

Abstract
Even in the era of personalized medicine and immunotherapy, temozolomide (TMZ), a small molecule DNA alkylating agent, remains the standard-of-care for glioblastoma (GBM). TMZ has an unusual mode-of-action, spontaneously converting to its active component via hydrolysis in vivo. While TMZ has been FDA approved for two decades, it provides little benefit to patients whose tumors express the resistance enzyme MGMT and gives rise to systemic toxicity through myelosuppression. TMZ was first synthesized in 1984, but certain key derivatives have been inaccessible due to the chemical sensitivity of TMZ, precluding broad exploration of the link between imidazotetrazine structure and biological activity. Here, we sought to discern the relationship between the hydrolytic stability and anticancer activity of imidazotetrazines, with the objectives of identifying optimal timing for prodrug activation and developing suitable compounds with enhanced efficacy via increased blood-brain barrier penetrance. This work necessitated the development of new synthetic methods to provide access to previously unexplored functionality (such as aliphatic, ketone, halogen, and aryl groups) at the C8 position of imidazotetrazines. Through synthesis and evaluation of a suite of compounds with a range of aqueous stabilities (from 0.5 to 40 h), we derive a predictive model for imidazotetrazine hydrolytic stability based on the Hammett constant of the C8 substituent. Promising compounds were identified that possess activity against a panel of GBM cell lines, appropriate hydrolytic and metabolic stability, and brain-to-serum ratios dramatically elevated relative to TMZ, leading to lower hematological toxicity profiles and superior activity to TMZ in a mouse model of GBM. This work points a clear path forward for the development of novel and effective anticancer imidazotetrazines.
Glioblastoma multiforme (GBM) is the most prevalent, infiltrative, and lethal primary malignant brain tumor with only 10% of patients surviving five years.1 The current standard-of-care for GBM is gross surgical resection followed by radiotherapy combined with temozolomide (TMZ, Scheme 1a), a small molecule DNA alkylating agent. The antitumor effect of TMZ is ultimately mediated through methylation of the O6-position of guanine residues and subsequent mismatch repair-dependent cell death.2−6 Among the beneficial properties of TMZ are favorable pharmacokinetics (including 100% oral bioavailability7), nonenzymatic prodrug activation, and some accumulation in the brain (cerebral spinal fluid:blood ratio of 17:83 in human cancer patients8,9). TMZ provides a significant therapeutic benefit to a subset of GBM patients; for example, in patients whose tumors do not express O6-methylguanine DNA methyltransferase (MGMT), an enzyme that removes O6-methylguanine lesions, TMZ extends median survival to approximately two years.10 Even in the era of personalized anticancer therapy, TMZ remains frontline therapy for oligodendrogliomas, diffuse astrocytic gliomas, and pleomorphic xanthoastrocytomas in addition to GBM.11 However, given the ineffectiveness of TMZ against tumors expressing MGMT and the inevitable recurrence of GBM after multimodal combination therapy, there remains a significant clinical need for better treatment strategies.
Scheme 1. (a) Mechanism of TMZ Activation in Aqueous Solution and (b) Favorable Stability of Compound 1 Relative to Related Versions (such as 2) Accounts for the Incorporation of the Amide at C8 of TMZ.

TMZ is a prodrug activated in aqueous solutions that ultimately releases methyl diazonium, the active alkylating component (Scheme 1a). The half-life of TMZ is ∼2 h in vivo and in aqueous solutions in vitro, and it has been suggested that the drug has an increased rate of hydrolysis in the more alkaline environment of gliomas, providing some selectivity for cancerous vs noncancerous cells.12−15 While this 2 h half-life enables TMZ to reach the central nervous system (CNS) and release methyldiazonium, there is scarce information on the relationship between half-life and anticancer activity; specifically, it is unclear if 2 h is optimal to maximize therapeutic efficacy or if shorter (or longer) half-lives may bolster its effect. Given the advantageous features of TMZ, we sought to understand the relationship between its structure, hydrolytic stability, and anticancer activity.
While the amide at C8 had been suggested in the past to be essential for activity,2,16 conflicting reports have since indicated that alternate functionality may be tolerated at this position.17−19 Indeed, our own analysis led us to believe that strategic substitutions at C8 could be used to tune the hydrolytic stability of imidazotetrazines, and that in doing so, a suite of compounds with a range of half-lives could be constructed. In addition to varying the stability of the prodrug, alterations at the C8 position could lead to compounds that retain the favorable pharmacokinetic properties of TMZ but have increased CNS penetrance. Conceivably, an imidazotetrazine with enhanced blood-brain barrier (BBB) penetrance may exhibit lower systemic toxicity and allow for higher and more efficacious dosing regimens because the dose-limiting toxicity of TMZ (myelosuppression) is not CNS-related.7,20,21 Herein, we describe the development of a model that accurately predicts the hydrolytic stability and half-life of imidazotetrazines, and we use this model to discover novel imidazotetrazines with exceptional BBB penetration and superior anticancer activity compared to that of TMZ, including in a murine model of GBM.
Results and Discussion
Construction of C8-Substituted Imidazotetrazines
The inclusion of an amide at the C8 position of TMZ is largely an artifact of the original synthesis of imidazotriazenes and imidazotetrazines. Both dacarbazine and TMZ are derived from precursor 4-diazoimidazole-5-carboxamide (1, Scheme 1b). The remarkable stability of this diazo species, reportedly >2.5 years at room temperature,22 permitted its use for exploratory chemistry where other diazoimidazole species (such as 4-diazoimidazole (2)) simply decomposed.23 Thus, the initial synthesis of dacarbazine in 1962 and TMZ in 1984 involved the quenching of 1 with dimethylamine24 or the cyclization of 1 with methyl isocyanate,25 respectively, and the primary amide moiety remained. Over time, there have been suggestions that this amide is critical for anticancer activity. Such claims were supported by theoretical studies suggesting that a hydrogen bond donor at C8 is required for activity,2,16 but clouding the picture is a conflicting structure–activity relationship (SAR) adopted from derivatives of a related compound (mitozolomide) in non-CNS cancer models.26 There are considerable challenges to establishing a general synthetic route that can be used to construct novel derivatives at the C8 position; these synthetic challenges have hindered the development of new imidazotetrazines, and in the absence of new compounds and biological data, the outdated SAR has persisted.
Key challenges to making novel imidazotetrazines include sensitivity to protic solvents or basic reagents, instability of intermediate diazo species, and the lack of efficient reagents to install the N3 methyl. The sensitivity of the prodrug to conditions involving base or water (at pH > 6) renders the tetrazinone unstable to many practical cross-coupling or reducing conditions. Another challenge, as alluded to above, is the instability of intermediate diazo species. The privileged 4-diazoimidazole-5-carboxamide (1, Scheme 1b) readily precipitates out of solution as a pure, stable compound; however, other 4-diazoimidazoles (such as 2) remain in aqueous solution, are exceptionally prone to decomposition, and are sensitive to heat, shock, and often light.23 Finally, installation of the N3-methyl group in the initial route to TMZ was achieved via cyclization with methyl isocyanate.25 Methyl isocyanate, however, is a poisonous gas and no longer commercially available. As such, alternate routes27 or less effective alternatives to methyl isocyanate such as N-succinimidyl N-methylcarbamate or N-methylcarbamic chloride must be used that reduce the yield of the cyclizations.
To provide access to certain derivatives of the C8 amide, we began by modifying an established route, largely developed for mitozolomide.28 This sequence begins with a hydrolysis of the amide of TMZ to carboxylic acid 3 (Scheme 2a), which can then be converted to the acid chloride. From this intermediate, various nucleophiles may be substituted in high yields. This route was used to synthesize amide, ester, and thioester derivatives 4–10 (Scheme 2a). Additionally, an established reaction was employed to install a cyano group (11) directly from TMZ (Scheme 2a).29 The creation of a structurally diverse panel of C8 analogues, however, would require novel synthetic routes, especially for those with aliphatic, ketone, halogen, and aryl groups; such substituents have not been described at this position in the ∼35 year history of TMZ. Thus, an aliphatic group at C8 was introduced via diazotization of 5-amino-4-methylimidazole 12 to diazo species 13 and subsequent cyclization with methyl isocyanate surrogate N-methylcarbamoyl chloride to afford C8-methyl derivative 14 (Scheme 2b). Although various amides, esters, and thioamides had been installed at C8, ketones were entirely absent, perhaps unsurprisingly because initial attempts to use Grignard or alkyllithium reagents led to complete degradation of the tetrazinone ring. Thus, a stepwise cyclization was utilized to synthesize methyl ketone derivative 17 from its disubstituted precursor 16, obtained upon hydrolytic degradation of 6-methylpurine N-oxide (15)30 (Scheme 2c). Bromine and chlorine substituents were directly incorporated at C8 in moderate yields upon a decarboxylative halogenation of intermediate 3 employing Dess–Martin periodinane and the respective tetraethylammonium salt (compounds 18 and 19, Scheme 2a). This strategy had not previously been applied to imidazoles and endows potential points of diversity in addition to representing novel derivatives themselves. Using 18 as a cross coupling partner, however, was unsuccessful due to the basic, aqueous conditions required. Instead, a Suzuki coupling fashioned 5-nitro-4-phenylimidazole (21) from the 5-nitro-4-bromoimidazole (20) precursor, which could be subsequently reduced to corresponding amine 22 and cyclized to the phenyl-substituted imidazotetrazine as above (Scheme 2d). This method supplied 23 as well as a small series of p-substituted aryl derivatives 24–26. Finally, heterocyclic compounds 27 and 28 (Scheme 2e) were synthesized upon cyclization of the C8 amide or thioamide, respectively (Supporting Information); an analogous route had been utilized to introduce bulkier 4-substituted oxazoles and thiazoles at the C8 position,19 but not smaller methyl groups.
Scheme 2. Synthesis of Novel C8-Substituted Imidazotetrazines.

DMP = Dess–Martin periodinane, SNMC = N-succinimidyl N-methylcarbamate.
Anticancer Activity of C8-Substituted Imidazotetrazines
With a suite of imidazotetrazines in hand, each compound was evaluated against a panel of human GBM cell lines (Table 1, Table S1). Cell lines were selected to include those expressing and lacking MGMT (Figure S1) and, consistent with literature reports, those with negligible MGMT expression were sensitive to TMZ (IC50 ∼ 50 μM or less), whereas those with significant MGMT expression were resistant (IC50 > 300 μM). Amide-substituted derivatives 4–8 as well as ester (9) and thioester (10) derivatives had activity comparable to TMZ in the MGMT-deficient U87 and D54 cell lines. Notably, the retention of activity for disubstituted amide (5–8) and ester (9) imidazotetrazines confirms that a hydrogen bond donor is not required at C8. In U118MG and T98G MGMT-expressing GBM cells, more potent activity was observed for these derivatives compared to TMZ. Ketone analogue 17 was also effective against MGMT-deficient cell lines, demonstrating that an amide is not required at the C8 position. Compounds completely lacking a carbonyl, such as 14, 19, 23, and 27 proved to be as (or more) potent than TMZ in the absence of MGMT and significantly more potent in cell lines expressing MGMT. Methyl (14) and phenyl (23) substitutions were the most active across all cell lines. Cyano derivative 11 and carboxylic acid derivative 3 were inactive in all tested cell lines (>7-fold less potent than TMZ), even in the absence of MGMT. In addition to these canonical adherent GBM cell lines, most analogues were more active than TMZ in the patient-derived U3054MG GBM cell line cultured under serum-free stem cell conditions.31
Table 1. Panel of C8-Substituted Imidazotetrazines and Associated IC50 Values (μM) in Multiple GBM Cell Linesa.

Cell lines were incubated with compound for 7 days, and then viability was assessed using the Alamar Blue assay. Error is SEM, n ≥ 3. Prl = pyrrolidine. A table with additional compounds (Table S1) and a Western blot for MGMT status of all cell lines used (Figure S1) can be found in the Supplementary Information.
Hydrolytic Stability of C8-Substituted Imidazotetrazines
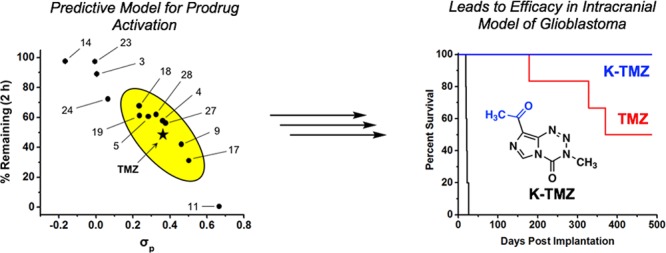
The principal aspect governing the anticancer activity of imidazotetrazines is the hydrolytic activation of the prodrug. As depicted in Scheme 1a, TMZ has a half-life of ∼2 h in humans.7 This timeline allows the intact prodrug to reach the brain and release the active methylating component prior to elimination. Beyond TMZ, the relationship between imidazotetrazine stability and anticancer activity is unknown; that is, while hydrolytic activation is required for cancer cell death, the optimal timing of this event is unclear both in vitro and in vivo. Toward this end, the hydrolytic stability of each new compound was assessed in buffered saline, which mimics in vivo conditions (in pH 7.4 PBS TMZ has a half-life of 119 min, Figure 1). An HPLC assay was developed to quantify the fraction of intact prodrug remaining in solution after 2 h at pH 7.4, 37 °C. The results of this experiment suggest that electronic substituent effects at C8 directly translate through the bicycle to C4, the site of hydrolysis. The magnitude of this effect was dramatic, with stabilities ranging from 0 to 97% remaining after 2 h depending on the substituent at the C8 position (Figure 1a). Because the group at C8 appeared to have such a clear influence on the aqueous stability of the prodrug, its Hammett constant (σp) was plotted against the percent remaining after 2 h. As shown in Figure 1a, an obvious relationship exists between these two parameters, suggesting that σp can be used to accurately predict the stability of C8-substituted imidazotetrazines. Among those compounds possessing substituents with similar electronics (0.23 < σp < 0.50) to a primary amide (σp = 0.36) were amide derivatives 4 and 5, ketone derivative 17, and chloro derivative 19. Each had measured half-lives within an hour of TMZ in PBS at pH 7.4 (Figure 1b). On either extreme were cyano analogue 11 (σp = 0.66), with a half-life of 0.5 h, and methyl derivative 14 (σp = −0.17), which remained in its prodrug form the longest with a half-life of 40 h. The same assay was used to confirm that hydrolysis remained pH-dependent for C8-substituted derivatives (e.g., K-TMZ 17, Figure S2).
Figure 1.

Hydrolytic stability of C8-substituted imidazotetrazines. (a) The percentage of compound remaining after 2 h plotted against the Hammett constant of its C8 substituent. Compounds with hydrolytic stability similar to TMZ are enclosed in the oval. (b) Half-lives of select C8 derivatives in PBS (pH 7.4, 37 °C).
Relationship between Hydrolytic Stability and Anticancer Activity
Methyl and phenyl derivatives 14 and 23 were consistently the most potent compounds in each of the tested cell lines (Table 1). Interestingly, they also possessed electron-donating substituents and, accordingly, the greatest aqueous stability (Figure 1), suggesting that a longer-lived prodrug is favorable for efficacy in cell culture. The opposite effect was observed for compound 11, which was the least stable in solution. Even in U87 cells lacking MGMT, it exhibited a 10-fold loss of activity compared to TMZ, suggesting that there is a critical threshold of aqueous stability below which hydrolysis occurs too quickly to methylate target DNA. Compounds with hydrolytic stabilities similar to TMZ such as 4, 5, 17, 19, and 27 retained activity in culture. Notably, ketone derivative 17 was equipotent to TMZ even with a shorter aqueous half-life, indicating that compounds with σp ∼ 0.50 can still retain marked anticancer activity.
Liver Microsome Stability
TMZ fortuitously possesses several ideal pharmacokinetic properties, including avoidance of primary metabolism.7 To assess whether modification or replacement of the amide at C8 would lead to significant metabolic liabilities, the stabilities of select compounds were assessed after 2 h in the presence of mouse liver microsomes. Prodrug hydrolysis was accounted for by including control runs that did not contain liver microsomes. The slightly acidic pH of the working solution resulted in enhanced stability of TMZ compared to incubation in PBS alone. Predictably, TMZ was insensitive to metabolic perturbation as its instability was entirely accounted for by hydrolysis (Table 2). The addition of methyl(s) to the amide (compounds 4 and 5) resulted in some susceptibility to the effects of the microsomes, and this effect was amplified for larger amide substitutions (compound 7), which demonstrated improved aqueous stability but markedly less stability in liver microsomes. Ketone 17 and chloro 19 were generally stable to oxidative metabolism, suggesting that for these compounds hydrolysis could drive the pharmacokinetics in vivo, similar to TMZ.
Table 2. Metabolic Stability, cLogBB, and CNS MPO Values for Relevant C8 Analoguesa.
| compound | stability (2 h, microsomes) (%) | stability (2 h, no microsomes) (%) | cLogBB | CNS MPO |
|---|---|---|---|---|
| propranolol | 68 ± 2 | 102 ± 3 | ND | ND |
| TMZ | 87 ± 6 | 86 ± 4 | –1.58 | 4.9 |
| 4 (Me-TMZ) | 86 ± 1 | 93 ± 1 | –1.34 | 5.7 |
| 5 (DiMe-TMZ) | 81 ± 2 | 92 ± 3 | –1.18 | 6.0 |
| 6 | 81 ± 1 | 95 ± 2 | –1.07 | 6.0 |
| 7 | 1 ± 1 | 98 ± 3 | –0.78 | 5.6 |
| 17 (K-TMZ) | 70 ± 1 | 77 ± 3 | –1.08 | 6.0 |
| 19 | 91 ± 3 | 91 ± 1 | –0.72 | 6.0 |
| 23 | 44 ± 2 | 103 ± 5 | –0.56 | 5.7 |
| 27 (Ox-TMZ) | 71 ± 1 | 95 ± 4 | –1.19 | 5.9 |
The metabolic stability was assessed in mouse liver microsomes. Compounds were incubated with microsomes for 2 h before the percentage remaining was quantified relative to t0. Experiments assessing stability in the absence of microsomes were identical but replaced liver microsomes with PBS. Error is SEM, n ≥ 2. Internal standard = N3-propyl TMZ. CNS MPO = central nervous system multiparameter optimization score.
Blood-Brain Barrier Penetrance
It has been reported that >98% of small molecule drugs do not penetrate the BBB,32 making TMZ unusual, especially among anticancer agents. In humans, TMZ is rapidly absorbed and reaches the brain in minutes with cerebral spinal fluid concentrations averaging 20% of those in the plasma;8,9 the accumulation of even more drug in the brain by increasing the BBB penetrance may be a viable strategy to increase efficacy against CNS-based tumors. To predict the BBB penetrance of the novel imidazotetrazines, logBB values were calculated (cLogBB) based on a formula utilizing cLogP and total polar surface area.33 When applied across a consistent drug scaffold, these types of in silico metrics have been used reliably to predict relative changes in BBB penetrance as well as other biological phenomenon,34−38 though not always reflective of absolute concentrations. The cLogBB value for TMZ is −1.58 (Table 2). Replacing the primary amide led to marked increases in the cLogBB and larger predicted brain:blood ratios relative to TMZ. Importantly, cLogBB does not account for molecular weight, making us wary of analogues with large, hydrophobic functionality (e.g., 7) even if they possess attractive predicted values. A more comprehensive metric, the CNS multiparameter optimization (MPO) tool39,40 was also employed to gauge prospective BBB permeabilities. CNS MPO scores span from 0 to 6.0 based on the optimal ranges of 6 physicochemical properties. Though TMZ has an agreeable MPO of 4.9, higher scores were achieved for the C8 analogues, which in several cases reached the top desirability value (Table 2). The more favorable cLogBB and CNS MPO values predicted for the panel suggests that certain derivatives may achieve drug concentrations in the brain higher than those of TMZ.
The BBB penetrance of top compounds (those with favorable anticancer activity, appropriate hydrolytic and liver microsome stability, and predicted BBB penetrance, Figure 2a) was thus assessed in vivo. In an initial experiment, Me-TMZ (4) and DiMe-TMZ (5) were tested head-to-head with TMZ to explore whether alkylation of the C8 amide could confer increased brain:blood ratios. Mice were administered 25 mg/kg drug intravenously and sacrificed 5 min after injection. The serum and perfused brain samples were immediately acidified to prevent prodrug degradation before the drug concentration within each compartment was quantitated by LC-MS/MS. After 5 min, drug concentrations in the brain were significantly elevated for analogues Me-TMZ and DiMe-TMZ versus TMZ, a >3-fold increase in brain:serum ratio for each compound (Figure 2b). The equivalent brain:serum ratios for Me-TMZ and DiMe-TMZ is likely due to the fast metabolism of the dimethylated amide to its monomethylated counterpart. This preliminary experiment suggested that other derivatives with higher predicted BBB penetrance may lead to greater brain permeability in vivo. Accordingly, compounds Ox-TMZ (27) and K-TMZ (17) were evaluated head-to-head with DiMe-TMZ and TMZ. After 5 min, each derivative had accumulated numerically higher concentrations in the brain than TMZ (Figure 2c). When paired with the corresponding serum concentrations (Figure 2d), TMZ had a relative brain:serum ratio of 0.23 ± 0.03 ng/g:ng/mL, comparable to the few other TMZ biodistribution experiments in murine systems.41,42 Assigning average mouse blood volumes to equate units (see Supporting Information), TMZ had an absolute brain:serum ratio of 8:92, while Ox-TMZ and K-TMZ boasted brain:serum ratios of 55:45 and 69:31, respectively. (Figure 2e). The dramatic differences in drug partitioning suggest that replacing the amide at C8 is a viable strategy to significantly increase local drug concentration in the brain relative to the blood, which may increase effectiveness against brain tumors and also reduce hematological toxicity.
Figure 2.

Blood-brain barrier permeability of imidazotetrazines. (a) Structures of lead C8-substituted compounds. (b) Relative brain:serum ratios of TMZ, Me-TMZ, and DiMe-TMZ (25 mg/kg) were measured 5 min after IV injection into mice. Values are the fold change of brain:serum ratio relative to TMZ. In a second experiment, brain (c) and serum (d) concentrations of TMZ and C8 analogues (25 mg/kg) were quantitated 5 min after IV injection into mice. (e) Brain:serum ratios were calculated based on (c) and (d) assuming a mouse blood volume of 58.5 mL/kg. Error is SEM, number of mice per cohort = 3. Statistical significance was determined by using a two-sample Student’s t test (two-tailed test, assuming equal variance).
Assessment of Hematological Toxicity
The elevated brain concentrations and dramatically decreased serum concentrations (Figures 2c and d) observed upon treatment with Ox-TMZ and K-TMZ compared to TMZ suggested that these C8-modified imidazotetrazines may attenuate the dose-limiting hematological toxicity observed for TMZ in the clinic. To test this hypothesis, mice were treated with a single dose of 125 mg/kg TMZ, Ox-TMZ, or K-TMZ intravenously; this dose of TMZ has been reported to induce nonlethal toxicity in mice.43,44 Seven days post-treatment, whole blood was collected and complete blood counts were obtained for each individual mouse. Expectedly, a dose of 125 mg/kg TMZ led to white blood cell (WBC) depletion relative to control mice (Figure 3a), suggestive of drug-induced myelosuppression. Both lymphocyte (Figure 3b) and neutrophil (Figure S3a) concentrations were decreased in TMZ-treated mice. Conversely, treatment with 125 mg/kg of Ox-TMZ or K-TMZ did not produce myelosuppression. Total WBC, lymphocyte, and neutrophil counts for mice treated with these compounds were equivalent to those of control mice. Notably, the novel imidazotetrazines did not give rise to other hematological symptoms such as red blood cell (RBC) toxicity (Figure S3b) or thrombocytopenia (Figure S3c) and did not lead to weight loss 7 days post-treatment (Figure S3d).
Figure 3.

Assessment of the hematological toxicity of imidazotetrazines in vivo. Mice were administered a single IV dose of 125 mg/kg imidazotetrazine. After 7 days, whole blood was collected, and a complete blood count was obtained for each individual mouse. (a) Total WBC count. Control vs Ox-TMZ: P = 0.7, Control vs K-TMZ: P = 0.9. (b) Lymphocyte concentrations. Control vs Ox-TMZ: P = 0.5, Control vs K-TMZ: P = 0.9. Error is SEM, number of mice per cohort = 4. Statistical significance was determined by using a two-sample Student’s t test (two-tailed test, assuming equal variance). The concentrations of other relevant blood constituents can be found in Supporting Information Figure S3.
Novel Imidazotetrazines Induce Alkylation-Mediated Cancer Cell Death
The cytotoxicity of TMZ is mediated by methylation of O6 guanine; subsequent single- and double-strand breaks and apoptosis are facilitated by the mismatch repair system.2−6 To assess if the novel imidazotetrazines kill through the same mechanism, O6-methylguanine adducts were quantitated in U87 cells treated with 100 or 1000 μM of each imidazotetrazine. After 8 h of incubation with compound, the genomic DNA was isolated, quantified, and hydrolyzed to its constituent deoxyribonucleosides, which were quantitated via LC-MS/MS analysis. Dose dependent increases in the concentration of O6-methylated deoxyguanosine were observed for TMZ as well as each of the lead compounds (Figure 4a), indicating that DNA methylation is occurring. Further confirmation that the novel compounds remain DNA alkylators was obtained upon pretreatment with MGMT inhibitor O6-benzylguanine (O6BG). O6BG is a pseudosubstrate for MGMT that quenches cellular stores of the enzyme, leading to the persistence of O6-methylguanine DNA adducts. Preincubation of MGMT-expressing T98G cells with O6BG (100 μM) led to an 8-fold enhancement in cytotoxicity for TMZ (Figure 4b), consistent with literature reports.45,46 Similarly, DiMe-TMZ, Ox-TMZ, and K-TMZ demonstrated a significant increase in activity when administered after O6BG, suggesting that O6-methylguanine lesions are the cause of cell death.
Figure 4.

(a) The concentration of O6-methylguanine was measured in U87 cells (10 μg DNA) after treatment with 100 or 1000 μM of imidazotetrazine for 8 h. (b) Imidazotetrazines were added to T98G cells with or without 3 h pretreatment of O6BG (100 μM). IC50 values after 7 day incubation and fold changes between (±) O6BG treatments are reported. P-values between IC50 values (±) O6BG < 0.02 for all compounds. Error is SEM, n ≥ 3. Statistical significance was determined by using a two-sample Student’s t test (two-tailed test, assuming equal variance).
Novel Imidazotetrazines Have Superior Activity in Mouse Models of GBM
The increased BBB penetrance observed for amide derivatives (Me-TMZ and DiMe-TMZ) relative to TMZ suggested that greater drug concentrations in the brain might lead to greater efficacy in an intracranial tumor model. GBM oncosphere lines were chosen for these studies as they more accurately recapitulate the genetic and histopathological features of human GBM than traditional adherent cell lines, which are passaged in serum and typically grow as compact masses in vivo.47 The Br23c GBM oncosphere cell line does not express MGMT, was sensitive to TMZ and the novel C8-substituted imidazotetrazines (Table S2), and was thus chosen as the model system. Mice implanted intracranially with these cells were administered 15 mg/kg TMZ or the equimolar equivalent of Me-TMZ or DiMe-TMZ once per day, 5×/week via oral gavage. As expected, TMZ significantly increased median survival compared with vehicle (Figure 5a). Mice treated with both Me-TMZ and DiMe-TMZ, however, outperformed TMZ and increased median survival by 24 and 46%, respectively, suggesting that increasing the BBB-permeability of imidazotetrazine prodrugs is a viable strategy to improve efficacy. In a second experiment, K-TMZ was selected for evaluation due to its most favorable brain:blood ratio (Figure 2e). Mice intracranially implanted with Br23c cells were treated with K-TMZ (via oral gavage), which led to an extended median survival of more than 50 days past TMZ-treated mice, and showed greater efficacy even compared to DiMe-TMZ (Figure 5b). Importantly, methyl derivative 14, which has excellent efficacy in cell culture but an extended (40 h) half-life in aqueous solution, had no effect in this in vivo model (Figure S4) suggesting that dramatically elongated half-lives are detrimental in vivo, likely due to compound clearance prior to hydrolytic activation.
Figure 5.

Evaluation of imidazotetrazines in intracranial mouse models of GBM. GBM Br23c oncospheres were intracranially implanted into female athymic nude mice. Treatment was started 5 days post implantation. (a) Mice were administered 15 mg/kg TMZ or an equimolar dose of Me-TMZ (16.1 mg/kg) or DiMe-TMZ (17.2 mg/kg) orally once per day, 5×/week for 7 weeks. Control vs TMZ: P = 0.0014, TMZ vs DiMe-TMZ: P = 0.061, TMZ vs Me-TMZ: P = 0.016. (b) Mice were administered 15 mg/kg TMZ or an equimolar dose of DiMe-TMZ (17.2 mg/kg) or K-TMZ (14.9 mg/kg) orally once per day for 5 total doses. Control vs TMZ: P = 0.0007, DiMe-TMZ vs TMZ: P = 0.7, K-TMZ vs TMZ: P = 0.055. Compounds were formulated in 10% PEG in PBS immediately prior to each treatment. Number of mice per treatment cohort ≥5. Survival curves were compared using log-rank test.
Conclusion
Despite being known since 1984, FDA approved since 1999, and reaching $1 billion in sales in 2009, TMZ remains the only approved imidazotetrazine anticancer drug; this likely stems from the lack of generalized syntheses for this class of compounds, prohibiting conventional medicinal chemistry campaigns. Here, we report new synthetic methods enabling the construction of novel C8-substituted imidazotetrazines that were previously inaccessible. Evaluation of these compounds in systematic, head-to-head assays allows us to definitively conclude that the C8 amide is not required for anticancer activity, and indeed compounds lacking an H-bond donor or acceptor (or both) at C8 can still retain activity comparable to that of TMZ against cancer cells in culture. Unmoored from the necessity of an amide at C8, a panel of imidazotetrazines was synthesized, varying this position. Strikingly, the electronic properties of the substituent at C8 has a dramatic effect on the activation of the corresponding prodrug, a previously undefined phenomenon. The relationship derived herein between the hydrolytic stability of imidazotetrazines and the electronics at C8 allows the stability of the prodrug to be tuned by employing easily accessible σp values, enabling the rational design of TMZ derivatives that have similar stabilities in vivo and facilitating investigation into the optimal timing of imidazotetrazine prodrug activation.
From this work, it appears that compounds with very short half-lives (such as 11, t1/2 = 0.57 h) simply hydrolyze too rapidly, releasing methyl diazonium prior to accumulation in the DNA microenvironment and diminishing anticancer activity. Thus, for activity against cancer cells in culture, a half-life of 1 h or greater is optimal. Conversely, compounds that have very long half-lives (such as 14 or 23, t1/2 > 20 h) can be distinctly more potent than TMZ in cell culture as the prodrug has ample time to distribute to the nucleus before conversion to the active methylating agent. However, these compounds with markedly increased hydrolytic stabilities are less likely to be useful in vivo as elimination through alternate pathways (excretion of the intact prodrug, oxidative metabolism, etc.) will occur before activation to the alkylating species. This hypothesis accounts for the lack of in vivo efficacy of compound 14.
A hallmark of GBM is its invasion into surrounding brain tissue at an early stage, making cure via surgical resection unachievable. As such, there is an obvious clinical need for improved compounds that can reach the entirety of the diffuse tumor in sufficient concentrations to be effective. Importantly, our data show that the BBB-penetrance of imidazotetrazines can be improved through modifications at the C8 position. The dramatically enhanced brain:serum distribution of Ox-TMZ and K-TMZ, in particular, could provide substantial improvement over TMZ for treatment of CNS cancers. Both of these compounds retain the favorable features of TMZ (timely prodrug activation, stability to liver microsomes) while also accumulating higher drug concentration in the brain and reduced concentration in the blood. We hypothesized that partitioning the imidazotetrazine more to the site of the tumor and less to the compartment responsible for adverse effects would expand the therapeutic window by enhancing anticancer activity while simultaneously reducing systemic toxicity. Myelotoxicity occurs in ∼20% of TMZ-treated patients, is the major dose-limiting toxicity,48 and is exacerbated in elderly and female GBM patients.49,50 Ox-TMZ and K-TMZ demonstrated significantly less in vivo toxicity to WBCs compared to TMZ, likely a direct result of the increased partitioning to the CNS. Imidazotetrazines such as these with lower toxicity profiles could permit elevated dosing schedules and additional anticancer efficacy and/or make this drug class accessible to more patients.
Other imidazotetrazines of various composition in the literature have failed to improve median survival head-to-head compared to TMZ in preclinical models despite promising results in cell culture.42,51,52 To our knowledge, only one derivative has outperformed TMZ in an intracranial murine model of GBM, bestowing a modest 10% increase in median survival.53 Clearly, the interplay between retaining the favorable properties that have kept TMZ as frontline treatment for GBM and modulating its structure is not trivial. The data reported herein now suggest that imidazotetrazines may be substantially modified without losing these advantages, and indeed, such new compounds can have dramatically enhanced in vivo efficacy. TMZ remains the gold standard for treating the most aggressive brain tumors, shows promise against brain metastases from other cancers,54 and its predictable activity (based on clinical biomarkers) has recently led to advocation for an expanded use of TMZ in the management of diverse cancer types.55 As such, the novel imidazotetrazines reported here could hold considerable promise for treatment of GBM and other cancers.
Acknowledgments
We thank L. Li (Metabolomics Center, Roy J. Carver Biotechnology Center, UIUC) for LC-MS/MS analysis and Charlie Pierce for preliminary stability data. We are grateful to the National Institutes of Health R21-CA195149 (to P.J.H. and G.J.R.) and R01-CA190223 (to G.J.R.) for support of this work. R.L.S. is a member of the NIH Chemistry-Biology Interface Training Grant (T32-GM070421). L.F. was a visiting student from the Department of Biomolecular Sciences, University of Urbino “Carlo Bo”, Piazza Rinascimento 6, I-61029 Urbino, Italy.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00864.
Supplementary Tables S1 and S2, Western blot for MGMT for all cell lines used, supplementary Figures S1–S3, experimental details for mouse liver microsome assay, in vivo BBB permeability and hematological toxicity studies, O6-MedG quantitation, in vivo mouse models, synthetic procedures, and characterization for C8 analogues (PDF)
The authors declare the following competing financial interest(s): The University of Illinois has filed patents on some of the compounds described herein.
Supplementary Material
References
- Stupp R.; Mason W.; van den Bent M. J.; Weller M.; Fisher B. M.; Taphoorn M. J. B.; Belanger K.; Brandes A. A.; Marosi C.; Bogdahn U.; Curschmann J.; Janzer R. C.; Ludwin S. K.; Gorlia T.; Allgeier A.; Lacombe D.; Cairncross J. G.; Eisenhauer E.; Mirimanoff R. O. (2005) Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 352, 987–996. 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Denny B. J.; Wheelhouse R. T.; Stevens M. F.; Tsang L. L.; Slack J. a. (1994) NMR and Molecular Modeling Investigation of the Mechanism of Activation of the Antitumor Drug Temozolomide and Its Interaction with DNA. Biochemistry 33, 9045–9051. 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- Karran P.; Macpherson P.; Ceccotti S.; Dogliotti E.; Griffin S.; Bignami M. (1993) O6-Methylguanine Residues Elicit DNA Repair Synthesis by Human Cell Extracts. J. Biol. Chem. 268, 15878–15886. [PubMed] [Google Scholar]
- Ceccotti S.; Aquilina G.; Macpherson P.; Yamada M.; Karran P.; Bignami M. (1996) Processing of O6-Methylguanine by Mismatch Correction in Human Cell Extracts. Curr. Biol. 6, 1528–1531. 10.1016/S0960-9822(96)00758-0. [DOI] [PubMed] [Google Scholar]
- Margison G. P.; Santibanez Koref M. F.; Povey A. C. (2002) Mechanisms of Carcinogenicity/Chemotherapy by O6-Methylguanine. Mutagenesis 17, 483–487. 10.1093/mutage/17.6.483. [DOI] [PubMed] [Google Scholar]
- Tisdale M. J. (1987) Antitumour Imidazotetrazines-XV. Role of Guanine O6 Alkylation in the Mechanism of Cytotoxicity of Imidazotetrazinones. Biochem. Pharmacol. 36, 457–462. 10.1016/0006-2952(87)90351-0. [DOI] [PubMed] [Google Scholar]
- Newlands E. S.; Blackledge G. R.; Slack J. A.; Rustin G. J.; Smith D. B.; Stuart N. S.; Quarterman C. P.; Hoffman R.; Stevens M. F.; Brampton M. H.; Gibson A. C. (1992) Phase I Trial of Temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 65, 287–291. 10.1038/bjc.1992.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann S.; Csajka C.; Buclin T.; Leyvraz S.; Lejeune F.; Decosterd L. a.; Stupp R. (2004) Plasma and Cerebrospinal Fluid Population Pharmacokinetics of Temozolomide in Malignant Glioma Patients. Clin. Cancer Res. 10, 3728–3736. 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- Portnow J.; Badie B.; Chen M.; Liu A.; Blanchard S.; Synold T. W. (2009) The Neuropharmacokinetics of Temozolomide in Patients with Resectable Brain Tumors: Potential Implications for the Current Approach to Chemoradiation. Clin. Cancer Res. 15, 7092–7098. 10.1158/1078-0432.CCR-09-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R.; Hegi M. E.; Mason W. P.; van den Bent M. J.; Taphoorn M. J.; Janzer R. C.; Ludwin S. K.; Allgeier A.; Fisher B.; Belanger K.; Hau P.; Brandes A. A.; Gijtenbeek J.; Marosi C.; Vecht C. J.; Mokhtari K.; Wesseling P.; Villa S.; Eisenhauer E.; Gorlia T.; Weller M.; Lacombe D.; Cairncross J. G.; Mirimanoff R. O. (2009) Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 10, 459–466. 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Reifenberger G.; Wirsching H.-G.; Knobbe-Thomsen C. B.; Weller M. (2016) Advances in the Molecular Genetics of Gliomas - Implications for Classification and Therapy. Nat. Rev. Clin. Oncol. 14, 434–452. 10.1038/nrclinonc.2016.204. [DOI] [PubMed] [Google Scholar]
- Vaupel P.; Kallinowski F.; Okunieff P. (1989) Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer Res. 49, 6449–6465. [PubMed] [Google Scholar]
- Rottenberg D. A.; Ginos J. Z.; Kearfott K. J.; Junck L.; Bigner D. D. (1984) In Vivo Measurement of Regional Brain Tissue PH Using Positron Emission Tomography. Ann. Neurol. 15, S98–S102. 10.1002/ana.410150718. [DOI] [PubMed] [Google Scholar]
- Arnold J. B.; Kraig R. P.; Rottenberg D. A. (1986) In Vivo Measurement of Regional Brain and Tumor pH Using [14 C]Dimethyloxazolidinedione and Quantitative Autoradiography. II: Characterization of the Extracellular Fluid Compartment Using PH-Sensitive Microelectrodes and [14 C]Sucrose. J. Cereb. Blood Flow Metab. 6, 435–440. 10.1038/jcbfm.1986.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean L. A.; Roscoe J.; Jorgensen N. K.; Gorin F. A.; Cala P. M. (2000) Malignant Gliomas Display Altered pH Regulation by NHE1 Compared with Nontransformed Astrocytes. Am. J. Physiol. Cell Physiol. 278, C676–88. 10.1152/ajpcell.2000.278.4.C676. [DOI] [PubMed] [Google Scholar]
- Lowe P. R.; Sansom C. E.; Schwalbe C. H.; Stevens M. F. G.; Clark A. S. (1992) Antitumor Imidazotetrazines. 25. Crystal Structure of 8-Carbamoyl-3-Methylimidazo[5,1-d]-1,2,3,5-Tetrazin-4(3H)-One (Temozolomide) and Structural Comparisons with the Related Drugs Mitozolomide and DTIC. J. Med. Chem. 35, 3377–3382. 10.1021/jm00096a013. [DOI] [PubMed] [Google Scholar]
- Suppasansatorn P.; Wang G.; Conway B. R.; Wang W.; Wang Y. (2006) Skin Delivery Potency and Antitumor Activities of Temozolomide Ester Prodrugs. Cancer Lett. 244, 42–52. 10.1016/j.canlet.2005.11.029. [DOI] [PubMed] [Google Scholar]
- Liu D.; Yang J. G.; Cheng J.; Zhao L. X. (2010) Synthesis and Antitumor Activity of 3-Methyl-4-Oxo-3,4-Dihydroimidazo [5,1-d][1,2,3,5]Tetrazine-8-Carboxylates and -Carboxamides. Molecules 15, 9427–9436. 10.3390/molecules15129427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummersone M. G., Stevens M. F. G., and Cousin D.. Preparation of 3-Substituted-3H-Imidazo[5,1-d][1,2,3,5]Tetrazin-4-One Compounds for Treating Proliferative Disorders. WO 2010149968, 2010.
- O’Reilly S. M.; Newlands E. S.; Brampton M.; Glaser M. G.; Rice-Edwards J. M.; Illingworth R. D.; Richards P. G.; Kennard C.; Colquhoun I. R.; Lewis P.; Stevens M. F. G. (1993) Temozolomide: A New Oral Cytotoxic Chemotherapeutic Agent with Promising Activity against Primary Brain Tumours. Eur. J. Cancer 29, 940–942. 10.1016/S0959-8049(05)80198-4. [DOI] [PubMed] [Google Scholar]
- Bleehen N. M.; Newlands E. S.; Lee S. M.; Thatcher N.; Selby P.; Calvert A. H.; Rustin G. J. S.; Brampton M.; Stevens M. F. G. (1995) Cancer Research Campaign Phase II Trial of Temozolomide in Metastatic Melanoma. J. Clin. Oncol. 13, 910–913. 10.1200/JCO.1995.13.4.910. [DOI] [PubMed] [Google Scholar]
- Shealy Y. F.; Struck R. F.; Holum L. B.; Montgomery J. A. (1961) Synthesis of Potential Anticancer Agents. XXIX. 5-Diazoimidazole-4-Carboxamide and 5-Diazo-v-Triazole-4-Carboxamide. J. Org. Chem. 26, 2396–2401. 10.1021/jo01351a060. [DOI] [Google Scholar]
- Shechter H.; Magee W. L.; Rao C. B.; Glinka J.; Hui H.; Amick T. J.; Fiscus D.; Kakodkar S.; Nair M. (1987) Dipolar Cycloaddition Reactions of Diazoazoles with Electron-Rich and with Strained Unsaturated Compounds. J. Org. Chem. 52, 5538–5548. 10.1021/jo00234a008. [DOI] [Google Scholar]
- Fulmer Shealy Y.; Krauth C. A.; Montgomery J. A. (1962) Imidazoles. I. Coupling Reactions of 5-Diazoimidazole-4-Carboxamide. J. Org. Chem. 27, 2150–2154. 10.1021/jo01053a060. [DOI] [Google Scholar]
- Stevens M. F.; Hickman J. A.; Stone R.; Gibson N. W.; Baig G. U.; Lunt E.; Newton C. G. (1984) Antitumor Imidazotetrazines. 1. Synthesis and Chemistry of 8-Carbamoyl-3-(2-Chloroethyl)Imidazo[5,1-d]-1,2,3,5-Tetrazin-4(3 H)-One, a Novel Broad-Spectrum Antitumor Agent. J. Med. Chem. 27, 196–201. 10.1021/jm00368a016. [DOI] [PubMed] [Google Scholar]
- Lunt E.; Newton C. G.; Smith C.; Stevens G. P.; Stevens M. F. G.; Straw C. G.; Walsh R. J. A.; Warren P. J.; Fizames C.; Lavelle F.; Langdon S. P.; Vickers L. M. (1987) Antitumor Imidazotetrazines. 14. Synthesis and Antitumor Activity of 6- and 8-Substituted Imidazo[5,l-d]-l,2,3,5-Tetrazinones and 8-Substituted Pyrazolo[5,l-d]-l,2,3,5-Tetrazinones. J. Med. Chem. 30, 357–366. 10.1021/jm00385a018. [DOI] [PubMed] [Google Scholar]
- Moseley C. K.; Carlin S. M.; Neelamegam R.; Hooker J. M. (2012) An Efficient and Practical Radiosynthesis of [11C]Temozolomide. Org. Lett. 14, 5872–5875. 10.1021/ol302791x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horspool K. R.; Stevens M. F. G.; Baig G. U.; Newton C. G.; Lunt E.; Walsh R. J. A.; Pedgrift B. L.; Lavelle F.; Fizames C. (1990) Antitumor Imidazotetrazines. 20. Preparation of the 8-Acid Derivative of Mitozolomide and Its Utility in the Preparation of Active Antitumor Agents. J. Med. Chem. 33, 1393–1399. 10.1021/jm00167a018. [DOI] [PubMed] [Google Scholar]
- Langnel D. A. F.; Arrowsmith J.; Stevens M. F. G. (2000) Antitumor Imidazotetrazines. 38. New 8-Substituted Derivatives of the Imidazo[5,1-d]-1,2,3,5-Tetrazines Temozolomide and Mitozolomide. ARKIVOC 421–437. [Google Scholar]
- Stevens M. A.; Giner-Sorolla A.; Smith H. W.; Brown G. B. (1962) Purine N-Oxides. X. The Effect of Some Substituents on Stability and Reactivity. J. Org. Chem. 27, 567–572. 10.1021/jo01049a053. [DOI] [Google Scholar]
- Xie Y.; Bergström T.; Jiang Y.; Johansson P.; Marinescu V. D.; Lindberg N.; Segerman A.; Wicher G.; Niklasson M.; Baskaran S.; Sreedharan S.; Everlien I.; Kastemar M.; Hermansson A.; Elfineh L.; Libard S.; Holland E. C.; Hesselager G.; Alafuzoff I.; Westermark B.; Nelander S.; Forsberg-Nilsson K.; Uhrbom L. (2015) The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine 2, 1351–1363. 10.1016/j.ebiom.2015.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. (2005) The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2, 3–14. 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norinder U.; Haeberlein M. (2002) Computational Approaches to the Prediction of the Blood-Brain Distribution. Adv. Drug Delivery Rev. 54, 291–313. 10.1016/S0169-409X(02)00005-4. [DOI] [PubMed] [Google Scholar]
- West D. C.; Qin Y.; Peterson Q. P.; Thomas D. L.; Palchaudhuri R.; Morrison K. C.; Lucas P. W.; Palmer A. E.; Fan T. M.; Hergenrother P. J. (2012) Differential Effects of Procaspase-3 Activating Compounds in the Induction of Cancer Cell Death. Mol. Pharmaceutics 9, 1425–1434. 10.1021/mp200673n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan W. J.; Merz K. M.; Baldwin J. J. (2000) Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 43, 3867–3877. 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- Tian S.; Wang J.; Li Y.; Li D.; Xu L.; Hou T. (2015) The Application of in Silico Drug-Likeness Predictions in Pharmaceutical Research. Adv. Drug Delivery Rev. 86, 2–10. 10.1016/j.addr.2015.01.009. [DOI] [PubMed] [Google Scholar]
- Daina A.; Zoete V. (2016) A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 11, 1117–1121. 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alifrangis L. H.; Christensen I. T.; Berglund A.; Sandberg M.; Hovgaard L.; Frokjaer S. (2000) Structure-Property Model-for Membrane Partitioning of Oligopeptides. J. Med. Chem. 43, 103–113. 10.1021/jm9910932. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. (2010) Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 1, 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. (2016) Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 7, 767–775. 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- Reyderman L.; Statkevich P.; Thonoor C. M.; Patrick J.; Batra V. K.; Wirth M. (2004) Disposition and Pharmacokinetics of Temozolomide in Rat. Xenobiotica 34, 487–500. 10.1080/00498250410001685737. [DOI] [PubMed] [Google Scholar]
- Rai R.; Banerjee M.; Wong D. H.; McCullagh E.; Gupta A.; Tripathi S.; Riquelme E.; Jangir R.; Yadav S.; Raja M.; Melkani P.; Dixit V.; Patil U.; Shrivastava R.; Middya S.; Olivares F.; Guerrero J.; Surya A.; Pham S. M.; Bernales S.; Protter A. A.; Hung D. T.; Chakravarty S. (2016) Temozolomide Analogs with Improved Brain/Plasma Ratios – Exploring the Possibility of Enhancing the Therapeutic Index of Temozolomide. Bioorg. Med. Chem. Lett. 26, 5103–5109. 10.1016/j.bmcl.2016.08.064. [DOI] [PubMed] [Google Scholar]
- Kim S. S.; Rait A.; Kim E.; DeMarco J.; Pirollo K. F.; Chang E. H. (2015) Encapsulation of Temozolomide in a Tumor-Targeting Nanocomplex Enhances Anti-Cancer Efficacy and Reduces Toxicity in a Mouse Model of Glioblastoma. Cancer Lett. 369, 250–258. 10.1016/j.canlet.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M. F.; Hickman J. A.; Langdon S. P.; Chubb D.; Vickers L.; Stone R.; Baig G.; Goddard C.; Gibson N. W.; Slack J. A. (1987) Antitumor Activity and Pharmacokinetics in Mice of 8-Carbamoyl-3-Methyl-Imidazo[5,1-d]-1,2,3,5-Tetrazin-4(3H)-One (CCRG 81045; M & B 39831), a Novel Drug with Potential as an Alternative to Dacarbazine. Cancer Res. 47, 5846–5852. [PubMed] [Google Scholar]
- Bobola M. S.; Tseng S. H.; Blank a.; Berger M. S.; Silber J. R. (1996) Role of O6-Methylguanine-DNA Methyltransferase in Resistance of Human Brain Tumor Cell Lines to the Clinically Relevant Methylating Agents Temozolomide and Streptozotocin. Clin. Cancer Res. 2, 735–741. [PubMed] [Google Scholar]
- Kanzawa T.; Bedwell J.; Kondo Y.; Kondo S.; Germano I. M. (2003) Inhibition of DNA Repair for Sensitizing Resistant Glioma Cells to Temozolomide. J. Neurosurg. 99, 1047–1052. 10.3171/jns.2003.99.6.1047. [DOI] [PubMed] [Google Scholar]
- Binder Z. A.; Wilson K. M.; Salmasi V.; Orr B. A.; Eberhart C. G.; Siu I. M.; Lim M.; Weingart J. D.; Quinones-Hinojosa A.; Bettegowda C.; Kassam A. B.; Olivi A.; Brem H.; Riggins G. J.; Gallia G. L. (2016) Establishment and Biological Characterization of a Panel of Glioblastoma Multiforme (GBM) and GBM Variant Oncosphere Cell Lines. PLoS One 11, e0150271. 10.1371/journal.pone.0150271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaringi C.; De Sanctis V.; Minniti G.; Enrici R. M. (2013) Temozolomide-Related Hematologic Toxicity. Onkologie 36, 444–449. 10.1159/000353752. [DOI] [PubMed] [Google Scholar]
- Thomas R. P.; Recht L.; Nagpal S. (2012) Advances in the Management of Glioblastoma: The Role of Temozolomide and MGMT Testing. Clin. Pharmacol.: Adv. Appl. 5, 1–9. 10.2147/CPAA.S26586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain M. C. (2010) Temozolomide: Therapeutic Limitations in the Treatment of Adult High-Grade Gliomas. Expert Rev. Neurother. 10, 1537–1544. 10.1586/ern.10.32. [DOI] [PubMed] [Google Scholar]
- Li R.; Tang D.; Zhang J.; Wu J.; Wang L.; Dong J. (2014) The Temozolomide Derivative 2T-P400 Inhibits Glioma Growth via Administration Route of Intravenous Injection. J. Neuro-Oncol. 116, 25–30. 10.1007/s11060-013-1255-7. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Sk U. H.; Zhang Y.; Ren X.; Zhang L.; Huber-Keener K. J.; Sun Y. W.; Liao J.; Amin S.; Sharma A. K.; Yang J. M. (2012) Rational Incorporation of Selenium into Temozolomide Elicits Superior Antitumor Activity Associated with Both Apoptotic and Autophagic Cell Death. PLoS One 7, e35104. 10.1371/journal.pone.0035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.-Y.; Wang W.; Jhaveri N.; Lee D. J.; Sharma N.; Dubeau L.; Schonthal A. H.; Hofman F. M.; Chen T. C. (2014) NEO212, Temozolomide Conjugated to Perillyl Alcohol, Is a Novel Drug for Effective Treatment of a Broad Range of Temozolomide-Resistant Gliomas. Mol. Cancer Ther. 13, 2004–2017. 10.1158/1535-7163.MCT-13-0964. [DOI] [PubMed] [Google Scholar]
- Owonikoko T. K.; Arbiser J.; Zelnak A.; Shu H.-K. G.; Shim H.; Robin A. M.; Kalkanis S. N.; Whitsett T. G.; Salhia B.; Tran N. L.; Ryken T.; Moore M. K.; Egan K. M.; Olson J. J. (2014) Current Approaches to the Treatment of Metastatic Brain Tumours. Nat. Rev. Clin. Oncol. 11, 203–222. 10.1038/nrclinonc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.; Tanaka M.; Trepel J.; Reinhold W. C.; Rajapakse V. N.; Pommier Y. (2017) Temozolomide in the Era of Precision Medicine. Cancer Res. 77, 823–826. 10.1158/0008-5472.CAN-16-2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.