

Abstract
Limb Girdle Muscular Dystrophy 2A (LGMD 2A) is the most common form of limb girdle muscular dystrophies caused by mutations in the calpain-3 gene (CAPN-3). The pattern of LGMD 2A can be clinically indistinguishable from that of Duchenne Muscular Dystrophy (DMD). We report a case of a 14-year-old boy which has the initial diagnosed as DMD at 6 years old, based on clinical features and very elevated serum creatine kinase levels. A muscle biopsy at the age of 10 showed atypical features which suggested a histiocytosis or neural damage. An MRI conducted 2 years later revealed fatty degeneration predominantly in the posterior region of the thigh and led the diagnosis to LGMD 2A, as well as the necessity to repeat the biopsy. Immunohistochemical analysis was normal for dystrophin, but the Western Blott showed a normal/borderline amount of calpain-3 in the muscle. We also performed a molecular analysis that identified a compound heterozygous mutation of the calpain 3 gene (CAPN 3). LGMD 2A was often misdiagnosed as DMD due to the similarities in clinical manifestations and technique limitations; the immunohistochemical examination, the magnetic resonance imaging examination and the molecular analysis are an essential tool for establishing a right diagnosis.
Keywords: DMD, LGMD 2A, differential diagnosis
Introduction
Duchenne Muscular Dystrophy an X-linked disease (Xp21) a type of dystrophinopathies (dystrophies caused by mutations in the gene DMD , that encodes the protein dystrophin), is the most common neuromuscular disorder presenting in early childhood.
DMD diagnosis is provided by clinical manifestation: (delayed motor milestones: - sitting, standing, and walking-), hypertrophy of calf, early contractures of Achille tendons, proximal legs muscle weakness, elevated creatin kinase levels, abnormal muscle biopsy (atrophy, hypertrophy, focal necrotic and regenerative fibers, negative immunohistochemical staining for dystrophin), and confirmed by molecular analysis. [1, 2]
Limb Girdle Muscular Dystrophy 2A (LGMD 2A), the most common form of limb girdle muscular dystrophies is an autosomal recessive disorder caused by mutations in the calpain-3 gene (CAPN-3) which encodes the enzyme calpain 3 (CAPN3), a protein with a more enzymatic rather than structural function [2, 3].
The diagnosis of LGMD 2A can be difficult due to the resemblance of its clinical features with other dystrophies (symmetrical involvement of proximal limb-girdle muscles, walking difficulty, motor clumsiness, rare pseudo hypertrophy of calf) [3], the lack of specificity of the histopathological examination (which show a typically dystrophic appearance that consists of necrosis, regeneration, fiber diameter variability, myofibrillar disorganization, and fibrosis) [4,5] and of the precision of protein analysis by immunoblotting (a normal or decreased quantity of calpain - 3 on an immunoblot does not exclude a defect in the gene), but also the genotypic variability in the CAPN3 gene (in 23% of patients believed to have LGMD2A only one mutation can be detected) [6]
Dystrophinopathy was diagnosed in about 17% of patients with a clinical diagnosis of Limb Girdle Muscular Dystrophy 2A (LGMD 2A) [7] because the clinical similarities between LGMD and dystrophinopathy.
Case Report
In our study we present the case of 14-year-old boy misdiagnosed as DMD at first presentation, 8 years ago, and show the reasons which led to this diagnosis instead of LGMD 2A. Also we exhibit the importance of supplementary tests in patients with muscular dystrophy features in order to establish a proper diagnosis, followed by accurate therapeutic protocol and competent genetic counseling.
The patient, age of 14 years, has presented to Genetic Department of UMF Craiova for diagnostic reassessment. He was diagnosed at the age of 6 as DMD, based on clinical features - muscle weakness, hypertrophic calf, toe walking, early primary contractures (Fig.1), moderate hyperlordosis - and high serum creatine kinase level (8000U/L). The familial history was unknown, the child being adopted, but family reported delay in motor and speech acquirements. At the age of 10 a muscle biopsy was performed, which showed inflammatory lesions and dystrophic features (atrophic and regenerated fibers) and a concurrent interference of a neurogenic factor was to be considered.
Figure 1.

Achileean retractions
Current physical examination showed almost the same features as in the onset, without physical worsening. Patient did not use wheelchairs, had no respiratory or cardiac problems and the severe and rapid evolution characteristic for DMD was not noticed. Accordingly, a diagnosis reassessment was required.
Repeated biopsy followed by immunohistochemical examination did not noticed a dystrophin deficiency .The immunohistochemical analysis of calpain wasn’t possible because of the lack of antibodies, but the Western Blott test showed a normal/borderline amount of calpain - 3 in the muscle.
The patient was evaluated by MRI imaging (T1, T2, STIR sequences). The results showed that the distribution of fibro-fatty replacement in lower limbs was more important in the posterior compartment of the thigh (mainly for the thigh adductors) and in the gluteus maximus (Fig. 2, 3) than the anterior compartment of the thigh, implying a LGMD 2A diagnosis.
Figure 2.
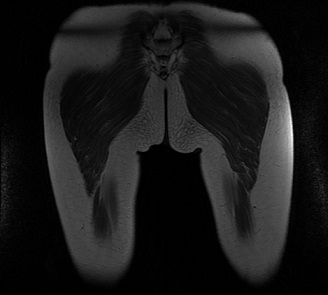
MRI evaluation of specific distribution of fibro-fatty replacement in the posterior compartment of the thigh (mainly for the thigh adductors)
Figure 3.

MRI evaluation of specific distribution of fibro-fatty replacement in the gluteus maximus
A following subsequent molecular analysis suggested that the patient was compound heterozygous for two mutations on calpain 3 (CAPN 3) gene: c. 380 - 1 G > A (exon 3) and c. 598 – 612 Del (exon 4), establishing the diagnosis of LGMD 2A.
Discussion
DMD is one of the most common neuromuscular disorders, starting in early childhood (3-6 years), with a prevalence of approximately 1 in 3500 live male birth [1,2].
Clinical manifestation is not usually apparent until the child starts walking. Motor milestones (sitting, standing, and walking) are delayed and about 50% of cases are late walking (beyond 18 month of age). Contractures of Achille tendons and hip flexors are early features and result in toe –walking. They have a characteristic waddling gait and lumbar lordosis. Patient is unable to jump or hop and they are prone to falls and get up with the typical Gowers manoeuvre. Muscle weakness is more proximal than distal and greater in the legs than the arms. Pseudohypertrophy of calf muscles resulting from an increase in fibrotic and adipose tissue is an early and specific feature [4, 8, 9, 10]
Some clinical features of LGMD 2A such as: rare pseudo hypertrophy of calf, walking difficulty (toe walking), early and severe contractures with early childhood onset can cause diagnosis difficulties with other muscular conditions [3].
After the initial suspicion of DMD diagnosis for a patient without familial history, but with evident clinical features of the disease, we screened for CK levels, which was elevated. HyperCKemia it is usually observed in young persons [11]. DMD is characterized by very high levels of CK, but normal concentrations have been reported in DMD - associated dilated cardiomyopathy [9]. The pattern of muscle weakness is clinically indistinguishable from that of Duchenne Muscular Dystrophy (DMD) and very high CK levels did not suggested the necessity of a differential diagnosis for the case.
The next step required for confirming the diagnosis - muscle biopsy- was not enlightening. The histopathological diagnosis for DMD is provided by characteristic features: abnormal variation in diameter of the muscle fibers (atrophy or hypertrophy), focal necrosis, regenerative fibers and replacement of muscle tissue with fat and connective tissue. Immunohistochemical staining with dystrophin antibodies should not detect dystrophin in the muscular fibers [12, 13]. For LGMD 2A, the patients with total protein loss displayed an active dystrophic process with an increase in fiber size variability, internalization of nuclei, necrotic fibers, regenerating and degenerating fibers. Patients with partial deficiency of protein displayed a relatively milder dystrophic process [6]. The histopathology examination of muscle biopsy of our patient showed dystrophic features of both types of fibers, I and II (atrophic fibers, regenerated fibers). Atrophic fibers grouped in small groups of 2-6 fibers and inflammatory lesions were considered concurrent interference of a neurogenic factor.
In the absence of possibility to perform immunohistochemical (IHC) analysis of calpainopathy on muscle biopsy cryosections or molecular testing neither for DMD nor for LGMD2A we kept the same diagnosis. It was a limitation due to the lack of technical possibilities at that moment.
Calpain-3 immunoblot analysis of muscle biopsy can be a helpful diagnosis in calpainopathy. The performed Western Blott test showed a normal/borderline amount of calpain - 3 in the muscle, thus being the time when we should consider alternative diagnosis. A normal or a decreased quantity of calpain - 3 on an immunoblot does not exclude a gene defect.
80 - 84% of individuals calpain-3 deficient have one or two pathogenic CAPN 3 mutations [6]. The severity of the phenotype and the amount of calpain-3 protein detected by immunoblot analysis are not correlated, symptomatic individuals with either no detectable protein or normal amounts of protein may have various degree of severity of the clinical phenotype [10, 14].
The LGMD 2A magnetic resonance imaging (MRI) examination reveals a selective and specific muscle involvement, predominantly from the posterior compartment of the thighs [15,16]. A distribution pattern of fibro-fatty replacement similar to specialty literature descriptions was exhibited, with a more important affectation in the posterior compartment of the thigh and in the gluteus than in the anterior compartment of the thigh. Clinical and paraclinical correlated results (CPK, immunoblotting and MRI exam) sustain the necessity of the molecular analysis for LGMD 2A, instead of DMD.
The mutations of CAPN 3 gene, which are dispersed throughout the entire length of the gene, fall into four types: missense (about 50%), small insertion/deletions, splice - site and non-sense mutations.
The lack of defined mutational hot spots, unidentified of 2 Th mutation in 23% of patients makes analysis of a 24 exon gene spanning a region of ∼40 kb laborious and expensive; on the other hand, some patients may carry mutations in more than one gene known to cause muscular dystrophy [15].
Mutation c. 380 - 1 G > A (exon 3) identification as a part of compound heterozygous genotype c. 380 - 1 G > A (exon 3)/c. 598 – 612 Del (exon 4) is responsible for the LGMD phenotype of our case and represents the particularity of this presentation because this mutation have never been previously described.
Genotype-phenotype correlation in calpainopathy is complicated because the most affected individuals are compound heterozygotes for two different pathogenic variants in CAPN3. A strong correlation exists between the genotype and the phenotype, especially when two null mutant alleles are detected (either homozygote or compound heterozygote); No definite correlations can be drawn from two missense mutant alleles (either homozygote or compound heterozygote): these patients may present a variable clinical phenotype at onset and also a variable level of calpain-3 protein (from 0–100%) [15].
Since c.380 - 1 G > A (exon 3) mutation have never been described in other patients, we cannot establish with certainty if its presence in heterozygous state is responsible for the similar phenotype of DMD.
Conclusion
Because of the variability of clinical phenotype, the difficulty to identify point mutations in a large gene and the reduced sensitivity and specificity of calpain-3 protein analysis, the LGMD2A diagnosis is difficult.
Confusion with other muscular dystrophy, especially DMD, should be considered in males with clinical and laboratory features that are common with calpainopathy: onset of weakness in the lower girdle muscles in adolescence or adulthood, achileean retractions, claf hypertrophy and elevated serum CK concentrations.
In our case, misdiagnosis was facilitated by the lack of technical posibilities. In the absence of possibility to perform immunohistochemical (IHC) analysis of calpainopathy, the muscle MRI can help in recognizing Limb Girdle Muscular Dystrophy 2A even the clinical presentation overlaps with DMD, and may therefore, be used as an additional investigation to target the appropriate biochemical and genetic tests.
Even the calpain 3 is highly susceptible to degradation, leading to loss of immunoreactivity of the protein and secondary calpain 3 deficiency can also be seen in other types of muscular dystrophy, calpain-3 immunoblot analysis of muscle biopsy can be a guided diagnosis tool in calpainopathy.
Molecular analysis represents the only way for a certainty diagnosis, and, besides the technical difficulties and corresponding high prices, is likely to become a standard tool in clinical genetics laboratories around the world.
Acknowledgments
This paper acknowledged financial support through the project “Excellence program for multidisciplinary doctoral and postdoctoral research in chronic diseases”, Grant No. POSDRU/159/1.5/S/133377, partially supported by the Sectoral Operational Programme Human Resources Development 2007–2013, financed from the European Social Fund.
References
- 1.Henriette JAR, Straub V, Bushby K, Guglier M. Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child. 2014;99(12):1074–1077. doi: 10.1136/archdischild-2014-306366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alan EHE. Neuromuscular disorders: clinical and molecular genetics. 1998. 1998;79-82:129–133. [Google Scholar]
- 3.Rosales XO, Tsao CY. Childhood Onset of Limb-Girdle Muscular Dystrophy. Pediatric Neurology. 2012;46(1):13–23. doi: 10.1016/j.pediatrneurol.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Chae J, Minami N, Jin Y, et al. Calpain 3 gene mutations: genetic and clinico-pathologic findings in limb-girdle muscular dystrophy. Neuromuscul Dis. 2001;11:547–555. doi: 10.1016/s0960-8966(01)00197-3. [DOI] [PubMed] [Google Scholar]
- 5.Hermanova M, Zapletalova E, Sedlackova J, et al. Analysis of histopathologic and molecular pathologic findings in Czech LGMD2A patients. Muscle Nerve. 2006;33:424–432. doi: 10.1002/mus.20480. [DOI] [PubMed] [Google Scholar]
- 6.Pathak P, Mehar CS, Chitra S, et al. Limb girdle muscular dystrophy type 2A in India: A study based on semi-quantitative protein analysis. with clinical and histopathological correlation. 2010;58(4):549–554. doi: 10.4103/0028-3886.68675. [DOI] [PubMed] [Google Scholar]
- 7.Arikawa E, Hoffman EP, Kaido M, et al. Neurology. 1991;41:1491–1496. doi: 10.1212/wnl.41.9.1491. [DOI] [PubMed] [Google Scholar]
- 8.Bushby K, Finkel R, Birnkrant D J, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis and pharmacological and psychosocial management. The Lancet Neurology. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 9.Mestroni L, Rocco C, Gregori D. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity, Heart Muscle Disease Study Group. J Am Coll Cardiol. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 10.Fanin M, Fulizio L, Nascimbeni AC, et al. Molecular diagnosis in LGMD2A: mutation analysis or protein testing. Hum Mutat. 2004;24:52–62. doi: 10.1002/humu.20058. [DOI] [PubMed] [Google Scholar]
- 11.Fanin M, Nascimbeni AC, Fulizio L, et al. Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am J Pathol. 2003;163:1929–1936. doi: 10.1016/S0002-9440(10)63551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biggar WD. Duchenne Muscular Dystrophy. Pediatrics in Review. 2006;27(3):83–88. doi: 10.1542/pir.27-3-83. [DOI] [PubMed] [Google Scholar]
- 13.Dubowitz V, Caroline AS. Muscle biopsy, a practical approach. 2013:250–276. [Google Scholar]
- 14.Fanin M, Nardetto L, Nascimbeni AC, et al. Correlations between clinical severity, genotype and muscle pathology in limb girdle muscular dystrophy type 2A. J Med Genet. 2007;44:609–614. doi: 10.1136/jmg.2007.050328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain. 1996;119:295–308. doi: 10.1093/brain/119.1.295. [DOI] [PubMed] [Google Scholar]
- 16.Fischer D, Walter MC, Kesper K, et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol. 2005;252:538–547. doi: 10.1007/s00415-005-0684-4. [DOI] [PubMed] [Google Scholar]