

Abstract
Single-molecule methods have been rapidly developing with the appealing prospect of transforming conventional ensemble-averaged analytical techniques. However, challenges remain especially in improving detection sensitivity and controlling molecular transport. In this article, we present a direct method for the fabrication of analytical sensors that combine the advantages of nanopores and field-effect transistors for simultaneous label-free single-molecule detection and manipulation. We show that these hybrid sensors have perfectly aligned nanopores and field-effect transistor components making it possible to detect molecular events with up to near 100% synchronization. Furthermore, we show that the transport across the nanopore can be voltage-gated to switch on/off translocations in real time. Finally, surface functionalization of the gate electrode can also be used to fine tune transport properties enabling more active control over the translocation velocity and capture rates.
Keywords: single-molecule sensing, nanopore, voltage gating, functionalized gate, tunable nanoscale transport, nanofluidic field-effect transistor
Introduction
The development of new analytical methods is currently at the forefront of healthcare research because of the need for improved performance compared to the current state-of-the-art. This is especially true for applications such as early-stage disease diagnosis and treatment.1,2 To achieve this, different techniques including fluorescence3−5 and optical6 and magnetic tweezers7,8 have been reported with sensitivities reaching the single-molecule limit. Even though such techniques could provide the sensitivity to detect and manipulate molecules one at a time, their broader application in a clinical setting is impeded by the complexities associated with sample preparation and processing. Therefore, the development of “single-pot” label-free approaches remains one of the most attractive avenues for single-molecule sensors. Among different label-free techniques, nanopores and field-effect transistors (FETs) have emerged as exceptionally promising methods because of their inherent sensitivity and ability for multiplexed detection.9−13
Nanopores operate by driving analytes electrokinetically across a nanoscale pore, allowing single-molecule identification by measuring characteristic modulations in the ionic current. By extracting the magnitude and duration of individual translocation events, one can obtain information such as molecular dimensions, concentration, and in some cases even charge and conformation.10,11,14 Although numerous advances have been made,10,11,15−18 fast analyte transport and analyte selectivity still represent substantial challenges.10,19−23 To this end, different strategies have recently been investigated including the use of FET-coupled nanopores. Nanoscale FETs are capable of label-free sensing of biological analytes in real time,12,24 and the sensing principle is based on monitoring the change of conductance between drain–source (DS) upon binding of biomolecules to the gate electrode. However, the sensing area is governed by the Debye screening length, which is relatively short under physiological conditions (∼1 nm).13 Unlike in nanopores, these limitations lower the possibility of actively transporting analyte to the sensing region and controlling the throughput.
It is no surprise that due to the distinct advantages offered by the two techniques, the development of combined nanopore–FET platforms has attracted an increasing amount of interest, both experimentally17,25−28 and theoretically.29−31 In a hybrid nanopore–FET design, the nanopore can be viewed as a drain–source channel for single-molecule translocation, while the FET is integrated by placing a gate electrode near the nanopore aperture. A distinctive advantage of such a platform is the ability to control the surface charge near the nanopore by varying the potential applied to the gate electrode, which, in turn, allows for the manipulation of the transport of single molecules across the nanopore. Furthermore, the possibility of simultaneously detecting the ionic current (drain–source) and gate current can offer an additional sensing channel and enrich the obtained single-molecule information. Previous works demonstrated simultaneous dual-channel single-molecule DNA detection by incorporating a solid-state nanopore with a silicon nanowire27 or graphene nanoribbon.17,32,33 More recently, Panday et al.34 proposed the integration of a carbon nanoelectrode within relatively large nanopores for the detection of 40 nm nanoparticles. Additionally, different approaches have been proposed and developed to control transport including programming voltage pulses,35,36 tuning the viscosity of solvents,37 changing the local temperature,38 and voltage gating.22 The variation of nanopore surface charge with gating potential is expected to play a key role in the manipulation of single-molecule translocation30,31,39 in improving or supplementing existing strategies for controlling single-molecule transport.
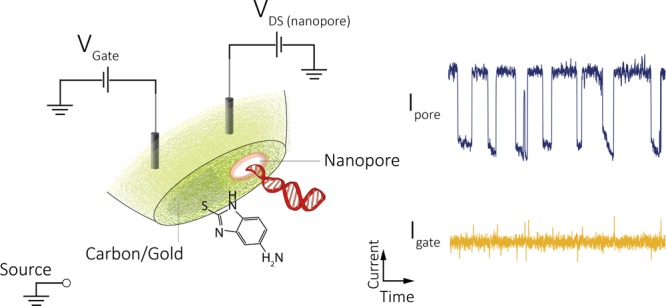
Building upon these advances, we present a new fabrication strategy for the facile design and alignment of a sub-30 nm nanopore to the gate electrode (Figure 1A) to control molecular transport at the single-molecule level. Rapid fabrication was achieved by controlled electrodeposition of gold onto a nanoscale carbon electrode formed inside one of the barrels of a double-barrel nanopore/nanopipette. Ion current feedback was used to fabricate the gold gate electrode, localized at the nanopore opening while monitoring and tuning the pore conductance in real time. We show that single-molecule events can be detected in both nanopore and gate channels with enhanced sensitivity and up to nearly 100% synchronization. Furthermore, voltage gating, as well as surface functionalization (Figure 1B,C), can be used to tune the nanopore surface charge, and in turn to switch on/off, or to accurately control the rate of single-molecule transport. The results demonstrate that these hybrid nanopore–FET devices can be readily used to control molecular transport by tuning nanopore charge both electrically and chemically.
Figure 1.

Conceptual design of the nanopore–FET using a double-barrel nanopipette. (A) Left barrel is filled with a carbon nanoelectrode fabricated via pyrolytic deposition of carbon. Gold is deposited on the tip of the nanoelectrode using feedback-controlled electrochemical deposition, which acts as the FET gate. The barrel on the right is filled with a buffer/analyte and can act as a conventional nanopore or as a drain–source in a FET configuration. (A, i) Gate (carbon/gold) can be used to detect and manipulate single-molecule translocations across the nanopore (drain−source, DS). (A, ii) Scanning electron microscopy (SEM) micrograph showing the deposited gold around the nanopore. (B) Gold on the gate could be further functionalized, for example, with a thiolated amine to tune the surface charge for gating control and (C) synchronized detection of single-molecule DNA translocation using the functionalized nanopore–FET.
Results and Discussion
The base platform used for the nanopore–FET is a double-barrel nanopipette, a class of solid-state nanopore sensors, fabricated via laser-assisted pulling of theta-shaped quartz glass capillaries. Double-barrel nanopipettes were used with a gold-coated carbon nanoelectrode in one barrel for gating and a hollow barrel for nanopore sensing; see Figure 1. The gating electrode was initially fabricated by pyrolysis of butane, Supporting Information (SI) Figure S1A–C, to selectively deposit carbon in one barrel, while leaving the second barrel open, according to well-established protocols,40,41 followed by electrochemical deposition of gold using the configuration shown in Figure 2A. The size of the carbon nanoelectrode formed at the tip of the nanopipette was estimated to be 50 ± 11 nm from the steady-state limiting current,40 as determined by cyclic voltammetry (CV) using 1 mM Ru(NH3)6Cl3 as a redox probe. Figure 2B (inset) shows a typical CV curve recorded at the carbon electrode upon cathodic polarization. The electrode dimensions, calculated from the nanopore conductance, were in good agreement with the initial nanopore diameter as measured by scanning electron microscopy (SEM) prior to carbon deposition (SI Figure S1D).
Figure 2.

Electrochemical fabrication of the nanopore–FET using real-time ionic current feedback. (A) Schematic of a bipotentiostatic configuration used to deposit and monitor gold deposition. The carbon nanoelectrode was used as a working electrode for the electrodeposition of gold (WEdeposition). To monitor the electrodeposition of gold around nanopore, another working electrode (WEfeedback) was inserted into the open barrel filled with 52 mM (NH4)2SO3 and used for real-time feedback. All potentials quoted are relative to a quasi-reference counter electrode (QRCE) placed in the plating bath filled with a 10 times diluted ECF64 gold plating solution. Potentials applied to the working electrodes were Vdeposition = −0.73 V, Vfeedback = −0.1 V. (B) To electrochemically characterize the gate electrode, cyclic voltammograms were recorded before (black) and after (orange) electrodeposition of gold, in the presence of 1 mM Ru(NH3)6Cl3 and 100 mM KCl, revealing an enhancement in the active electrode area after gold deposition. The inset confirms the formation of a carbon nanoelectrode on the tip of double-barrel nanopipettes. (C) Transmission electron microscopy micrograph showing the deposition of gold at the tip of the nanopipette and around the nanopore. d is the diameter of the nanopore. (D) Both the feedback current in the nanopore and the amount of gold deposited could be monitored in real time. (E) Ionic current feedback could be stopped at a given threshold to (F) control the pore conductance. This is shown for three threshold currents of −1.5, −1, and −0.5 nA, giving final pore conductances of 7.2 ± 1.4, 3.5 ± 1.9, and 2.0 ± 0.4 nS, respectively, as revealed by I–V characterization and histograms of nanopore conductance before and after feedback-controlled deposition of gold. The average pore conductance before gold deposition was 17.3 ± 6.4 nS (N = 90).
A gold gate electrode was made by electrochemically depositing gold from a sulfite gold plating solution42 (ECF64, Metalor Technologies) onto the carbon nanoelectrode. To achieve controlled deposition, real-time ionic feedback was implemented using a bipotentiostatic configuration,43 with a quasi-reference counter electrode (QRCE) in the plating bath, as shown in Figure 2. This electrode configuration allowed us to simultaneously deposit gold on the gate electrode and monitor the conductance decrease in the nanopore channel in real time, which in turn is related to the pore dimensions.
Typical current–time (I–t) traces for deposition and feedback are shown in Figure 2D, and the growth pattern of gold was imaged by dark-field microscopy (SI Figure S2), which revealed a gradual deposition outward of the carbon nanoelectrode and toward the nanopore (Figure 2C). In the feedback (nanopore) channel, the current remained constant, until the nanopore started to close, as indicated by a sharp transition regime and a rapid current decrease (Figure 2D). Although the initial current level and the time required to reach this regime varied from pipette to pipette, approximately 90% of all devices (N = 374) followed such electrodeposition trends. By monitoring the nanopore current, it was, therefore, possible to turn off the deposition at a predefined threshold, and in the process to precisely control the nanopore conductance. Three representative chronoamperometric traces at feedback current thresholds of −1.5, −1, and −0.5 nA are shown in Figure 2E. Lower feedback current thresholds resulted in nanopore devices with lower-than-average conductance (indicative of smaller nanopore dimensions) and very similar (current–voltage) I–V characteristics. Figures 2F and SI S3 show corresponding I–V curves and histograms of the nanopore conductance before and after deposition (N = 90), yielding values of 7.2 ± 1.4, 3.5 ± 1.9, and 2.0 ± 0.4 nS, respectively (in a solution consisting of 52 mM (NH4)2SO3). The conductance for unmodified (carbon only) nanopores was significantly larger with a broader distribution (17.3 ± 6.4 nS), than the deposited devices, indicating bigger initial nanopores with larger device-to-device variation. CV curves recorded at the gold-deposited electrodes showed a significant increase in the limiting current when compared to those at bare carbon electrodes, mainly due to the larger surface area introduced by the gold deposition (Figure 2B). The deposition of the gold at the nanopore was further confirmed by SEM, indicating that nanopore openings were reduced down to ∼20 nm (Figure 1A-ii). At this size regime, nanopores have been shown to possess ion selectivity,44 which was confirmed for our gold-coated nanopores by measuring reversal potential using KCl concentration gradients. Prior to gold deposition, nanopores showed cation selectivity (SI Figure S4), as a result of the negatively charged glass surface. For gold-coated nanopores, the cation selectivity decreased, indicating a reduction of the negative surface charge on the nanopore.
Single-molecule sensing functionality of the hybrid nanopore–FET device was validated using a three-electrode setup with the gate electrode and the nanopore connected to independent patch electrodes using a Multiclamp 700B patch-clamp amplifier (Molecular Devices) and the reference/ground electrodes placed in the bath. It should be noted that measurements were performed both with two independent and linked reference/ground electrodes. No noticeable differences were observed in the experimental outcomes likely due to the solution/electrode resistance being several orders of magnitude smaller than those of the nanopore and the gate (SI Figure S5).
In terms of conventional nanopore detection, single-molecule sensitivity was initially confirmed using plain quartz/carbon devices without gold electrodeposition. To this end, 400 pM of 10 kbp ds DNA in 100 mM KCl was added inside the nanopore barrel of the pipette. In this configuration, under applied negative voltage, translocations occur from the inside of the nanopipette (cis) to the bath (trans), resulting in an overall increase in the conductance when using 100 mM KCl at pH 8.35 Across all devices, we found dwell times and peak currents for 10 kbp ds DNA to be in good agreement with those in previous studies using unmodified (bare quartz) single-barrel nanopipettes,45−47 indicating that the incorporation of a carbon nanoelectrode adjacent to a quartz nanopore caused negligible effect on single-molecule DNA translocation (SI Figure S6).
After gold electrodeposition, we observed a substantial (up to 200%) increase in the mean peak current, as can be seen in SI Figure S6. Smaller Au-coated nanopores likely resulted in enhanced interactions between DNA and nanopore surface, leading to larger peak currents, longer dwell times, and lower capture rates. Furthermore, due to the change of nanopore surface charge after gold deposition, the direction of the nanopore current changed at 100 mM KCl; current enhancement was observed with carbon-/glass-coated nanopipettes, while the current blockade was recorded with gold-coated ones.
When accessing the additional FET sensing modality in our nanopore devices, simultaneous chronoamperometric measurements at the gating channel revealed synchronized biphasic events, correlating to the DNA translocation events measured in the nanopore ionic current channel (Figure 3A–C). The origin of synchronization between the nanopore and the gate detection channel is to some extent debated in the literature, albeit most examples use differing platforms.17,27,33,34,48 The working mechanism is usually correlated with the device architecture (e.g., material and geometry of the pore and gate electrode) as well as physicochemical parameters such as the type and the concentration of the electrolyte across the nanopore and FET channel. In our case, the observed synchronization across FET and nanopore detection channels can be attributed to a change of local potential around the gate electrode due to capacitive coupling between the nanopore and the gold-deposited gate electrode. To confirm this, the capacitance and the resistance components of our devices were measured, and circuit simulations were performed, as shown in SI Figures S7–S9, similar to measurements described by Puster et al.33 The simulated translocation events in the gate channel with a biphasic shape were very similar to the measured DNA translocations (Figure 3A–C). A detailed analysis of these simulated events revealed that they are time derivatives of the nanopore ionic current and linked to the change of potential at the gate electrode resulting in transient currents, confirming capacitive coupling between the nanopore and the gold-deposited gate electrode.
Figure 3.

Synchronized nanopore–FET of 10 kbp DNA. Salt-concentration-dependent translocations for 1 M, 100 mM, and 50 mM KCl are shown in (A)–(C) for both the nanopore (upper) and gate (lower) channels. At lower KCl concentrations, the amplitude of biphasic peaks on the gate (Igate) was more pronounced relative to blockade current from the nanopore (Ipore). Asymmetric salt concentrations were also used across the nanopore (D–F) to tune the Debye screening length. For asymmetric concentrations ([cis] = 100 mM for all cases), both blockade current and peak amplitude on the gate were shown to decrease with a greater dilution of KCl in the bath. More importantly, the ratio between Igate and Ipore was increased. Igate was extracted by summing up the absolute values of negative amplitude with positive amplitude. Experimental conditions: 400 pM 10 kbp ds DNA was added into the nanopore and translocated from the inside (cis) of the open barrel to the outside (trans); in all cases, Vpore = −700 mV, Vgate = 0 mV.
In FET devices, the gate response is largely dependent on the electrolyte concentration. In nanopore–FETs, solutions with different ionic strength will influence the electric field gradient across the nanopore.27,33,34,49,50 We tested this by performing electrolyte-concentration-dependent experiments. First, the concentration of the electrolyte was decreased from 1 M to 50 mM KCl for both the inside and the outside of the nanopore, and the number of events (N) and their peak amplitudes (I) detected from the nanopore and the gate, respectively, were analyzed. Interestingly, at 1 M, the Debye screening length was substantially reduced, thus resulting in the synchronization ratio (Ngate/Npore = 0.52) being less than 1 (Figure 3A). At 100 mM and 50 mM KCl, the synchronization ratio is 1 (Figure 3B,C), with perfect synchronization between the nanopore and the gate. Moreover, we found that with decreasing electrolyte concentration, the current amplitudes of the biphasic peaks (Igate) became more pronounced relative to blockade current from the nanopore (Ipore).
Second, diluting the electrolyte concentration outside of the nanopipette (trans side) leads to a decrease in the solution resistance around the gate electrode. With increasing [cis]/[trans] ratio, a more gradual electrolyte concentration gradient across the nanopore was created. The resistance of the trans side becomes comparable to the nanopore resistance, while the resistance of the cis side becomes negligible.27 In this case, due to the extended screening length across the nanopore, a greater fraction of the gate electrode is exposed to this potential gradient during DNA translocation across the nanopore, causing larger current modulation in the gate channel because of translocation and increasing the ratio of Igate/Ipore (Figure 3D–F).27,33
Importantly, the gate channel can be used to modulate DNA transport across the nanopore when a voltage is applied to the gate electrode (Figure 4). The variation of nanopore surface charge with gating potentials can be used to turn on and off the transport through the nanopore by applying a bias at or near the potential of zero charge (pzc), which could be measured from differential capacitance measurements. From these measurements, we found that the pzc in our devices was located at (−90 ± 10 mV), Figure 4C, in excellent agreement with the value of (−90 ± 5 mV) in 100 mM KCl reported in the literature.51 On the basis of this value and the electrochemical window, 10 kbp DNA translocation studies were performed at varying Vgate. Importantly, DNA transport through the nanopore could be switched off at Vgate ≈ −100 mV. The transport could then be turned on at Vgate ≈ −100 mV or higher (Figure 4D). It should be noted that this on/off behavior is reproducible with multiple cycles. Previous simulation studies by Sugimoto et al.52 suggested that the use of voltage gating to control DNA translocation is mainly influenced by (i) electrophoretic force under an applied Vpore, (ii) electroosmotic flow, and (iii) electrostatic interactions between DNA and the nanopore surface. In this case, molecular transport across the nanopore is switched off when the gold electrode possesses a net negative charge (Vgate < −100 mV). The electrostatic repulsion between DNA and the nanopore surface effectively shuts off DNA translocations.
Figure 4.

Voltage gating of the nanopore. (A) I–V characterization of the nanopore–FET before and after gold deposition and (B) after gold deposition at varying Vgate. (C) Variation of nanopore surface charge with gating potentials (Vgate) was probed by differential capacitance measurements on an ultramicro-gold-deposited electrode, showing a capacitance minimum at −100 mV, which is attributed to the potential of zero charge (pzc). (D) This correlates well with the gating of 10 kbp DNA in 100 mM KCl, 10 mM Tris–HCl, and 1 mM ethylenediaminetetraacetic acid (EDTA) buffer at pH 8. DNA transport could be switched off or on depending on whether the potential applied to Vgate was above or below the pzc. By controlling the gate potential, translocations can be switched on and off in real time as shown in (E).
Single-molecule transport can be further tuned by chemical functionalization of the gate electrode. Changes in the gate potential allow for tuning of the surface charge of the functional groups bound to the electrode surface53−56 and enable a route to dynamically tune the interactions between the gate/nanopore surface and transported molecules. In our system, the gold locally deposited at the nanopore can be easily functionalized via thiol–gold self-assembly. The fabrication scheme for functionalized devices and detailed characterization after each stage are presented in SI Figure S4. We used a thiolated amine with a net positive charge at pH 857 (SI Figure S4). Functionalization was performed by immersing gold-deposited nanopipettes in the thiol solution for 12 h, followed by rinsing with methanol and water to remove any excess thiolated amine. Positively charged amine coating on the gate electrode was confirmed via an increase in anion selectivity after functionalization. In the functionalized nanopore, the rectification inverted from negative to positive with a 98% change of the rectification ratio (r = |I–400 mV/I400 mV|) from 4.41 ± 0.17 to 0.11 ± 0.01 by lowering the pH from 8 to 4, indicating a higher degree of protonation (SI Figure S4). In contrast, in gold-coated pipettes, the change of rectification ratio was much smaller (4.84 ± 0.14 to 2.02 ± 0.09). Further confirmation of the successful functionalization was obtained via differential capacitance measurements on an amine-modified electrode where an additional pzc peak appeared at a more positive potential of 150 mV (SI Figure S10).
Translocations with these surface-modified nanopores were performed using 200 pM 10 kbp DNA in 100 mM KCl at pH 8 (Figure 5). An operating range Vgate = (−400, 400 mV) was selected to minimize Faradaic processes at the functionalized surface (SI Figure S10). By increasing Vgate from −400 to 400 mV, translocations across the functionalized nanopore can be switched on/off, and more importantly, the translocation velocity and frequency can be controlled. A decrease in mean translocation velocity (Figure 5B-i) from 16.1 to 1.6 bp/μs and a controllable increase in event frequency (Figure 5B-iii) from 0 to 147 ± 29 s–1 were achieved via a stepwise tuning of Vgate from −400 to 400 mV. The reproducibility of gating control was verified by repeating the experiment using different functionalized nanopipettes (SI Figure S11). Interestingly, synchronized detection from the functionalized gold electrode can be still achieved, as revealed by elongated separation of biphasic peaks (SI Figure S12) with a lower Igate/Ipore, because of the additional presence of an amine layer that slows down DNA translocation through a functionalized nanopipette.
Figure 5.

Gating of an amine-functionalized nanopore–FET. Voltage gating was achieved via a stepwise increase of Vgate from −400 to 400 mV at 200 mV intervals. (A) Current–time traces showing the gating control of 10 kbp DNA translocation at applied Vgate of −400, −200, 0, 200, and 400 mV. (B) Dependence on (i) dwell time, (ii) equivalent area, and (iii) event frequency is shown as a function of the gating potentials. All data were expressed as percentage change with respect to Vgate = 0 mV, and in all cases, Vpore = −500 mV.
Conclusions
In conclusion, we demonstrated a new class of nanopore–FETs for double-channel synchronized detection and gating manipulation of single molecules. The use of ion current feedback during electrochemical fabrication enabled us to easily place the gold gate electrode in or near the nanopore in a highly controlled manner. Owing to the flexibility and reproducibility offered by feedback-controlled deposition of gold, the pore ionic conductance can be tailored to suit the need for different analytes, bringing more diversity into this nanopore–FET sensing system. Subsequently, the fabricated devices were not only able to detect single-molecule translocations using conventional ionic current blockade, but also from electrical signals at the gate electrode. Voltage gating was implemented into this system by tuning both electrophoretic and electroosmotic flow, as well as electrostatic interactions with analytes, allowing to switch on/off DNA transport in real time. The platform allows for easy surface functionalization, which allows us to further tune molecular gate transport through the nanopore and including dwell time and capture rates. The performance of nanopore–FET devices presented in this article has implications in enhancing the controllability, sensitivity, and selectivity of nanopore sensing. Facile chemical functionalization of gate electrode opens up the possibility for using this platform to detect and manipulate a wide range of biological analytes with improved sensitivity and selectivity.
Methods
Nanopipette Fabrication
Nanopipettes were fabricated from double-barrel theta quartz capillaries (Friedrich & Dimmock, Inc.; 1.2 mm outside diameter × 0.90 mm inside diameter × 100 mm length) using a P-2000 laser puller (Sutter Instruments). A custom two-line protocol was used: (1) heat: 870, filament: 4, velocity: 30, delay: 160, pull: 100. (2) Heat: 900, filament: 3, velocity: 20, delay: 130, pull: 160. The established protocol can be used to fabricate double-barrel nanopipettes with a diameter ranging from 50 to 100 nm across each barrel. It should be noted that the pulling parameters are device specific and are sensitive toward the variation of humidity and temperature.
Selective Pyrolysis of Butane
Pyrolytic deposition of carbon was performed using the setup shown in SI Figure S1, adapted from the procedure described previously by our group and collaborators.20,40,41 Butane was passed through one barrel through a silicon tubing. Another barrel of the nanopipette was closed using Blu Tack (Bostick). Therefore, carbon deposition happened only inside the open barrel. A butane torch was used to heat the tip of the pipette for pyrolytic deposition of the carbon end, which was maintained under an argon flow to prevent further oxidation of the deposited carbon. A heating time of 35 s was chosen to produce an extensive filling of amorphous carbon through the one barrel while leaving another barrel open. The flame temperature of the butane torch is lower than the softening temperature of quartz. Moreover, the end of the nanopipette tip was protected under an argon flow. At the tip of the open barrel, the nanopore is not closed as there is not enough temperature for quartz to soften and carbon deposition is not occurring in the open barrel.20,41 All of the nanopipettes were fabricated freshly on the day and stored in a sealed Petri dish until use to minimize any contamination. The size of carbon electrode was characterized by linear sweep voltammetry from 0 to −0.5 V (vs Ag/AgCl) at a scan rate of 0.05 V/s and a sampling interval of 0.002 V, in an aqueous solution containing 1 mM Ru(NH3)6Cl3 and 100 mM KCl.
Feedback-Controlled Electrodeposition of Gold
A bipotentiostatic configuration (Figure 2A) was used to perform electrodeposition of gold on the carbon electrode using a CHI 760 C potentiostat (CH Instruments). A 10 times diluted ECF64D (Metalor Technologies) plating solution containing 4.4 mM NH4AuSO3 in 52 mM (NH4)2SO3 was filled into the plating bath. To establish electrical contact with the carbon electrode, a 0.25 mm diameter copper wire (Goodfellow) was inserted into the carbon barrel. Because of the instability of AgCl in ECF64 (amine complexation),42 a 0.125 mm diameter Ag wire (Goodfellow) was chosen for use as both the working electrode inside the open barrel and the quasi-reference counter electrode (QRCE). A potential of −0.73 V (vs QRCE) was held at the carbon electrode to reduce the gold from the plating solution. Meanwhile, a potential difference of −0.1 V was applied between the second WE and the QRCE, to monitor in real time the ionic current flowing through the open barrel. Notably, a low voltage was chosen to minimize the possible interference with the deposition process. Before and after deposition, cyclic voltammetry (−0.5–0.5 V, scan rate 0.05 V/s, sampling interval 0.02 V) of the second WE was performed to extract the change of conductance. After deposition, the open barrel was rinsed with water to minimize contamination. To avoid salt crystallization at the tip, all nanopipettes were stored in a sealed vial filled with deionized water until use.
Differential Capacitance Measurements
Capacitance measurements were performed using a three-electrode configuration on a Gamry Reference 600 potentiostat, with deposited gold being the working electrode. A Ag/AgCl wire was used as the reference electrode. The counter electrode was a Pt wire (Goodfellow). Differential capacitance versus potential curves was obtained by impedance measurements at varying direct current potentials in the presence of a small 10 mV, peak-to-peak alternating current perturbation at a constant frequency of 10 Hz for the gold-deposited electrode and 500 Hz for the amine-functionalized electrode
Surface Functionalization of the Deposited Gold
Nanopipettes were rinsed with acetone and methanol to remove surface contaminants on the deposited gold. The open barrel was filled with the corresponding solvent during rinsing. Then, the resulting nanopipettes were immersed into a 5 mM solution of 5-amino-2-mercaptobenzimidazole (Sigma-Aldrich) in methanol for 12 h. Prior to DNA translocation experiments, the open barrels of functionalized nanopipettes were washed with methanol and water to remove any remaining thiol diffused into the open barrels during functionalization. All nanopipettes were freshly functionalized one day before use and used only once for translocation experiments.
DNA Solutions
Double-stranded 10 kbp DNA (New England Biolabs) was diluted from a stock concentration of 500 μg/mL in 10 mM Tris–HCl and 1 mM EDTA. Different concentrations of DNA solutions were prepared by serial dilution using the same buffer solution. All DNA solutions were stored in a freezer until use and freshly prepared before the experiments.
Dual-Channel Recordings
Both ionic current and current from the carbon electrode were measured by a Multiclamp 700B (Molecular Devices) in a voltage clamp mode with 10 kHz bandwidth and 100 kHz sampling frequency. The resulting signal was digitized by Axon Digidata 1550B. Data recordings were achieved using pClamp 10.6 software (Molecular Devices). Data analysis was performed using a custom-written MATLAB code developed in-house. A baseline current was calculated for every five data points, and the baseline threshold was set equal to or higher than six standard deviations. Any peaks above the threshold were identified as DNA translocation events.
Circuit Simulation
Circuit simulations were completed by an open-access simulation software LTspice (Linear Technologies). Both the nanopore and the gate were modeled as a resistor and a capacitor in parallel. The resistance of solution (Rsoln) was approximated by measuring the resistance between two Ag/AgCl electrodes as a function of their distance. The values for the resistance of the nanopore and the gate were obtained by fitting their I–V measurements. The capacitances of the nanopore and the gate were estimated using a triangular wave method suggested by Balan et al.58 Single-molecule translocation events were modeled by a change of nanopore resistance with a 10 ns step size for 3 ms, with the response from the gate simulated by the software.
Acknowledgments
J.B.E. has been funded in part by an ERC starting (NanoP) and consolidator (NanoPD) grants. A.P.I. and J.B.E. acknowledge support from EPSRC grant EP/P011985/1 and BBSRC grant no. BB/R022429/1. A.P.I. acknowledges the support of the IC Research Fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b13721.
Pyrolytic deposition of the carbon nanoelectrode; electrochemical deposition of gold; validation and simulation of synchronized detection; reproducibility and stability of the gating system (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Turner A. P. F. Biosensors: Sense and Sensibility. Chem. Soc. Rev. 2013, 42, 3184–3196. 10.1039/c3cs35528d. [DOI] [PubMed] [Google Scholar]
- Newman J. D.; Turner A. P. F. Home Blood Glucose Biosensors: A Commercial Perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. 10.1016/j.bios.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Eid J.; Fehr A.; Gray J.; Luong K.; Lyle J.; Otto G.; Peluso P.; Rank D.; Baybayan P.; Bettman B.; Bibillo A.; Bjornson K.; Chaudhuri B.; Christians F.; Cicero R.; Clark S.; Dalal R.; DeWinter A.; Dixon J.; Foquet M.; Gaertner A.; Hardenbol P.; Heiner C.; Hester K.; Holden D.; Kearns G.; Kong X.; Kuse R.; Lacroix Y.; Lin S.; Lundquist P.; Ma C.; Marks P.; Maxham M.; Murphy D.; Park I.; Pham T.; Phillips M.; Roy J.; Sebra R.; Shen G.; Sorenson J.; Tomaney A.; Travers K.; Trulson M.; Vieceli J.; Wegener J.; Wu D.; Yang A.; Zaccarin D.; Zhao P.; Zhong F.; Korlach J.; Turner S. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Juette M. F.; Terry D. S.; Wasserman M. R.; Zhou Z.; Altman R. B.; Zheng Q.; Blanchard S. C. The Bright Future of Single-Molecule Fluorescence Imaging. Curr. Opin. Chem. Biol. 2014, 20, 103–111. 10.1016/j.cbpa.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.; Romero-Ruiz M.; Castell O. K.; Bayley H.; Wallace M. I. High-Throughput Optical Sensing of Nucleic Acids in a Nanopore Array. Nat. Nanotechnol. 2015, 10, 986–997. 10.1038/nnano.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulushev R. D.; Marion S.; Petrova E.; Davis S. J.; Maerkl S. J.; Radenovic A. Single Molecule Localization and Discrimination of DNA-Protein Complexes by Controlled Translocation Through Nanocapillaries. Nano Lett. 2016, 16, 7882–7890. 10.1021/acs.nanolett.6b04165. [DOI] [PubMed] [Google Scholar]
- Peng H.; Ling X. S. Reverse DNA Translocation through a Solid-State Nanopore by Magnetic Tweezers. Nanotechnology 2009, 20, 185101 10.1088/0957-4484/20/18/185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman K. C.; Nagy a Single-Molecule Force Spectroscopy: Optical Tweezers, Magnetic Tweezers and Atomic Force Microscopy. Nat. Methods 2008, 5, 491–505. 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanunu M.; Meller A. Chemically Modified Solid-State Nanopores. Nano Lett. 2007, 7, 1580–1585. 10.1021/nl070462b. [DOI] [PubMed] [Google Scholar]
- Miles B. N.; Ivanov A. P.; Wilson K.; Doğan F.; Japrung D.; Edel J. B. Single Molecule Sensing with Solid-State Nanopores: Novel Materials, Methods, and Applications. Chem. Soc. Rev. 2013, 42, 15–28. 10.1039/C2CS35286A. [DOI] [PubMed] [Google Scholar]
- Venkatesan B. M.; Bashir R. Nanopore Sensors for Nucleic Acid Analysis. Nat. Nanotechnol. 2011, 6, 615–624. 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- Cui Y.; Wei Q.; Park H.; Lieber C. M. Nanowire Nanosensors for Highly Sensitive and Selective Detection of Biological and Chemical Species. Science 2001, 293, 1289–1292. 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- Zhang A.; Lieber C. M. Nano-Bioelectronics. Chem. Rev. 2016, 116, 215–257. 10.1021/acs.chemrev.5b00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C.; Li M.-Y.; Cirauqui N.; Wang Y.-Q.; Dal Peraro M.; Tian H.; Long Y.-T. Mapping the Sensing Spots of Aerolysin for Single Oligonucleotides Analysis. Nat. Commun. 2018, 9, 2823 10.1038/s41467-018-05108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman K. J.; Otto L. M.; Ivanov A. P.; Barik A.; Oh S.-H.; Edel J. B. Nanopore Sensing at Ultra-Low Concentrations Using Single-Molecule Dielectrophoretic Trapping. Nat. Commun. 2016, 7, 10217 10.1038/ncomms10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.; Wu H.; Jayasinghe L.; Patel A.; Reid S.; Bayley H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol. 2009, 4, 265–270. 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Traversi F.; Raillon C.; Benameur S. M.; Liu K.; Khlybov S.; Tosun M.; Krasnozhon D.; Kis A.; Radenovic A. Detecting the Translocation of DNA through a Nanopore Using Graphene Nanoribbons. Nat. Nanotechnol. 2013, 8, 939–945. 10.1038/nnano.2013.240. [DOI] [PubMed] [Google Scholar]
- Ivanov A. P.; Instuli E.; McGilvery C. M.; Baldwin G.; McComb D. W.; Albrecht T.; Edel J. B. DNA Tunneling Detector Embedded in a Nanopore. Nano Lett. 2011, 11, 279–285. 10.1021/nl103873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze J. Y. Y.; Ivanov A. P.; Cass A. E. G.; Edel J. B. Single Molecule Multiplexed Nanopore Protein Screening in Human Serum Using Aptamer Modified DNA Carriers. Nat. Commun. 2017, 8, 1552 10.1038/s41467-017-01584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R.; Zhang Y.; Nadappuram B. P.; Akpinar B.; Klenerman D.; Ivanov A. P.; Edel J. B.; Korchev Y. Nanopore Extended Field-Effect Transistor for Selective Single-Molecule Biosensing. Nat. Commun. 2017, 8, 586 10.1038/s41467-017-00549-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerema S. J.; Dekker C. Graphene Nanodevices for DNA Sequencing. Nat. Nanotechnol. 2016, 11, 127–136. 10.1038/nnano.2015.307. [DOI] [PubMed] [Google Scholar]
- Pulizzi F. Building a Better Nanopore. Nat. Nanotechnol. 2016, 11, 105. 10.1038/nnano.2016.15. [DOI] [PubMed] [Google Scholar]
- Lindsay S. The Promises and Challenges of Solid-State Sequencing. Nat. Nanotechnol. 2016, 11, 109–111. 10.1038/nnano.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patolsky F.; Zheng G.; Hayden O.; Lakadamyali M.; Zhuang X.; Lieber C. M. Electrical Detection of Single Viruses. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 14017–14022. 10.1073/pnas.0406159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi I.; Oura T.; Haga T.; Ando M.; Yamamoto J.; Mine T.; Ishida T.; Hatano T.; Akahori R.; Yokoi T.; Anazawa T. Side-Gated Ultrathin-Channel Nanopore FET Sensors. Nanotechnology 2016, 27, 115501 10.1088/0957-4484/27/11/115501. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Clausmeyer J.; Babakinejad B.; López Córdoba A.; Ali T.; Shevchuk A.; Takahashi Y.; Novak P.; Edwards C.; Lab M.; Gopal S.; Chiappini C.; Anand U.; Magnani L.; Coombes R. C.; Gorelik J.; Matsue T.; Schuhmann W.; Klenerman D.; Sviderskaya E. V.; Korchev Y. Spearhead Nanometric Field-Effect Transistor Sensors for Single-Cell Analysis. ACS Nano 2016, 10, 3214–3221. 10.1021/acsnano.5b05211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P.; Xiong Q.; Fang Y.; Qing Q.; Lieber C. M. Local Electrical Potential Detection of DNA by Nanowire–nanopore Sensors. Nat. Nanotechnol. 2011, 7, 119–125. 10.1038/nnano.2011.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. B.; Nam S. W.; Rooks M. J.; Rossnagel S. M. Ionic Field Effect Transistors with Sub-10 Nm Multiple Nanopores. Nano Lett. 2009, 9, 2044–2048. 10.1021/nl900309s. [DOI] [PubMed] [Google Scholar]
- Luan B.; Stolovitzky G.; Martyna G. Slowing and Controlling the Translocation of DNA in a Solid-State Nanopore. Nanoscale 2012, 4, 1068–1077. 10.1039/C1NR11201E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik K. H.; Liu Y.; Tabard-Cossa V.; Waugh M. J.; Huber D. E.; Provine J.; Howe R. T.; Dutton R. W.; Davis R. W. Control of DNA Capture by Nanofluidic Transistors. ACS Nano 2012, 6, 6767–6775. 10.1021/nn3014917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Tsutsui M.; Fan C.; Taniguchi M.; Kawai T. Controlling DNA Translocation through Gate Modulation of Nanopore Wall Surface Charges. ACS Nano 2011, 5, 5509–5518. 10.1021/nn201883b. [DOI] [PubMed] [Google Scholar]
- Heerema S. J.; Vicarelli L.; Pud S.; Schouten R. N.; Zandbergen H. W.; Dekker C. Probing DNA Translocations with Inplane Current Signals in a Graphene Nanoribbon with a Nanopore. ACS Nano 2018, 12, 2623–2633. 10.1021/acsnano.7b08635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puster M.; Balan A.; Rodríguez-Manzo J. A.; Danda G.; Ahn J. H.; Parkin W.; Drndić M. Cross-Talk Between Ionic and Nanoribbon Current Signals in Graphene Nanoribbon-Nanopore Sensors for Single-Molecule Detection. Small 2015, 11, 6309–6316. 10.1002/smll.201502134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panday N.; Qian G.; Wang X.; Chang S.; Pandey P.; He J. Simultaneous Ionic Current and Potential Detection of Nanoparticles by a Multifunctional Nanopipette. ACS Nano 2016, 10, 11237–11248. 10.1021/acsnano.6b06307. [DOI] [PubMed] [Google Scholar]
- Ivanov A. P.; Actis P.; Jönsson P.; et al. On-Demand Delivery of Single DNA Molecules Using Nanopipets. ACS Nano 2015, 9, 3587–3595. 10.1021/acsnano.5b00911. [DOI] [PubMed] [Google Scholar]
- Gershow M.; Golovchenko J. A. Recapturing and Trapping Single Molecules with a Solid-State Nanopore. Nat. Nanotechnol. 2007, 2, 775–779. 10.1038/nnano.2007.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesa C.; van Loo N.; Dekker C. DNA Nanopore Translocation in Glutamate Solutions. Nanoscale 2015, 7, 13605–13609. 10.1039/C5NR02793D. [DOI] [PubMed] [Google Scholar]
- Crick C. R.; Albella P.; Ng B.; Ivanov A. P.; Roschuk T.; Cecchini M. P.; Bresme F.; Maier S. A.; Edel J. B. Precise Attoliter Temperature Control of Nanopore Sensors Using a Nanoplasmonic Bullseye. Nano Lett. 2015, 15, 553–559. 10.1021/nl504536j. [DOI] [PubMed] [Google Scholar]
- Karnik R.; Fan R.; Yue M.; Li D.; Yang P.; Majumdar A. Electrostatic Control of Ions and Molecules in Nanofluidic Transistors. Nano Lett. 2005, 5, 943–948. 10.1021/nl050493b. [DOI] [PubMed] [Google Scholar]
- Actis P.; Tokar S.; Clausmeyer J.; Babakinejad B.; Mikhaleva S.; Cornut R.; Takahashi Y.; López Córdoba A.; Novak P.; Shevchuck A. I.; Dougan J. A.; Kazarian S. G.; Gorelkin P. V.; Erofeev A. S.; Yaminsky I. V.; Unwin P. R.; Schuhmann W.; Klenerman D.; Rusakov D. A.; Sviderskaya E. V.; Korchev Y. E. Electrochemical Nanoprobes for Single-Cell Analysis. ACS Nano 2014, 8, 875–884. 10.1021/nn405612q. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Shevchuk A. I.; Novak P.; Zhang Y.; Ebejer N.; MacPherson J. V.; Unwin P. R.; Pollard A. J.; Roy D.; Clifford C. A.; Shiku H.; Matsue T.; Klenerman D.; Korchev Y. E. Multifunctional Nanoprobes for Nanoscale Chemical Imaging and Localized Chemical Delivery at Surfaces and Interfaces. Angew. Chem., Int. Ed. 2011, 50, 9638–9642. 10.1002/anie.201102796. [DOI] [PubMed] [Google Scholar]
- Rutkowska A.; Freedman K.; Skalkowska J.; Kim M. J.; Edel J. B.; Albrecht T. Electrodeposition and Bipolar Effects in Metallized Nanopores and Their Use in the Detection of Insulin. Anal. Chem. 2015, 87, 2337–2344. 10.1021/ac504463r. [DOI] [PubMed] [Google Scholar]
- Ayub M.; Ivanov A.; Hong J.; Kuhn P.; Instuli E.; Edel J. B.; Albrecht T. Precise Electrochemical Fabrication of Sub-20 Nm Solid-State Nanopores for Single-Molecule Biosensing. J. Phys.: Condens. Matter 2010, 22, 454128 10.1088/0953-8984/22/45/454128. [DOI] [PubMed] [Google Scholar]
- Rollings R. C.; Kuan A. T.; Golovchenko J. A. Ion Selectivity of Graphene Nanopores. Nat. Commun. 2016, 7, 11408 10.1038/ncomms11408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb T. R.; Ivanov A. P.; Edel J. B.; Albrecht T. Single Molecule Ionic Current Sensing in Segmented Flow Micro Fluidics. Anal. Chem. 2014, 86, 1864–1871. 10.1021/ac403921m. [DOI] [PubMed] [Google Scholar]
- Gong X.; Patil A. V.; Ivanov A. P.; Kong Q.; Gibb T.; Dogan F.; Demello A. J.; Edel J. B. Label-Free in-Flow Detection of Single DNA Molecules Using Glass Nanopipettes. Anal. Chem. 2014, 86, 835–841. 10.1021/ac403391q. [DOI] [PubMed] [Google Scholar]
- Crick C. R.; Sze J. Y. Y.; Rosillo-Lopez M.; Salzmann C. G.; Edel J. B. Selectively-Sized Graphene-Based Nanopores for In-Situ Single Molecule Sensing. ACS Appl. Mater. Interfaces 2015, 7, 18188–18194. 10.1021/acsami.5b06212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R.; Ying Y. L.; Li Y. J.; Hu Y. X.; Yu R. J.; Lin Y.; Long Y. T. A 30 Nm Nanopore Electrode: Facile Fabrication and Direct Insights into the Intrinsic Feature of Single Nanoparticle Collisions. Angew. Chem., Int. Ed. 2018, 57, 1011–1015. 10.1002/anie.201710201. [DOI] [PubMed] [Google Scholar]
- Wanunu M.; Morrison W.; Rabin Y.; Grosberg A. Y.; Meller A. Electrostatic Focusing of Unlabelled DNA into Nanoscale Pores Using a Salt Gradient. Nat. Nanotechnol. 2010, 5, 160–165. 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin W. M.; Drndic M. Signal and Noise in FET-Nanopore Devices. ACS Sensors 2018, 3, 313–319. 10.1021/acssensors.7b00708. [DOI] [PubMed] [Google Scholar]
- Andersen T. N.; Perkins R. S.; Eyring H. Zero-Charge Potentials of Solid Metals. J. Am. Chem. Soc. 1964, 86, 4496 10.1021/ja01074a060. [DOI] [Google Scholar]
- Sugimoto M.; Kato Y.; Ishida K.; Hyun C.; Li J.; Mitsui T. DNA Motion Induced by Electrokinetic Flow near an Au Coated Nanopore Surface as Voltage Controlled Gate. Nanotechnology 2015, 26, 065002 10.1088/0957-4484/26/6/065502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Kapinos L.; Blosser T. R.; Magalhães T.; van Nies P.; Lim R. Y. H.; Dekker C. Single-Molecule Transport across an Individual Biomimetic Nuclear Pore Complex. Nat. Nanotechnol. 2011, 6, 433–438. 10.1038/nnano.2011.88. [DOI] [PubMed] [Google Scholar]
- Harrington L.; Alexander L. T.; Knapp S.; Bayley H. Angewandte Pim Kinase Inhibitors Evaluated with a Single-Molecule Engineered Nanopore Sensor. Angew. Chem., Int. Ed. 2015, 54, 8154–8159. 10.1002/anie.201503141. [DOI] [PubMed] [Google Scholar]
- Yusko E. C.; Johnson J. M.; Majd S.; Prangkio P.; Rollings R. C.; Li J.; Yang J.; Mayer M. Controlling Protein Translocation through Nanopores with Bio-Inspired Fluid Walls. Nat. Nanotechnol. 2011, 6, 253–260. 10.1038/nnano.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mitta G.; Peinetti A. S.; Cortez M. L.; Toimil-Molares M. E.; Trautmann C.; Azzaroni O. Highly Sensitive Biosensing with Solid-State Nanopores Displaying Enzymatically Reconfigurable Rectification Properties. Nano Lett. 2018, 18, 3303–3310. 10.1021/acs.nanolett.8b01281. [DOI] [PubMed] [Google Scholar]
- Walba H.; Isensee R. W. Acidity Constants of Some Arylimidazoles and Their Cations. J. Org. Chem. 1961, 26, 2789–2791. 10.1021/jo01066a039. [DOI] [Google Scholar]
- Balan A.; Machielse B.; Niedzwiecki D.; Lin J.; Ong P.; Engelke R.; Shepard K. L.; Drndic M. Improving Signal-to-Noise Performance for DNA Translocation in Solid-State Nanopores at MHz Bandwidths. Nano Lett. 2014, 14, 7215–7220. 10.1021/nl504345y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.