cell line overexpressing furin.
This work describes a method that uses a directed evolution approach to prepare pore-forming immunotoxins with caged activity. Our design contains three separate elements that can be easily exchanged: a water-soluble pore-forming toxin that oligomerizes on lipid membranes, a membrane targeting unit, and protein trigger for in situ activation. The targeting element and the protein trigger allows a two-degree control of the toxin activity. The pore-forming ability of the immunotoxin may offer advantages, compared to conventional toxins. The activity can be easily assayed on red blood cells, which, in turn, allows the use of random mutagenesis approaches to improve the synthesis of the proteins in E. coli cells and to fine-tune the properties of the protein conjugate. In addition, the oligomeric nature of the assembled cytotoxic pores will allow conjugating the protein drug with multiple membrane targeting motives, which will allow further fine-tuning of the cytotoxicity toward targeted cells.
Methods
Hemolytic Activity Assay
Defibrinated sheep blood (ThermoFisher Scientific) was washed with 150 mM NaCl, 15 mM Tris.HCl (pH 7.5) until the supernatant was clear. The erythrocytes were then resuspended with the same buffer to ∼1% concentration (OD650 = 0.6–0.8). The suspension (120 μL) was then mixed with the solutions containing immunotoxin. Hemolytic activity was measured by monitoring the decrease in OD650 using the MultiskanTM GO Microplate spectrophotometer (ThermoFisher Scientific). The hemolysis rate was calculated as the inverse of the time elapsed until a 50% decrease in turbidity was attained. To determine hemolytic activity of caged FraC, toxin was mixed with the appropriate protease and incubated for 5 min before application to the erythrocyte solutions.
Cytotoxicity Assay
Cell viability after the addition of immunotoxin was assessed with the WST-8 cell proliferation assay. First, 20 000 cells were seeded per well in a 96-well plate in the corresponding medium and incubated for 24 h. Cells were then treated in triplicate with varying concentrations of immunotoxins, toxins, epidermal growth factor and folate. After 24 or 48 h, 5 μL of the CCK-8 solution was added per well, and the plates were incubated for 2 h under standard conditions. Lastly, the absorption was measured at 450 nm in a MultiskanTM GO Microplate spectrophotometer. IC50 values were calculated by using sigmoidal fitting with Origin (OriginLab, Northampton, MA). Cell viability of reference wells with untreated cells was set to 100%.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00720.
Detailed information on construction of mutants and libraries, protein productions and purifications, hemolytic activity assays, used cell lines, and cultivation conditions; toxicity of ClyA variants in the presence and absence of 15 nM EGF on A431 epidermoid carcinoma cells; selection of evolved immunotoxins; time and concentration dependence of ClyA cytotoxicity toward A431 cells; FraC targeting cancer cells; cytotoxicity of DHFR-ClyA fusion; cytotoxicity of DHFR-FraC fusion (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lagassé H. A. D.; Alexaki A.; Simhadri V. L.; Katagiri N. H.; Jankowski W.; Sauna Z. E.; Kimchi-Sarfaty C. (2017) Recent advances in (therapeutic protein) drug development. F1000Research 6, 113. 10.12688/f1000research.9970.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A. M.; Wolchok J. D.; Old L. J. (2012) Antibody therapy of cancer. Nat. Rev. Cancer 12, 278. 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- Samaranayake H.; Wirth T.; Schenkwein D.; Räty J. K.; Ylä-Herttuala S. (2009) Challenges in monoclonal antibody-based therapies. Ann. Med. 41, 322. 10.1080/07853890802698842. [DOI] [PubMed] [Google Scholar]
- Bandaranayake A. D.; Almo S. C. (2014) Recent advances in mammalian protein production. FEBS Lett. 588, 253–260. 10.1016/j.febslet.2013.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingermann T. (2008) Recombinant therapeutic proteins: Production platforms and challenges. Biotechnol. J. 3, 90–97. 10.1002/biot.200700214. [DOI] [PubMed] [Google Scholar]
- Liu H. F.; Ma J.; Winter C.; Bayer R. (2010) Recovery and purification process development for monoclonal antibody production. MAbs 2, 480–99. 10.4161/mabs.2.5.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchinton A. I.; Tannock I. F. (2006) Drug penetration in solid tumours. Nat. Rev. Cancer 6, 583. 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Rosenberg A. S.; Sauna Z. E. (2018) Immunogenicity assessment during the development of protein therapeutics. J. Pharm. Pharmacol. 70, 584–594. 10.1111/jphp.12810. [DOI] [PubMed] [Google Scholar]
- Baudino T. A. (2015) Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discovery Technol. 12, 3–20. 10.2174/1570163812666150602144310. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Teicher B. A.; Hassan R. (2016) Antibody–drug conjugates for cancer therapy. Lancet Oncol. 17, e254–e262. 10.1016/S1470-2045(16)30030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J. M.; Berkenblit A. (2018) Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 69, 191–207. 10.1146/annurev-med-061516-121357. [DOI] [PubMed] [Google Scholar]
- Pastan I.; Hassan R.; Fitzgerald D. J.; Kreitman R. J. (2006) Immunotoxin therapy of cancer. Nat. Rev. Cancer 6, 559–65. 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- Alewine C.; Hassan R.; Pastan I. (2015) Advances in anticancer immunotoxin therapy. Oncologist 20, 176–85. 10.1634/theoncologist.2014-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasche N.; Neri D. (2012) June) Immunocytokines: A novel class of potent armed antibodies. Drug Discovery Today 17, 583. 10.1016/j.drudis.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Kitson S. L.; Cuccurullo V.; Moody T. S.; Mansi L. (2013) Radionuclide antibody-conjugates, a targeted therapy towards cancer. Curr. Radiopharm. 6, 57–71. 10.2174/1874471011306020001. [DOI] [PubMed] [Google Scholar]
- Williams D. P.; Parker K.; Bacha P.; Bishai W.; Borowski M.; Genbauffe F.; Strom T. B.; Murphy J. R. (1987) Diphtheria toxin receptor binding domain substitution with interleukin-2: genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein Eng., Des. Sel. 1, 493–498. 10.1093/protein/1.6.493. [DOI] [PubMed] [Google Scholar]
- Mansfield E.; Amlot P.; Pastan I.; FitzGerald D. J. (1997) Recombinant RFB4 immunotoxins exhibit potent cytotoxic activity for CD22-bearing cells and tumors. Blood 90, 2020–2026. [PubMed] [Google Scholar]
- Du X.; Beers R.; FitzGerald D. J.; Pastan I. (2008) Differential Cellular Internalization of Anti-CD19 and -CD22 Immunotoxins Results in Different Cytotoxic Activity. Cancer Res. 68, 6300–6305. 10.1158/0008-5472.CAN-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirie C. M.; Hackel B. J.; Rosenblum M. G.; Wittrup K. D. (2011) Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 286, 4165–72. 10.1074/jbc.M110.186973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito L.; Mercatelli D.; Bortolotti M.; Maiello S.; Djemil A.; Battelli M.; Bolognesi A. (2017) Two Saporin-Containing Immunotoxins Specific for CD20 and CD22 Show Different Behavior in Killing Lymphoma Cells. Toxins 9, 182. 10.3390/toxins9060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy H. (2016) Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 8, 659–671. 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasarao M.; Low P. S. (2017) Ligand-Targeted Drug Delivery. Chem. Rev. 117, 12133–12164. 10.1021/acs.chemrev.7b00013. [DOI] [PubMed] [Google Scholar]
- Grawunder U., and Barth S. (2017) Next Generation Antibody Drug Conjugates (ADCs) and Immunotoxins; Springer: Cham, Switzerland. [Google Scholar]
- Al-Yahyaee S. A. S.; Ellar D. J. (1996) Cell targeting of a pore-forming toxin, CytA δ-endotoxin from Bacillus thuringiensis subspecies israelensis, by conjugating CytA with anti-Thy 1 monoclonal antibodies and insulin. Bioconjugate Chem. 7, 451–460. 10.1021/bc960030k. [DOI] [PubMed] [Google Scholar]
- Tejuca M.; Anderluh G.; Dalla Serra M. (2009) Sea anemone cytolysins as toxic components of immunotoxins. Toxicon 54, 1206–1214. 10.1016/j.toxicon.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Wan L.; Zeng L.; Chen L.; Huang Q.; Li S.; Lu Y.; Li Y.; Cheng J.; Lu X. (2006) Expression, purification, and refolding of a novel immunotoxin containing humanized single-chain fragment variable antibody against CTLA4 and the N-terminal fragment of human perforin. Protein Expression Purif. 48, 307–313. 10.1016/j.pep.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Avila A. D.; de Acosta C. M.; Lage A. (1988) A new immunotoxin built by linking a hemolytic toxin to a monoclonal antibody specific for immature T lymphocytes. Int. J. Cancer 42, 568–571. 10.1002/ijc.2910420417. [DOI] [PubMed] [Google Scholar]
- Avila A. D.; Mateo Acosta C. De; Lage A. (1989) A carcinoembryonic antigen-directed immunotoxin built by linking a monoclonal antibody to a hemolytic toxin. Int. J. Cancer 43, 926–929. 10.1002/ijc.2910430533. [DOI] [PubMed] [Google Scholar]
- Avila A. D.; Calderón C. F.; Pérez R. M.; Pons C.; Pereda C. M.; Ortiz A. R. (2007) Construction of an immunotoxin by linking a monoclonal antibody against the human epidermal growth factor receptor and a hemolytic toxin. Biol. Res. 40, 173–183. 10.4067/S0716-97602007000200008. [DOI] [PubMed] [Google Scholar]
- Tejuca M.; Díaz I.; Figueredo R.; Roque L.; Pazos F.; Martínez D.; Iznaga-Escobar N.; Pérez R.; Alvarez C.; Lanio M. E. (2004) Construction of an immunotoxin with the pore forming protein StI and ior C5, a monoclonal antibody against a colon cancer cell line. Int. Immunopharmacol. 4, 731–744. 10.1016/j.intimp.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan M.; Sathyanarayana P.; Maiti P. K.; Visweswariah S. S.; Ayappa K. G. (2014) Lysis dynamics and membrane oligomerization pathways for Cytolysin A (ClyA) pore-forming toxin. RSC Adv. 4, 4930. 10.1039/c3ra45159c. [DOI] [Google Scholar]
- Soletti R. C.; de Faria G. P.; Vernal J.; Terenzi H.; Anderluh G.; Borges H. L.; Moura-Neto V.; Gabilan N. H. (2008) Potentiation of anticancer-drug cytotoxicity by sea anemone pore-forming proteins in human glioblastoma cells. Anti-Cancer Drugs 19, 517–25. 10.1097/CAD.0b013e3282faa704. [DOI] [PubMed] [Google Scholar]
- Koblinski J. E.; Ahram M.; Sloane B. F. (2000) Unraveling the role of proteases in cancer. Clin. Chim. Acta 291, 113. 10.1016/S0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- Jaaks P.; Bernasconi M. (2017) The proprotein convertase furin in tumour progression. Int. J. Cancer 141, 654–663. 10.1002/ijc.30714. [DOI] [PubMed] [Google Scholar]
- Weldon J. E.; Skarzynski M.; Therres J. A.; Ostovitz J. R.; Zhou H.; Kreitman R. J.; Pastan I. (2015) Designing the furin-cleavable linker in recombinant immunotoxins based on Pseudomonas exotoxin A. Bioconjugate Chem. 26, 1120–8. 10.1021/acs.bioconjchem.5b00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandooren J.; Opdenakker G.; Loadman P. M.; Edwards D. R. (2016) Proteases in cancer drug delivery. Adv. Drug Delivery Rev. 97, 144–155. 10.1016/j.addr.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Walker B.; Bayley H. (1994) A pore-forming protein with a protease-activated trigger. Protein Eng., Des. Sel. 7, 91–97. 10.1093/protein/7.1.91. [DOI] [PubMed] [Google Scholar]
- Panchal R. G.; Cusack E.; Cheley S.; Bayley H. (1996) Tumor protease-activated, pore-forming toxins from a combinatorial library. Nat. Biotechnol. 14, 852–856. 10.1038/nbt0796-852. [DOI] [PubMed] [Google Scholar]
- Mueller M.; Grauschopf U.; Maier T.; Glockshuber R.; Ban N. (2009) The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature 459, 726–730. 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; De Maeyer M.; Maglia G. (2013) Tuning the size and properties of ClyA nanopores assisted by directed evolution. J. Am. Chem. Soc. 135, 13456–13463. 10.1021/ja4053398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers R. C.; Vosjan M. J. W. D.; Laeremans T.; El Khoulati R.; De Bruin R. C. G.; Ferguson K. M.; Verkleij A. J.; Van Dongen G. A. M. S.; Van Bergen En Henegouwen P. M. P. (2011) A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int. J. Cancer 129, 2013–2024. 10.1002/ijc.26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers R. C.; Laeremans T.; Huang L.; De Taeye S.; Verkleij A. J.; Revets H.; De Haard H. J.; Van Bergen En Henegouwen P. M. P. (2007) Efficient inhibition of EGFR signalling and of tumour growth by antagonistic anti-EGFR Nanobodies. Cancer Immunol. Immunother. 56, 303–317. 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umekita Y.; Ohi Y.; Sagara Y.; Yoshida H. (2000) Co-expression of epidermal growth factor receptor and transforming growth factor-?? predicts worse prognosis in breast-cancer patients. Int. J. Cancer 89, 484–487. . [DOI] [PubMed] [Google Scholar]
- Nicholson R. I.; Gee J. M.; Harper M. E. (2001) EGFR and cancer prognosis. Eur. J. Cancer 37 (Suppl 4), 9–15. 10.1016/S0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Ullrich A.; Coussens L.; Hayflick J. S.; Dull T. J.; Gray A.; Tam A. W.; Lee J.; Yarden Y.; Libermann T. A.; Schlessinger J.; Downward J.; Mayes E. L. V.; Whittle N.; Waterfield M. D.; Seeburg P. H. (1984) Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425. 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Caaveiro J. M. M.; Morante K.; González-Manãs J. M.; Tsumoto K. (2015) Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 6, 6337. 10.1038/ncomms7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbikay M.; Sirois F.; Yao J.; Seidah N. G.; Chrétien M. (1997) Comparative analysis of expression of the proprotein convertases furin, PACE4, PC1 and PC2 in human lung tumours. Br. J. Cancer 75, 1509–14. 10.1038/bjc.1997.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried G.; Basak A.; Cromlish J. A.; Benjannet S.; Marcinkiewicz J.; Chrétien M.; Seidah N. G.; Khatib A. M. (2003) The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J. Clin. Invest. 111, 1723–1732. 10.1172/JCI200317220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazar A. P.; Henkin J.; Goldfarb R. H. (1999) The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis 3, 15–32. 10.1023/A:1009095825561. [DOI] [PubMed] [Google Scholar]
- Mohamed M. M.; Sloane B. F. (2006) Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 6, 764–775. 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Bassi D. E.; Mahloogi H.; Al-Saleem L.; Lopez De Cicco R.; Ridge J. A.; Klein-Szanto A. J. (2001) Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol. Carcinog. 31, 224–32. 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- Bassi D. E.; Zhang J.; Renner C.; Klein-Szanto A. J. (2017) Targeting proprotein convertases in furin-rich lung cancer cells results in decreased in vitro and in vivo growth. Mol. Carcinog. 56, 1182–1188. 10.1002/mc.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schaeybroeck S.; Kyula J.; Kelly D. M.; Karaiskou-McCaul A.; Stokesberry S. A.; Van Cutsem E.; Longley D. B.; Johnston P. G. (2006) Chemotherapy-induced epidermal growth factor receptor activation determines response to combined gefitinib/chemotherapy treatment in non-small cell lung cancer cells. Mol. Cancer Ther. 5, 1154–1165. 10.1158/1535-7163.MCT-05-0446. [DOI] [PubMed] [Google Scholar]
- Van Der Linden R. H. J.; Frenken L. G. J.; De Geus B.; Harmsen M. M.; Ruuls R. C.; Stok W.; De Ron L.; Wilson S.; Davis P.; Verrips C. T. (1999) Comparison of physical chemical properties of llama V(HH) antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1431, 37–46. 10.1016/S0167-4838(99)00030-8. [DOI] [PubMed] [Google Scholar]
- Bannas P.; Well L.; Lenz A.; Rissiek B.; Haag F.; Schmid J.; Hochgräfe K.; Trepel M.; Adam G.; Ittrich H.; Koch-Nolte F. (2014) In vivo near-infrared fluorescence targeting of T cells: Comparison of nanobodies and conventional monoclonal antibodies. Contrast Media Mol. Imaging 9, 135–142. 10.1002/cmmi.1548. [DOI] [PubMed] [Google Scholar]
- Cortez-Retamozo V.; Lauwereys M.; Hassanzadeh Gh G.; Gobert M.; Conrath K.; Muyldermans S.; De Baetselier P.; Revets H. (2002) Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 98, 456–462. 10.1002/ijc.10212. [DOI] [PubMed] [Google Scholar]
- Thomas G. (2002) Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766. 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y.; Doody A. M.; Chen D. J.; Cremona G. H.; Shuler M. L.; Putnam D.; DeLisa M. P. (2008) Engineered Bacterial Outer Membrane Vesicles with Enhanced Functionality. J. Mol. Biol. 380, 51–66. 10.1016/j.jmb.2008.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.